

Abstract
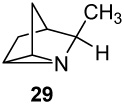
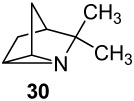
A series of seven cyclopent-3-en-1-ylmethylamines bearing one, two, or three methyl substituents at the C2, C3, C4, or Cα positions, including the unsubstituted parent, was accessed by ring-closing metatheses of α,α-diallylacetonitrile (or methallyl variants) and α,α-diallylacetone followed by hydride reductions, or by Curtius degradations of α,α-dimethyl- and 2,2,3-trimethylcyclopent-3-enylacetic acids. Oxidation of the primary amines with Pb(OAc)4 in CH2Cl2, CHCl3 or benzene in the presence of K2CO3 effected efficient intramolecular aziridinations, in all cases except the α-methyl analogue (16), to form the corresponding 1-azatricyclo[2.2.1.02,6]heptanes, including the novel monoterpene analogues, 1-azatricyclene and the 2-azatricyclene enantiomers. The cumulative rate increases of aziridination reactions observed by 1H NMR spectroscopy in CDCl3 resulting from the presence of one or two methyl groups on the cyclopentene double bond, in comparison to the rate of the unsubstituted parent amine (1 : 17.5 : >280), indicate a highly electrophilic intermediate as the nitrene donor and a symmetrical aziridine-like transition state. A mechanism is outlined in which the amine displaces an acetate ligand from Pb(OAc)4 to form a lead(IV) amide intermediate RNHPb(OAc)3 proposed as the actual aziridinating species.
Introduction
Protonated aziridines (pKa 8.0)1 are nitrogen analogues of protonated cyclopropanes and protonated epoxides (Fig. 1), species commonly invoked as initiating groups in polyene cyclizations, or in the former case as carbocation intermediates in biosynthetic mechanisms leading to isoprenoid natural products.2,3 The obvious structural similarities have stimulated the synthesis and evaluation of many isoprenoid aziridines as inhibitors of terpene biosynthesis. For example, 2,3-epiiminosqualene, the aziridine analogue of 2,3-oxidosqualene, is an effective inhibitor of oxidosqualene cyclases.4 Sterol analogues bearing an aziridine ring in the side chain at the 24, 25 position are potent inhibitors of cholesterol biosynthesis in rat hepatoma cells,5a and they block sterol methyl transferase activity essential for plant development5b and fungal cell growth.5c,5d The epiimino analogue of 24,28-epoxyfucosterol, an intermediate in phytosterol side chain cleavage, retards the growth and development of the silk worm, Bombyx mori.5e Aziridine analogues of the Cecropea juvenile hormones (JH) potentiate the JH activity of the natural epoxide metamorphosis regulators.6
Figure 1.

Illustration of the structural similarity of a generic protonated aziridine to protonated cyclopropanes and protonated epoxides.
The cyclopropane rings frequently found in terpene natural products7 (eg presqualene diphosphate and cycloartenol) and marine steroid side chains (eg gorgosterol)8 are presumably formed by proton eliminations from various types of protonated cyclopropanes or related bridged carbocation intermediates. In accord with this analogy, aziridine analogues of presqualene diphosphate were found to be inhibitors of squalene synthase at low micromolar concentrations.9 The pentacyclic aziridine aza-trachylobane in the presence of inorganic pyrophosphate is a potent competitive inhibitor (IC50 0.02 µM) of recombinant kaurene synthase from Arabidopsis thaliana.10 These isoprenoid aziridines presumably exert their inhibitory effects by selective binding in the active sites and by specific interactions with catalytic residues. Since aziridinium ions are potential electrophiles, they may also undergo covalent reactions with active site residues. These transition state mimics have applications as active site probes and aids to assist crystallization of recombinant terpene synthases and to locate the active sites in co-crystals with these enzymes.11
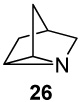
We became interested in the synthesis of 1-azatricyclo[2.2.1.02, 6]heptanes12 as aziridinium analogues of bridged ions in Wagner-Meerwein rearrangements that occur in biosynthetic mechanisms leading to bicyclic and tricyclic monoterpenes (Fig 2),2b, 13 and in the evaluation of the bridged aziridines as active site probes for monoterpene synthases. In this paper we explore the scope of the intramolecular aziridination of methyl-substituted cyclopent-3-enylmethylamines to the corresponding methyl-bearing 1-azatricyclo[2.2.1.02, 6]heptanes, including the aza-tricyclenes, by means of lead tetraacetate oxidations.14 The rates of the cyclizations afford insights into the mechanism and structure of the nitrenoid intermediate in the reactions.
Figure 2.

Non-classical bridged bornyl carbonium (1) and its azatricyclo[2.2.1.02,6]heptane (azatricyclene) analogues 2 and 3.
Considerable current interest in the synthesis, reactions, and applications of aziridines is evident from numerous recent reviews15, 16 and a monograph on aziridines and epoxides.17 In principle bicyclic aziridines might be accessible by intramolecular reactions of unsaturated nitrenes or nitrenium ions.18,19 However, a more practical approach for intramolecular aziridination involves the cyclization of nitrene equivalents bearing electron-withdrawing groups generated by oxidation of amides and sulfonamides, either presence or absence of Cu and Rh mediators.20 Although oxidations of N-amino quinazolones and N-amino phthalimide with Pb(OAc)4 or PhI(OAc)2 generate nitrenoids capable of intermolecular aziridination of C=C double bonds,15,21,22 these approaches are not readily adaptable to intramolecular aziridinations. A homologue of azatricylene 2 (1,3,3-trimethyltricyclo[2.2.2.02,6]octane) was prepared by reaction of terpinylamine with NBS in acetone.23 We decided to apply Nagata's direct synthesis of bridged, polycyclic aziridines by Pb(OAc)4 oxidation of unsaturated primary amines,14 as reported previously by Portoghese and Sepp for the parent 1-aza-nortricyclene,12 and employed previously in the synthesis of azatrachylobane.10
Synthesis of Cyclopentenylmethylamines
The parent amine (6)12 was obtained from diallylacetonitrile (4)24 by ring-closing metathesis (RCM) using Grubbs’ 1st-generation ruthenium catalyst,25a followed by LiAlH4 reduction of cyclopentenyl nitrile 5 as summarized in Scheme 1. Similar metatheses of α-allyl-α-methallylacetonitrile and bis-(α-methallyl)acetonitrile using the Grubbs 2nd-generation ruthenium catalyst (8 → 9 and 11 → 12)25b and subsequent hydride reductions afforded the homologues bearing one and two methyl groups on the cyclopentene double bond (10 and 13). The modest 31% yield of 12 reflects the slower, incomplete ring-closing metathesis at the low concentration necessary to minimize the competing polymerization. The two methallyl acetonitrile derivatives (8 and 11) were prepared by alkylations of the corresponding cyanoacetate precursors (ethyl α-allylcyanoacetate (7) and ethyl cyanoacetate) with methallyl chloride followed by chloride-induced de-ethylation-decarboxylations (NaCl, DMSO, 150 °C).26
scheme 1.

The syntheses of substrates bearing one or two methyl substituents on the exocyclic aminomethyl position (16 and 20) are outlined in Scheme 2. De-ethylation-decarboxylation (LiCl, DMF, reflux) of α,α-diallylacetoacetate gave α,α-diallylacetone (14, 44%) which underwent metathesis and reductive amination27 to 1-(cyclopenten-3-yl)ethylamine (15 → 16). The low purity of the crude product necessitated purification by conversion to the t-Boc derivative followed by TFA hydrolysis-decarboxylation (35% over 3 steps). Alkylation of the lithium enolate of methyl isobutyrate with homoallylic cyclopentenyl tosylate 1728 in THF and subsequent hydrolysis of the resulting methyl ester intermediate gave α-cyclopentenyl isobutyric acid 18 in high overall yield. Curtius rearrangement with diphenylphosphoryl azide,29 conversion of the resulting isocyanate to the carbamate 19 (An = p-anisyl) with p-methoxybenzyl alcohol, and alkaline hydrolysis afforded geminal dimethyl analogue 20.
scheme 2.

The long known retro-Prins fragmentation of (S)-(+)-camphorsulfonic acid (21) by KOH fusion at 200–220 °C for 5 min afforded (+)-α-campholenic acid 22, in accord with the literature procedures30 (Sch 3). The intermediacy of the expected exocyclic isomer (+)-isocampholenic acid (23), observed as a trace impurity (ca 2%) in 22, was verified by interrupting the fusion reaction after 1 min, at which time mixtures of 22 and 23 were present. Selective epoxidation of the former in a 2:1 isomer mixture and chromatographic separation of the relatively polar epoxide enabled isolation of pure 23 having optical rotation and NMR spectral data matching the literature values.31 The exo/endo ratios varied from 1:1 to 1:9 (yields 75–90%) in 10 runs differing in the proportion of water (0–30%) added to 85% KOH in preparation of the fusion reagent. Nor-campholenyl amine 25 was obtained by Curtius rearrangement29 of acid 22, carbamate derivatization (24, An = p-anisyl), and alkaline hydrolysis as described previously for the α,α-dimethyl amine 20 in Scheme 2. The same reaction sequence with (R)-(−)-camphorsulfonic acid (ent-21) provided (−)-α-campholenic acid (ent-22), the related carbamate intermediate ent-24, and the enantiomeric nor-campholenyl amine ent-25 bearing the three ring methyl groups.
scheme 3.

Intramolecular Aziridinations
The preparative aziridination reactions with the seven cyclopentenylmethylamines were conducted with 1–2 equivalents of Pb(OAc)4 in CH2Cl2 at room temp in the presence of anhydrous K2CO3 as base to neutralize the acetic acid released according to similar procedures described by Nagata, Portoghese and their co-workers. (See Sch 4 and Table 1).10,12,14 The reactions were terminated by addition of satd aq K2CO3, and the aziridines were extracted into ether and converted to the stable trifluoroacetate (TFA) salts, at which point yields were calculated. The free aziridine bases were later liberated as needed for characterization by reactions with solid K2CO3 in CH2Cl2, CDCl3, or other solvents. The aziridines proved to be stable in dilute solutions (CH2Cl2, CDCl3, frozen benzene) at −20 °C. However, as noted previously,10,12,14 the strained heterocycles decomposed readily as pure liquids, presumably undergoing polymerization to insoluble, uncharacterized material.
scheme 4.

Table 1.
Yields, Half-lives, and Relative Rates of Intramolecular Aziridination of 3-Cyclopentenylmethylamine (6) and its Methyl Derivatives with Pb(OAc)4 in CH2Cl2 (Preparative Runs) or CDCl3 (kinetic Runs) at 25 °C.
| Entry | Amine No. |
Aziridine No. |
Yielda (%) |
t1/2 (min, ave) |
krel |
|---|---|---|---|---|---|
| 1 | 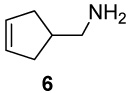 |
 |
65 | 280 | (1.0) |
| 2 |  |
 |
78 | 16, 19 (17.5) |
17.5 |
| 3 | 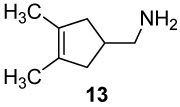 |
 |
81 | < 1b | > 280 |
| 4 | 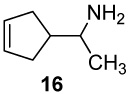 |
 |
NDc | 100e | 2.8 |
| 5 |  |
 |
68d | 125 | 2.2 |
| 6 | 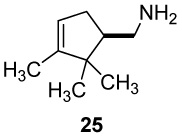 |
 |
88 | 8, 11 (9.5) |
29 |
| 7 | 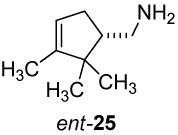 |
 |
80 | NDc | NDc |
Yield of TFA salt.
Complete conversion at 4 min.
ND = not determined.
Yield based on methiodide salt.
Time at ca 50% conversion.
The proton and carbon-13 NMR chemical shifts for the aziridine ring protons and carbons, the C-7 N-CH2, and C-CH3 attached to the aziridines in the free base and TFA salt forms are collected in Table 2. The assignments are based on chemical shift trends, and in some cases on the carbon multiplicities from DEPT experiments.
Table 2.
Selected Proton and Carbon-13 NMR Chemical Shifts for 1-Azabicyclo[2.2.1.02,6]heptanes and Their Trifluoroacetate (TFA) Salts in CDCl3.
| Aziridine No. |
CH3 substituents |
Aziridine N-C-H |
Aziridine N-C-CH3 |
C-7 N-CH2(CH) |
C-8 N-C-CH3 |
|---|---|---|---|---|---|
| 26 | none | δH 2.22 δC 31.8 |
— | 2.18 55.8 |
— |
| 26 • TFA | none | δH 3.70 δC 38.1 |
— | 2.92 51.0 |
— |
| 27 | 2-CH3 | δH 2.10 δC 38.4, 39.1 |
1.35 16.0 |
2.25,2.39 56.7 |
— |
| 27 • TFA | 2-CH3 | δH 3.63 δC 43.9, 49.8 |
1.71 11.8 |
2.92, 3.05 51.6 |
— |
| 28 | 2,6-(CH3)2 | δH— δC 45.1 |
1.38 12.5 |
2.53 56.3 |
— |
| 28 • TFA | 2,6-(CH3)2 | δH— δC 53.2 |
1.65 9.6 |
3.01 52.2 |
— |
| 30a | 7,7-(CH3)2 | δH 2.19 δC 36.8 |
— | 45.4 | 1.02 22.9 |
| 30 • TFAa | 7,7-(CH3)2 | δH 3.72 δC 39.1 |
— | — 45.2 |
1.37 20.6 |
| 32 | 2,3,3-(CH3)3 | δH 2.03 δC 41.2, 45.2 |
1.16 19.7 |
2.11, 2.80 52.2 |
— |
| 32 • TFA | 2,3,3-(CH3)3 | δH 3.66 δC 45.5, 49.6 |
1.55 7.0 |
2.85, 3.43 55.3 |
— |
Peaks from impurities appear in the 13C NMR spectrum.
Some interesting trends are apparent from the data in Table 2. The chemical shifts for the aziridine ring C-H protons are consistently in the range 2.03 to 2.22 ppm, and they are shifted downfield (Δδ = +1.48 to +1.63 ppm) in the protonated forms, reflecting the positive charge and hybridization changes in the internal C-N bonds. The values for the C-7 N-CH2 moieties are spread over a considerable range, δ2.11–2.80 ppm. A shift to lower field is evidently caused by CH3 substitution on the aziridine ring carbons. In contrast downfield shifts arising from N-protonation are reasonably consistent at Δδ = 0.49–0.74 ppm. The higher field position of aziridine C-CH3 in aza-tricyclene 32 (δH 1.16) as compared to the aziridine ring methyl derivatives 27 and 28 (δH 1.35 and 1.38) is attributable to C-C bond anisotropy arising from the adjacent CH3 groups. The protonation shifts for the ring CH3 vary from Δδ +0.36 (27) to Δδ +0.27 ppm (28).
The 13C NMR chemical shifts for the aziridine ring carbons (C-2 and C-6) move from 31.8 (26) to 38.4 or 39.1 for one CH3 (27) and to 45.1 for the dimethyl case (28), and they undergo downfield shifts by Δδ +5.5 to + 10.7 ppm in the corresponding TFA salts. In contrast the aziridine ring methyl resonances (δC 16.0 and 12.5 ppm) appear at higher field positions in the TFA salts (Δδ −4.2 to −2.9 ppm for 27 and 28). Similar protonation shieldings for carbons β to nitrogen in acyclic amines (generally Δδ −3 to −5 ppm) are attributed to "electric field effects and C-H bond polarizations."32b,c
First-order rates for the aziridinations reactions were determined by integration of 1H NMR spectra carried out in NMR tubes with the free amines and 2 equivalents of Pb(OAc)4 in CDCl3 (Table 1). An initial "burst phase" apparent in the slower reactions is attributed to warming resulting from the exothermic interactions of the amines with Pb(OAc)4. The proton NMR peaks for the α-methyl groups of 16 and 20 immediately moved somewhat downfield upon addition of Pb(OAc)4 (Δδ H +0.118 and +0.156 ppm, respectively). Reaction progress was monitored by the consistent decrease in the vinyl proton integrations, or in the case of 13 by the integration changes accompanying conversion of the vinyl CH3 to aziridine CH3. Log plots (Fig 3 below) were linear over approximately 0.5 to 1 half-lives.
Figure 3.

Pseudo first-order kinetic plots (t1/2 – ln (2)/k) for Pb(OAc)4 oxidations of cyclopentenyl amines 6, 10, 20, and 24 by 1H NMR integrations of peaks for the corresponding aziridine products 26, 27, 30, and 32.
The proton NMR spectra acquired during oxidation of α-methylamine 16 showed extraneous peaks considered to be impurities formed competitively with aziridine 29. We suppose that these byproducts arise from competing dehydrogenation to the ketimine and its subsequent condensation and/or oxidations of the enamine tautomers.33a Although the presence of aziridine 29 was evident from its characteristic 1H NMR signals (eg new methyl peak δH 1.18 (d, J = 6.5 Hz, 3H)), the numerous impurities present after high conversions prevented isolation of the aziridine TFA salt in pure form, and the isolated yield was not determined in this case. The intramolecular aziridination of amine 13 bearing two CH3 groups on the double bond was essentially complete within 4 min, and the half-life in this case could only be estimated to be < 1 min. The half-lives varied approximately ± 15 % in duplicate runs, and were independent of the amount of Pb(OAc)4 reagent added.
The rates of several aziridinations in the same heterogeneous medium used in the preparative reactions, ie TFA salts in the presence of 5 equivalents of solid K2CO3, were also measured by NMR analyses. The reactions of the TFA amine salts bearing methyl groups on the cyclopentene double bond proceeded considerably more slowly (13·TFA t1/2 9 min vs < 1 min, and 10·TFA 95 min vs 16 min), whereas the rate of the parent cyclopentenylmethylamine (6) TFA salt was unchanged. Evidently liberation of the free amines 13 and 10 from the TFA salts becomes the rate-limiting step under these heterogeneous conditions.
The oxidative aziridination with α,α-dimethyl amine 20 was also effected with 2 equivalents of PhI(OAc)2 21 in CH2Cl2 with a half-life of 6 h, ie about 3 times more slowly than the reaction with Pb(OAc)4. In this case the methyl singlet shifted from 1.058 to 1.176 and then to 1.252 over 30 min, and remained at 1.252 as the peaks for the aziridine grew more intense.
The solubility and stability of Pb(OAc)4 in many organic solvents33 provided an opportunity to evaluate the effects of additives and medium on the aziridination process. No reaction occurred with 2 equiv of Pb(OAc)4 and α, α-dimethylamine 20 in CD3CN after 10 days or in CD3CO2H after 2 days, and the aziridination was very slow in CD3OH. The aziridination with 20 proceeded ca 4 times more slowly in benzene-d6 (t1/2 530 min vs 125 min in CDCl3). No aziridine formation was observed when 2 equiv of the sterically hindered tertiary amine base diisopropylethylamine was present in CDCl3, although shifts of the NMR peaks for the N-isopropyl and N-ethyl groups were noticeable. The aziridination rate with the TFA salt of 10 was about the same (t1/2 16 and 15 min) when effected by 1.1 or 2.0 equivalents of Pb(OAc)4, 2 equiv of pyridine, and 3 equiv of K2CO3. Much faster rates were observed in aziridinations conducted with free amines 6, 10, and 20 and 2 equiv of Pb(OAc)4 in pyridine-d5 (t1/2 <15 min vs 280 min, ca 6 min vs 16 min, and <2 min vs 125 min, respectively). However, the product from the unsubstituted amine 6 was a 1:1 mixture of aziridine 26 and nitrile 5 resulting from double dehydrogenation. The oxidation of primary amine to nitriles with Pb(OAc)4 has been reported.34
Scope and Mechanism of Aziridinations
The intramolecular aziridinations proceeded in satisfactory to high yields (65 – 88%) with all amines except α-methyl amine 16, despite the large ring strain of the 1-azatricyclo[2.2.1.02,6]heptane products estimated to be in the neighborhood of 47 kcal/mol. This figure is based on the known strain energy of nortricyclene (47.0 kcal/mol)35a together with the very similar values recorded for cyclopropane (27.5 and 28.13 kcal/mol)35a and aziridine (27.1 kcal/mol).35b We presume that our inability to isolate aziridine 29 is attributable to competing dehydrogenation to a labile ketimine and its relatively rapid oxidation and/or self-condensation to a mixture of amine by-products having properties similar to those of the aziridine. The various tricyclo[3.2.1.02,7]octanes prepared in Nagata's pioneering investigation of aziridinations with Pb(OAc)4 are further indications of the scope of this little used reaction.14
The increased rates (krel 2.8 and 2.2) observed with amines bearing one or two α-methyl groups (16 and 20) clearly indicates the insensitivity of the aziridination reaction to steric hindrance around the nitrogen center. In fact the cyclization reactions of all methyl-substituted derivatives proceeded faster than that of the parent amine 6. The rates of these formal [2+1] heterocycloadditions increased dramatically and cumulatively with one or two methyl groups situated on the cyclopentenyl double bond (10 krel 17.5 and 13 krel 280), demonstrating that aziridination is the slow step in the mechanism, and revealing the highly electrophilic nature of the nitrenoid intermediate and the apparent symmetry of the transition state.
A wide range of mechanisms and types of nitrenoid intermediates has been considered for the well-known aziridinations of olefins with N-amino heterocycles (primarily N-aminophthalimides and N-aminoquinazolones). 21a, 22 Plausible intermediates include N-acetoxyamines, nitrenes, nitrenium ions, aminyl radicals, hypervalent iodine (III) species (amino- or imino-iodinanes) with PhI(OAc)2 as oxidant, and in the case of Pb(OAc)4 lead (IV) species (lead amides or lead imines). Although Atkinson advocated the intermediacy of N-acetoxyamino heterocycles,22 the selectivity variations with Pb(OAc)4 and PhI(OAc)2 as oxidant are considered inconsistent with a common intermediate.21a,21b
We decided to test the possibility of an N-acetoxy intermediate by independent synthesis of the hydroxylamino ester 35 from the parent cyclopentenylmethylamine 6 as shown in Scheme 5.36 However, the reaction of 35 with Pb(OAc)4 under the usual conditions with monitoring by 1H NMR spectroscopy proceeded slowly with the development of numerous peaks, of which none were the characteristic signals of aziridine 26. No evidence for a detectable buildup of the characteristic NMR absorptions for 35 could be seen in spectra taken during the kinetic runs of 6 with Pb(OAc)4. Furthermore the 3-fold faster aziridination of α,α-dimethylamine 20 observed with Pb(OAc)4 as oxidant (t1/2 125 min) compared to that with PhI(OAc)2 (t1/2 360 min) is inconsistent with a common intermediate.
scheme 5.

It seems reasonable to expect the amines to undergo ligand substitution with Pb(OAc)4 to generate lead(IV) amides (36) or possibly lead imines (37 and 38) by the equilibrations shown in Scheme 6. One driving force for the exchange would be the subsequent formation of an acetate salt between the remaining amine and the equivalent of acetic acid released. The slight exotherm observed upon adding Pb(OAc)4 to the amine as well the slight downfield of the proton NMR signals for the geminal methyls in 20 are attributable to the exchange process. Although the Pb(IV)-N bond is very weak (ED Pb-N ~ 20–30 kcal/mol),37 many trialkyllead amides (eg Et3PbNEt2) and trityllead amides and imides were reported in the literature by 1980.37 A similar exchange equilibrium was proposed as the first step in the Steglitz rearrangement of tritylamine to benzophenone N-phenylimine (Pb(OAc)4, benzene, reflux).38
scheme 6.

We propose that the aziridinations effected with Pb(OAc)4 occur by intramolecular electrophilic attack of lead(IV) amides onto the ring double bonds with simultaneous formation of the aziridinium acetate salt (41) and Pb(OAc)2 as depicted in the mechanism 39 → 40 → 41 (Sch 7). The large and cumulative rate-accelerating effects of methyl groups on the double bonds (10 krel 17.5 and 13 krel > 280, k10/k13 >16) clearly signify substantial and symmetrical positive charge accumulation at carbon in the transition state. Major driving forces for this formal [2+1]cycloaddition are the exchange of the weak Pb-N bond and for the stronger C-N bonds in the product, as well as the exothermic Pb(IV) → Pb(II) 2-electron reduction (E° at 25 °C, Pb+4 + 2 e− = Pb+2 1.65V).39 This mechanism is similar to that proposed by Richardson et al21a for intermolecular aziridinations of olefins with N-aminoheterocycles and aryl iodinanes ArI(OAc)2 in the absence of transition metal catalysts. Transition state [40] bears a resemblance to one proposed for the Pb(OAc)4-induced Steglitz rearrangement of tritylamines mentioned above.38
scheme 7.

The transition state proposed for the intramolecular aziridinations [40] is similar to those associated with peroxy acid epoxidations of C=C double bonds (42 → [43] → 44)40, 41 and with the solvolytic cyclizations of cyclopentenylethyl sulfonates to symmetrically-bridged norbornyl carbonium ions (45 → [46] → 47)42 as illustrated in Scheme 7. The cumulative, rate-enhancing effects of alkyl substituents on olefin epoxidations (ca 10–20 times per alkyl substituent),43 and the accelerating influence of one and two methyl groups on the cyclopentenyl double bond of 45 (increases of 7 and 38.5 times)42 are taken to indicate analogous transition structures [43] and [46] having substantial positive charge accumulation simultaneously at both C=C carbons.
Transition state 40 may be likened to an intramolecular SN2 reaction taking place at nitrogen, with the C=C double bond as nucleophile and Pb(OAc)2 as the leaving group. This analogy suggests aziridination via lead imine 38 would be disfavored by the presumed sp2 hybridization at the C=N nitrogen, in accord with the highly suppressed rates of SN2 substitutions at vinylic carbon. Furthermore, to our knowledge lead(IV) imines of this type are unknown in the literature.
The solvent effects observed on the aziridinations seem generally consistent with the mechanism and transition state in Scheme 6 and Scheme 7. The somewhat faster rates observed in CDCl3 compared to those in C6D6 are attributable to transition state stabilization by the dipole of the former. However, the slower rates in CD3CN and CD3OH can be rationalized by competing ligation of Pb(IV) with the excess solvents. The rate suppression observed in CD3CO2H is probably caused by formation of an ammonium acetate salt and the resulting reduced concentration of free amine. Similarly the faster rates in pyridine probably arise from the scavenging of acetic acid, and the resulting higher concentrations of free amine and key intermediate 36.
Conclusion
This work has established the generality of the Pb(OAc)4-induced intramolecular aziridination of cyclopentenylmethylamines to 1-azatricyclo[2.2.1.02,6]heptanes12 and has provided evidence indicating the highly electrophilic nature of the key nitrene donor 39 and a symmetrical aziridine-like transition state 40. The strained, bridged aziridine products, or their N-alkyl or N-acyl aziridinium derivatives, and others available by analogous intramolecular aziridinations of related types of unsaturated primary amines,14 would undergo predictable nucleophilic and electrophilic ring cleavage reactions,12, 15–17,44 affording access to various heterocyclic structures. A noteworthy distinction of the intramolecular aziridinations with Pb(OAc)4 12,14 is the use of unsaturated primary amines lacking the commonly employed N-acyl or N-sulfonyl linkages.15–17, 45 The compatibility of Pb(OAc)4 with a wide range of functional groups and solvents,33 together with the many synthetic approaches to unsaturated amines, point to a broad generality for the intramolecular aziridination-ring opening route to nitrogen heterocycles.
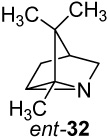
The azatricyclenes reported are novel aza analogues of the bridged bornyl carbonium ion (Fig 2) and potentially useful transition state inhibitors and active site probes for monoterpene cyclases. The enantiomeric 2-aza-tricyclenes ((+)-32 and (−)-32) provide a means to evaluate the stereoselectivity of interactions with the biosynthetic enzymes that produce enantiomeric monoterpenes, eg (+)- and (−)-bornyl diphosphate synthases, and (+)- and (−)-pinene cyclases.2b,13 Furthermore, ring opening reactions of the azatricyclenes afford a simple route to aza-monoterpenes such as 6-azaborneol and 2-azacamphane.46
Experimental Section
4-Methoxybenzyl 2-(Cyclopent-3-en-1-yl)prop-2-ylcarbamate (19)
In the same manner as described below for urethane 24, compound 19 was prepared from acid 18 (2.8 g, 18.18 mmol). Carbamate 19 (4.2 g, 80%) was obtained as an oil: 1H-NMR (CDCl3, 400 MHz) δ 7.30 (d, 2H, J = 8.8 Hz), 6.89 (d, 2H, J = 8.8 Hz), 5.65 (s, 2H), 4.97 (br s, 2H), 4.64 (br s, 1H), 3.81 (s, 3H), 2.70 (app quintet, 1H, Japp = 8.4 Hz),47 2.36 (br dd, 2H, J = 14.8, 9.6 Hz), 2.18 (br dd, 2H, J = 14.8, 8.5 Hz), 1.30 (br s, 6H). 13C NMR (CDCl3, 100.6 MHz) δ 159.7, 159.3, 130.2 129.6, 128.9, 114.1, 71.6, 66.0, 55.5, 46.5, 34.1, 24.6; HRMS (ESI) m/z calcd for C17H27NO3 (M + H)+ 290.1756, found: 290.1752.
2-(Cyclopent-3-en-1-yl)propan-2-amine (20)
As described below for amine 25, carbamate 19 (4.2 g, 14.53 mmol) was hydrolyzed by refluxing in 40% methanolic KOH (25 mL). Amine 20 (1.75 g, 98%) was obtained as a colorless liquid: 1H NMR (CDCl3, 500 MHz) δ 5.66 (s, 2H), 2.40–2.26 (m, 3H), 2.16 (br dd, 2H, J = 13.5, 8. Hz), 1.06 (s, 6H); 13C NMR (CDCl3, 125 MHz) δ 129.6, 50.9, 49.5, 34.0, 28.6; HRMS (ESI) m/z calcd for C8H15N (M + H)+ 125.1205, found: 125.1202.
(+)-(1S)-4-Methoxybenzyl (2,2,3-Trimethylcyclopent-3-enyl)methylcarbamate ((+)-24)
Yamada's modified procedure29 for the Curtius rearrangement was followed. A solution of acid (+)-22 (3.0 g, 17.9 mmol) and Et3N (4.2 mL, 3.0 g, 30.1 mmol) in 1,2-dichloroethane (40 mL) was stirred under N2 as neat diphenylphosphoryl azide (3.9 mL, 5.0 g, 18.1 mmol) was added. The solution was heated at reflux for 100 min, p-methoxybenzyl alcohol (3.7 mL, 4.1 g, 30.0 mmol) was added via syringe, and heating was resumed for 12 h. The solution was concentrated under reduced pressure, and the resulting yellow, oily residue was purified by flash chromatography using a gradient of 5–10% EtOAc/hexane to give 4.5 g (83%) of urethane (+)-24 as a colorless oil, which crystallized at −15 °C. Data for crystalline (+)-24: TLC Rf 0.75 (10% EtOAc-hexane); mp 43–44 °C; [α]25 D +5.67° (c 1.1, CHCl3); 1H NMR (CDCl3, 500 MHz) as a 5:2 mixture of two rotamers: δ 7.30 (d, 2H, J = 8.5 Hz), 6.88 (d, 2H, J = 8.5 Hz), 5.20 (s, 1H), 5.03 (s, 2H), 4.68 (br s, 1H), 3.80 (s, 3H,), 3.36 (m, 1H), 3.15 (m, 1H), 2.30 (m, 1H), 1.94 (m, 2H), 1.58 (s, 3H), 1.03 (s, 3H,), 0.83 (s, 3H,); 13C NMR (CDCl3, 125 MHz) δ 159.7(s), 156.7(s), 148.5 (s), 130.2 (d, 2C), 128.9 (s), 121.4 (d), 114.1 (d, 2C), 66.6 (t), 55.4 (q), 49.7 (d), 46.6 (s), 42.4 (t), 34.3 (t), 26.5 (q), 19.9 (q), 12.5 (q); HRMS (ESI) m/z calcd for C18H26NO3 (M + H)+ 304.1913, found: 304.1910.
(−)-(1R)-4-Methoxybenzyl (2,2,3-Trimethylcyclopent-3-enyl)methylcarbamate ((−)-24)
Crystalline urethane ent-22 (6.2 g, 84%) was prepared as described above for (+)-22 starting from (−)-α-campholenic acid (−)-ent-22 (4.1 g, 24.4 mmol): mp 42–44 °C; [α]25 D −5.23° (c 2.9, CHCl3); the 1H and 13C NMR spectra of (−)-24 were identical to those previously recorded for carbamate (+)-24; HRMS (ESI) m/z calcd for C18H26NO3 (M + H)+ 304.1913, found: 304.1912.
(±)-4-Methoxybenzyl (2,2,3-Trimethylcyclopent-3-enyl)methylcarbamate ((±)-24)
Urethane (±)-24 (oil, 2.7 g, 83%) was prepared as described above for (+)-24 starting from (±)-α-campholenic acid (±)-22 (1.8 g, 10.7 mmol). The 1H and 13C NMR spectra of compound (±)-24 were identical to those previously recorded for carbamate (+)-24; HRMS (ESI) m/z calcd for C18H26NO3 (M + H)+ 304.1913, found: 304.1910.
(+)-(1S)-(2,2,3-Trimethylcyclopent-3-enyl)methylamine ((+)-25)
Hydrolysis of the (S)-carbamate (+)-24 was accomplished following a literature procedure.48 A solution of (+)-24 (4.0 g, 13.20 mmol) and KOH (10.0 g, 178.6 mmol) in 30 mL of 95% EtOH (4% H2O) was heated at reflux for 2.5 h, cooled to 0 °C, and acidified with concd HCl. The solution was partially concentrated under reduced pressure, diluted with water (150 mL), and extracted with CH2Cl2 (2 × 25 mL). The aq phase was cooled again to 0 °C, basified with KOH pellets, and extracted with CH2Cl2 (3 × 15 mL). The combined organic extracts were dried (K2CO3) and evaporated at atmospheric pressure to yield 1.7 g (93%) of amine (+)-25 as a clear oil: TLC Rf 0.24 (alumina, 1% MeOH-CHCl3); [α]25 D +9.11° (c 1.1, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 5.21 (br s, 1H), 2.84 (dd, 1H, J = 12, 4.5 Hz), 2.62 (dd, 1H, J = 12, 10 Hz), 2.37 (m, 1H), 1.93-1.79 (m, 2H), 1.58 (br s, 3H), 1.49 (br s, 2H), 1.02 (s, 3H), 0.77 (s, 3H). 13C NMR (CDCl3, 125 MHz) δ 48.7, 121.5, 53.6, 46.6, 43.3, 34.5, 26.6, 19.9, 12.5.
(−)-(1R)-[2,2,3-Trimethylcyclopent-3-enyl)]methylamine ((−)-25)
Hydrolysis of urethane (−)-24 (4.2 g, 13.86 mmol) following the procedure described above for (+)-25, gave the enantiomeric amine ent-25 (1.8 g, 93%). Data for (−)-25: [α]25 D −9.16° (c 3.8, CHCl3). The 1H and 13C NMR spectra of (−)-25 were identical to those previously recorded for (+)-25.
(±)-[2,2,3-Trimethylcyclopent-3-enyl)]methylamine ((±)-25)
Hydrolysis of urethane (±)-24 (2.2 g, 7.26 mmol) following the procedure described above for (+)-25, gave the racemic amine (±)-25 (0.91 g, 89%). The 1H and 13C NMR spectra of (±)-25 were identical to those previously recorded for (+)-25.
Aziridination Reactions
1-Azatricyclo [2.2.1.02,6]heptane (26)
A suspension of anhyd K2CO3 (460 mg, 3.33 mmol) in a solution of amine 6 as its trifluoracetic acid (TFA) salt (129 mg, 0.611 mmol) in 40 mL of dry CH2Cl2 was stirred magnetically as 580 mg (1.27 mmol) of solid Pb(OAc)4 was added according to literature procedures10, 12, 14 except that CH2Cl2 was used as solvent instead of benzene. TLC analyses (CHCl3 : MeOH = 10 : 1) showed complete reaction after 13 h. Satd aq K2CO3 (20 mL) was added, and the product was extracted with ether (2 × 15 mL). The organic layers were combined, dried with MgSO4, and treated with TFA (90 mg, 0.79 mmol). Evaporation of the solvent provided the pure TFA salt of 1-azatricyclo [2.2.1.02,6]-heptane (26) as a yellow oil: yield, 83.0 mg (0.406 mmol, 67%); 1H NMR (CDCl3, 400 MHz)47 δ 10.30–11.30 (m, 1H), 3.70 (s, 2H), 2.92 (s, 2H), 2.72 (s, 1H), 1.89 (app d, 2H, Japp = 12.4 Hz), 1.74 (app dd, 2H, Japp = 13.2, 1.6 Hz); 13C NMR (CDCl3, 100 MHz) δ 161.0 (q, J = 38.4 Hz), 115.8 (q, J = 286.6 Hz), 51.0, 38.1, 31.0, 29.1. Free aziridine: 1H NMR (CDCl3, 400 MHz)47 δ 2.22 (s, 2H), 2.18 (s, 2H), 2.08 (s, 1H), 1.34 (app d, 2H, Japp = 11.2 Hz), 1.21 (app dd, 2H, Japp = 11.2, 1.2 Hz); 13C NMR (CDCl3, 100 MHz) δ 55.8, 31.8, 31.5, 28.5; IR (NaCl): 3027, 2924, 1446, 1393; HRMS (ESI-HRMS) m/z calcd for (M + H+) 96.0813, found 96.0717.
The following preparative aziridinations were conducted as described above for the unsubstituted 1-azatricyclo [2.2.1.02,6]heptane (26).
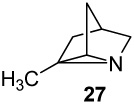
2-Methyl-1-azatricyclo [2.2.1.02,6]heptane (27)
Amine 10 as the TFA salt (105 mg, 0.47 mmol), K2CO3 (350 mg, 2.5 mmol), CH2Cl2 (20 mL), Pb(OAc)4 (440 mg, 1.00 mmol), time (2 h), TFA (70 mg, 0.61 mmol), yield of aziridine 27 TFA salt (81 mg, 78%): 1H NMR (CDCl3, 400 MHz)47 δ 10.50–11.00 (m, 1H), 3.63 (s, 1H), 3.05 and 2.91 (ABdd, JAB = 9.2 Hz, 2H), 2.67 (s, 1H), 1.90 (app ABd1, Japp = 12.4 Hz, 1H), 1.85 (app ABXdd1, Japp = 12.4, 1.2 Hz, 1H), 1.78 (app ABd2, Japp = 12.4 Hz, 1H), 1.71 (app ABXdd2, Japp = 12.4, 1.2 Hz, 1H), 1.71 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 159.5 – 162.0 (m), 112.0 – 123.0 (m), 112.0 – 123.0 (m), 51.6, 49.8, 43.9, 36.4, 32.4, 31.1, 11.8. Free aziridine: 1H NMR (CDCl3, 500 MHz)47 δ 2.39 and 2.25 (app ABdd, Japp = 9.9 Hz, 2H), 2.00–2.11 (br m, 1H), 2.08 (s, 1H), 1.32–1.40 (m, 2H), 1.35 (s, 3H), 1.16–1.24 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 56.7, 39.1, 38.5, 37.0, 33.1, 31.6, 16.0; IR (NaCl): 3289, 2960, 1557, 1455, 1377; HRMS (ESI-HRMS) m/z calcd for (M + H+) 110.0970, found: 110.0968.
2,6-Dimethyl-1-Azatricyclo [2.2.1.02,6]heptane (28)
Amine 13 TFA salt (100 mg, 0.42 mmol), K2CO3 (310 mg, 2.24 mmol), CH2Cl2 (20 mL), Pb(OAc)4 (380 mg, 0.84 mmol), time 1 h, TFA (60 mg, 0.53 mmol), yield of aziridine 28 TFA salt (80 mg, 81 %): 1H NMR (CDCl3, 500 MHz)47 δ 10.30–10.70 (m, 1H), 3.01 (s, 2H), 2.60 (s, 1H), 1.83 (app dd, Japp = 13.0, 1.0 Hz, 2H), 1.79 (app dd, Japp = 12.5, 1.5 Hz, 2H), 1.65 (s, 6H); 13C NMR (CDCl3, 125 MHz) δ 160.9 (q, J = 38.1 Hz), 116.0 (q, J = 287.9 Hz), 53.2, 52.2, 37.8, 31.4, 9.6. Free aziridine: 1H NMR (CDCl3, 500 MHz) δ 2.53 (s, 2H), 2.16 (s, 1H), 1.35–1.44 (m, 4H), 1.38(s, 6H); 13C NMR (CDCl3, 125 MHz): δ 56.3, 45.1, 38.4, 32.3, 12.5; IR (NaCl): 3252, 2949, 1557, 1455, 1386; HRMS (ESI-HRMS) m/z calcd for (M + H+) 124.1126, found: 124.1121.
7,7-Dimethyl-1-azatricyclo[2.2.1.02,6]heptane (30) and its Methiodide Salt (31)
Following Nagata’s procedure for the synthesis of bridged aziridines.14 To a stirred suspension of primary amine 20 (894 mg, 7.3 mmol) and anhyd K2CO3 (4.02 g, 29 mmol) in 150 mL of CH2Cl2 was added Pb(OAc)4 (6.45 g, 14.5 mmol) in 2-g portions. After 20 h, the solids were filtered, and the resulting filtrate was passed through a small column of basic alumina (Aldrich, 50.0 g, 150 mesh, 58 Ǻ) under vacuum. The column was washed using a gradient of 1–2% MeOH/CH2Cl2 as eluent. Fractions containing the aziridine (TLC alumina, 1% MeOH/CH2Cl2 single spot at Rf 0.7 when visualized with I2 vapors) were combined and washed with 10% aq HCl. The aq layer was cooled to 0 °C, basified with NaOH pellets, extracted with Et2O (total volume 250 mL), and dried over anhyd K2CO3. For characterization purposes, a small portion of this solution was evaporated (0 °C) using a stream of nitrogen to give aziridine 30 as a colorless and highly volatile liquid: 1H NMR (CDCl3, 500 MHz) δ 2.19 (s, 2H), 1.79 (d, 2H, J = 11 Hz), 1.59 (s, 1H), 1.13 (d, 2H, J = 11 Hz), 1.02 (s, 6H). Methyl iodide (905 µL, 14.5 mmol) was added to the above dry ethereal solution. After 20 h at room temp, the white crystals that formed were filtered, washed with ether, and recrystallized from EtOH-Et2O to afford the methiodide salt 31 (1.3 g, 68% over two steps). Data for recrystallized 31: mp 278–280 °C; 1H NMR (CDCl3, 500 MHz) δ 4.51 (s, 2H), 3.34 (s, 3H,), 2.45 (d, 2H, J = 13 Hz), 2.34 (s, 1H), 1.77 (d, 2H, J = 13 Hz), 1.40 (s, 6H); 13C NMR (CDCl3, 125 MHz) δ 49.3, 39.8, 34.7, 30.7, 19.8, 8.7; HRMS (ESI) calcd for m/z C9H16N 138.1283, found, 138.1282.
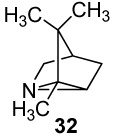
(+)-2,3,3-Trimethyl-1-azatricyclo[2.2.1.02,6]heptane ((+)-2-Azatricyclene, (+)-32)
A suspension of primary amine (+)-25 (308 mg, 2.2 mmol) and K2CO3 (1.23 g, 8.9 mmol) in 150 mL of CH2Cl2 was stirred and cooled in an ice bath as Pb(OAc)4 (1.97 g, 4.44 mmol) was added in 0.5-g portions over 5 min. After 20 min, the solids were removed by filtration, and the resulting filtrate was passed through basic alumina (Aldrich, 50.0 g, 150 mesh, 58 Ǻ) under vacuum using a gradient of 1–2% MeOH/CH2Cl2 as eluent. Fractions containing aziridine (+)-32 (TLC alumina Rf 0.6, using 1% MeOH-CHCl3, single yellow spot when visualized with I2 vapors) were combined and washed with 10% aq HCl (3 × 25 mL). The aq layer (containing the HCl salt of the aziridine) was cooled at 0 °C, basified with NaOH pellets, extracted with Et2O (3 × 100 mL), and dried over anhyd K2CO3. For characterization purposes, a small portion of this solution was evaporated using a steam of nitrogen to give aziridine (+)-32 as a colorless and volatile liquid: [α]25 D +3.67° (c 0.33, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 2.80 (d, 1H, J = 10 Hz), 2.11 (d, 1H, J = 10 Hz), 2.03 (s, 1H), 1.74 (dt, 1H, J = 12, 1.6 Hz), 1.62 (s, 1H), 1.24 (d, 1H, J = 12 Hz), 1.16 (s, 3H), 0.93 (s, 3H), 0.91 (s, 3H); 1H-NMR (D2O, 500 MHz) δ 2.64 (d, 1H, J = 10 Hz), 2.10 (s, 1H), 1.96 (d, 1H, J = 10 Hz), 1.67 (d, 1H, J = 12 Hz), 1.60 (s, 1H), 1.11 (d, 1H, J = 12 Hz), 0.99 (s, 3H), 0.74 (s, 3H), 0.73 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 54.1, 45.2, 42.4, 41.2, 40.0, 30.1, 19.7 (2C), 10.5. HRMS (ESI) m/z calcd for C9H16N (M + H) + 138.1283, found: 138.1281. The dry ethereal solution thus obtained was filtered and kept at −20 °C for storage. The aziridine readily polymerizes when stored as a neat liquid.
(+)-2,3,3-Trimethyl-1-azatricyclo[2.2.1.02,6]heptane ((+)-2-Azatricyclene, (+)-32), Trifluoroacetate Salt
To a cold (0 °C) and stirred solution of aziridine (+)-32 (152.1 mg, 1.1 mmol) in 150 mL of ether was added TFA (122.8 mg, 83 µL, 1.11 mmol). Removal of the solvents under reduced pressure afforded the aziridinium TFA salt of (+)-32 (246 mg, 88% for two steps) as an oil. A small sample was crystallized from ether/pentane. Data for the recrystallized salt: mp 117–118 °C; [α]25 D +1.26° (c 1.35, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 3.66 (1H, s), 3.43 (d, 1H, J = 10 Hz), 2.85 (d, 1H, J = 10 Hz), 2.22 (s, 1H), 2.17 (d, 1H, J = 13.2 Hz), 1.79 (d, 1H, J = 13.2 Hz), 1.55 (s, 3H), 1.14 (s, 3H), 1.05 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ 160.6 (q, J = 39.6 Hz), 115.4 (q, J = 288.2 Hz), 55.3, 49.6, 45.5, 43.6, 40.1, 29.5, 18.4, 18.3, 7.0; 19F NMR (376 MHz) δ −76.01 (br s).
(−)-2,3,3-Trimethyl-1-azatricyclo[2.2.1.02,6]heptane ((−)-2-Azatricyclene, (−)-32)
Pb(OAc)4 oxidation of amine (−)-25 (540 mg, 3.88 mmol) following the procedure given above for (+)-25 gave the bridged aziridine ent-32, [α]25 D −3.52° (c 0.8, CHCl3). The 1H and 13C NMR spectra of (−)-32 were identical to those previously recorded for the enantiomer (+)-32. HRMS (ESI) m/z calcd for C9H16N (M + H)+ 138.1283, found: 138.1280.
(−)-2,3,3-Trimethyl-1-azatricyclo[2.2.1.02,6]heptane [(−)-2-Azatricyclene] TFA Salt
The TFA salt of ent-25 (386 mg, 80% for two steps), prepared as described for (+)-32, showed mp 116–117 °C; [α]25 D −1.31° (c 1.6, CHCl3). The 1H, 13C and 19F NMR spectra of the TFA salt were identical to those previously recorded for the salt of the enantiomeric (+)-32.
(±)-2,3,3-Trimethyl-1-azatricyclo[2.2.1.02,6]heptane ((±)-2-Azatricyclene, (±)-32) and its Picrate Salt
Pb(OAc)4 oxidation of racemic amine (±)-25 (122 mg, 0.86 mmol) following the procedure given above for (+)-25 gave the bridged aziridine (±)-32. The 1H and 13C NMR spectra of (±)-32 were identical to those previously recorded for aziridine (+)-32. HRMS (ESI) m/z calcd for C9H16N (M + H) + 138.1283, found: 138.1282.
The picrate salt of (±)-2-azatricyclene ((±)-32) was prepared by adding a solution of amine (±)-32 (55.1 mg, 0.40 mmol) in 1 mL of 95% ethanol to a warm solution of picric acid (91.0 mg, 0.40 mmol) in 1 mL of 95% EtOH. Recrystallization from absolute ethanol gave the picrate salt (133 mg, 93%): mp 234–236 °C; 1H NMR (CDCl3, 500 MHz) δ 8.88 (s, 2H), 3.49 (s, 1H), 3.48 (d, 1H, J = 10 Hz), 2.93 (d, 1H, J = 10 Hz), 2.23 (s, 1H), 2.19 (d, 1H, J = 13.5 Hz), 1.81 (d, 1H, J = 12 Hz), 1.59 (s, 3H), 1.17 (s, 3H), 1.07 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 161.5, 141.8, 126.5 (4C), 54.4, 49.9, 44.8, 43.7, 40.2, 29.7, 18.9, 18.6, 7.3.
(±)-2,3,3-Trimethyl-1-azatricyclo[2.2.1.02,6]heptane ((±)-2-Azatricyclene) TFA Salt
The TFA salt of (±)-32 (18.5 mg, 83% over two steps), prepared as described for (+)-32, showed mp 111–113 °C. The 1H, 13C and 19F NMR spectra were identical to those previously recorded for TFA salt of (+)-32.
Representative Procedures and Data for Kinetic Runs
3-Methylcyclopent-3-enylmethylamine (10) was liberated from its TFA salt (15.8 mg, 0.0702 mmol) by reaction with excess anhyd K2CO3 in CDCl3 (0.70 mL), the supernatant solution was transferred to an NMR tube, and solid Pb(OAc)4 (60.0 mg, 0.138 mmol) was added. The progress of the aziridination reaction (10 → 27) was monitored by integration of the vinyl CH3 signal in 1H NMR spectra taken at 5- and 3-minute intervals. Figure 3 (orange circles) shows a plot of -ln [(integral 10)/(integral 10 + 27)] at each time point. The half-life for this run was 19 min. Spectra taken at shorter times indicate faster conversion to the aziridine owing to warming of the solution caused by the initial exothermic reaction of Pb(OAc)4 with the amine.
The kinetics of the other aziridination reactions were conducted similarly by NMR monitoring of CH3 signals, or in the case of the unsubstituted amine (6), by integration of the vinyl protons. The plots from three other kinetic run are shown in Figure 3. No peaks for side products were observed in the NMR spectra, except after extended time in aziridination 16 → 29. The half-lives and relative rates are collected in Table 1.
Supplementary Material
Acknowledgment
We thank Dr. Bindu Santhamma for conducting the initial aziridination reactions with Pb(OAc)4. This work was supported by National Institutes of Health grant GM 13956 to R.M.C.
Footnotes
Supporting Information Available: General aspects, instrumentation, preparative procedures, and characterization data for cyanocyclopentenes, cyclopentenylmethylamines, and related intermediates (4–6 and 8–16); N-(cyclopentenyl)methylhydroxylamines (33 – 35); cyclopentenyl tosylate (17) and α,α-dimethylcyclopentenylacetic acid (18); and campholenic acids 22 and 23; and 1H and 13C NMR spectra of most compounds reported. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.(a) Lange's Handbook of Chemistry. 15th Ed. New York: McGraw-Hill; 1992. Table 8.8. [Google Scholar]; (b) Kallies B, Mitzner R. J. Phys. Chem. B. 1997;101:2959–2967. [Google Scholar]
- 2.(a) Porter JW, Spurgeon SL, editors. Biosynthesis of Isoprenoid Compounds. Vol 1. New York: Wiley; 1983. [Google Scholar]; (b) Davis EM, Croteau R. Top. Curr. Chem. 2000;209:53–95. [Google Scholar]
- 3.Barton D, Nakanishi K, Meth-Cohn O, editors. Comprehensive Natural Product Chemistry. Oxford: Elsevier Science; 1999. [Google Scholar]; Cane DE, editor. Isoprenoids Including Carotenoids and Steroids. Vol. 2 [Google Scholar]
- 4.Corey EJ, Ortiz de Montellano PR, Lin K, Dean PDG. J. Am. Chem. Soc. 1967;89:2797–2798. doi: 10.1021/ja00987a089. [DOI] [PubMed] [Google Scholar]
- 5.(a) Popjak G, Meenan A, Parish EJ, Nes WD. J. Biol. Chem. 1989;264:6230–6238. [PubMed] [Google Scholar]; (b) Nes WD, Janssen GG, Norton RA, Kalinowska M, Crumley FG, Tal B, Bergenstrahle A, Jonsson L. Biochem. Biophys. Res. Commun. 1991;177:566–574. doi: 10.1016/0006-291x(91)92021-b. [DOI] [PubMed] [Google Scholar]; (c) Nes WD, Hanners PK, Parish EJ. Biochem. Biophys. Res. Commun. 1986;139:410–415. doi: 10.1016/s0006-291x(86)80006-7. [DOI] [PubMed] [Google Scholar]; (d) Maia C, Attias M, Urbina J, Gilbert I, Magaraci F, De Souza W. Biochem. Biophys. Res. Commun. 2007;363:310–316. doi: 10.1016/j.bbrc.2007.08.174. [DOI] [PubMed] [Google Scholar]; (e) Fujimoto Y, Morisaki M, Ikekawa N, Horie Y, Nakasone S. Steroids. 1974;24:367–375. doi: 10.1016/0039-128x(74)90034-8. [DOI] [PubMed] [Google Scholar]
- 6.(a) Corey EJ, Riddiford LM, Ajami AM, Yamamoto H, Anderson JE. J. Am. Chem. Soc. 1971;93:1815–1816. [Google Scholar]; (b) Anderson RJ, Henrick CA, Siddall JB. J. Org. Chem. 1972;37:1266–1268. [Google Scholar]
- 7.(a) Dev S, editor. CRC Handbook of Terpenoids. Boca Raton, FL: CRC Press; pp. 1982–1989. [Google Scholar]; (b) Nakanishi K, Goto T, Ito S, Natori S, Nozoe S, editors. Natural Products Chemistry. Vols 1 and 3. Tokyo: Kodansha; 1974 and 1983. [Google Scholar]; (c) Connolly JD, Hill RA. Dictionary of Terpenoids. Vols 1–3. London: Chapman & Hall; 1991. [Google Scholar]; (d) Glasby JS. Encyclopedia of the Terpenoids. Vols. 1 and 2. Chichester: Wiley; 1982. 1982. [Google Scholar]; (e) Buckingham J, editor. Dictionary of Natural Products. London: Chapman and Hall; 1994. [Google Scholar]; (f) Buckingham J. Dictionary of Natural Products (on-line web edition) London: Chapman & Hall/CRC Press; 2002. [Google Scholar]
- 8.(a) Giner JL, Djerassi C. Acta Chem. Scand. 1992;46:678–679. doi: 10.3891/acta.chem.scand.46-0678. [DOI] [PubMed] [Google Scholar]; (b) Ling NC, Hale RL, Djerassi C. J. Am. Chem. Soc. 1970;92:5281–5282. doi: 10.1021/ja00720a082. [DOI] [PubMed] [Google Scholar]
- 9.Koohang A, Coates RM, Owen D, Poulter CD. J. Org. Chem. 1999;64:6–7. doi: 10.1021/jo981833z. [DOI] [PubMed] [Google Scholar]
- 10.Roy A, Roberts FG, Wilderman PR, Zhou K, Peters RJ, Coates RM. J. Am. Chem. Soc. 2007;129:12453–12460. doi: 10.1021/ja072447e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christianson DW. Chem. Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 12.Portoghese PS, Sepp DT. Tetrahedron. 1973;29:2253–2256. [Google Scholar]
- 13.In ref 2a Croteau R. Biosynthesis of Monoterpenes. :225–282. Chap. 5. ; In ref 3 Wise ML, Croteau R. Monoterpene Biosynthesis. :97–153. Chap. 2.05.
- 14.(a) Nagata W, Hirai S, Kawata K, Aoki T. J. Am. Chem. Soc. 1967;89:5045–5046. [Google Scholar]; (b) Narisada M, Watanabe F, Nagata W. Synth. Commun. 1972;2:21–26. [Google Scholar]; (c) Nagata W, Wakabayashi T, Haga N. Synth. Commun. 1972;2:11–19. [Google Scholar]
- 15.(a) Duan P-W, Chiu C-C, Lee W-D, Pan LS, Venkatesham U, Tzeng Z-H, Chen K. Tetrahedron: Asymmetry. 2008;19:682–690. [Google Scholar]; (b) Watson IDG, Yu L, Yudin AK. Acc. Chem. Res. 2006;39:194–206. doi: 10.1021/ar050038m. [DOI] [PubMed] [Google Scholar]; (c) Padwa A, Murphree SS. Ark. Kemi. 2006:6–33. [Google Scholar]; (d) Halfen JA. Curr. Org. Chem. 2005;9:657–669. [Google Scholar]; (e) Müller P, Fruit C. Chem. Rev. 2003;103:2905–2919. doi: 10.1021/cr020043t. [DOI] [PubMed] [Google Scholar]; (f) Dahanukar VH, Zavialov IA. Curr. Opin. Drug Dis. Dev. 2002;5:918–927. [PubMed] [Google Scholar]; (g) McCoull W, Davis FA. Synthesis. 2000:1347–1365. [Google Scholar]; (h) Mitchinson A, Nadin A. J. Chem. Soc., Perkin Trans. 2000;1:2862–2892. [Google Scholar]; (i) Osborn HMI, Sweeney J. Tetrahedron: Asymmetry. 1997;8:1693–1715. [Google Scholar]; (j) Li A-H, Dai L-X, Aggarwal VK. Chem. Rev. 1997;97:2341–2372. doi: 10.1021/cr960411r. [DOI] [PubMed] [Google Scholar]; (k) Tanner D. Angew. Chem. Int. Ed. Engl. 1994;33:599–619. [Google Scholar]
- 16.Padwa A. Aziridines and Azirines: Monocyclic. In: Katritsky AR, Ramsden CA, Scriven EFV, Taylor RJK, editors. Comprehensive Heterocyclic Chemistry III. Vol. 1. Amsterdam: Elsevier; 2008. pp. 1–104. [Google Scholar]
- 17.Yudin AK, editor. Aziridines and Epoxides in Organic Synthesis. Weinheim: Wiley-VCH; 2006. [Google Scholar]
- 18.Moss RA, Platz MS, Jones M Jr, editors. Reactive Intermediate Chemistry. Hoboken, NJ: Wiley-Interscience; 2004. pp. 501–650. Chaps 11–13. [Google Scholar]
- 19.Moody CJ, Whitham GH. Reactive Intermediates. Oxford UK: Oxford University Press; 1992. [Google Scholar]
- 20.(a) Padwa A, Flick AC, Leverett CA, Stengel T. J. Org. Chem. 2004;69:6377–6386. doi: 10.1021/jo048990k. [DOI] [PubMed] [Google Scholar]; (b) Lebel H, Lectard S, Parmentier M. Org. Lett. 2007;9:4797–4800. doi: 10.1021/ol702152e. [DOI] [PubMed] [Google Scholar]; (c) Liu R, Herron SR, Fleming SA. J. Org. Chem. 2007;72:5587–5591. doi: 10.1021/jo0705014. [DOI] [PubMed] [Google Scholar]
- 21.(a) Richardson RD, Desaize M, Wirth T. Chem. Eur. J. 2007;13:6745–6754. doi: 10.1002/chem.200700306. [DOI] [PubMed] [Google Scholar]; (b) Li J, Chan PWH, Che C-M. Org. Lett. 2005;7:5801–5804. doi: 10.1021/ol052293c. [DOI] [PubMed] [Google Scholar]
- 22.Atkinson RS. Tetrahedron. 1999;55:1519–1559. [Google Scholar]
- 23.Carman RM, Derbyshire RPC. Aust. J. Chem. 2003;56:319–322. [Google Scholar]
- 24.Lin QJ, Zhang YM, Zhang CR, Song WZ, Qiu Q. Chin. Chem. Lett. 1991;2:517–520. [Google Scholar]
- 25.(a) Kirkland TA, Grubbs RH. J. Org. Chem. 1997;62:7310–7318. doi: 10.1021/jo970877p. [DOI] [PubMed] [Google Scholar]; (b) Berlin JM, Campbell K, Ritter T, Funk TW, Chlenov A, Grubbs RH. Org. Lett. 2007;9:1339–1342. doi: 10.1021/ol070194o. [DOI] [PubMed] [Google Scholar]
- 26.Ergün Y, Patir S, Okay G. J. Heterocycl. Chem. 2002;39:315–317. [Google Scholar]
- 27.Miriyala B, Bhattacharyya S, Williamson JS. Tetrahedron. 2004;60:1463–1471. [Google Scholar]
- 28.(a) Winstein S, Allred EL, Sonnenberg J. J. Am. Chem. Soc. 1959;81:5833. [Google Scholar]; (b) Allred EL, Sonnenberg J, Winstein S. J. Org. Chem. 1960;25:26–29. [Google Scholar]
- 29.(a) Shioiri T, Ninomiya K, Yamada S-i. J. Am. Chem. Soc. 1972;94:6203–6205. doi: 10.1021/ja00772a052. [DOI] [PubMed] [Google Scholar]; (b) Kedrowski BL. J. Org. Chem. 2003;68:5403–5406. doi: 10.1021/jo034170g. [DOI] [PubMed] [Google Scholar]; (c) Shioiri T, Yamada S-i. Org. Synth. 1990;Vol. 7:206–207. Coll. [Google Scholar]
- 30.(a) Bredt J, Houben J, Levy P. Chem. Ber. 1902;35:1286–1292. [Google Scholar]; (b) Sauers RR. J. Am. Chem. Soc. 1959;81:925–927. [Google Scholar]; (c) Crist BV, Rodgers SL, Lightner DA. J. Am. Chem. Soc. 1982;104:6040–6045. [Google Scholar]
- 31.Hutchinson JH, Money T, Piper SE. Can. J. Chem. 1986;64:854–860. [Google Scholar]
- 32.(a) Olah GA, Szilagyi PJ. J. Am. Chem. Soc. 1969;91:2949–2955. [Google Scholar]; (b) Breitmaier E, Voelter W. Carbon-13 NMR Spectroscopy. Weinheim: VCH; 1987. pp. 236–237. [Google Scholar]; (c) Sarneski JE, Surprenant HL, Molen FK, Reilly CN. Anal. Chem. 1975;47:2116–2124. [Google Scholar]
- 33.(a) Butler RN. Chem. Rev. 1984;84:249–276. [Google Scholar]; (b) Mihailović MLj, Čecović Ž. "Lead (IV) Acetate, ". In: Paquette LA, editor. Encyclopedia of Reagents for Organic Synthesis. Vol. 5. Chichester: Chief; Wiley; 1995. pp. 2949–2954. [Google Scholar]
- 34.(a) Mihailović MLj, Stojiljković A, Adrejević V. Tetrahedron Lett. 1965;6:461–464. [Google Scholar]; (b) Stojiljković A, Adrejević V, Mihailović MLj. Tetrahedron. 1967;23:721–732. [Google Scholar]
- 35.(a) Osawa E, Szalontai G, Tsurumoto A. J. Chem. Soc., Perkin Trans. 1983;2:1209–1216. [Google Scholar]; (b) Banks HD. J. Org.Chem. 2006;71:8089–8097. doi: 10.1021/jo061255j. [DOI] [PubMed] [Google Scholar]
- 36.(a) Phanstiel O, Wang QX, Powell DH, Ospina MP, Leeson BA. J. Org. Chem. 1999;64:803–806. doi: 10.1021/jo981613l. [DOI] [PubMed] [Google Scholar]; (b) Eriksson C, Sjödin K, Schlyter F, Högberg H-E. Tetrahedron: Asymmetry. 2006;17:1074–1080. [Google Scholar]; (c) Cheng Y-S, Lupo AT, Jr, Fowler FW. J. Am. Chem. Soc. 1983;105:7696–7703. [Google Scholar]
- 37.Lappert MF, Power PP, Sanger AR, Srivastava RC. Metal and Metalloid Amides. Chichester, UK: Ellis Horwood; 1980. pp. 271–272. 352–355. [Google Scholar]
- 38.Sisti AJ, Milstein SR. J. Org. Chem. 1974;39:3932–3936. [Google Scholar]
- 39.Dean JA. Lange's Handbook of Chemistry, 15th Ed. New York: McGraw-Hill; 1999. pp. 8–129. Table 8.27. [Google Scholar]
- 40.Bartlett PD. Rec. Chem. Progr. 1950;11:47–51. [Google Scholar]
- 41. Swern D. Organic Peroxides. Vol. II. New York: Wiley; 1971. Chap. V. Dryuk VG. Tetrahedron. 1976;32:2855–2866. Finn MG, Sharpless KB. In: Asymmetric Synthesis. Morrison JD, editor. Vol. 5. Orlando, FL: Academic Press; 1985. Chap. 8. Bach RD, Canepa C, Winter JE, Blanchette PE. J. Org. Chem. 1997;62:5191–5197. Bach RD, Glukhovtsev MN, Gonzalez C, Marquez M, Estevez CM, Baboul AG, Schlegel HB. J. Phys. Chem. A. 1997;101:6092–6100. (f) And relevant references cited in (a) – (e).
- 42.Bartlett PD, Sargent GD. J. Am. Chem. Soc. 1965;87:1297–1307. [Google Scholar]
- 43.Swern D. J. Am. Chem. Soc. 1947;69:1692–1698. doi: 10.1021/ja01195a603. [DOI] [PubMed] [Google Scholar]
- 44.(a) Stamm H. J. Prakt. Chem., Chem. Ztg. 1999;341:319–331. [Google Scholar]; (b) Hu XE. Tetrahedron. 2004;60:2701–2743. [Google Scholar]; (c) Watson IDG, Yudin AK. J. Org. Chem. 2003;68:5160–5167. doi: 10.1021/jo0343578. [DOI] [PubMed] [Google Scholar]; (d) Goujon J-Y, Gueyrard D, Compain P, Martin OR, Ikeda K, Kato A, Asano N. Bioorg. Med. Chem. 2005;13:2313–2324. doi: 10.1016/j.bmc.2004.12.043. [DOI] [PubMed] [Google Scholar]
- 45.The intramolecular aziridination (77%) of a t-alkyl primary amine with 1 equiv of NBS in acetone has been reported (see Introduction).23 However the scope of this potential alternative method is unclear
- 46.Faraldos JA, Coates RM. unpublished results. [Google Scholar]
- 47.The abbreviation "app" (apparent) in NMR data means that the interpretation was based on a first-order analysis of the multiplet in question, and the data give the appearance, positions, and spacings of the signals. If the multiplet is actually part of a second-order spin system, the “apparent” chemical shift(s), multiplicity, and J values reported may be different than the true values
- 48.Angle SR, Arnaiz DO. Tetrahedron Lett. 1989;30:515–518. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.