

Mutations of a single gene, fragile x mental retardation 1 (FMR1), give rise to a family of disorders occurring throughout the entire life span, including the most common heritable form of intellectual disability, fragile X syndrome, and premature menopause (primary ovarian insufficiency). Moreover, mutations of FMR1 are the cause of one of the most common single-gene, late-onset neurodegenerative disorders, fragile X–associated tremor/ataxia syndrome (FXTAS). Some clinicians might assume they will rarely if ever encounter one of these disorders; that assumption would be both false and unwise.
Because of the risks associated with these disorders, newborn screening is being considered. The recent development of an accurate and inexpensive blood spot screening test capable of identifying both premutation and full mutation alleles1 is facilitating at least 2 large-scale newborn screening studies to move forward in the United States. New-born screening for expanded alleles of the FMR1 gene will lead to earlier intervention for individuals with developmental delays in both full mutation and premutation categories; genetic counseling before the birth of a second child with fragile X syndrome; elimination of the prolonged diagnostic odyssey families experience prior to the correct diagnosis; and identification of a range of fragile X–associated disorders among adults and across the extended family, thus permitting earlier treatments for many individuals.2
FMR1 belongs to a growing family of genes that lead to disease through trinucleotide repeat expansions; DMPK (myotonic dystrophy) and HD (Huntington disease) are 2 examples. For individuals with FMR1 (CGG repeat) expansions in the full mutation range (>200 CGG repeats), the gene is generally silenced, resulting in the absence of a protein (FMRP) important for synaptic function. More recently, it has become clear that clinical involvement also can occur in carriers of smaller, premutation alleles (55-200 CGG repeats), in which disease pathogenesis occurs by a completely different mechanism involving increased expression and RNA toxicity (Figure).
Figure. Expression of the FMR1 Gene and Associated Clinical Disorders.
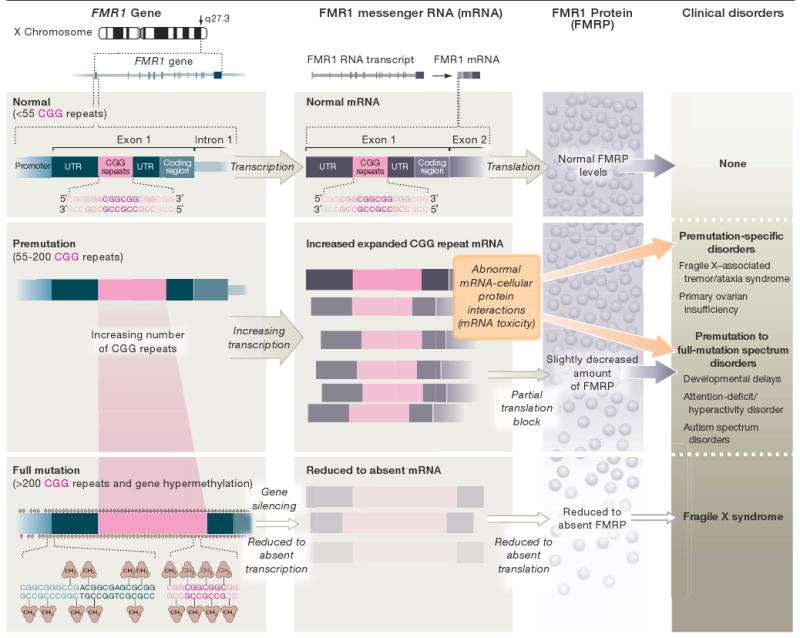
Expression of the fragile X mental retardation 1 (FMR1) gene depends on the length of the CGG repeat. Premutation alleles result in elevated mRNA levels (increased RNA synthesis). Within this CGG repeat range, the expanded CGG repeat in FMR1 mRNA partially blocks translation to an extent that depends on the size of the repeat. Full mutation alleles are generally hypermethylated and silenced, thus producing little or no mRNA or protein. The excess premutation mRNA is now believed to give rise to the premutation-specific disorders, fragile X–associated tremor/ataxia syndrome and primary ovarian insufficiency, and may also contribute to developmental delays, attention-deficit/hyperactivity disorder, or autism spectrum disorders in some children with the premutation. By contrast, fragile X syndrome is caused by the absence of the FMR1 mRNA and protein (FMRP). In the upper portion of the premutation CGG repeat range, mRNA toxicity and reduced FMRP levels may both contribute to clinical involvement. UTR indicates 5′ untranslated region.
Individuals who carry a full mutation form of the gene experience a broad range of intellectual and developmental disability, and clinical involvement can include psychiatric or emotional difficulties even in those with a normal or borderline IQ. Autistic behavior is seen in the majority of boys with fragile X syndrome and approximately 30% fulfill all Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) diagnostic criteria for autism.3 Autisticlike behavior also is common in girls with the full mutation; however, shyness and social anxiety are even more prevalent in girls. Although the majority of children with the premutation have normal cognitive abilities, evidence is emerging that they also may have developmental problems, particularly deficits in attention and social communication in boys.4
Premutation alleles are relatively common in the general population, occurring in approximately 1 in 130 to 250 women and 1 in 250 to 810 men.5 Premutation expansions give rise to enhanced production (≤8-fold) of the expanded CGG-repeat messenger RNA, resulting in a messenger RNA–dependent dysregulation of numerous proteins (eg, nuclear lamin A/C) and cellular stress response proteins (eg, αB-crystallin, Hsp27).6
An important clinical consequence of premutation-associated messenger RNA toxicity is the development of progressive neurological dysfunction in older adults, which includes peripheral neuropathy, cognitive decline or dementia, psychiatric problems, parkinsonism, tremor, and gait ataxia. Indeed, it was the clinical association between the neurodegenerative features in grandfathers of children with fragile X syndrome and excess gene activity that led to the messenger RNA toxicity hypothesis for FXTAS, which occurs in 40% of male carriers and a smaller portion of female carriers who have been ascertained through families with fragile X syndrome.7
For carriers who have not developed the core features of FXTAS, a variety of neurological and psychiatric problems may occur, including hypertension, orthostatic hypotension, neuropathy, migraines, anxiety, shyness or social deficits, and depression.8,9 These symptoms reflect the molecular and cellular changes that occur in neural cells in the central nervous system as well as in autonomic ganglia and nonneural cells outside the central nervous system.10 Because carriers of premutation alleles are common in the general population, clinical involvement associated with the premutation will likely contribute to clinical populations with other initial diagnoses,11 and will be comorbid with Alzheimer disease, Parkinson disease with Lewy bodies, and multiple sclerosis. Among younger adults, nearly one-fifth of female premutation carriers will experience primary ovarian insufficiency. Moreover, the premutation is the leading genetic cause of primary ovarian insufficiency, present in 7% to 12% of familial cases and in 2% to 7% of sporadic cases.12 In addition, female carriers with FXTAS are at higher risk for hypothyroidism (50% of cases) and fibromyalgia (42% of cases) compared with the general population.8
It is time to seriously consider routine testing for fragile X in some patient groups, particularly in children with intellectual and developmental disabilities, developmental delays or autistic behaviors, adult women with features of early menopause, or older adults with gait ataxia, intention tremor, or both. It also is important to test those individuals with cognitive deficits or dementia, peripheral neuropathy, parkinsonism, atypical multiple sclerosis, and anxiety disorders, particularly if there is a family history of intellectual and developmental disability, autistic behavior, or both. Testing is important to diagnose both premutation and full-mutation disorders because knowledge of genetic status is critical for early intervention across the spectrum of disorders. Such interventions include genetic counseling for the entire family, direct behavioral and medical intervention for the developmentally disabled child, improved reproductive options for women who are at risk of developing primary ovarian insufficiency, treatment for adults with the premutation who are starting to show signs of FXTAS, and early treatment of anxiety or depression.13,14 Newborn screening will facilitate the early intervention process for the children and the genetic counseling, early identification, and treatment for many other family members.
One of the most exciting advances in the treatment of fragile X syndrome was the discovery that the absence of FMRP leads to enhanced long-term depression of synaptic function of glutamatergic neurons, and that inhibition of the metabotropic (mGluR5) glutamate receptor at least partially normalizes long-term depression.15 This discovery has led to the study of a number of mGluR5 antagonists that have demonstrated significant benefit in animal models of fragile X syndrome, leading in turn to the development of newer mGluR5 antagonists that are currently undergoing clinical trials in patients with fragile X syndrome. These trials, if successful, make the case of newborn screening more compelling and raise the prospect of future intervention during the developmental period to prevent long-term disability.
Acknowledgments
Funding/Support: This work was supported by National Institutes of Health grants HD036071 and HD02274; National Institute of Dental and Craniofacial Research grant DE19583; National Institute on Aging grants AG032119 and AG032115; and the Administration of Developmental Disabilities grant 90DD05969 for the Center of Excellence in Developmental Disabilities.
Role of the Sponsor: Our sponsors did not have a role in the design and conduct of our studies or in the preparation, review, or approval of the manuscript.
Footnotes
Financial Disclosures: Dr R. Hagerman reported serving as a consultant for Neuropharm, Seaside Therapeutics, Roche, and Novartis regarding targeted treatments in fragile X syndrome. Dr R. Hagerman also reported that Neuropharm has funded a treatment study.
References
- 1.Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn. 2008;10(1):43–49. doi: 10.2353/jmoldx.2008.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailey DB, Jr, Skinner D, Sparkman KL. Discovering fragile X syndrome: family experiences and perceptions. Pediatrics. 2003;111(2):407–416. doi: 10.1542/peds.111.2.407. [DOI] [PubMed] [Google Scholar]
- 3.Loesch DZ, Bui QM, Dissanayake C, et al. Molecular and cognitive predictors of the continuum of autistic behaviours in fragile X. Neurosci Biobehav Rev. 2007;31(3):315–326. doi: 10.1016/j.neubiorev.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farzin F, Perry H, Hessl D, et al. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006;27(2 suppl):S137–S144. doi: 10.1097/00004703-200604002-00012. [DOI] [PubMed] [Google Scholar]
- 5.Song FJ, Barton P, Sleightholme V, Yao GL, Fry-Smith A. Screening for fragile X syndrome: a literature review and modeling study. Health Technol Assess. 2003;7(16):1–106. doi: 10.3310/hta7160. [DOI] [PubMed] [Google Scholar]
- 6.Iwahashi CK, Yasui DH, An HJ, et al. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129(pt 1):256–271. doi: 10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]
- 7.Jacquemont S, Hagerman RJ, Leehey MA, et al. Penetrance of the fragile X–associated tremor/ataxia syndrome in a premutation carrier population. JAMA. 2004;291(4):460–469. doi: 10.1001/jama.291.4.460. [DOI] [PubMed] [Google Scholar]
- 8.Coffey SM, Cook K, Tartaglia N, et al. Expanded clinical phenotype of women with the FMR1 premutation. Am J Med Genet A. 2008;146(8):1009–1016. doi: 10.1002/ajmg.a.32060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts JE, Bailey DB, Jr, Mankowski J, et al. Mood and anxiety disorders in females with the FMR1 premutation. Am J Med Genet B Neuropsychiatr Genet. doi: 10.1002/ajmg.b.30786. published online ahead of print June 13, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Greco CM, Soontarapornchai K, Wirojanan J, Gould JE, Hagerman PJ, Hagerman RJ. Testicular and pituitary inclusion formation in fragile X associated tremor/ataxia syndrome. J Urol. 2007;177(4):1434–1437. doi: 10.1016/j.juro.2006.11.097. [DOI] [PubMed] [Google Scholar]
- 11.Hall DA, Berry-Kravis E, Jacquemont S, et al. Initial diagnoses given to persons with the fragile X-associated tremor/ataxia syndrome (FXTAS) Neurology. 2005;65(2):299–301. doi: 10.1212/01.wnl.0000168900.86323.9c. [DOI] [PubMed] [Google Scholar]
- 12.Sullivan AK, Marcus M, Epstein MP, et al. Association of FMR1 repeat size with ovarian dysfunction. Hum Reprod. 2005;20(2):402–412. doi: 10.1093/humrep/deh635. [DOI] [PubMed] [Google Scholar]
- 13.Hagerman RJ, Berry-Kravis E, Kaufmann WE, et al. Advances in the treatment of fragile X syndrome. Pediatrics. doi: 10.1542/peds.2008-0317. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hagerman RJ, Hall DA, Coffey S, et al. Treatment of fragile X-associated tremor ataxia syndrome (FXTAS) and related neurological problems. Clin Interv Aging. 2008;3(2):251–262. doi: 10.2147/cia.s1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bear MF. Therapeutic implications of the mGluR theory of fragile X mental retardation. Genes Brain Behav. 2005;4(6):393–398. doi: 10.1111/j.1601-183X.2005.00135.x. [DOI] [PubMed] [Google Scholar]