

Abstract
Computer-based docking screens are now widely used to discover new ligands for targets of known structure; in the last two years alone, the discovery of ligands for over 20 proteins have been reported. Recently, investigators have also turned to predicting new substrates and for enzymes of unknown function, taking docking in a wholly new direction. Increasingly, the hit-rates, the true- and the false-positives from the docking screens are being compared to those from empirical, high-throughput screens, revealing the strengths, weaknesses and complementarities of both techniques. The recent efflorescence of GPCR structures has made these quintessential drug targets available to structure-based approaches. Consistent with their “druggability”, the docking screens have returned high hit-rates and potent molecules. Finally, in the last several years, an approach almost exactly opposite to docking has also appeared; this pharmacological network approach begins not with the structure of the target but rather those of drug molecules and asks, given a pattern of chemistry in the ligands, what targets may a particular drug bind to? This method, which returns to an older, pharmacology logic, has been surprisingly successful in predicting new “off-targets” for established drugs.
Since the work of Goodford in the mid-1970’s [1], protein structures have held the promise of guiding the design of drugs. As is often true, the early potential of the field was largely unmet, and in the early 1990s there was a sense that “structure-based drug design” was not pragmatic. In the last decade, however, the use of structure-based computational design has made steady, if often unspectacular [2] technical progress, to the point where most pharmaceutical organizations have substantial groups devoted to its application. If one asks, “How many drugs have been discovered entirely by structure-based methods?”, the answer will be “none”, as these methods largely contribute only to the discovery and early optimization of leads for drug design. If one asks, conversely, “To the development of how many drugs have structure-based methods been critical?” then the answer will be close to ten. Whereas this number must seem small, it is put into perspective by the number of drugs that owe their origin to empirical high throughput screening (as of this writing, only two or three [3], though many more are now in trials or awaiting approval).
Perhaps the area where structure-based methods have had the most quantifiable impact is in computational screens for of large compound libraries, looking for new chemical matter that will bind to and modulate a protein of known structure. These “virtual” or “structure-based” screens typically use molecular docking programs to fit small organic molecules into protein structures, evaluating them for structural and chemical complementarity. Several million molecules may be docked into the structure of the target protein, and those that fit best, according to the docking scoring function, will be tested experimentally (Figure 1). Although these scoring functions retain substantial inaccuracies, the focus on commercially available molecules has made failure cheap—since one can always just purchase and test the next set of compounds—and so pragmatic. In the last two years alone, over twenty papers have appeared in which docking screens were used to predict a ligand which was then subsequently confirmed by experiment (Table 1). Most of these studies emerged from academic research groups; many others that have been conducted in industrial groups remain unpublished.
Figure 1.

Structure-based screens for novel ligands. Large libraries of commercially or otherwise available compounds are fit into target structures by a docking computer program. Each molecule is sampled in thousands to millions of possible configurations and scored based on structural complementarity to the target protein. Of the perhaps millions of molecules in the library, tens to hundreds of top-scoring hits are subsequently tested for activity in an experimental assay.
Table 1.
Docking predictions subsequently confirmed by experiments: 2007 to mid 2009.
| Target | Docking program | Lead inhibitor IC50 (µM) |
|---|---|---|
| AdoMetDC [4] | Glide | 12 |
| AHAS [5] | DOCK 4/AutoDock | 15.2 |
| Aldose reductase [6] | N/A | 0.53 |
| CDC25 phosphatase [7] | FRED/Surflex/LigandFit | 13 |
| DNA gyrase [8] | DOCK 5 | 50 |
| EphB4 [9] | DAIM-SEED-FFLD | 1.5 |
| FFAR1 [10] | Glide | 3.6 |
| Histamine H4 [11] | FlexX | 95.8 |
| Human Pregnane X [12] | Surflex | 0.049 |
| MCH-R1 [13] | ICM | 7.5 |
| Pim-1 kinase [14] | Glide | 0.091 |
| PNP [15] | GOLD | 18.9 |
| PPAR-γ [16] | Glide/IFD | 2.9 |
| Tm0936 [17] | DOCK 3.5 | 105 M−1S−1 (Kcat/ Km) |
| TRH-R1/TRH-R2 [18] | FlexX | 0.29 |
| β2-adrenergic receptor [19] | DOCK 3.5.54 | 0.009 |
| β-lactamase [20] | DOCK 3.5.54 | 140 |
| SHP2 [21] | DOCK | 100 |
| Al-2 quorum sensing [22] | DOCK 5 | 35 |
| Anthrax edema factor [23] | HINT/AutoDock | 1.7 |
| hPRMT1 [24] | GOLD | 12 |
Here we focus on research directions in structure-based screening that, even very recently, would have been hard to anticipate. We begin with efforts to combine virtual and high-throughput screens. These campaigns enable the discovery of false positives and false negatives for both techniques, and their combination reveals complementary strengths. We then turn to the use of structure-based screens against membrane bound receptors, the structures for which have only recently become available. Finally, we consider methods that turn the structure-based paradigm on its head, asking not what ligands might be discovered based on the structure of the targets, but rather what targets might be discovered from the identity of the ligands.
Virtual versus high-throughput screening
A stringent test of a docking screen is to compare it to a high-throughput screen (HTS), where every single molecule in a library is empirically tested, not just the top scoring fifty or several hundred suggested by docking. This could reveal not only docking false positives but also false negatives, which would otherwise always remain opaque, and whether docking hit rates were high enough to justify the focus on only a relatively small number of candidate ligands. In studies beginning in the early 2000’s, this turned out to be the case, with docking hit rates being 10 to 1000-fold higher than those returned by HTS against the same target [25–28]. Whereas these early studies were encouraging, the full set of docking predictions was not compared to the HTS results, and sometimes different compound libraries were used.
In recent studies exactly the same molecules have been docked and screened empirically, against exactly the same target. In a quantitative HTS (qHTS, where each compound is tested in 7 point dose response) of over 70,000 compounds against AmpC β-lactamase, qHTS yielded 1274 hits initial hits. A detergent counter-screen revealed 95% of these to be colloidal aggregators, which are typically largest source of false-positives in biochemical assays [29]. Of the 70 “hits” that remained, 25 were β-lactams, which are known to covalently bind to β-lactamase, and so are uninteresting except as controls, 24 were irreproducible on re-testing, nine were aggregators that resisted the low levels of detergent used in the counter-screen, and 9 were promiscuous covalent inhibitors [20]. No reversible, specific, competitive inhibitors were found by HTS whatsoever.
This result was unexpected—a goal of the exercise had been to reveal docking false negatives, but with no new molecules discovered by the qHTS, this was impossible. To explore whether the empirical screen suffered from false-negatives, 16 high-ranking docking hits were retested in low throughput at concentrations higher than used in the qHTS campaign (where the maximum concentration of compounds was 30 µM). Two mid-micromolar competitive inhibitors were found, one that had ranked 80th and one that had ranked 200th of the 66,000 library molecules docked. These compounds had Ki values of 37 and 55 µM, and IC50 values higher still, which explains why they were missed by the qHTS. Subsequently, a crystal structure of AmpC in complex with one of these inhibitors showed close correspondence between the x-ray structure and that predicted by docking (Figure 2) [20].
Figure 2.
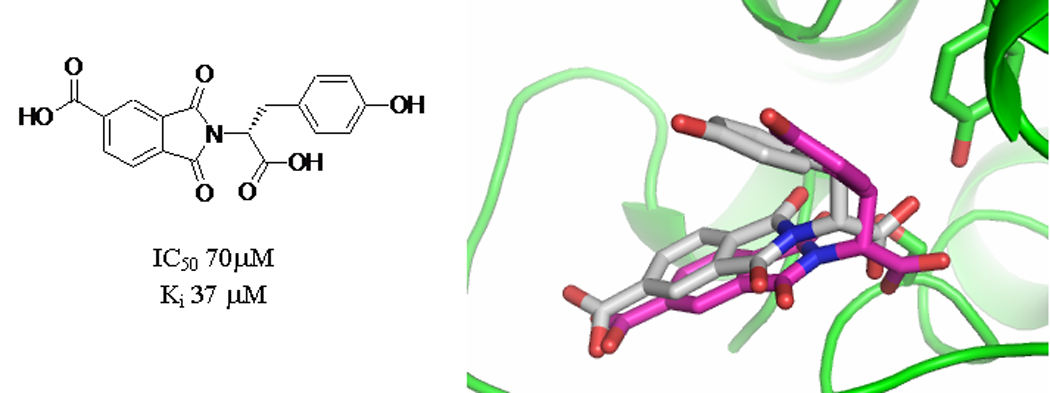
A docking-derived novel β-lactamase inhibitor. Structure of the new inhibitor and overlay of the crystal structure (white carbons) to the initial pose proposed by DOCK (magenta carbons). The RMS deviation of the heavy atoms between the two poses was 0.9 Å.
In a search for formylpeptide receptor ligands, approximately the top 1% of the library ranked by virtual screening was tested by HTS, and nine chemical families of hits were confirmed [30]. In another study, a combination of 2D and 3D similarity approaches was used to screen 10,000 compounds against the estrogen receptor GPR30. Biochemical screen of the top 100 ranked compounds identified of a potent ligand (Ki = 5.7 nM) [31]. These studies illustrate how prioritization of compounds to be tested can lead to identification of true hits. However, they don’t answer the question of how many scaffolds were missed by virtual screening. To do so one needs to screen exactly the same library by both methods and compare the hits. A good example of such a study was reported in a search for GSK-3β in which four out of six scaffolds found by HTS were found in the top 1% by docking [28], indicating the potential for using virtual screening as a guide in the follow up of HTS hits. Unfortunately, few direct comparisons between these methods are currently reported, and a bigger number of similar studies would help to draw conclusions on how frequently such success is attained by virtual screening.
At this early point, three tentative lessons may be drawn: first, both docking and HTS suffer from false-positives and false negatives; second, that an intense amount of work is required to follow up and confirm screening hits; and third, that the two techniques may complement each other. These conclusions are supported by a new docking and qHTS campaign of now 198,000 molecules against the enzyme cruzain, with the proviso that here, finally, screening revealed docking false-negatives (unpublished results). How reliable these conclusions are, and whether we may expect docking and HTS to be combined as a standard procedure, must await the outcome of further studies now underway.
Docking to membrane proteins
Structure-based approaches to ligand design for membrane proteins have been hampered by a lack of x-ray structures. This was especially grave in the case of guanine nucleotide-binding protein (G protein)-coupled receptors (GPCRs), where, until recently, only the structure of rhodopsin was known [32]. This absence was keenly felt, as 25% of drug targets are GPCRs, and 40% of drugs bind to these targets [33]. To overcome this gap, protein structure modeling was widely used to generate 3D structures as scaffolds to which to dock. In the past two years, modeling was mainly concerned with the refinement of the “raw” modeled structures. These structures were then successfully used in several docking screens, although the hit rates remained low [13,18,34].
The recent determinations of the structures of the β2-adrenergic (β2AR) [35,36], the β1-adrenergic [37], and the adenosine A2A [38] GPCRs reveals why these receptors are wonderful targets for small molecules. The binding sites almost entirely enclose the ligands, ensuring close complementarity. Each site combines a mixture of polar groups, allowing for specificity, and non-polar ones, for affinity (Figure 3). The use of an x-ray structure for docking should be a substantial advantage over modeled structures, not least because of how the structure reveals the precise layouts of the receptor binding sites and their interactions with the ligand. With these new structures, can we anticipate an efflorescence of ligand discovery, will they be better templates for discovery than the homology models that preceded them, and how will the new ligands compare to those discovered, over the last 50 years, by traditional ligand-based methods, which progressed without the advantage of a crystal structure?
Figure 3.
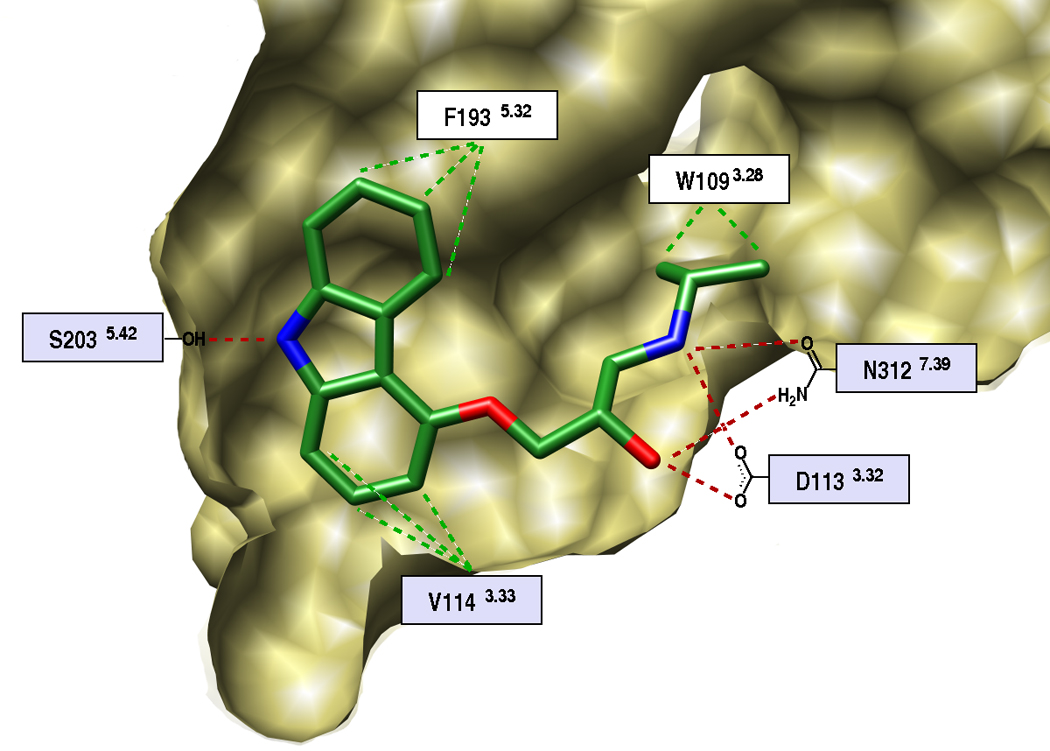
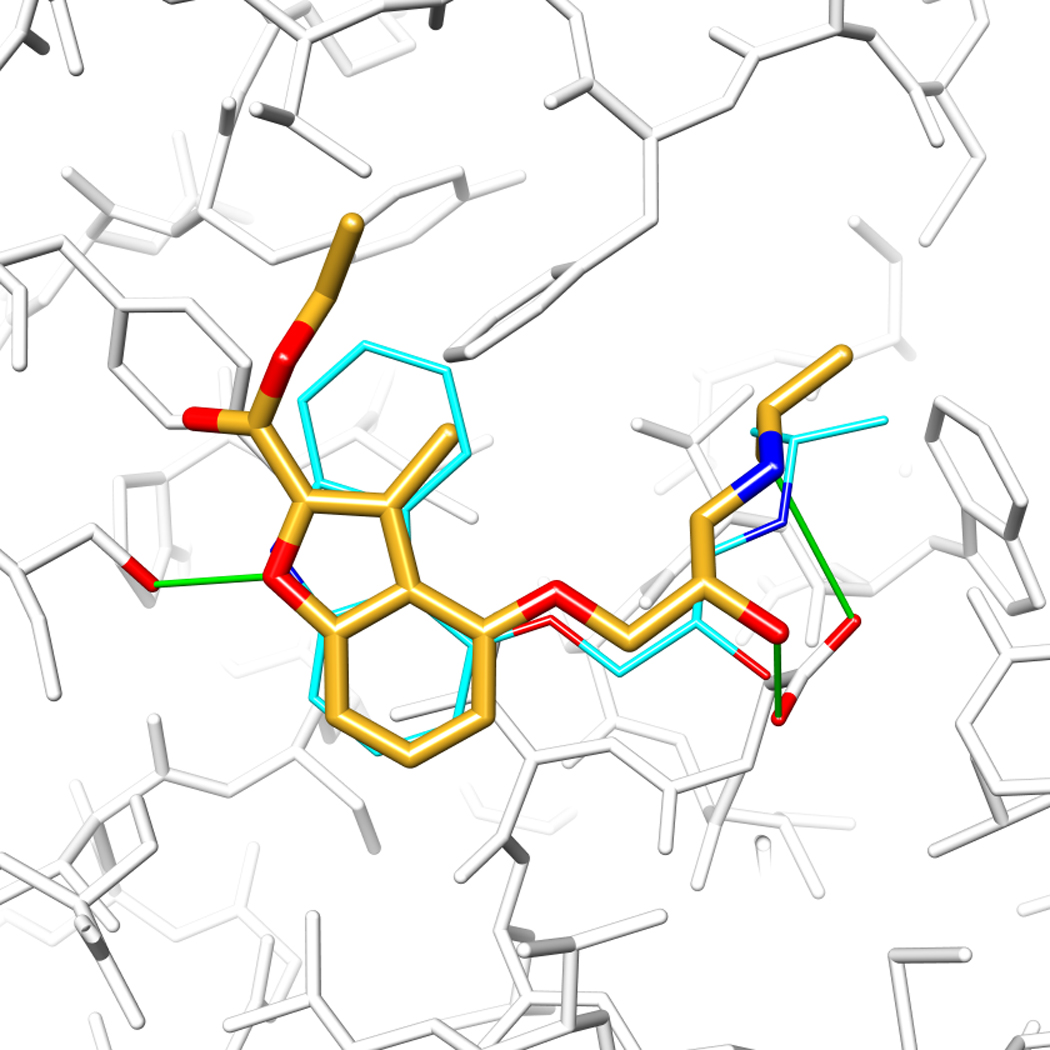
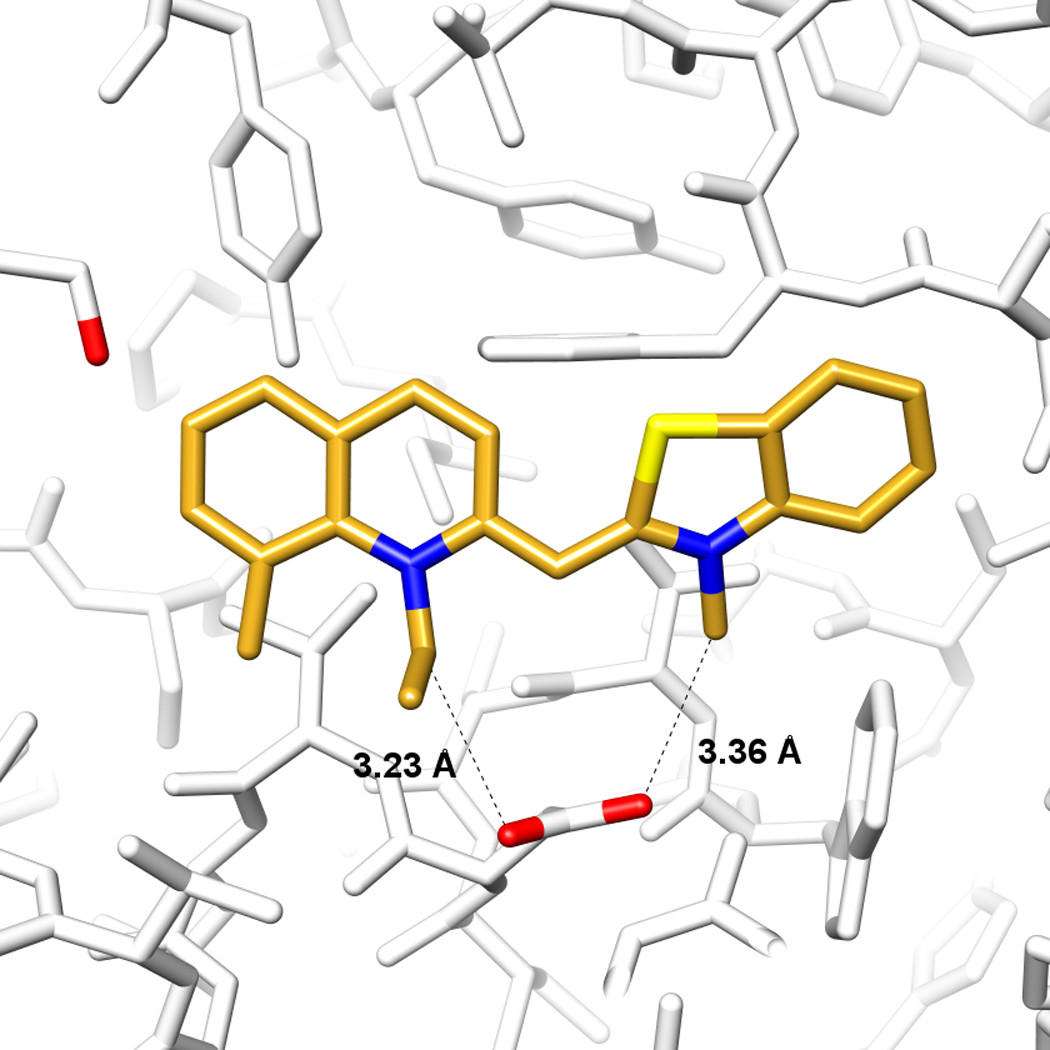
The GPCR β2AR as a target for structure-based screening. A. Side view of the orthosteric site, the proximal portion of the protein has been cut away for clarity. Several key interacting residues are marked. Red and green dashed lines indicate polar and hydrophobic contacts, respectively. Residues in a light-blue box are essential for agonist and antagonist binding. B. Comparison of the docked pose of a docking-derived, 9 nM inverse agonist [19] (gold carbons), with the x-ray structure of carazolol (cyan). Hydrogen bonds are shown as green sticks, and residues Asp-113 and Ser-203 are emphasized with red oxygens. C. Predicted binding mode of a docking-derived novel ligand ref. [19] (gold carbons), chemically distinct from previously known βAR ligands. The distances between the alkyl substituents and the respective closest oxygen of Asp-113 are shown as dashed lines.
In a screen against the β2AR, about a million “lead-like” molecules from the ZINC database (http://zinc.docking.org) were screened against the structure of the target. Twenty-five were chosen from the top 400 docking-ranked molecules and tested in ligand displacement assays. Six modulated β2AR with affinities between 9 nM and 4 µM, a hit rate of 24% [19] (Figure 3). Intriguingly, five of these were inverse agonists, as was the ligand bound in the x-ray structure, carazolol, against which the screen occurred. In a similar study, an in-house database of 400,000 ligands was screened against β2AR [39]. 36% of the molecules tested were active, with the best having a 0.114 nM Ki. An innovative experiment was to dock for pharmaceutical chaperones of misfolded rhodopsin [40]. About 24,000 compounds were docked from the NCI database against the x-ray structure of the P23H mutation of opsin, which is associated with retinitis pigmentosa. Five docking prioritized compounds were tested, one of which was weakly active as an inhibitor of opsin regeneration.
The relatively high hit rates and high potencies of the ligands to emerge against the x-ray structures supports the idea that these are better templates for discovery and design, compared to the earlier homology models. This is borne out in a community-wide, blind assessment (GPCR Dock 2008 [41]) of the prediction of the structure of the human adenosine A2A receptor in complex with the ligand ZM241385 [38]. Twenty-nine groups submitted a total of 206 structural models before the actual publication of the x-ray structure. The best model had a ligand RMSD of 2.8 Å and a Cα RMSD of 3.0 Å, but ranked only second among the models submitted by that participant. Slightly more discouragingly, the average RMSD of the ligand predictions was 9.5 Å, despite an average Cα RMSD of only 4.2 Å between protein model and x-ray structure. This indicates that docking didn’t work well and is sensitive to changes in protein structure, a point that has been made before.
By docking standards—where a 5% hit rate is considered substantial, and where a “hit” might have a mid-micromolar affinity, the results in the GPCR docking screens seem extraordinary. This is partly explained by the substantial bias in even our commercial libraries toward GPCR-like ligands, the product of 50 years of intense medicinal chemistry in this area. On the other hand, the β2AR and opsin docking screens suggest that despite the attention lavished on these targets, novel chemotypes, with arguably new biology, may yet be found. Both observations support the idea that further structure-based screens against GPCRs, both those now determined and the new structures that are eagerly anticipated, will merit the effort. A challenge for the future will be leveraging the often antagonist, or inverse-agonist, bound complexes to discover agonists; recent work from the Rognan and Abagyan labs suggest that this will be possible [42,43]
Predicting the activities for enzymes of unknown function
In the early years of docking several investigators entertained the idea of predicting not only what would inhibit an enzyme or a receptor, but also their true physiological substrates and agonists. For two reasons the idea was put aside. First, we could not then (early 1990’s) imagine many proteins whose structures had been determined but whose functions remained unknown—why would anyone go to such trouble? Second, predicting activity seemed much harder than predicting inhibitors of activity, notwithstanding the relevance of both for drug discovery and for biological understanding. This idea thus remained little more than idle conversation for two decades.
With the advent of the Structural Genomics projects, however, an increasing number of proteins of unknown and function have had their structures determined, over 50 such are catalogued by the Protein Databank (PDB) alone. This has inspired several groups to return to the challenge of predicting the enzyme activity. Whereas the original technical concerns remain germane, not least because of the difficulties in modeling conformational changes to which activity is often coupled, there are preliminary signs of progress. Thornton and colleagues have investigated docking a limited set of metabolites against the structures of short-chain dehydrogenases/reductases, exploring different docking protocols in retrospective studies [44]. An interesting aspect of this work is the use of representative metabolites to represent a class of molecules, reducing computational costs. In more prospective work, Hermann and co-workers and Xiang and co-workers have predicted and tested the activities of two targets from structural genomics—the amidohydrolases Tm0936 [17] and Dr0930 [45]. Docking the high-energy intermediate, transition-state-like forms of the KEGG metabolites, Tm0936 was correctly predicted to act as a deaminase of adenosine and S-adenosine homocysteine, while Dr0930 was correctly predicted to act as a lactonase. Whereas both of these predictions were confirmed experimentally, close inspection of the results reveals the strengths and weaknesses of the approach. For Tm0936 the docking was almost entirely correct—not only was the right substrate predicted, but so was its docked geometry compared to a crystal structure of the product of the reaction that was subsequently determined [17]. With Dr0930, conversely, whereas the docking identified lactones as a general class of substrate, the precise preferences among lactones was not captured, as δ-lactones were predicted to be as good as, often better than γ-lactones, when in fact the reverse was found to be true. Thus docking the high-energy intermediate forms of the metabolites was grossly successful, but missed important particulars.
Thus it is too early to tell whether a structure-based approach to function prediction has yet overcome the concerns that kept it on the sidelines for the last two decades. What can be said is that it is no longer so sidelined, and is being pursued actively by several groups. Hopefully, two years from now when the next version of this review becomes timely, we will have a better sense if this approach has advanced to the point of reliability—for now, it remains intriguing enough, with enough preliminary successes, to justify a focused research effort.
Polypharmacology and the chemical view of biology
The logic used by classical pharmacology inverts the target-oriented approach of molecular biology—in pharmacology, biology is characterized by the actions of organic small molecules. Even now, most receptors are characterized by more or less specific ligands that, for instance, distinguish α-adrenergic from β-adrenergic receptors, and β1 subtypes from β2. Recently, computational chemists and biologists have returned to this older chemical view, quantifying the relationships among receptors based not on their structures or sequences, but on chemical patterns among the ligands that bind to them. This has revealed connections among targets that to a traditional pharmacologist—now largely extinct—might seem entirely reasonable, but to the now dominant molecular view will be surprising.
A seminal paper in this area, by Hopkins and colleagues when at Pfizer, mapped the links among targets as articulated by shared ligands. Many targets that were unrelated by sequence or structure nevertheless had common ligands [46]. Similarly, Vidal and colleagues adapted network techniques developed for protein-protein association to drug-target associations [47]. Many drugs shared multiple targets, and unsurprisingly the converse was also true. Older drugs were often distinguished from newer, sometimes investigational drugs by the greater specificity of the latter, which the authors attributed to the influence of target-based or “rational” design. A comparison of drug targets to those directly involved in disease etiology suggested that most drugs bound to proteins distant from the etiological targets, implying that most drugs did not work on the cause of the disease, but were rather palliative.
In contrast to the trends toward specificity inferred by Vidal, Roth has argued that polypharmacology is not only common but that, especially in the CNS, it is often essential for efficacy [48] (polypharmacology is taken to mean the action of one drug on multiple targets at physiologically relevant concentrations). Effective CNS drugs typically target at least two targets, such as serotonin and dopamine subtypes, and often more; genuine specificity correlates with lack of efficacy. Roth has established an NIH Roadmap Psychoactive Drug Screening Program (PDSP, http://pdsp.med.unc.edu/indexR.html) to screen pharmacologically active agents against a broad spectrum of targets, including over 300 GPCRs and, increasingly, ion channels, transporters and kinases. Recently, systematic screening has revealed that the histamine H1 receptor can be responsible for weight gain under antipsychotic treatment, an enormous problem for drugs such as Zyprexa, that the kappa opioid receptor is the target of the potent hallucinogen salvinorin A, and that the clozapine metabolite desmethylclozapine activates M1 muscarinic receptors, which may contribute to the clinical efficacy of the parent antipsychotic [49].
In collaboration with Roth’s group, we have tried to predict and test new off target effects of established drugs. Drawing on a systematic map relating drug targets by their ligands, using BLAST-derived algorithms to normalize for random chemical similarity, we found that targets related by ligands quantitatively differed from those related by sequence [50]. For instance, the serotonergic 5HT-3 receptor was closely related to the 5HT-4 receptor, even though the former is an ion channel and the second a GPCR; both bind congeners of serotonin. Similar relationships among the ligands for multiple ionotropic (ion channels) and metabotropic (GPCRs) receptors, and even transporters, were observed. Based on these observations, new off-targets were predicted, including the antagonistic effects of the µ-opioiod methadone on M3 muscarinic receptors, the activity of the antiparasitic emetine on α-adrenergic receptors, and the activity of the gut µ-opioiod loperamide on NK2 receptors. When tested in the Roth lab, all three had low or sub-micromolar activities on their predicted off targets. Similarly, Bork and colleagues at the EMBL have analyzed the shared side effect profiles of 746 drugs using a text-based analysis of drug inserts, correlating these with chemoinformatic similarities. A network of 1018 drug-drug relations was revealed, 261 of which were formed among dissimilar drugs from different indications. Twenty specific drug off-targets were tested, and 13 were confirmed experimentally [51]. Other studies, which return to a more structure-based method for predicting off-target effects, including docking, have also revealed new off-targets, though these efforts remain at an earlier stage the chemoinformatics approaches [52].
Drug polypharmacology is common [53], occasionally essential, often has unwanted side effects [54], and can cross classic molecular categorizations. The most successful methods to characterize and, occasionally, predict [50,51] such off target effects have been those that are strictly ligand-based, but is there a way to understand these effects from a molecular target view? In a fascinating recent paper, Klebe and colleagues compared the structural similarities of binding sites among often unrelated targets, and find that the similarity among these is often unrelated to the sequence identities of the proteins that harbor them [55]. It may be that as the ligand-bound structures or more and more targets become available, we will be able to understand these cross target activities. For now, the older, pharmacological organization is, paradoxically, the more generative for predicting and classifying polypharmacology than the structure-based approaches which have been our primary focus here [56].
Conclusions
With all their weaknesses, docking screens are now common in molecular discovery. The ongoing growth of molecular structures, not least among membrane targets, will continue to widen the remit of this technique. In upcoming research one can look not only for the discovery of new molecular entities, but their genuine application to pragmatic problems in drug discovery and chemical biology. In the short term, the highest impact studies may well come from the chemoinformatics approaches that return to an older, classical pharmacology view of drug action, as these work with drugs themselves, whose impact on human health and biology is already clear.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goodford PJ. Drug Design by the Method of Receptor Fit. J. Med. Chem. 1984;27:557–564. doi: 10.1021/jm00371a001. [DOI] [PubMed] [Google Scholar]
- 2.Leach AR, Shoichet BK, Peishoff CE. Prediction of protein-ligand interactions. Docking and scoring: successes and gaps. J Med Chem. 2006;49:5851–5855. doi: 10.1021/jm060999m. [DOI] [PubMed] [Google Scholar]
- 3.Hert J, Irwin JJ, Laggner C, Keiser MJ, Shoichet BK. Quantifying biogenic bias in screening libraries. Nat Chem Biol. 2009;5:479–483. doi: 10.1038/nchembio.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brooks WH, McCloskey DE, Daniel KG, Ealick SE, Secrist JA, Waud WR, Pegg AE, Guida WC. In silico chemical library screening and experimental validation of a novel 9-aminoacridine based lead-inhibitor of human S-adenosylmethionine decarboxylase. Journal of Chemical Information and Modeling. 2007;47:1897–1905. doi: 10.1021/ci700005t. [DOI] [PubMed] [Google Scholar]
- 5.Wang JG, Xiao YJ, Li YH, Ma Y, Li ZM. Identification of some novel AHAS inhibitors via molecular docking and virtual screening approach. Bioorganic & Medicinal Chemistry. 2007;15:374–380. doi: 10.1016/j.bmc.2006.09.050. [DOI] [PubMed] [Google Scholar]
- 6.Steuber H, Heine A, Klebe G. Structural and thermodynamic study on aldose reductase: nitro-substituted inhibitors with strong enthalpic binding contribution. Journal of Molecular Biology. 2007;368:618–638. doi: 10.1016/j.jmb.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Montes M, Braud E, Miteva MA, Goddard ML, Mondesert O, Kolb S, Brun MP, Ducommun B, Garbay C, Villoutreix BO. Receptor-based virtual ligand screening for the identification of novel CDC25 phosphatase inhibitors. Journal of Chemical Information and Modeling. 2008;48:157–165. doi: 10.1021/ci700313e. [DOI] [PubMed] [Google Scholar]
- 8.Ostrov DA, Prada JAH, Corsino PE, Finton KA, Le N, Rowe TC. Discovery of novel DNA gyrase inhibitors by high-throughput virtual screening. Antimicrobial Agents and Chemotherapy. 2007;51:3688–3698. doi: 10.1128/AAC.00392-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolb P, Berset Kipouros C, Huang D, Caflisch A. Structure-based tailoring of compound libraries for high-throughput screening: Discovery of novel EphB4 inhibitors. Proteins: Structure, Function, and Bioinformatics. 2008 doi: 10.1002/prot.22028. [DOI] [PubMed] [Google Scholar]
- 10.Tikhonova IG, Sum CS, Neumann S, Engel S, Raaka BM, Costanzi S, Gershengorn MC. Discovery of novel Agonists and antagonists of the free fatty acid receptor 1 (FFAR1) using virtual screening. Journal of Medicinal Chemistry. 2008;51:625–633. doi: 10.1021/jm7012425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kiss R, Kiss B, Konczol A, Szalai F, Jelinek I, Laszlo V, Noszal B, Falus A, Keseru GM. Discovery of Novel Human Histamine H4 Receptor Ligands by Large-Scale Structure-Based Virtual Screening. Journal of Medicinal Chemistry. 2008 doi: 10.1021/jm7014777. [DOI] [PubMed] [Google Scholar]
- 12.Lemaire G, Benod C, Nahoum V, Pillon A, Boussioux AM, Guichou JF, Subra G, Pascussi JM, Bourguet W, Chavanieu A, et al. Discovery of a highly active ligand of human pregnane x receptor: a case study from pharmacophore modeling and virtual screening to "in vivo" biological activityDiscovery of a highly active ligand of human pregnane x receptor: a case study from pharmacophore modeling and virtual screening to "in vivo" biological activity. Mol Pharmacol. 2007;72:572–581. doi: 10.1124/mol.106.033415. [DOI] [PubMed] [Google Scholar]
- 13.Cavasotto CN, Orry AJW, Murgolo NJ, Czarniecki MF, Kocsi SA, Hawes BE, O’Neill KA, Hine H, Burton MS, Voigt JH, et al. Discovery of Novel Chemotypes to a G-Protein-Coupled Receptor through Ligand-Steered Homology Modeling and Structure-Based Virtual Screening. Journal of Medicinal Chemistry. 2008;51:581–588. doi: 10.1021/jm070759m. [DOI] [PubMed] [Google Scholar]
- 14.Pierce AC, Jacobs M, Stuver-Moody C. Docking study yields four novel inhibitors of the protooncogene Pim-1 kinase. J Med Chem. 2008;51:1972–1975. doi: 10.1021/jm701248t. [DOI] [PubMed] [Google Scholar]
- 15.Pereira HM, Berdini V, Cleasby A, Garratt RC. Crystal structure of calf spleen purine nucleoside phosphorylase complexed to a novel purine analogue. FEBS Letters. 2007;581:5082–5086. doi: 10.1016/j.febslet.2007.09.051. [DOI] [PubMed] [Google Scholar]
- 16.Salam NK, Huang THW, Kota BP, Kim MS, Li YH, Hibbs DE. Novel PPAR-gamma agonists identified from a natural product library: A virtual screening, induced-fit docking and biological assay study. Chemical Biology & Drug Design. 2008;71:57–70. doi: 10.1111/j.1747-0285.2007.00606.x. [DOI] [PubMed] [Google Scholar]
- 17.Hermann JC, Marti-Arbona R, Fedorov AA, Fedorov E, Almo SC, Shoichet BK, Raushel FM. Structure-based activity prediction for an enzyme of unknown function. Nature. 2007;448:775–779. doi: 10.1038/nature05981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engel S, Skoumbourdis AP, Childress J, Neumann S, Deschamps JR, Thomas CJ, Colson AO, Costanzi S, Gershengorn MC. A virtual screen for diverse ligands: Discovery of selective G protein-coupled receptor antagonists. Journal of the American Chemical Society. 2008;130:5115–5123. doi: 10.1021/ja077620l. [DOI] [PubMed] [Google Scholar]
- 19.Kolb P, Rosenbaum DM, Irwin JJ, Fung J, Kobilka BK, Shoichet BK. Structure-based discovery of β2-adrenergic receptor ligands. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6843–6848. doi: 10.1073/pnas.0812657106.One of the first docking screens against the recent GPCR x-ray structures. Both potent and novel chemotypes were found.
- 20.Babaoglu KSA, Irwin J, Nelson ME, Feng B, Thomas CJ, Cancian L, Costi P, Maltby DA, Jadhav A, Inglese J, Austin C, Shoichet BK. Comprehensive mechanistic analysis of hits from high-throughput and docking screens against b-lactamase. J. Med. Chem. 2008;51:2502–2511. doi: 10.1021/jm701500e.The mechanism of every hit from a qHTS campaign is determined, revealing the sources of artifacts and their prevalence. Whereas no true inhibitors were ultimately discovered by the qHTS, reinvestigation of the hits from a docking screen, run in parallel, revealed an HTS false-negative true inhibitor. The pose predicted by the docking was confirmed by crystallography.
- 21.Yu WM, Guvench O, Mackerell AD, Jr, Qu CK. Identification of Small Molecular Weight Inhibitors of Src Homology 2 Domain-Containing Tyrosine Phosphatase 2 (SHP-2) via in Silico Database Screening Combined with Experimental Assay. Journal of Medicinal Chemistry. 2008 doi: 10.1021/jm800229d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li M, Ni N, Chou HT, Lu CD, Tai PC, Wang B. Structure-based discovery and experimental verification of novel AI-2 quorum sensing inhibitors against Vibrio harveyi. ChemMedChem. 2008;3:1242–1249. doi: 10.1002/cmdc.200800076. [DOI] [PubMed] [Google Scholar]
- 23.Chen D, Misra M, Sower L, Peterson JW, Kellogg GE, Schein CH. Novel inhibitors of anthrax edema factor. Bioorganic and Medicinal Chemistry. 2008;16:7225–7233. doi: 10.1016/j.bmc.2008.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinke R, Spannhoff A, Meier R, Trojer P, Bauer I, Jung M, Sippl W. Virtual Screening and Biological Characterization of Novel Histone Arginine Methyltransferase PRMT1 Inhibitors. ChemMedChem. 2008;4:69–77. doi: 10.1002/cmdc.200800301. [DOI] [PubMed] [Google Scholar]
- 25.Doman TNMS, Witherbee BJ, Kasten TP, Kurumbail R, Stallings WC, Connolly DT, Shoichet BK. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J. Med. Chem. 2002;45:2213–2221. doi: 10.1021/jm010548w. [DOI] [PubMed] [Google Scholar]
- 26.Paiva AMVD, Blanchard JS, Kozarich JW, Williamson JM, Kelly TM. Inhibitors of dihydrodipicolinate reductase, a key enzyme of the diaminopimelate pathway of Mycobacterium tuberculosis. Biochimica et Biophysica Acta. 2001;1545:67–77. doi: 10.1016/s0167-4838(00)00262-4. [DOI] [PubMed] [Google Scholar]
- 27.Oshiro CBE, Eksterowicz J, Evensen E, Lamb ML, Lanctot JK, Putta S, Stanton R, Grootenhuis PJD. Performance of 3D-Database Molecular Docking Studies into Homology Models. J Med Chem. 2004;47:764–767. doi: 10.1021/jm0300781. [DOI] [PubMed] [Google Scholar]
- 28.Polgar TBA, Szendrei GI, Keseru GM. Comparative Virtual and Experimental High-Throughput Screening for Glycogen Synthase Kinase-3b Inhibitors. J Med Chem. 2005;48:7946–7959. doi: 10.1021/jm050504d. [DOI] [PubMed] [Google Scholar]
- 29.Feng BYSA, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. A high-throughput screen for aggregation-based inhibition in a large compound library. J. Med. Chem. 2007;50:2385–2390. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- 30.Edwards BSBC, Young SM, Balakin KV, Prossnitz ER, Savchuck NP, Sklar LA, Oprea TI. Integration of virtual screening with high-throughput flow cytometry to identify novel small molecule formylpeptide receptor antagonists. Mol Pharmacol. 2005;68:1301–1310. doi: 10.1124/mol.105.014068. [DOI] [PubMed] [Google Scholar]
- 31.Bologa CGRC, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- 32.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Trong IL, Teller DC, Okada T, Stenkamp RE, et al. Crystal Structure of Rhodopsin: A G Protein-Coupled Receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 33.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 34.Schlegel B, Laggner C, Meier R, Langer T, Schnell D, Seifert R, Stark H, Holtje HD, Sippl W. Generation of a homology model of the human histamine H-3 receptor for ligand docking and pharmacophore-based screening. Journal of Computer-Aided Molecular Design. 2007;21:437–453. doi: 10.1007/s10822-007-9127-x.This paper goes beyond the usual model building in that a model of the histamine h3 receptor is generated and afterwards refined with molecular dynamics simulations in a lipid membrane. Also, the docking results are directly compared to a pharmacophore-based screening.
- 35.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, et al. GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609.The first x-ray crystal structure of a pharmaceutically relevant GPCR.
- 37.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AGW, Tate CG, Schertler GFX. Structure of a β1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101.The x-ray crystal structure of the β1AR
- 38.Jaakola V-P, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, Ijzerman AP, Stevens RC. The 2.6 Angstrom Crystal Structure of a Human A2A Adenosine Receptor Bound to an Antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772.The x-ray crystal structure of the adenosine receptor, the third pharmaceutically relevant GPCR structure to be determined
- 39.Sabio M, Jones K, Topiol S. Use of the X-ray structure of the β2-adrenergic receptor for drug discovery. Part 2: Identification of active compounds. Bioorganic & Medicinal Chemistry Letters. 2008;18:5391–5395. doi: 10.1016/j.bmcl.2008.09.046. [DOI] [PubMed] [Google Scholar]
- 40.Noorwez SM, Ostrov DA, McDowell JH, Krebs MP, Kaushal S. A high-throughput screening method for small-molecule pharmacologic chaperones of misfolded rhodopsin. Investigative Ophthalmology & Visual Science. 2008;49:3224–3230. doi: 10.1167/iovs.07-1539. [DOI] [PubMed] [Google Scholar]
- 41.Michino M, Abola E, Brooks CL, Dixon JS, Moult J, Stevens RC. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nature Reviews Drug Discovery. 2009;8:455–463. doi: 10.1038/nrd2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Graaf C, Rognan D. Selective Structure-Based Virtual Screening for Full and Partial Agonists of the β2 Adrenergic Receptor (vol 51, pg 4978, 2008) Journal of Medicinal Chemistry. 2008;51:6620–6620. doi: 10.1021/jm800710x. [DOI] [PubMed] [Google Scholar]
- 43.Reynolds KA, Katritch V, Abagyan R. Identifying conformational changes of the beta(2) adrenoceptor that enable accurate prediction of ligand/receptor interactions and screening for GPCR modulators. J Comput Aided Mol Des. 2009;23:273–288. doi: 10.1007/s10822-008-9257-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Favia AD, Nobeli I, Glaser F, Thornton JM. Molecular docking for substrate identification: The short-chain dehydrogenases/reductases. Journal of Molecular Biology. 2008;375:855–874. doi: 10.1016/j.jmb.2007.10.065. [DOI] [PubMed] [Google Scholar]
- 45.Xiang DF, Kolb P, Fedorov AA, Meier MM, Fedorov LV, Nguyen TT, Sterner R, Almo SC, Shoichet BK, Raushel FM. Functional Annotation and Three-Dimensional Structure of Dr0930 from Deinococcus radiodurans, a Close Relative of Phosphotriesterase in the Amidohydrolase Superfamily. Biochemistry. 2009;48:2237–2247. doi: 10.1021/bi802274f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paolini GV, Shapland RH, van Hoorn WP, Mason JS, Hopkins AL. Global mapping of pharmacological space. Nat Biotechnol. 2006;24:805–815. doi: 10.1038/nbt1228. [DOI] [PubMed] [Google Scholar]
- 47.Yildirim MA, Goh KI, Cusick ME, Barabasi AL, Vidal M. Drug-target network. Nat Biotechnol. 2007;25:1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- 48.Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov. 2004;3:353–359. doi: 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- 49.Jensen NH, Roth BL. Massively parallel screening of the receptorome. Comb Chem High Throughput Screen. 2008;11:420–426. doi: 10.2174/138620708784911483. [DOI] [PubMed] [Google Scholar]
- 50.Keiser MJ, Roth BL, Armbruster BN, Ernsberger P, Irwin JJ, Shoichet BK. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007;25:197–206. doi: 10.1038/nbt1284.Applying statistical models derived from the BLAST algorithms, drug targets are related to each other by the similarity of their ligands. Targets unrelated by sequence or structure are found to be highly related by the chemistry of their ligands. Three drugs are predicted and shown to have new and unexpected off-targets, based on these similarities.
- 51.Campillos M, Kuhn M, Gavin AC, Jensen LJ, Bork P. Drug target identification using side-effect similarity. Science. 2008;321:263–266. doi: 10.1126/science.1158140.Over 1000 drug-target pairs are related by textual analyses of drug inserts, describing side effect profiles. Twenty new targets for established drugs are tested experimentally, and 13 are confirmed in experimental assays. This approach differs substantially from the chemoinformatic methods employed in Keiser, but the motivations of the two papers are related.
- 52.Kinnings SL, Liu N, Buchmeier N, Tonge PJ, Xie L, Bourne PE. Drug discovery using chemical systems biology: repositioning the safe medicine Comtan to treat multi-drug and extensively drug resistant tuberculosis. PLoS Comput Biol. 2009;5:e1000423. doi: 10.1371/journal.pcbi.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mestres J, Gregori-Puigjane E, Valverde S, Sole RV. Data completeness--the Achilles heel of drug-target networks. Nat Biotechnol. 2008;26:983–984. doi: 10.1038/nbt0908-983. [DOI] [PubMed] [Google Scholar]
- 54.Scheiber J, Jenkins JL, Sukuru SC, Bender A, Mikhailov D, Milik M, Azzaoui K, Whitebread S, Hamon J, Urban L, et al. Mapping adverse drug reactions in chemical space. J Med Chem. 2009;52:3103–3107. doi: 10.1021/jm801546k. [DOI] [PubMed] [Google Scholar]
- 55.Weskamp N, Hullermeier E, Klebe G. Merging chemical and biological space: Structural mapping of enzyme binding pocket space. Proteins. 2008;76:317–330. doi: 10.1002/prot.22345. [DOI] [PubMed] [Google Scholar]
- 56.Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4:682–690. doi: 10.1038/nchembio.118.A survey and call to arms of what may be an emerging field.