

Summary
The CXCR2 chemokine receptor is a G-protein-coupled receptor that undergoes clathrin-mediated endocytosis upon ligand binding. The trafficking of CXCR2 is crucial for cells to maintain a proper chemotactic response. The mechanisms that regulate the recycling/degradation sorting decision are unknown. In this study, we used dominant-negative (T19N) and GTPase-deficient activated (Q63L) RhoB mutants, as well as RhoB small interfering RNA (siRNA) to investigate the role of RhoB in CXCR2 trafficking. Expression of either of the RhoB mutants or transfection of RhoB siRNA impaired CXCR2-mediated chemotaxis. Expression of RhoB T19N and transfection of RhoB siRNA impaired sorting of CXCR2 to the lysosome after 3 hours of CXCL8 stimulation and impaired CXCL8-induced CXCR2 degradation. In cells expressing the RhoB Q63L mutant, CXCR2 recycling through the Rab11a recycling compartment was impaired after 30 minutes of CXCL8 stimulation as was CXCL8-induced CXCR2 degradation. For cells expressing activated RhoB, CXCR2 colocalized with Rab4, a marker for the rapid recycling pathway, and with the mannose-6-phosphate receptor, which traffics between the trans-Golgi network and endosomes. These data suggest that CXCR2 recycles through alternative pathways. We conclude that oscillation of RhoB GTPase activity is essential for appropriate sorting decisions, and for directing CXCR2 degradation and recycling – events that are required for optimal chemotaxis.
Keywords: RhoB, CXCR2, Trafficking, Recycling, Chemotaxis
Introduction
A major mechanism by which chemokine receptors undergo ligand-induced internalization is through clathrin-mediated endocytosis (Signoret et al., 2000; Venkatesan et al., 2003; Vila-Coro et al., 1999; Weber et al., 2004; Yang et al., 1999). The binding of ligand results in phosphorylation of Ser and Thr residues in the intracellular loops and the C-terminus of the chemokine receptor by G-protein-coupled receptor kinases (GRKs) (Ferguson, 2001; Ferguson et al., 1998; Freedman and Lefkowitz, 1996; Krupnick and Benovic, 1998). Receptor phosphorylation results in the uncoupling of the G protein subunits from the receptor and receptor desensitization in some cases (Ferguson, 2001; Ferguson et al., 1996; Krupnick and Benovic, 1998). In addition, phosphorylation of these residues and/or the presence of di-leucine motifs in the C-terminal domain of chemokine receptors are important for the recruitment of adaptor molecules that link the receptor to a lattice of clathrin that facilitates receptor internalization. The association of receptors with these adaptor molecules results in recruitment of clathrin and formation of clathrin-coated pits which pinch-off from the membrane through the action of dynamin and become clathrin-coated vesicles (Barlic et al., 1999; Colvin et al., 2004; Droese et al., 2004; Jimenez-Sainz et al., 2003; Orsini et al., 1999; van der Bliek et al., 1993; Vila-Coro et al., 1999; Weber et al., 2004; Yang et al., 1999). Indeed, chemokine receptors may concentrate and internalize through preformed clathrin lattices where endocytic machinery is accumulated (Signoret et al., 2005). The clathrin-coated vesicle is uncoated and the receptor-ligand complex enters the early endosome. The receptor can either enter the recycling compartment and traffic back to the plasma membrane to bind ligand, or enter the late endosome where it will be sorted to the lysosome for degradation. The factors that mediate the trafficking fate of internalized chemokine receptors are largely unknown.
Prior studies show that the length of stimulation with ligand plays a role in the recycling/degradation sorting decision (Fan et al., 2003; Neel et al., 2005; Signoret et al., 2000). At early time periods after CXCL8 stimulation of CXCR2, the receptor enters the recycling compartment; conversely, following extended periods of stimulation, the receptor enters the late endosome and lysosome (Fan et al., 2003). The ability of internalized CXCR2 to recycle is crucial for continued gradient sensing and chemotactic response to ligand. When CXCR2 recycling is inhibited, chemotaxis and signaling are impaired (Fan et al., 2003; Fan et al., 2004; Zaslaver et al., 2001).
Rabs are small GTPases that are post-translationally modified with geranylgeranyl groups at their C-termini (Pereira-Leal and Seabra, 2000; Zerial and McBride, 2001). This modification allows Rabs to associate with particular intracellular membrane-bound compartments and regulate a number of cellular trafficking events. Rab5, a mediator of the early endocytic response, is important for CXCR2 trafficking. CXCR2 localizes to Rab5-positive endosomes during early time points of ligand stimulation and expression of a Rab5 dominant-negative mutant attenuates CXCR2 internalization (Fan et al., 2003). The recycling pathway involving Rab11a is important in the intracellular trafficking of chemokine receptors and the responses mediated by these receptors. Following ligand stimulation, CXCR2 localizes to the Rab11a-positive compartment. Expression of a dominant-negative mutant reduces CXCR2 recycling (Fan et al., 2003). Rab7 appears to be involved in the lysosomal sorting of chemokine receptors. Expression of dominant-negative Rab7 decreases localization of CXCR2 to the lysosome (LAMP-1-positive) and increases localization to Rab5- and Rab11a-positive endosomes after prolonged ligand stimulation (Fan et al., 2003). These data suggest that Rab7 regulates the transfer of CXCR2 to the lysosome.
Although the small GTPase RhoB has nearly 85% sequence similarity to the extensively characterized RhoA, a number of studies suggest a unique function for RhoB (Ridley, 2001; Wennerberg and Der, 2004; Wheeler and Ridley, 2004). RhoB can be prenylated with either a farnesyl or a geranylgeranyl group, where RhoA and RhoC are only geranylgeranylated. These differences in post-translational modification may be responsible for the unique cellular functions and localization of RhoB from other isoforms. The majority of RhoB is localized to endosomes but its precise localization remains controversial. Studies have reported it to be localized to the plasma membrane (Michaelson et al., 2001), late endosomes (Wherlock et al., 2004), and more recently to early endosomes (Rojas et al., 2004). Despite the ambiguity of its localization, it does appear that RhoB plays an important role in intracellular trafficking (Gampel et al., 1999; Mellor et al., 1998).
In the current study, we used dominant-negative (T19N) and GTPase-deficient activated (Q63L) RhoB mutants as well as siRNA directed against RhoB to investigate the potential role of RhoB in CXCR2 chemokine receptor trafficking. We found that the RhoB T19N mutant, RhoB Q63L mutant and small interfering RNA (siRNA) directed against RhoB impair CXCR2-mediated chemotaxis, and disrupt the intracellular trafficking of CXCR2. In addition, we show for the first time that CXCR2 may recycle through alternative pathways when RhoB function is disrupted. These data suggest that RhoB plays a key role in the recycling/degradation sorting decision in CXCR2 receptor trafficking.
Results
RhoB is activated upon CXCL8 stimulation
In order to determine whether RhoB is activated upon stimulation with the CXCR2 ligand CXCL8, we used the Rho-binding domain of the rhotekin (TRBD) GST-fusion protein to isolate GTP-bound (active) RhoB from HEK293 cells stably expressing CXCR2. It has been previously demonstrated that epidermal growth factor (EGF) activates RhoB (Gampel et al., 1999); therefore EGF was used as a positive control for RhoB activation. The GTPase-deficient activated (Q63L) RhoB mutant was used as an additional positive control for GTP-bound RhoB. Cells were transfected with WT Myc-RhoB or with Q63L Myc-RhoB and either stimulated with vehicle (untreated), EGF for 60 minutes, or CXCL8 for 5 minutes, 30 minutes, or 60 minutes. Following stimulation, GTP-bound RhoB was isolated using GST-TRBD and analyzed by SDS-PAGE and western blot with detection using RhoB antibody. Activation of exogenous RhoB was detected after CXCL8 stimulation (Fig. 1). These experiments were repeated six times and the activation of RhoB after 5 minutes of CXCL8 stimulation was statistically significant (P<0.05, Student’s t-test). Activation of RhoB after 30 minutes and 60 minutes of CXCL8 stimulation was also observed in some experiments. It is expected that the localized activation of RhoB is stronger and biologically more relevant than the overall detection observed by the pull-down assay. Discrepancies in specific time point of activation may be due to the two differentially localized forms of RhoB and variations among different preparations of the GST-TRBD fusion protein.
Fig. 1.
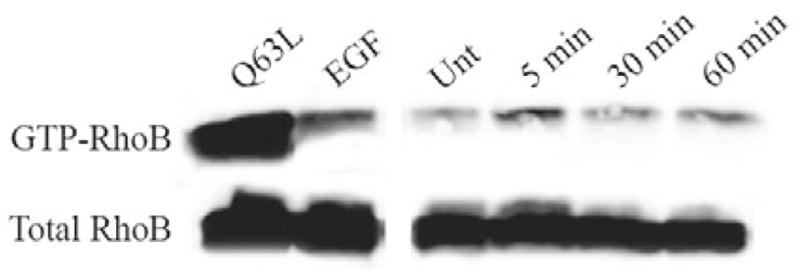
RhoB is activated upon CXCL8 stimulation. Western blot analysis with anti-RhoB antibody of input lysates (Total RhoB) and GST-TRBD bound RhoB (GTP-RhoB) separated by SDS-PAGE. HEK293 cells stably expressing CXCR2 were transiently transfected with Myc-WT RhoB or Myc-Q63L RhoB mutant. Cells transfected with Myc-Q63L RhoB were stimulated with vehicle. Cells transfected with Myc-WT RhoB were stimulated with 100ng/ml EGF for 60 minutes (EGF), vehicle (Unt), or 100 ng/ml CXCL8 for 5 minutes, 30 minutes or 60 minutes. Lysates were incubated with GST-TRBD (rhotekin Rho-binding domain) to isolate GTP-bound RhoB. Data shown are representative from three separate experiments. Induction at 5-minute and 60-minute time points when normalized to total RhoB was 1.6-fold and 1.1-fold, respectively, in the experiment shown here.
Dominant-negative (T19N) and GTPase-deficient activated (Q63L) RhoB mutants impair CXCR2-mediated chemotaxis
To assess whether CXCR2 function is regulated by RhoB, CXCR2-mediated chemotaxis was investigated using the Boyden chamber chemotaxis assay in cells stably expressing CXCR2. Cells were transiently transfected with empty pcDNA3 vector, Myc-RhoB T19N (dominant negative) or Myc-RhoB Q63L mutants (activated). Cells transfected with the empty vector demonstrate a characteristic bell-shaped chemotactic response. By contrast, cells expressing either the Myc-RhoB T19N or Myc-RhoB Q63L mutants exhibit a twofold reduction in the number of migrated cells (Fig. 2A,B). We also examined CXCR2-mediated chemotaxis in cells expressing Myc-RhoB WT and found that there was no effect on chemotaxis when WT RhoB is expressed (Fig. 2C). To investigate whether the decrease in chemotaxis was a result of impaired actin polymerization, we examined F-actin by phalloidin staining and confocal microscopy, prepared Triton X-100-soluble and -insoluble fractions, and performed western blot analysis for actin. Expression of RhoB T19N or Q63L did not alter the appearance of F-actin staining or actin levels in Triton X-100-soluble and -insoluble fractions (supplementary material Fig. S1). These data suggest that the effect of altering RhoB GTPase activity on CXCR2 function is not due to a more general impairment of actin polymerization. The RhoB T19N mutant is GTP-binding-deficient and acts dominant negative by binding and sequestering guanine nucleotide exchange factors (GEFs), and by making them unavailable to endogenous RhoB. Therefore, in order to rule out the possibility that the phenotype associated with this mutant is nonspecific, we examined the effect of knocking down endogenous RhoB on chemotaxis. Knocking down endogenous RhoB expression using siRNA also results in impaired CXCR2-mediated chemotaxis (supplementary material Fig. S2).
Fig. 2.

RhoB T19N and Q63L mutants impair CXCR2-mediated chemotaxis. (A-C) Boyden chamber assay assessing chemotaxis of HEK293 cells stably expressing CXCR2 and transiently transfected with empty vector, Myc-RhoB T19N or Myc-RhoB Q63L. A twofold reduction in CXCR2-mediated chemotaxis was observed when Myc-RhoB T19N and Myc-RhoB Q63L mutants were expressed. No effect on chemotaxis was observed when Myc-RhoB WT was expressed. CXCR2-mediated chemotaxis of cells expressing Myc-RhoB T19N (A), Myc-RhoB Q63L (B) and Myc-RhoB WT (C). The graphs display numbers of cells from twenty separate fields ± s.e.m., using the 20× objective lens. + and * indicate significant differences (P<0.01 and P<0.005; Student’s t-test) of vector-transfected cells versus cells transfected with Myc-RhoB T19N or Myc-RhoB Q63L. Data shown are representative from three separate experiments.
Expression of dominant-negative (T19N) RhoB alters trafficking of CXCR2 following 3 hours of CXCL8 stimulation
Because RhoB activation occurs upon EGF stimulation and is involved in the intracellular trafficking of the EGF receptor (Gampel et al., 1999; Wherlock et al., 2004), we sought to investigate whether RhoB similarly regulates the trafficking of CXCR2. Therefore, we transiently expressed empty vector or Myc-RhoB T19N in HEK293 cells stably expressing CXCR2. trafficking of CXCR2 was evaluated by immunofluorescence staining and confocal microscopy. Previous studies in our laboratory have revealed that the majority of CXCR2 enters the perinuclear Rab11a-positive recycling compartment and recycles back to the plasma membrane after periods of CXCL8 stimulation longer than 30 minutes and shorter than 1 hour. However, upon longer periods of stimulation with saturating concentrations of CXCL8, the majority of CXCR2 no longer enters the recycling compartment and instead traffics to the late endosomal compartment where it is then transferred to the lysosome for degradation (Fan et al., 2003). In our current study, CXCR2 substantially colocalized with the lysosomal marker LAMP-1 upon 3 hours of CXCL8 stimulation in vector transfected cells (44.4±15.8%) and cells transfected with Myc-RhoB WT (56.6±22.6%) (Fig. 3A,C, supplementary material Fig. S3). By contrast, HEK293 cells expressing the Myc-RhoB T19N mutant exhibited minimal colocalization of CXCR2 with the lysosomal marker LAMP-1 (12.9±8%) after 3 hours of CXCL8 stimulation (Fig. 3B,C). Similarly, knocking down endogenous RhoB with siRNA also resulted in a significant decrease in colocalization of CXCR2 with LAMP-1 after 3 hours of CXCL8 stimulation (12.8±3% colocalization) when compared with control siRNA (36.5±7.6% colocalization; P<0.005; supplementary material Fig. S4). Furthermore, there was minimal colocalization of CXCR2 with Rab11a after 3 hours of CXCL8 stimulation in cells expressing empty vector (24.8±11.6%) or Myc-RhoB WT (18.7±6.8%) (Fig. 3D,F, supplementary material Fig. S3). In contrast to cells transfected with vector and Myc-RhoB WT, HEK293 cells expressing Myc-RhoB T19N exhibited extensive CXCR2 accumulation in the Rab11a compartment (70.7±9.9%; Fig. 3E,F). At earlier time periods of CXCL8 stimulation there was no difference observed in the localization of CXCR2 in cells transfected with Myc-RhoB T19N compared to vector-transfected cells.
Fig. 3.

Expression of dominant-negative (T19N) RhoB alters trafficking of CXCR2 following 3 hours of CXCL8 stimulation. Confocal images of immunofluorescence staining of HEK293 cells stably expressing CXCR2. (A,B) Staining of CXCR2 and LAMP-1 in cells transfected with vector (A) or Myc-RhoB T19N (B) and stimulated with vehicle (Untreated) or 100 ng/ml CXCL8 for 3 hours. Transfected cells were identified by staining with anti-Myc antibody. Overlay images are pseudocolored; red is CXCR2, green is LAMP-1, blue is Myc-RhoB T19N. Bars, 10 μm. (C) Quantification of colocalization of CXCR2 with LAMP-1 in cells transfected with vector, Myc-RhoB WT or Myc-RhoB T19N. Values shown are the mean ± s.e.m. *P<0.005, significant differences (Student’s t-test) of vector or Myc-RhoB WT versus Myc-RhoB T19N transfected cells. (D,E) CXCR2 and Rab11a staining in cells transfected with empty vector (D) or Myc-RhoB T19N (E) stimulated with vehicle (Untreated) or 100 ng/ml CXCL8 for 3 hours. Transfected cells were identified by staining with anti-Myc antibody. Overlay images are pseudocolored; red is CXCR2, green is Rab11a, blue is Myc-RhoB T19N. Bars, 10 μm. (F) Quantification of colocalization of CXCR2 with Rab11a in cells transfected with vector, Myc-RhoB WT, or Myc-RhoB T19N. Values shown are the mean ± s.e.m. *P≤0.05, significant differences (Student’s t-test) of vector or cells transfected with Myc-RhoB WT versus cells transfected with Myc-RhoB T19N. Quantification of the percentage of CXCR2 colocalized with LAMP-1 or Rab11a in 20 fields was performed using the MetaMorph Imaging system (Universal Imaging). Images were processed using Photoshop (Adobe Systems). Data shown are representative from three separate experiments.
To determine whether expression of RhoB T19N effects a change in the movement of CXCR2 through endosomal compartments that can be observed biochemically, iodixanol-density gradients were used to fractionate endosomal compartments of HEK293 cells stably expressing CXCR2 and transiently expressing either empty vector or Myc-RhoB T19N mutant following ligand stimulation. We found that a substantial amount of CXCR2 co-fractionates with Rab11a after 3 hours of CXCL8 stimulation in cells expressing Myc-RhoB T19N (data not shown). These data suggest that RhoB activity is required for the movement of CXCR2 into the lysosomal compartment after prolonged ligand stimulation.
Expression of RhoB T19N does not alter Rab11a-positive endosome motility
To evaluate whether the accumulation of CXCR2 in the Rab11a compartment is related to endosome motility and actin polymerization, the effects of actin-disrupting agents on CXCR2 trafficking were examined. Indeed, it has been previously demonstrated that cytochalasin D impairs CXCR2 recycling (Zaslaver et al., 2001). HEK293 cells stably expressing CXCR2 were stimulated with vehicle or CXCL8 for 30 minutes and treated with either latrunculin B or cytochalasin D. CXCR2 and Rab11a were then examined by immunofluorescence staining and confocal microscopy. Both latrunculin B and cytochalasin D treatment resulted in accumulation of CXCR2 in the Rab11a compartment (supplementary material Fig. S5). These data suggest that actin polymerization is not required for movement of CXCR2 into the perinuclear recycling compartment but it might be required when the receptor exits that compartment.
We hypothesized that the accumulation of CXCR2 in the Rab11a compartment in cells expressing RhoB T19N is a result of a defect in actin-dependent endosome motility. In order to test this hypothesis, we examined Rab11a-GFP endosomes in live cells using time-lapse confocal microscopy. HEK293 cells stably expressing CXCR2 were either transiently co-transfected with a EGFP-Rab11a construct and the Myc-RhoB T19N mutant or the EGFP-Rab11a construct and the empty vector. Cells were then stimulated with vehicle or 100 ng/ml CXCL8 for 30 minutes and examined using time-lapse confocal microscopy. The movement of the EGFP-Rab11a-positive endosomal structures was manually tracked as described in Materials and Methods. The average velocities and distances traveled are displayed in Table 1. No detectable differences in Rab11a-positive endosome velocity or maximum distance traveled from the point of origin were observed in untreated cells or in cells stimulated for 30 minutes with CXCL8, and vector-transduced (supplementary material Movie 1) and Myc-RhoB-T19N-transduced (supplementary material Movie 2) cells, respectively. These results indicate that RhoB GTPase activity is not required for Rab11a-positive endosome motility.
Table 1.
Average velocities and maximum distance traveled from origin of Rab11a-positive endosomes in untreated and CXCL8 stimulated cells
| Average velocity (nm/second) | Maximum distance from origin (nm) | ||
|---|---|---|---|
| Untreated | Vector | 81.3±6.9 | 846.9±162.6 |
| T19N RhoB | 72.1±18.6 | 773.5±384.9 | |
| CXCL8* | Vector | 90.1±12.5 | 972.6±165.1 |
| T19N RhoB | 86.8±15.5 | 823.3±359.3 |
CXCL8 treatment was for 30 minutes.
We also examined receptor recovery to the plasma membrane after ligand stimulation using radioligand binding to determine wether expression of Myc-RhoB T19N impaired the return of CXCR2 to the plasma membrane. We found that the reappearance of CXCR2 at the plasma membrane following ligand stimulation in the presence of cycloheximide was not impaired in cells expressing Myc-RhoB T19N (Fig. 4). Receptor recycling was also assessed using FACS analysis, and no defect in recycling was observed using this method (data not shown). These data suggest that the expression of the RhoB T19N mutant did not result in the accumulation of CXCR2 in the Rab11a compartment, but rather in the enhanced recycling of CXCR2 even after long-term ligand stimulation.
Fig. 4.

Expression of Myc-RhoB T19N does not impair CXCR2 recycling. 125I-CXCL8 binding in HEK293 cells stably expressing CXCR2 and transfected with empty vector or Myc-RhoB T19N. Cells were pretreated with 20 μg/ml cycloheximide for 30 minutes and placed in the presence of 20 μg/ml cycloheximide throughout the experiment. Cells were stimulated with vehicle (total binding) or 100 ng/ml CXCL8 for 30 minutes, CXCL8 was removed, and cells were allowed to recover for 0 minutes, 30 minutes or 60 minutes. Cells were incubated with 0.1 nM 125I-CXCL8 (specific activity =2200 Ci/mmol) for 2 hours, washed to remove non-specific binding, and subjected to γ-counting as described in Materials and Methods. Values represent three independent experiments and are shown as percent of total binding ± s.e.m. Data shown are representative from three separate experiments.
Expression of RhoB T19N impairs CXCR2 degradation following 3 hours of CXCL8 stimulation
Upon long-term stimulation with CXCL8, CXCR2 traffics to the lysosome and is degraded. Because very little CXCR2 colocalized with LAMP-1 in HEK293 cells expressing Myc-RhoB T19N after 3 hours of CXCL8 stimulation, we suspected that degradation of the receptor was impaired. To examine whether expression of Myc-RhoB T19N impairs CXCL8-induced CXCR2 degradation, we examined CXCR2 protein levels following 30 minutes, 180 minutes and 360 minutes of CXCL8 stimulation in the presence of cycloheximide. The CXCR2 band runs at a slightly higher molecular mass following stimulation due to phosphorylation. In addition, CXCR2 is differentially phosphorylated on several serine residues and these various phosphorylated forms are visible by western blot analysis. In vector transfected cells, a significant amount of CXCR2 is degraded after 180 minutes (69.1±6.3% remaining) and 360 minutes of CXCL8 stimulation (48.9±9.2% remaining; P<0.05). Expression of Myc-WT RhoB did not inhibit CXCL8-induced CXCR2 degradation (supplementary material Fig. S6). Expression of the RhoB T19N mutant severely impairs CXCR2 degradation after 180 minutes (94±7.3% remaining) and 360 minutes of CXCL8 stimulation (97.1±3.1% remaining) (Fig. 5). This is presumably because of enhanced CXCR2 recycling and marked reduction in receptor trafficking to the lysosome.
Fig. 5.

Expression of Myc-RhoB T19N impairs CXCL8-induced CXCR2 degradation following 3 hours of CXCL8 stimulation. Representative western blot analysis of CXCR2 expression in cells transfected with vector or Myc-RhoB T19N in the presence of 20 μg/ml cycloheximide following stimulation with vehicle (Unt) or 100 ng/ml CXCL8 for 30 minutes, 180 minutes or 360 minutes. Actin is shown as a control to monitor for equal loading of protein. Data shown are representative from three separate experiments.
Expression of GTPase-deficient activated (Q63L) RhoB mutant alters trafficking of CXCR2 following 30 minutes of CXCL8 stimulation
We have previously demonstrated that CXCR2 is recycled back to the plasma membrane when stimulated with CXCL8 for short periods of time (Fan et al., 2003). To characterize more extensively the role of RhoB in CXCR2 trafficking, we examined the effects of a GTPase-deficient RhoB-mutant (Q63L) on CXCR2 trafficking using immunofluorescence and confocal microscopy. CXCR2 colocalizes with the recycling endosomal marker Rab11a following 30 minutes of CXCL8 stimulation in CXCR2-expressing HEK293 cells transfected with empty vector (53.3±17.6%) or Myc RhoB WT (40.9±1.0%) (Fig. 6A,C, supplementary material Fig. S7). When cells were transfected with the Myc-RhoB Q63L mutant, less colocalization with Rab11a is observed (18.2±8.2%) (Fig. 6B,C). There is very little colocalization with the EGFP-Rab7, a late endosomal marker, in vector transfected cells (5.4±8.1%) (Fig. 7D,F). By contrast, there is substantial colocalization with EGFP-Rab7 (39.7±16.7%) (Fig. 6E,F) in cells transfected with Myc-RhoB Q63L.
Fig. 6.

Expression of GTPase-deficient RhoB (Q63L) mutant alters trafficking of CXCR2 following 30 minutes of CXCL8 stimulation. Confocal images of immunofluorescence-stained HEK293 cells stably expressing CXCR2. (A,B) CXCR2 and Rab11a staining in cells transfected with empty vector (A) or Myc-RhoB Q63L (B) stimulated with vehicle (Untreated) or 100 ng/ml CXCL8 for 30 minutes. Transfected cells were identified by staining with anti-Myc antibody. Overlay images are pseudocolored; red is CXCR2, green is Rab11a, blue is Myc-RhoB Q63L. Bars, 10 μm. (C) Quantification of colocalization of CXCR2 with Rab11a in cells transfected with vector, Myc-RhoB WT or Myc-RhoB Q63L. Values shown are the mean ± s.e.m. *P<0.05, significant differences (Student’s t-test) of vector or cells transfected with Myc-RhoB WT versus cells transfected with Myc-RhoB Q63L. (D,E) CXCR2 staining and EGFP-Rab7 in cells transfected with empty vector (D) and Myc-RhoB Q63L (E) and stimulated with vehicle (Untreated) or 100 ng/ml CXCL8 for 30 minutes. Transfected cells were identified by staining with anti-Myc antibody. Overlay images are pseudocolored; red is CXCR2, green is EGFP-Rab7, blue is Myc-RhoB Q63L. Bars, 10 μm. (F) Quantification of colocalization of CXCR2 with EGFP-Rab7. Values shown are the mean ± s.e.m. *P<0.05, significant differences (Student’s t-test) of cells transfected with Myc-RhoB Q63L versus vector-transfected cells. Quantification of the percentage of CXCR2 colocalized with Rab11a or EGFP-Rab7 in 20 fields was performed using the MetaMorph Imaging system (Universal Imaging). Images were processed using Photoshop (Adobe Systems). Data shown are representative from three separate experiments.
Fig. 7.

Expression of Myc-RhoB Q63L does not impair CXCR2 recycling. 125I-CXCL8 binding in HEK293 cells stably expressing CXCR2 and transfected with empty vector or Myc-RhoB Q63L. Cells were pretreated with 20 μg/ml cycloheximide for 30 minutes and placed in the presence of 20 μg/ml cycloheximide throughout the experiment. Cells were stimulated with vehicle (total binding), 100 ng/ml CXCL8 for 30 minutes, CXCL8 was removed, and cells were allowed to recover for 0 minutes, 30 minutes or 60 minutes. Cells were incubated with 0.1 nM 125I-CXCL8 (specific activity =2200 Ci/mmol) for 2 hours, washed to remove non-specific binding, and subjected to γ-counting as described in Materials and Methods. Values represent three independent experiments and are shown as percent of total binding ± s.e.m. Data shown are representative from three separate experiments.
One interpretation of our data, showing altered trafficking of CXCR2 in RhoB T19N or RhoB Q63L transfected cells, is that RhoB plays a general role in the sorting of internalized receptors and alteration of its activity disturbs the composition and/or function of the respective endosomal compartments. For example, it is possible that the Rab7 endosomal compartment is mislocalized in cells expressing Myc-RhoB Q63L. To clarify these issues, we investigated whether expression of the Myc-RhoB-Q63L mutant alters the trafficking of the transferrin receptor (TfnR), which normally recycles through the Rab11a compartment and is not sorted to the late endosome. We used immunofluorescence and confocal microscopy to observe the colocalization of TfnR with EGFP-Rab7 in cells transfected with either empty vector or Myc-RhoB Q63L. Expression of Myc-RhoB Q63L did not result in colocalization of TfnR with EGFP-Rab7 (supplementary material Fig. S8). These results suggest that CXCR2 sorting occurs in a RhoB-dependent manner, whereas TfnR sorting occurs through a RhoB-independent mechanism. Moreover, these results argue against a generalized effect that mutant RhoB alters the localization of various Rabs in the endosomal compartments.
Expression of RhoB Q63L does not impair CXCR2 recycling
Because CXCR2 traffics to the late endosomal compartment following 30 minutes of CXCL8 stimulation in cells transfected with RhoB Q63L, we expected the receptor would not efficiently recycle back to the plasma membrane in these cells. We examined receptor recovery to the plasma membrane after ligand stimulation using a modified radioligand binding assay. Surprisingly, we found that the reappearance of CXCR2 at the plasma membrane following ligand stimulation in the presence of cycloheximide was not impaired in cells expressing Myc-RhoB Q63L (Fig. 7). Receptor recycling was also examined using FACS analysis and results were similar (data not shown). These data indicate that CXCR2 can return to the plasma membrane without entering the Rab11a perinuclear recycling compartment.
Expression of RhoB Q63L impairs CXCR2 degradation and colocalization with lysosomal markers
To determine whether CXCR2 degradation occurs at early time periods of CXCL8 stimulation in RhoB Q63L transfected cells where it is prematurely sorted to the late endosomal compartment, CXCR2 levels were determined by western blot in the presence of cycloheximide in vehicle-treated cells and cells stimulated with CXCL8 for 30 minutes, 180 minutes or 360 minutes. These experiments revealed that CXCR2 is degraded upon 180 minutes (69.1±6.3% remaining) and 360 minutes (48.9±9.2% remaining) of CXCL8 stimulation in vector transfected cells. Unexpectedly, CXCR2 degradation was actually impaired in cells expressing Myc-RhoB Q63L after 180 minutes (97.5±8% remaining) and 360 minutes (94.2±5% remaining) of CXCL8 stimulation (Fig. 8A). These results suggest that CXCR2 traffics to the late endosomal (Rab7-positive) compartment in cells expressing Myc-RhoB Q63L, but it is unable to transfer to the lysosome for degradation. The ability of CXCR2 to enter the lysosome in cells expressing the RhoB Q63L mutant was also examined by immunofluorescence staining of CXCR2, and the LAMP-1 and CD63 lysosomal markers. CXCR2 did not colocalize with the LAMP-1 or CD63 lysosomal markers in cells transfected with RhoB Q63L (Fig. 8B).
Fig. 8.

Expression of Myc-RhoB Q63L impairs CXCL8-induced CXCR2 degradation and does not result in colocalization with lysosomal markers. (A) Western blot analysis of CXCR2 expression in cells transfected with vector or Myc-RhoB Q63L in the presence of 20 μg/ml cycloheximide following stimulation with vehicle (Unt) or 100 ng/ml CXCL8 for 30 minutes, 180 minutes or 360 minutes. Actin is shown as a control to monitor for equal loading of protein. (B) Confocal images from three independent experiments of immunofluorescence-stained HEK293 cells stably expressing CXCR2 and transiently transfected with Myc-RhoB Q63L. Transfected cells were identified by staining with anti-Myc antibody. CXCR2 and LAMP-1 (top panels) or CD63 (bottom panels) staining in cells stimulated with vehicle (Untreated) or 100 ng/ml CXCL8 for 30 minutes. Overlay images are pseudocolored; red is CXCR2, green is LAMP-1 or CD63, blue is Myc-RhoB Q63L. Images were processed using Photoshop (Adobe Systems). Bars, 10 μm. Data shown are representative from three separate experiments.
Expression of Q63L RhoB results in colocalization of CXCR2 with Rab4 and mannose-6-phosphate receptor
Our data, showing that CXCR2 is unable to enter the Rab11a-positive perinuclear recycling compartment or the lysosome in cells expressing Myc-RhoB Q63L, suggest that internalized CXCR2 uses an alternative recycling pathway to return to the plasma membrane. Therefore, we sought to investigate the mechanism by which CXCR2 returns to the plasma membrane in cells expressing Myc-RhoB Q63L. There are two main endosomal recycling pathways to which Rab11a and Rab4 can contribute, a slow and a rapid recycling process, respectively. The second pathway bypasses the Rab11a-positive perinuclear endosomes and mediates rapid recycling of receptors through Rab4-positive endosomes (Sheff et al., 1999; Sonnichsen et al., 2000). We examined whether CXCR2 colocalized with Rab4 following 30 minutes of CXCL8 stimulation in vector-transfected cells and cells transfected with Myc-RhoB Q63L. There was only 4.1±2.4% colocalization of CXCR2 with Rab4 in vector transfected cells (Fig. 9A,C). By contrast, there was substantial colocalization of CXCR2 with Rab4 in cells transfected with Myc-RhoB Q63L (49.7±18% colocalization) following 30 minutes of CXCL8 stimulation (Fig. 9B,C). These data suggest that, in part, cells transfected with Myc-RhoB Q63L use the rapid recycling pathway to return CXCR2 to the membrane following ligand stimulation.
Fig. 9.

Expression of Q63L RhoB results in colocalization of CXCR2 with Rab4 and mannose-6-phosphate receptor (MPR). Confocal images of immunofluorescence-stained HEK293 cells stably expressing CXCR2. (A,B) CXCR2 and Rab4 staining in cells transfected with empty vector (A) or Myc-RhoB Q63L (B) and stimulated with vehicle (Untreated) or 100 ng/ml CXCL8 for 30 minutes. Transfected cells were identified by staining with anti-Myc antibody. Overlay images are pseudocolored; red is CXCR2, green is Rab4, blue is Myc-RhoB Q63L. Bars, 10 μm. (C) Quantification of colocalization of CXCR2 with Rab4. Values shown are the mean ± s.e.m. *P<0.0005, significant differences (Student’s t-test) of cells transfected with Myc-RhoB Q63L versus vector-transfected cells. (D,E) CXCR2 and MPR staining in cells transfected with empty vector (D) or Myc-RhoB Q63L (E) and stimulated with vehicle (Untreated) or 100 ng/ml CXCL8 for 30 minutes. Transfected cells were identified by staining with anti-Myc antibody. Overlay images are pseudocolored; red is CXCR2, green is MPR, blue is Myc-RhoB Q63L. Bars, 10 μm. (F) Quantification of colocalization of CXCR2 with MPR. Values shown are the mean ± s.e.m. *P<0.0005, significant differences (Student’s t-test) of cells transfected with Myc-RhoB Q63L versus vector-transfected cells. Quantification of the percentage of CXCR2 colocalized with Rab4 or MPR in 20 fields was performed using the MetaMorph Imaging system (Universal Imaging). Images were processed using Photoshop (Adobe Systems). Data shown are representative from three separate experiments.
An alternative but hypothetical pathway for recycling to the plasma membrane might involve the trans-Golgi network. The cation-dependent mannose-6-phosphate receptor (MPR) delivers acid hydrolases from the trans-Golgi network to the late endosomal system. The receptor binds its cargo and traffics from the Golgi complex to the endosome and is then recycled back after the release of the hydrolases into late endosomes (reviewed in Ghosh et al., 2003; Hille-Rehfeld, 1995; Le Borgne and Hoflack, 1998). We examined whether CXCR2 colocalized with this receptor in cells expressing Myc-RhoB Q63L. In vector-transfected cells CXCR2 only minimally colocalizes with MPR after 30 minutes of CXCL8 stimulation (13.2±7.8% colocalization) (Fig. 9D,F). Surprisingly, CXCR2 significantly colocalized with MPR in cells expressing Myc-RhoB Q63L after 30 minutes of CXCL8 stimulation (74.1±11.1% colocalization, P<0.0005) (Fig. 9E,F). Therefore, it is possible that in cells transfected with Myc-RhoB Q63L CXCR2 enters the Golgi network from the sorting endosome and returns to the plasma membrane from the trans-Golgi network. These data suggest that CXCR2 may use alternative recycling pathways when it is unable to enter the Rab11a perinuclear recycling pathway in cells expressing Myc-RhoB Q63L.
Discussion
Early studies revealed that RhoB regulates the transport of the EGF receptor from late endosomes to the lysosome (Mellor et al., 1998). Because RhoB interacts with a number of proteins that are not isoform-specific, the function of RhoB has been difficult to establish. One possibility is that RhoB regulates signaling events by recruiting effectors to the endosomal compartments. For example, protein kinase C (PKC)-related kinase 1 (PRK1) colocalizes with RhoB on endosomal compartments and expression of the kinase-dead mutant PRK1 antagonizes the effect of RhoB on EGF receptor trafficking (Gampel et al., 1999; Mellor et al., 1998). Recent work identified RhoB-positive endosomes as a source of new actin polymerization through the action of Scar1 (WAVE1) in a Src-dependent manner (Sandilands et al., 2004). We have shown here that RhoB plays a role in the intracellular CXCR2 sorting decision and that interference with RhoB function alters CXCR2-mediated chemotaxis.
The RhoB T19N and Myc-RhoB Q63L mutants and also RhoB siRNA severely impair CXCR2-mediated chemotaxis. Although the membranes were coated with collagen IV for these experiments, it does not appear that differences in invasion contribute significantly to the inhibition. We did not observe any difference in the migration of vector-transduced cells and cells transduced with Myc-RhoB T19N in the absence of CXCL8, suggesting that there is no difference in the ligand-independent invasiveness of these two cell lines. Although, when Myc-RhoB Q63L is expressed, there is a decrease in the number of migrated cells in the absence of CXCL8, this would not account for the significant inhibition of CXCL8-mediated chemotaxis. When either mutant was expressed, we did not observe any significant differences in the activation of MAPK or PI 3-kinase signaling pathways over a 30-minute time course, as measured by phosphorylation of ERK1/2 and Akt, respectively. However, it is possible that localized signaling is affected when these mutants are expressed.
Expression of the dominant-negative (T19N) RhoB mutant results in accumulation of CXCR2 in the perinuclear recycling compartment and in enhanced recycling to the plasma membrane after long-term ligand stimulation (Fig. 10). Also, CXCR2 degradation is impaired when this mutant is expressed. These results suggest that RhoB GTPase activity is required for CXCR2 entry to the lysosome after long-term ligand stimulation. However, this accumulation of CXCR2 in the perinuclear recycling compartment and the failure to enter the lysosome and degrade are not a result of the inability of CXCR2 to exit the recycling compartment and return to the cell surface, because 125I-CXCL8-binding studies reveal that the receptor does return to the plasma membrane and bind ligand. We hypothesize that the accumulation of CXCR2 in the Rab11a compartment is a result of a ‘system overload’ because virtually all CXCR2 in the cells is passing through this compartment to return to the plasma membrane. CXCR2 is probably rapidly re-internalized upon return to the plasma membrane, reducing the time it resides at the plasma membrane. This may contribute to the inability to visualize the receptor at the plasma membrane by microscopy. Moreover, there are no differences in EGFP-Rab11a endosome motility, so differences in the rate of movement of the endosomes cannot account for the failure to enter the lysosome. Thus, it appears that expression of Myc-RhoB T19N impairs the ability of CXCR2 to enter the lysosome and causes prolonged CXCR2 recycling.
Fig. 10.

Schematic representation of the effects the T19N and Q63L RhoB mutants have on CXCR2 trafficking. After 30 minutes of CXCL8 stimulation CXCR2 traffics to the Rab11a perinuclear recycling compartment and recycles back to the plasma membrane. CXCR2 traffics to the lysosome and is degraded after 3 hours of CXCL8 stimulation. Expression of T19N RhoB mutant results in constitutive recycling of CXCR2 through the Rab11a perinuclear recycling compartment after 3 hours of CXCL8 stimulation (blue arrows). Expression of the Q63L RhoB mutant results in the inability of CXCR2 to enter the Rab11a perinuclear recycling compartment after 30 minutes of CXCL8 stimulation and leads to CXCR2 recycling to the plasma membrane by alternative pathways (red arrows).
Because expression of the RhoB T19N mutant results in decreased degradation and increased recycling of CXCR2, we expected that chemotaxis might be enhanced in cells expressing this mutant. Surprisingly, the opposite effect was observed, and chemotaxis was impaired in cells expressing RhoB T19N. Although CXCR2 is efficiently recycled in these cells, the recycled receptor may not be functional upon its return to the plasma membrane. For example, the dephosphorylation of the receptor is thought to be necessary for subsequent resensitization of CXCR2. Prior studies have shown that, when the CCR5 receptor is stimulated with an N-terminally modified CCL5 ligand, the receptor fails to efficiently respond to subsequent ligand challenge (Mack et al., 1998; Proudfoot et al., 1999). Further investigation revealed that, when stimulated with this modified ligand, CCR5 is not dephosphorylated and therefore has only a very brief period of residency at the plasma membrane before being re-internalized (Signoret et al., 2000). In addition, studies examining the interaction of protein phosphatase 2A (PP2A) and CXCR2 demonstrated that inhibition of PP2A activity by treatment with okadaic acid impairs CXCR2-mediated chemotaxis and Ca2+ mobilization in response to CXCL8 (Fan et al., 2001). These studies suggest that receptor dephosphorylation may not be required for recycling but for establishing a functional receptor back at the plasma membrane. Interestingly, a novel interaction between RhoB and the catalytic subunit of PP2A was recently identified, further suggesting a potential link between dephosphorylation and RhoB (Lee et al., 2007). It is not clear where in the endosomal trafficking pathway CXCR2 is dephosphorylated. In the future it will be of interest to examine whether CXCR2 dephosphorylation is affected in cells expressing RhoB T19N.
Treatment of cells with the actin-disrupting drugs cytochalasin D and latrunculin B resulted in a similar accumulation of CXCR2 in the Rab11a compartment as the RhoB T19N mutant. Since RhoB coordinates actin polymerization on endosomes (Fernandez-Borja et al., 2005; Sandilands et al., 2004), we suspected that RhoB regulates the motility of the Rab11a-positive endosomes in an actin-dependent manner. To explore this possibility we examined the motility of EGFP-Rab11a-positive endosomes using time-lapse confocal microscopy. The overall velocity of these endosomes and maximum distance traveled did not significantly differ between the vector-transfected cells and cells transfected with RhoB T19N. These results indicate that Rab11a-positive endosomes are able to exit the perinuclear region and return to the plasma membrane when RhoB T19N is expressed.
It is not clear whether the RhoB T19N mutant actually promotes trafficking of CXCR2 to the Rab11a compartment or merely inhibits the ability of the receptor to enter the lysosome for degradation, which results in receptor recycling by default. Moreover, it is unclear whether the RhoB Q63L mutant inhibits entry into the perinuclear recycling compartment or actually promotes accumulation in the sorting endosome and colocalization with Rab4, mannose-6-phosphate receptor and Rab7. Similarly, expression of activated RhoB Q63L enhances localization of rhophilin-2 to late endosomes but not to the lysosome (Steuve et al., 2006). Studies using the EGF receptor combined with our results suggest a broad role for RhoB in receptor sorting, which is not limited to a specific sorting decision. Alternatively, RhoB may play a variant role in the trafficking of receptors, such as CXCR2, that can be differentially sorted to the recycling endosome or the lysosome compared with those receptors, such as the EGF receptor, that are predominantly sorted to the lysosome. RhoB can recruit proteins that may be involved in intracellular trafficking to endosomal membranes such as Dia1 (Fernandez-Borja et al., 2005) and PRK1 (Mellor et al., 1998). The recruitment of different effectors to RhoB may mediate the various responses elicited by RhoB.
Interestingly, the RhoB Q63L mutant impairs the ability of CXCR2 to enter the perinuclear recycling compartment but the receptor is still able to recycle back to the plasma membrane (Fig. 10). The degradation of CXCR2 and colocalization of CXCR2 with lysosomal markers is also impaired when the Q63L mutant is expressed. This finding implicates not only the ability of RhoB to exchange GDP for GTP but also the ability of RhoB to hydrolyze GTP for proper function. It appears that GDP-bound RhoB specifies sorting to the Rab11a perinuclear recycling compartment, whereas GTP-bound RhoB and subsequent GTP hydrolysis is necessary for lysosomal sorting.
The colocalization of CXCR2 with Rab4 and the mannose-6-phosphate receptor indicate that CXCR2 enters the sorting compartment, and recycles back to the plasma membrane through alternative recycling pathways. It is interesting that CXCR2 may enter the trans-Golgi network and recycle back to the plasma membrane through this compartment. Our data indicate that CXCR2 recycling is a default pathway, whereas degradation is largely mediated through the activation of CXCL8 or RhoB.
In summary, the dominant-negative (T19N) RhoB mutant, the GTPase-deficient activated (Q63L) RhoB mutant and siRNA directed against RhoB all impair CXCR2-mediated chemotaxis. This impairment is accompanied by the failure of CXCR2 to traffic appropriately. Normal activity of RhoB is essential for CXCR2 degradation in the lysosome and recycling through the perinuclear Rab11a-positive compartment. We therefore propose that RhoB plays a role in the CXCR2 sorting decision. The ability of the cell to differentially sort chemokine receptors is crucial for the chemotactic response. These results establish for the first time that RhoB plays a role in the differential sorting of a chemokine receptor.
Materials and Methods
Materials and antibodies
Anti-CXCR2 affinity-purified polyclonal antibody was generated in our laboratory and described previously (Mueller et al., 1994). Anti-CXCR2, anti-CD63 monoclonal and anti-Actin rabbit polyclonal antibodies were purchased from Santa Cruz, Biotechnology, Inc. (Santa Cruz, CA). Anti-RhoB rabbit polyclonal antibody was acquired from Bethyl Laboratories, Inc. (Montgomery, TX). Anti-EEA-1 and Anti-LAMP-1 (CD107a) monoclonal antibodies were obtained from Becton Dickinson Biosciences (Franklin Lakes, NJ). Anti-Rab11a polyclonal antibody was used for immunofluorescence staining. Anti-Calreticulin polyclonal antibody and anti-Rab4 rabbit polyclonal antibody were purchased from Abcam (Cambridge, MA). Anti-Golgin-97 and anti-CD71 monoclonal antibodies, and Oregon Green 488 phalloidin were obtained from Invitrogen (Carlsbad, CA). Anti-Na+/K+ ATPase monoclonal antibody was purchased from Upstate (α-1, clone 464.6, Lake Placid, NY). Anti-mannose-6-phosphate polyclonal antibody was a generous gift from Lisa Matovcik and has been described previously (Goldenring et al., 1999). Species-specific Cy2-conjugated (donkey anti-rabbit), Cy5-conjugated (donkey anti-mouse), and Cy3-conjugated (donkey anti-goat) secondary antibodies were acquired from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). Anti-β-tubulin monoclonal antibody was purchased from Sigma-Aldrich (St Louis, MO). 125I-CXCL8 was purchased from PerkinElmer Life Sciences (Boston, MA).
Plasmids
The human CXCR2 plasmid has been constructed and described previously (Mueller et al., 1997). The human Myc-tagged wild-type, T19N and Q63L RhoB constructs were a generous gift from Harry Mellor (University of Bristol, Bristol, UK). The EGFP-Rab7 construct was described previously (Fan et al., 2003) and was a gift from Angela Wandinger-Ness (University of New Mexico School of Medicine, Albuquerque, NM). The EGFP-Rab11a construct has been described previously (Lapierre et al., 2001).
Cell culture and transfection
Human embryonic kidney (HEK) 293 cells were cultured in DMEM (Dulbecco’s modified Eagle’s medium) supplemented with penicillin (50 units/ml)/streptomycin (50 μg/ml), 3 mM glutamine, 10% heat-inactivated fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA) at 37°C, 5% CO2 and transfected with human CXCR2 plasmid using Fugene6 transfection reagent following the manufacturer’s protocol (Roche Applied Science, Indianapolis, IN). Neomycin-resistant cells were selected and surface expression of CXCR2 was confirmed using FACS analysis. Cells were transfected with Fugene6 for transient transfections and experiments were performed after 48 hours. All experiments conducted using Myc-tagged RhoB mutants and EGFP-Rab7 or –Rab11a were transiently transfected into cells stably expressing hCXCR2.
RhoB RNA interference
Pre-designed Cy-3-labeled siRNA oligomers containing 21 nucleotides were purchased from Ambion (Austin, TX). siRNA identification numbers 42060 or 120362 were used to specifically target human RhoB with sequences 5′-GCCUACGACUACCUCGAGUTT-3′ and 5′-GCAUGAACAGGACUUGACCTT-3′, respectively. Nonspecific Cy-3-labeled siRNA (no. 4621) was also purchased from Ambion and used as a negative control in these studies. Transfections were performed using Oligofectamine reagent (Invitrogen, Carlsbad, CA). Experiments were performed 48 hours after transfection.
Isolation of GST-TRBD
GST-TRBD fusion protein was isolated from E. coli as previously described (Ren and Schwartz, 2000). Briefly, cells were grown in LB medium with ampicillin and protein expression was induced with 0.5 mM isopropyl b-D-thiogalactopyranoside (IPTG) for 2 hours at 37°C. Cells were harvested, resuspended in 50 mM Tris-HCl pH 7.4, 0.5% Triton X-100, 150 mM NaCl, 5 mM MgCl2 containing bacterial protease inhibitor cocktail (Sigma-Aldrich), disrupted by sonication and debris was removed by centrifugation. GST-TRBD protein was purified by incubating lysates with Glutathione-agarose (Sigma-Aldrich).
RhoB activation assay
GST-TRBD expression construct and pull-down assays have been previously described (Ren and Schwartz, 2000) and were adapted for RhoB (Gampel and Mellor, 2002). Approximately 1.5×107 HEK293 cells stably expressing CXCR2 were transiently transfected with Myc-RhoB WT or Myc-RhoB Q63L 48 hours prior to experiment. Cells were serum-starved overnight, stimulated with vehicle (0.1% BSA in PBS) for 60 minutes, 100 ng/ml EGF for 60 minutes or 100 ng/ml CXCL8 for 5 minutes, 30 minutes or 60 minutes, and lysed in 50 mM Tris-HCl pH 7.4, 1% Triton X-100, 0.1% SDS, 500 mM NaCl, 10 mM MgCl2 containing mammalian protease inhibitor cocktail and phosphatase inhibitor cocktails I and II (Sigma-Aldrich). Lysates were cleared by centrifugation, and an aliquot was removed from each and used as total RhoB for western blot. GST-TRBD agarose beads were incubated with lysates at 4°C and washed three times with 50 mM Tris-HCl pH 7.4, 1% Triton X-100, 150 mM NaCl, 10 mM MgCl2.
Immunofluorescence and confocal microscopy
Cells were grown on glass coverslips coated with 0.1 mg/ml poly-L-lysine (Sigma-Aldrich) and transfected with indicated constructs. Cells were serum-starved for 4 hours and stimulated with vehicle (0.1% BSA in PBS) or 100 ng/ml CXCL8 at 37°C for indicated times. Cells were fixed in 4% paraformaldehyde for 10 minutes, permeabilized in 0.2% Triton X-100 in PBS for 5 minutes, blocked in 10% normal donkey serum for 30 minutes (Jackson ImmunoResearch Laboratories), and incubated with primary antibodies for 2 hours at room temperature. After washing three times with 0.1% Tween 20 in PBS, the coverslips were incubated with fluorescence-conjugated secondary antibodies for 1 hour. After three washes with 0.1% Tween 20 in PBS, coverslips were mounted with ProLong Gold antifade reagent (Invitrogen). Confocal images were acquired using a LSM-510 Meta laser scanning microscope (Carl Zeiss, Thornwood, NY) with a 63× 1.3 numerical aperture oil immersion lens and images were processed by Photoshop software (Adobe Systems, San Jose, CA).
Quantitation of colocalization in confocal images
Colocalization of CXCR2 with endosomal markers was quantified using Metamorph Imaging System software package (Molecular Devices Corporation, Sunnyvale, CA). Threshold levels for all images were kept consistent among vector and mutant transduced cells. At least twenty fields were quantified for each time point. The percent colocalization is indicative of the area of CXCR2-stained fluorescent pixels overlapping that of endocytic markers.
Measurement of endosome motility
Velocity and distance traveled by individual endosomes were measured using Metamorph Imaging System software package (Molecular Devices Corporation, Sunnyvale, CA). A series of 60 images was taken at 1-second intervals for cells stimulated with vehicle or CXCL8 for 30 minutes. Because it is difficult to distinguish directional linear movement with random movement of endosomes, the maximum distance traveled from point of origin (frame 1 of the time-lapse series) was also measured. The average velocities and distances were measured for at least 30 individual endosomes in at least ten randomly selected cells.
Chemotaxis assay
A 96-well chemotaxis chamber (Neuroprobe, Gaithersburg, MD) was used for chemotaxis assays as described previously (Fan et al., 2004). The number of cells were counted in 20 microscopic fields (20× objective).
CXCR2 degradation by western blot analysis
Cells were transfected 48 hours prior to experiments with vector, Myc-RhoB WT, T19N or Q63L. Cells were pretreated for 30 minutes at 37°C with 20 μg/ml cycloheximide to inhibit new receptor synthesis, stimulated with vehicle (0.1% BSA in PBS) or 100 ng/ml CXCL8 for 30 minutes, 180 minutes or 360 minutes, and lysed in 50 mM Tris-Cl pH 7.5, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.5% sodium deoxylcholate. Lysates were subjected to SDS-PAGE, and CXCR2 and actin were detected by western blot analysis. The densities of the CXCR2 and actin protein bands were measured using Odyssey 2.1 software package (Li-COR Biosciences, Lincoln, NE). The densities of the CXCR2 bands were normalized to total protein by dividing them by the densities of the actin bands. Values of remaining receptor (in percent) were calculated by dividing the normalized densities of the treated samples by the normalized density of the vehicle-treated samples multiplied by 100.
CXCR2 resurfacing to the plasma membrane analyzed by modified 125I-CXCL8 binding assay
Cells were transfected 48 hours prior to experiments with vector, Myc-RhoB WT, T19N or Q63L. Cells were pretreated for 30 minutes at 37°C with 20 μg/ml cycloheximide to inhibit new receptor synthesis, stimulated with vehicle (0.1% BSA in PBS) or 100 ng/ml CXCL8 for 30 minutes, washed twice with 10% FBS in DMEM, and CXCR2 surface expression was recovered by placing cells in 0% FBS in DMEM containing 20 μg/ml cycloheximide for 0 minutes, 30 minutes or 60 minutes at 37°C. Cells were washed three times with ice-cold binding buffer (1% BSA in DMEM). Then, 0.1 nM 125I-CXCL8 prepared in binding buffer containing 20 μg/ml cycloheximide was added and incubated for 2 hours on ice. Following binding, cells were washed three times with ice-cold binding buffer and lysed in 1% SDS in NaOH (0.1 M), collected, and the level of radioactivity quantified by γ-counter. Experiments were performed in triplicate and total binding (in percent) was calculated by dividing the average radioactive counts for each recovery time by the radioactive counts for the vehicle treated cells (total binding).
Preparation of Triton-soluble and -insoluble fractions
Fractions soluble and insoluble in Triton X-100 were prepared as described previously (Minamide et al., 1997). Briefly, HEK293 cells stably expressing CXCR2 and transiently transfected with empty vector, Myc-RhoB T19N or Myc-RhoB Q63L were washed four times with ice-cold PBS and lysed in 10 mM Tris-HCl pH 7.5, 2 mM MgCl2, 0.5 mM DTT, 2 mM EGTA, 1% Triton X-100 and 7.5% glycerol containing protease and phosphatase inhibitors. Soluble and insoluble fractions were prepared by centrifugation at 170,000 g for 20 minutes. Actin in each fraction was detected by western blot analysis.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We acknowledge Sam Wells, Dawn Kilkenny, and the Vanderbilt University Medical Center Cell Imaging Shared Resource with which confocal images were acquired and quantification was performed (National Institutes of Health grants DK-20593, DK-58404, HD-15052, DK-59637 and EY-08126). We thank Dayanidhi Raman and Paige Baugher for reading and editing this manuscript. This work was supported by grant CA-34590 (to A.R.), DK48370 (to J.G.), and DK070856 (to J.G.) from the National Institutes of Health, Career Scientist Award from the Department of Veterans Affairs (to A.R.), Multidisciplinary Basic Research Training in Cancer Grant T32CA09592, and Vanderbilt-Ingram Cancer Center Support grant CA68485.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/120/9/1559/DC1
References
- Barlic J, Khandaker MH, Mahon E, Andrews J, DeVries ME, Mitchell GB, Rahimpour R, Tan CM, Ferguson SS, Kelvin DJ. beta-arrestins regulate interleukin-8-induced CXCR1 internalization. J Biol Chem. 1999;274:16287–16294. doi: 10.1074/jbc.274.23.16287. [DOI] [PubMed] [Google Scholar]
- Colvin RA, Campanella GS, Sun J, Luster AD. Intracellular domains of CXCR3 that mediate CXCL9, CXCL10, and CXCL11 function. J Biol Chem. 2004;279:30219–30227. doi: 10.1074/jbc.M403595200. [DOI] [PubMed] [Google Scholar]
- Droese J, Mokros T, Hermosilla R, Schulein R, Lipp M, Hopken UE, Rehm A. HCMV-encoded chemokine receptor US28 employs multiple routes for internalization. Biochem Biophys Res Commun. 2004;322:42–49. doi: 10.1016/j.bbrc.2004.07.076. [DOI] [PubMed] [Google Scholar]
- Fan GH, Yang W, Sai J, Richmond A. Phosphorylation-independent association of CXCR2 with the protein phosphatase 2A core enzyme. J Biol Chem. 2001;276:16960–16968. doi: 10.1074/jbc.M009292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan GH, Lapierre LA, Goldenring JR, Richmond A. Differential regulation of CXCR2 trafficking by Rab GTPases. Blood. 2003;101:2115–2124. doi: 10.1182/blood-2002-07-1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan GH, Lapierre LA, Goldenring JR, Sai J, Richmond A. Rab11-family interacting protein 2 and myosin Vb are required for CXCR2 recycling and receptor-mediated chemotaxis. Mol Biol Cell. 2004;15:2456–2469. doi: 10.1091/mbc.E03-09-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- Ferguson SS, Downey WE, 3rd, Colapietro AM, Barak LS, Menard L, Caron MG. Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science. 1996;271:363–366. doi: 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- Ferguson SS, Zhang J, Barak LS, Caron MG. Molecular mechanisms of G protein-coupled receptor desensitization and resensitization. Life Sci. 1998;62:1561–1565. doi: 10.1016/s0024-3205(98)00107-6. [DOI] [PubMed] [Google Scholar]
- Fernandez-Borja M, Janssen L, Verwoerd D, Hordijk P, Neefjes J. RhoB regulates endosome transport by promoting actin assembly on endosomal membranes through Dia1. J Cell Sci. 2005;118:2661–2670. doi: 10.1242/jcs.02384. [DOI] [PubMed] [Google Scholar]
- Freedman NJ, Lefkowitz RJ. Desensitization of G protein-coupled receptors. Recent Prog Horm Res. 1996;51:319–351. discussion 352–353. [PubMed] [Google Scholar]
- Gampel A, Mellor H. Small interfering RNAs as a tool to assign Rho GTPase exchange-factor function in vivo. Biochem J. 2002;366:393–398. doi: 10.1042/BJ20020844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gampel A, Parker PJ, Mellor H. Regulation of epidermal growth factor receptor traffic by the small GTPase rhoB. Curr Biol. 1999;9:955–958. doi: 10.1016/s0960-9822(99)80422-9. [DOI] [PubMed] [Google Scholar]
- Ghosh P, Dahms NM, Kornfeld S. Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol. 2003;4:202–212. doi: 10.1038/nrm1050. [DOI] [PubMed] [Google Scholar]
- Goldenring JR, Ray GS, Lee JR. Rab11 in dysplasia of Barrett’s epithelia. Yale J Biol Med. 1999;72:113–120. [PMC free article] [PubMed] [Google Scholar]
- Hille-Rehfeld A. Mannose 6-phosphate receptors in sorting and transport of lysosomal enzymes. Biochim Biophys Acta. 1995;1241:177–194. doi: 10.1016/0304-4157(95)00004-b. [DOI] [PubMed] [Google Scholar]
- Jimenez-Sainz MC, Fast B, Mayor F, Jr, Aragay AM. Signaling pathways for monocyte chemoattractant protein 1-mediated extracellular signal-regulated kinase activation. Mol Pharmacol. 2003;64:773–782. doi: 10.1124/mol.64.3.773. [DOI] [PubMed] [Google Scholar]
- Krupnick JG, Benovic JL. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol. 1998;38:289–319. doi: 10.1146/annurev.pharmtox.38.1.289. [DOI] [PubMed] [Google Scholar]
- Lapierre LA, Kumar R, Hales CM, Navarre J, Bhartur SG, Burnette JO, Provance DW, Jr, Mercer JA, Bahler M, Goldenring JR. Myosin vb is associated with plasma membrane recycling systems. Mol Biol Cell. 2001;12:1843–1857. doi: 10.1091/mbc.12.6.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Borgne R, Hoflack B. Protein transport from the secretory to the endocytic pathway in mammalian cells. Biochim Biophys Acta. 1998;1404:195–209. doi: 10.1016/s0167-4889(98)00057-3. [DOI] [PubMed] [Google Scholar]
- Lee WJ, Kim DU, Lee MY, Choi KY. Identification of proteins interacting with the catalytic subunit of PP2A by proteomics. Proteomics. 2007;7:206–214. doi: 10.1002/pmic.200600480. [DOI] [PubMed] [Google Scholar]
- Mack M, Luckow B, Nelson PJ, Cihak J, Simmons G, Clapham PR, Signoret N, Marsh M, Stangassinger M, Borlat F, et al. Aminooxypentane-RANTES induces CCR5 internalization but inhibits recycling: a novel inhibitory mechanism of HIV infectivity. J Exp Med. 1998;187:1215–1224. doi: 10.1084/jem.187.8.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor H, Flynn P, Nobes CD, Hall A, Parker PJ. PRK1 is targeted to endosomes by the small GTPase, RhoB. J Biol Chem. 1998;273:4811–4814. doi: 10.1074/jbc.273.9.4811. [DOI] [PubMed] [Google Scholar]
- Michaelson D, Silletti J, Murphy G, D’Eustachio P, Rush M, Philips MR. Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J Cell Biol. 2001;152:111–126. doi: 10.1083/jcb.152.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamide LS, Painter WB, Schevzov G, Gunning P, Bamburg JR. Differential regulation of actin depolymerizing factor and cofilin in response to alterations in the actin monomer pool. J Biol Chem. 1997;272:8303–8309. doi: 10.1074/jbc.272.13.8303. [DOI] [PubMed] [Google Scholar]
- Mueller SG, Schraw WP, Richmond A. Melanoma growth stimulatory activity enhances the phosphorylation of the class II interleukin-8 receptor in non-hematopoietic cells. J Biol Chem. 1994;269:1973–1980. [PubMed] [Google Scholar]
- Mueller SG, White JR, Schraw WP, Lam V, Richmond A. Ligand-induced desensitization of the human CXC chemokine receptor-2 is modulated by multiple serine residues in the carboxyl-terminal domain of the receptor. J Biol Chem. 1997;272:8207–8214. doi: 10.1074/jbc.272.13.8207. [DOI] [PubMed] [Google Scholar]
- Neel NF, Schutyser E, Sai J, Fan GH, Richmond A. Chemokine receptor internalization and intracellular trafficking. Cytokine Growth Factor Rev. 2005;16:637–658. doi: 10.1016/j.cytogfr.2005.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini MJ, Parent JL, Mundell SJ, Benovic JL, Marchese A. Trafficking of the HIV coreceptor CXCR4. Role of arrestins and identification of residues in the c-terminal tail that mediate receptor internalization. J Biol Chem. 1999;274:31076–31086. doi: 10.1074/jbc.274.43.31076. [DOI] [PubMed] [Google Scholar]
- Pereira-Leal JB, Seabra MC. The mammalian Rab family of small GTPases: definition of family and subfamily sequence motifs suggests a mechanism for functional specificity in the Ras superfamily. J Mol Biol. 2000;301:1077–1087. doi: 10.1006/jmbi.2000.4010. [DOI] [PubMed] [Google Scholar]
- Proudfoot AE, Buser R, Borlat F, Alouani S, Soler D, Offord RE, Schroder JM, Power CA, Wells TN. Amino-terminally modified RANTES analogues demonstrate differential effects on RANTES receptors. J Biol Chem. 1999;274:32478–32485. doi: 10.1074/jbc.274.45.32478. [DOI] [PubMed] [Google Scholar]
- Ren XD, Schwartz MA. Determination of GTP loading on Rho. Meth Enzymol. 2000;325:264–272. doi: 10.1016/s0076-6879(00)25448-7. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Rho proteins: linking signaling with membrane trafficking. Traffic. 2001;2:303–310. doi: 10.1034/j.1600-0854.2001.002005303.x. [DOI] [PubMed] [Google Scholar]
- Rojas R, Ruiz WG, Wang E, Kinlough CL, Poland PA, Hughey RP, Dunn KW, Apodaca G. RhoB-dependent modulation of early endocytic traffic in Madin-Darby canine kidney cells. J Biol Chem. 2004 doi: 10.1074/jbc.M408387200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandilands E, Cans C, Fincham VJ, Brunton VG, Mellor H, Prendergast GC, Norman JC, Superti-Furga G, Frame MC. RhoB and actin polymerization coordinate Src activation with endosome-mediated delivery to the membrane. Dev Cell. 2004;7:855–869. doi: 10.1016/j.devcel.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Sheff DR, Daro EA, Hull M, Mellman I. The receptor recycling pathway contains two distinct populations of early endosomes with different sorting functions. J Cell Biol. 1999;145:123–139. doi: 10.1083/jcb.145.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signoret N, Pelchen-Matthews A, Mack M, Proudfoot AE, Marsh M. Endocytosis and recycling of the HIV coreceptor CCR5. J Cell Biol. 2000;151:1281–1294. doi: 10.1083/jcb.151.6.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signoret N, Hewlett L, Wavre S, Pelchen-Matthews A, Oppermann M, Marsh M. Agonist-induced endocytosis of CC chemokine receptor 5 is clathrin dependent. Mol Biol Cell. 2005;16:902–917. doi: 10.1091/mbc.E04-08-0687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnichsen B, De Renzis S, Nielsen E, Rietdorf J, Zerial M. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J Cell Biol. 2000;149:901–914. doi: 10.1083/jcb.149.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steuve S, Devosse T, Lauwers E, Vanderwinden JM, Andre B, Courtoy PJ, Pirson I. Rhophilin-2 is targeted to late-endosomal structures of the vesicular machinery in the presence of activated RhoB. Exp Cell Res. 2006;312:3981–3989. doi: 10.1016/j.yexcr.2006.08.028. [DOI] [PubMed] [Google Scholar]
- van der Bliek AM, Redelmeier TE, Damke H, Tisdale EJ, Meyerowitz EM, Schmid SL. Mutations in human dynamin block an intermediate stage in coated vesicle formation. J Cell Biol. 1993;122:553–563. doi: 10.1083/jcb.122.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan S, Rose JJ, Lodge R, Murphy PM, Foley JF. Distinct mechanisms of agonist-induced endocytosis for human chemokine receptors CCR5 and CXCR4. Mol Biol Cell. 2003;14:3305–3324. doi: 10.1091/mbc.E02-11-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila-Coro AJ, Mellado M, Martin de Ana A, Martinez AC, Rodriguez-Frade JM. Characterization of RANTES- and aminooxypentane-RANTES-triggered desensitization signals reveals differences in recruitment of the G protein-coupled receptor complex. J Immunol. 1999;163:3037–3044. [PubMed] [Google Scholar]
- Weber M, Blair E, Simpson CV, O’Hara M, Blackburn PE, Rot A, Graham GJ, Nibbs RJ. The chemokine receptor D6 constitutively traffics to and from the cell surface to internalize and degrade chemokines. Mol Biol Cell. 2004;15:2492–2508. doi: 10.1091/mbc.E03-09-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennerberg K, Der CJ. Rho-family GTPases: it’s not only Rac and Rho (and I like it) J Cell Sci. 2004;117:1301–1312. doi: 10.1242/jcs.01118. [DOI] [PubMed] [Google Scholar]
- Wheeler AP, Ridley AJ. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res. 2004;301:43–49. doi: 10.1016/j.yexcr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Wherlock M, Gampel A, Futter C, Mellor H. Farnesyltransferase inhibitors disrupt EGF receptor traffic through modulation of the RhoB GTPase. J Cell Sci. 2004;117:3221–3231. doi: 10.1242/jcs.01193. [DOI] [PubMed] [Google Scholar]
- Yang W, Wang D, Richmond A. Role of clathrin-mediated endocytosis in CXCR2 sequestration, resensitization, and signal transduction. J Biol Chem. 1999;274:11328–11333. doi: 10.1074/jbc.274.16.11328. [DOI] [PubMed] [Google Scholar]
- Zaslaver A, Feniger-Barish R, Ben-Baruch A. Actin filaments are involved in the regulation of trafficking of two closely related chemokine receptors, CXCR1 and CXCR2. J Immunol. 2001;166:1272–1284. doi: 10.4049/jimmunol.166.2.1272. [DOI] [PubMed] [Google Scholar]
- Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2:107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.