

Summary
Interleukin-12 (IL-12) and IL-23 share a common chain. Yet, their production in response to pathogens is differentially regulated, and their functions are distinct and often antithetic. IL-12 is involved in the induction or amplification of the T-helper type 1 (Th1) response, whereas IL-23 has been associated with the generation of the Th17 response and IL-17 production. Mycobacterium tuberculosis and yeast zymosan induce IL-23, but in absence of other stimuli, no IL-12 is induced in human dendritic cells (DCs). The stimulation of IL-23 by M. tuberculosis was mostly explained by the triggering of Toll-like receptor 2 (TLR2) and the cytoplasmic receptor nucleotide oligomerization domain-containing protein 2 (NOD2), whereas zymosan induces IL-23 primarily by stimulating the β-glucan receptor dectin-1 alone or in combination with TLR2. IL-23, IL-6, transforming growth factor-β (TGF-β), and IL-1β in supernatants from activated human DCs induce human naive CD4+ T cells to produce IL-17. These data are consistent with various recent reports that TGF-β is an inducer of IL-17 production both in human and mouse cells. However, IL-1 is necessary in combination with some or all of the other cytokines to induce IL-17 production in human T cells. The ability of various stimuli to induce Th17 cells depends not only on their induction of IL-23, IL-6, and TGF-β production in DCs but also on their ability to activate directly or indirectly the inflammasome and to induce IL-1β.
Keywords: interleukin-1, interleukin-12, interleukin-17, interleukin-23, T-helper 17, Mycobacterium tuberculosis, zymosan
Introduction
Dendritic cells (DCs) and other pro-inflammatory accessory cells represent an important bridge between innate resistance and immunity. When exposed to pathogens, DCs produce an array of pro-inflammatory cytokines that set the stage for the antigen-specific immune response to which DCs also contribute as efficient antigen-presenting cells (APCs). The Toll-like receptor (TLR) family has a central role in the recognition of pathogens’ molecular patterns and in the induction of inflammation/innate resistance (1). The triggering of TLR in combination with other pattern recognition receptors (PRRs) induces pro- and anti-inflammatory cytokines that regulate not only the resistance to infection but also autoimmunity as well as tumor initiation, progression, and metastasis formation (1–3). These inflammatory and innate cytokines create the environment in which antigen-specific adaptive T cells expand and differentiate. Thus, the innate response to infections and to other exogenous or endogenous inflammatory stimuli determines the antimicrobial, immunoregulatory, and immunopathogenic functions of the effector cells of the adaptive immune response (4–6).
Mossman and Coffman (7) showed over 20 years ago that effector CD4+ T cells differentiate into two subsets, the T-helper 1 (Th1) and Th2 cells, that are characterized by distinct patterns of cytokine and receptor expression modulating the effect of the subsets against various types of pathogens and also their ability to mediate distinct immunoregulatory and immunopathogenic mechanisms. In addition to Th1 and Th2, various subsets of T cells with regulatory activities have been identified, and more recently a subset named Th17 has been defined. This subset is primarily characterized by secretion of the cytokine interleukin-17 (IL-17). IL-17 was identified in the mouse, rat, and humans (8–11) in the mid-1990s and has been shown to induce stromal cell production of pro-inflammatory and hematopoietic cytokines, including various chemokines. It was soon recognized that IL-17 had an important role in tissue inflammation and immune damage. However, the complexity of the regulation of tissue inflammatory and immune responses had been difficult to dissect, and it is of interest in the light of present knowledge to reflect on the question originally poised by Hal E. Broxmeyer (12) in commenting on the discovery of human IL-17: ‘Is interleukin 17, an inducible cytokine that stimulates production of other cytokines, merely a redundant player in a sea of other biomolecules?’
Although the production of IL-17 by human and mouse T cells had already been reported in these early studies, it was not until three years ago that Th17 cells were recognized as a different subset of T cells that, in addition to the prototype IL-17A, also produce other cytokines, including IL-17F and IL-22 (13). There is now an extensive body of literature in mice and in humans showing that Th17 cells are not only important for the resistance to fungal pathogens (14) and for mucosal immunity, but they are also instrumental for the induction of various types of organ-specific autoimmunity (15), a function previously attributed to Th1 cells. Th17 cells might also create an environment that suppresses tumor immunosurveillance and favors tumor growth, progression, and spreading (16, 17). It is also becoming more and more evident that although several distinct subsets of Th cells have now been described and well characterized, this distinction is not absolute, and T-cell subsets that produce intermediate patterns of cytokines exist. The ability of Th1 cells in certain conditions to produce high levels of IL-10, a cytokine originally described as Th2 restricted, is a clear example (18). The production of the cytokines forming the distinctive Th17 pattern is not always regulated in a concordant way (19), as discussed in more detail later. This variation is most likely due to the fact that while certain groups of cytokines are molecularly co-regulated, as in the case of many of the prototypic Th2-cytokines, the expression of other cytokines is induced by different primary transcription factors or modulated by secondary transcriptional or post-transcriptional mechanisms.
The differentiation of the various Th subsets is optimal in a microenvironment in which several cytokines are produced during innate responses and some of the cytokines sustain the differentiation of multiple subsets, depending on their combination with other cytokines. Cytokines produced by T cells as well as by other lymphocyte subsets, such as natural killer (NK) cells, B cells, and NKT cells, also play an important role by affecting cytokine production from the APCs or by directly participating in the induction or maintenance of the Th responses. However, cytokines specifically and primarily involved in the induction of given Th subsets have been described, particularly IL-4 for Th2, IL-12 for Th1, and IL-23 for Th17 cells. Although IL-4 was originally reported to be produced by non-T non-B cells (20), it is not produced during innate responses by APCs but rather by basophils (21). Unlike IL-4, the two heterodimeric cytokines IL-12 and IL-23 are produced during innate response by APCs such as DCs and macrophages. The role of IL-12 for Th1 induction and of IL-23 for Th17 induction is complex, because the receptors for these two cytokines are not well expressed on naive T cells. Thus, these cytokines are thought to expand, fix, or maintain the phenotype and the number of the Th1 and Th17 cells, whereas other cytokines are recognized to initiate the response and to induce the expression of the IL-12/IL-23 receptors (22). In addition to the innate inflammatory environment, the T-cell decision among the different subsets also depends on T-cell intrinsic mechanisms, e.g. the strength of the T-cell receptor (TCR) signal or the presence of costimulation, that affect the way T cells respond to the innate environment, in part by modulating the expression of cytokine receptors or their signaling pathways (23). However, the in vivo production of IL-12 and IL-23 is very important for optimal Th1 and Th17 responses, respectively, and in their absence, the Th responses are significantly reduced. The production of IL-12 and IL-23 by APCs is quite differently regulated, even if these two heterodimeric cytokines share one common chain. Thus, the innate stimuli that differentially regulate IL-12 and IL-23 production are expected to have important effects on the type of Th response generated.
In this review, we summarize the present knowledge of the mechanisms regulating the production of IL-12, IL-23, and other innate cytokines affecting Th17 differentiation by human APCs and their effect on the induction of Th17 cells. In this young but fast moving field, there has been controversy on whether Th17 immune responses are similarly or differently regulated in mice and in humans. In a period of less than a year, several studies have been published with completely opposite results. Although the experimental mouse models have provided irreplaceable tools for the understanding of the human immune system, it is also clear that irrefutable inter-species differences exist. The most obvious situation is that in which gene-encoding molecules involved in innate resistance or in adaptive immunity are not present in one of the two species. However, many of the differences that have been reported through the years have been due either to different experimental conditions (e.g. the study of blood in humans versus lymphoid organs in the mice) or to a superficial analysis. These factors have made it difficult to fully appreciate the value of the mouse as a model for the human immune system and to determine how much and when the mouse models can be used to predict human immune responses and to efficiently guide their study before costly clinical mistakes are made. It is understandable that in a highly competitive scientific field such as that of Th17 cells ‘who acts in haste, repents in leisure’, but let us also not forget that ‘fools rush in where angels fear to tread’.
IL-12, its brothers, and cousins
When it was originally described in 1989 (24), IL-12 was the only known heterodimeric cytokine. IL-12 is now part of a family of six cytokines, and it shares important but distinct functions in the regulation of both innate resistance and adaptive immunity with three of these family members: IL-23, IL-27, and IL-35 (25–27). IL-12 and IL-23, two heterodimeric cytokines produced by APCs, are composed of a specific polypeptide characterized by a four-α helix motif, namely p35 for IL-12 (IL-12A) and p19 for IL-23 (IL-23A), disulfide-linked to a common p40 chain (IL-12B) to form the biologically active molecules (24, 28). Whereas p19 and p35 are not secreted in the absence of the p40 chain, a consistent amount of free p40, whose biological functions are still debated (29), is also produced, often in large excess over the complete IL-12 and IL-23 heterodimeric proteins (30).
The membrane receptor complex of IL-12 is formed by two chains, IL-12Rβ1 and IL-12Rβ2, that are homologous to gp130. IL-12Rβ1 is associated with the Janus kinase (Jak) Tyk2 and binds IL-12 p40; IL-12Rβ2 is associated with Jak2 and binds either the heterodimer or the p35 chain. Signaling through the IL-12 receptor complex induces phosphorylation, dimerization, and nuclear translocation of several signal transducer and activator of transcription (STAT) family members (STAT1, 3, 4, 5), but most of the biological responses to IL-12 have been attributed to STAT4 (22). The IL-23 membrane receptor complex is composed of IL-12Rβ1 and by a distinct gp130-like chain, the IL-23R (31). IL-23 signaling activates Tyk2, Jak2, and STAT 1, 3, 4. However, the activation of STAT4 is not as predominant as it is for IL-12, and STAT3/4 heterodimers, rather than STAT4 homodimers, are induced by IL-23.
IL-12 plays important roles in both innate resistance and acquired immunity, since it induces interferon-γ (IFN-γ) production from NK and NKT cells in the early phases of the immune response, and it is involved in the differentiation of Th1 cells, important in the anti-infective and anti-tumor responses (22). The protective role for the response generated by IL-12/IL-23 has been well established in the study of Mendelian susceptibility to mycobacterial and other infectious diseases in children with genetic defects of the IL-12/23-IFN-γ circuit (32). Studies using p19−/− or p35−/− mice in experimental models of autoimmunity (25–27), tumors (33), and inflammatory bowel disease (34–37) have identified IL-23, rather than IL-12, as the major factor responsible for lesions due to chronic inflammation. IL-23 mediates tissue pathology by inducing a Th17 response as well as through other less well characterized mechanisms of innate resistance. In humans, a role for IL-23 in chronic inflammation has been proposed based on the association of polymorphisms in the IL23R locus with Crohn’s disease (38). Indeed, after the nucleotide oligomerization domain containing protein 2 (NOD2) polymorphism, the IL-23R locus polymorphism shows the highest significant association to Crohn’s disease (38). In addition, polymorphisms in the IL-23R locus (and in some cases the IL12B locus) have been found to be significantly associated with ulcerative colitis, celiac disease, multiple sclerosis, psoriasis, and ankylosing spondylitis (39–44), but not with rheumatoid arthritis (45).
In 2002 it was shown that Epstein Barr virus-induced protein 3 (EBI3), an α-receptor-like soluble chain homologous to IL-12p40, allowed the secretion of a novel cytokine-like chain, p28, together with which it formed a non-covalently linked heterodimeric cytokine termed IL-27 (46). One chain of the IL-27 receptor complex is the orphan receptor WSX-1/TCCR, whereas the second chain of the IL-27 receptor complex is gp130, the common receptor chain of the IL-6 family. IL-27 activates Jak1, STAT1, and STAT3. IL-27 is produced by human phagocytic cells and DCs rapidly after activation, possibly earlier than the IL-12 heterodimer. Although IL-27 synergizes with IL-12 in inducing IFN-γ production by T and NK cells and is a very potent mitogen for anti-CD3-activated naive T cells, its major physiological roles appear to be the downregulation of Th1, Th2, and Th17 responses and the induction of production of IL-10 by all Th subsets (47, 48). EBI3 was discovered several years before IL-27 was described, and it was originally shown that similarly to IL-12p40, it allows the secretion of the cytoplasmic IL-12p35 chain by forming with it a non-covalently linked heterodimer (49, 50). However, the physiologic relevance of this observation remained unclear, and no biological functions were associated with the EBI3-p35 heterodimer until recently, when it was shown that T-regulatory cells (Tregs) produce this heterodimer, now referred to as IL-35 (51). The precise functions of IL-35 and the nature of its receptor remain to be studied, but this cytokine appears to be able both to activate Tregs and to mediate their regulatory functions (51, 52).
Two other members of the heterodimeric family of cytokines with no known immunological functions are represented by the neuropoietic cytokines of the IL-6 family. Cyliary neutrotrophic factor (CNTF) associates with the soluble extracellular part of its specific receptor (sCNTFR) to form a biologically active heterodimer signaling through a receptor complex composed of gp130 and leukemia inhibitory factor receptor (LIFR). CNTFR induces the secretion and is required for functional responses to the cardiotrophin-like cytokine (CLC) (53). CLC can be secreted either in association with sCNTFR or with another solublelα receptor-homologue, the cytokine-like factor-1 (CLF-1), thus forming two different heterodimeric cytokines. sCNTFR/CLC is active on cells expressing gp130 and LIFR, whereas CLC/CLF-1 activity also requires the transmembrane CNTFR (53).
Regulation of IL-12 and IL-23 production
Whereas the production of IL-12 has been extensively characterized in many in vitro and in vivo situations, much less is known about IL-23. This cytokine was only recently described, and specific immunoassays to measure it have been available only for the last couple of years.
Although IL-12 was purified from the supernatants of EBV-transformed cell lines (24), it was soon shown that it is produced prevalently by myeloid cells and DCs in response to microbial stimuli, such as those mediated by bacteria, fungi, viruses, and intracellular parasites (30). The early production of IL-12 during the innate response to infections (30) and the ability of IL-12 to sustain Th1 adaptive responses (54) have provided evidence for the role of IL-12 as a bridge between innate resistance and adaptive immunity.
IL-12 and IL-23 are produced in response to many infectious or inflammatory stimuli. Both genes encoding IL-12 or IL-23 need to be coordinately expressed in the same cells to produce the biologically active heterodimers (55). In the absence of IL-12p35 or IL-23p19, p40 is secreted as a monomer or a homodimer, whereas p35 and p19 can be secreted only when associated with p40 (55) (Fig. 1). IL-12p35 mRNA is present in many cell types, including T cells, that are not known to produce IL-12, but it has never been detected in the cell-free supernatant by a free p35-specific radioimmunoassay, whereas accumulation of p40 mRNA is mostly restricted to cells that produce the IL-12 heterodimer (30). The recent description of the ability of IL-12p35 to associate with the new cytokine IL-35 in a subset of T cells (51) may provide an explanation for the observed expression of p35 in cells not producing IL-12. However, the almost ubiquitous expression of p35 has made it difficult to analyze its regulation when mixed populations of cells were used; when purified populations were used (e.g. monocytes or DCs), it became evident that upon cell activation the transcription of both genes is induced (56). Because of the lower abundance of p35 mRNA than p40 in IL-12-producing cells, it is generally accepted that p35 expression is the rate-limiting element for the heterodimer production (57), although the formation of the IL-12 heterodimers is also controlled by post-transcriptional mechanisms (58). A similar situation also exists for IL-23, and the accumulation of p35 and p19 transcripts rather than p40 is in general a better predictor of IL-12 and IL-23 production, respectively.
Fig. 1. IL-12 and IL-23 are heterodimeric cytokines sharing a common chain.

(A). IL-12 p35 and IL-23 p19 are secreted by human DCs and monocytes only as high molecular weight complexes with IL-12/23 p40, whereas the IL-12/23 p40 is secreted free or complexed with either IL-12 p35 or IL-23 p19. IFN-γ-treated or untreated human monocyte-derived DCs were [35S]-methionine labeled and stimulated with LPS (1 yg/ml). Supernatants were immunoprecipitated after 18 h with anti-IL-12 p40 antibodies (left panel). Supernatants from the IFN-γ-primed cells (right panel) were also immunoprecipitated with anti-IL-12 p35 antibodies. The sample was split in two equal parts, immunoprecipitated with anti-IL-23 p19 or anti-IL-12 p40 antibodies, and then analyzed by SDS-PAGE under non-reducing conditions. (B). IL-23 heterodimers have various isoforms from 51 kDa to 61 kDa, mostly due to variable glycosylation of the p40 chain, whereas the p19 chain is homogenous and not N-glycosylated. IL-4- and IFN-γtreated human peripheral blood monocytes were [35S]-methionine labeled and stimulated with LPS (1 yg/ml). Supernatants (SN) and cell lysates (CL) were immunoprecipitated after 7 h with anti-IL-23 p19 antibodies and purified in non-reducing SDS-PAGE (left panel). The CL61 kDa, CL57 kDa, SN61 kDa, SN57 kDa and SN51 kDa bands were separately cut out from the gel, reduced, alkylated, and resolved in SDS-PAGE under reducing conditions (right panel). (C). Two-dimensional-peptide mapping shows that the bands corresponding to the p19, p35, and p40 chains are molecularly distinct proteins. The supernatants from the same cells used in (B) were immunoprecipitated with anti-IL-12 p35 (left gel, left lane) and, after exhaustive depletion for p35, sequentially immunoprecipitated with anti-IL-12 p40 antibodies (left gel, right lane) and resolved in SDS-PAGE under reducing conditions. The 35 kDa and the 33 kDa bands derived from the anti-IL-12 p35 immunoprecipitate corresponding to the p35 glycosylation isoforms of p35 derived from the IL-12 heterodimer, the 41 kDa and 34 kDa corresponding to glycosylation isoforms of IL-12/23 p40, and the 20 kDa band corresponding to p19 chain derived from the IL-23 heterodimer immunoprecipitated material were separately cut out from the gel. After reduction, alkylation, and pepsin digestion, the samples were analyzed in two dimensional-peptide mapping (TLC with electrophoresis in the first dimension and ascending chromatography in the second dimension). Modified from Ref. (63).
Innate effector cells detect foreign microorganisms through a limited number of germ-line-encoded PRRs that recognize conserved molecular patterns essential for the survival of the microorganisms (1, 59). The simultaneous engagement of distinct PRRs often cooperates in the innate response to microorganisms or their products, influencing both the magnitude and the quality of the immune response (47). Products from microorganisms, including bacteria, intracellular parasites, viruses, fungi, double-stranded RNA, bacterial DNA, and CpG-containing oligonucleotides, are strong inducers of IL-12 and IL-23 production in macrophages, monocytes, neutrophils, and DCs (56). The relative efficiency of the various inducers depends on the differential expression of the TLRs and other PRRs on phagocytic cells and DC subsets that these products engage (60, 61). Ligands for single TLRs are not very efficient in inducing production of IL-12 heterodimers, and they often induce only low levels of p40. However, as also observed with other pro-inflammatory cytokines, IL-12 is synergistically induced when more than one PRR is engaged. This synergy was observed when two or more TLRs were simultaneously triggered, particularly when a TLR (e.g. TLR7, TLR8, TLR9, TLR11) coupled with the adapter myeloid differentiation factor 88 (MyD88) and a TLR (e.g. TLR3, TLR4) coupled with the adapter TRIF [Toll-IL-1R (TIR)-domain-containing adapter-inducing IFN-β] were stimulated (62). However, synergy was also observed between TLRs and other PRRs, such as the intracellular NOD-like receptors or membrane-activating receptors (63). Protein secretion and gene expression of tumor necrosis factor (TNF), IL-1β, IL-6, IL-10, IL-12, and IL-23 were shown to be several-fold higher in DCs stimulated with combinations of TLR ligands compared with cells stimulated by a single agonist (62, 64). However, microarray analysis showed that this synergy does not apply to all genes induced by TLR stimulation and that only 1% of the genes induced by a single TLR ligand were synergistically increased when a combination of TLR ligands was used (64). Thus, the regulation of genes by combining TLR ligands is selective and seems to affect prevalently gene products involved in the regulation of the innate and adaptive immune response to pathogens that often express multiple TLR ligands.
In addition to the synergistic interaction between various PRRs, various cytokines such as IFN-γ and IL-4 also enhance IL-12 production (65, 66). Although IFN-γ enhances the transcription of both p40 and p35 genes, it dramatically increases the level of secreted IL-12 heterodimer and the ratio of heterodimers versus free p40. Whether IFN-γ accomplishes this by inducing an optimal ratio between the p40 and p35 transcripts or possibly by affecting post-transcriptional mechanisms remains to be determined (66). The ability of IFN-γ to enhance IL-12 production represents a positive feedback mechanism during inflammatory and Th1 responses. The effect of IFN-γ on the production of IL-23 is still incompletely characterized, and IFN-γ in some conditions has been reported to be ineffective or even to inhibit IL-23 production (63, 67).
The two Th2 cytokines, IL-4 and IL-13, are surprisingly also potent enhancers of IL-12 production (68). The effect of IL-4 and IL-13 on the expression of the p40 gene is bimodal: at early times (< 24 h) they inhibit p40 production, whereas at later times they strongly enhance it. IL-4 and IL-13 act by amplifying the transcription of both the p40 and, particularly, the p35 gene, and they are even more efficient than IFN-γ in augmenting the production of the heterodimer (68, 69). The effect of these two cytokines is particularly noteworthy in the case of DCs, because they are often used for the generation of DCs in vitro from monocytes or hematopoietic precursor cells, thus explaining the ability of DCs obtained in these culture systems to produce more IL-12 than ex vivo-purified DCs (70, 71).
T cells enhance IL-12 production not only through production of cytokines such as IFN-γ and IL-4 but also through direct cell-to-cell interaction, mostly through receptor ligands of the TNF family, best exemplified by the interaction of CD40L on activated T cells with CD40 on DCs or macrophages (72–74). Priming with a bacterial stimulus induces CD40 expression on DCs and makes them responsive to CD40L for optimal IL-12 production (73). CD40L has also been shown to be able to synergize with TLR signaling for IL-23 production (75). The second signals for IL-12 and possibly IL-23 production may come not only from activated T cells in adaptive immunity but also from effector cells of innate resistance such as NK cells, both through secretion of IFN-γ and IL-13 and through cell-to-cell interactions (76).
When DCs are stimulated by an individual TLR ligand, they produce negligible if any IL-12, unless they are also costimulated by either secreted (for example, IFNγ) or membrane-bound (for example, CD40L) signals derived from T cells or NK cells (66, 77). Thus, the question arises whether IL-12 is the initial signal for innate IFN-γ production and Th1 response to pathogens, or whether it is produced only at later times during an infection when NK cells or T cells are already activated and simply acts as an amplifier of the initial response (74). When purified DCs are simultaneously stimulated with ligands for two TLRs, they produce levels of IL-12 as high as or higher than those produced in response to a single TLR ligand and costimulation (62, 78). Fig. 2 summarizes the major regulatory mechanisms likely involved in the regulation of IL-12 production during an infection.
Fig. 2. Regulation of IL-12 production in DCs during infection.
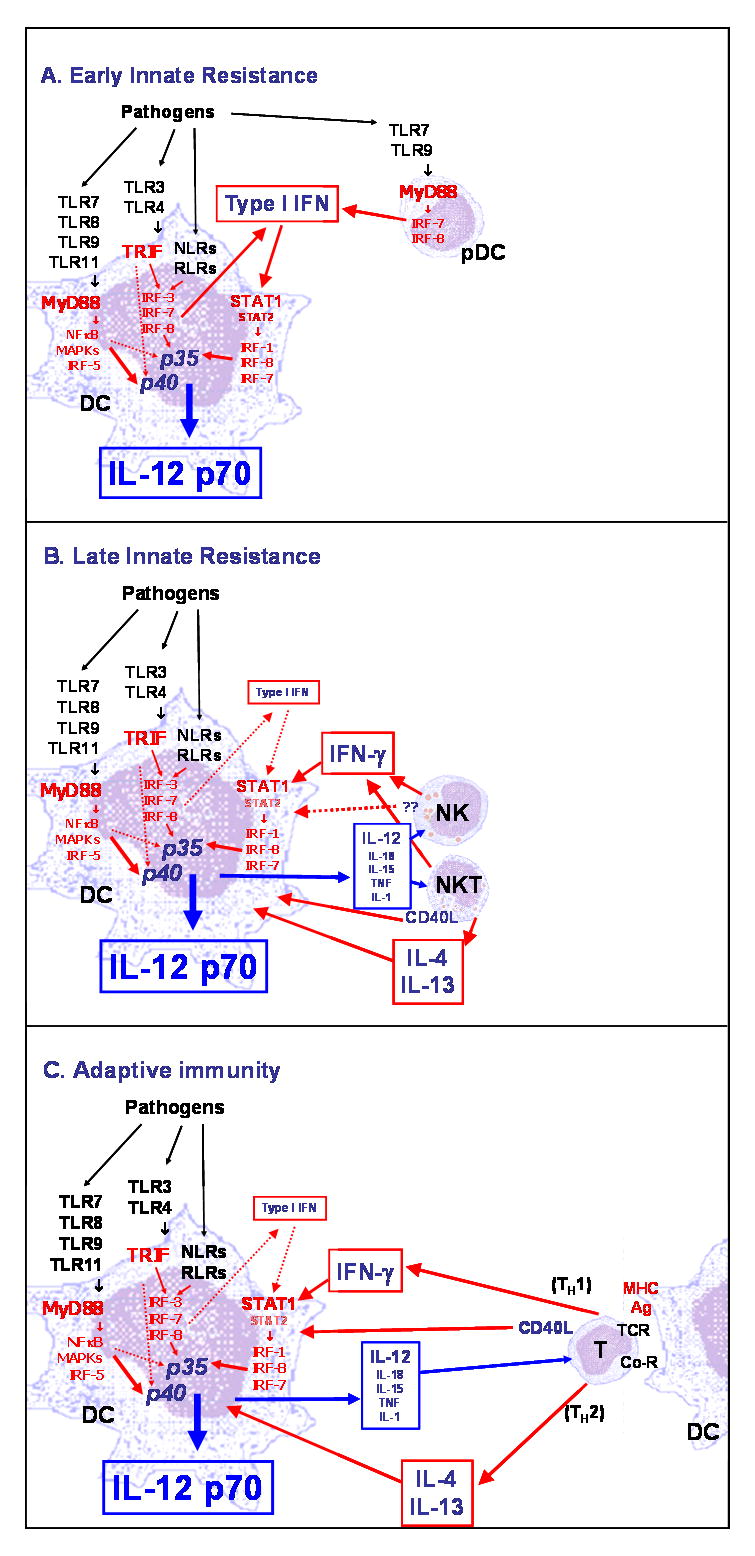
(A). Early innate resistance. DCs exposed during infections to ligands of a single PRR produce IL12 p40 but negligible levels of the biologically active IL-12 heterodimer. However, significant amounts of IL-12 p70 are produced when pathogens simultaneously present ligands for different PRRs (62, 64). The synergy could be due in part to a preferential induction of IL-12 p40 transcription by MyD88-linked TLR and of IL-12 p35 by TRIF-linked TLR but also likely be due to an interaction among the different signaling pathways affecting the production of IL-12 p70 at the transcriptional and post-transcriptional levels. Optimal transcription of the gene encoding IL-12 p35 and production of IL-12 p70 requires phosphorylation of STAT1 and endocrine production of type I IFN that is induced by stimulation of TRIF-associated TLR and nucleic acid-recognizing cytoplasmic receptors, but also at very low level by MyD88-associated TLRs with the exception of TLR2 (62). Type I IFN upregulates transcriptional factors such as IRF1, IRF8, and possibly IRF7 that are required for transcription of the p35 gene and that are then activated by TLR signaling (191, 192). Type I IFN is produced by DCs by stimulation of TRIF-coupled TLRs as well as NOD-like receptors (NRLs) and RIG-I-like receptors (RLRs). However, the levels of endogenous type I IFN required for optimal IL-12 production are very low, and the amount of type I IFN is not a significant limiting factor for IL-12 p70 production (62). Thus, early production of IL-12 p70 by DCs is likely to be cell autonomous. In response to TLR7 and 9 ligands, expressed by viruses and other pathogens, plasmacytoid DCs produce very high levels of type I IFN (193) within a few hours of infection, which may replace the need for endocrine production of type I IFN by conventional DCs. However, high levels of type I IFN inhibit transcription of the IL-12p40 gene (194), indicating a complex role of type I IFN in the regulation of IL-12 p40, p35, and p70. (B). Late innate resistance. IL-12 and other pro-inflammatory cytokines such as IL-18, IL-15, TNF, and IL-1 produced by DCs rapidly induce NK, NKT, and activated/effector T cells to produce IFN-γ and, in the case of NKT cells, IL-4 and IL-13. IFN-γ as well as IL-4 and IL-13 enhance the production of IL-12 by DCs and overcome the need for endogenous type I IFN. Activated NK and NKT cells also increase IL-12 production in DCs by cellular contacts through membrane receptor/ligand interaction that, in the case of NKT cells, may include CD40/CD40L interaction (73, 76, 195). (C). Adaptive immunity. When adaptive immunity is established at 5 to 7 days following infection, the IFN-γ produced by antigen-specific Th1 cells is important for amplifying DC production of IL-12 that is required for maximal production of IFN-γ and optimal Th1 responses. In addition, activated T cells also increase IL-12 p70 production by interacting with DCs through CD40L-CD40 interaction (73). IL-4 and IL-13 produced by Th2 cells also are important costimulators of IL-12 p70 production in DCs (68).
In response to a single or combination of TLR ligands, the production of IL-12 as well as the accumulation of IL-12p35 mRNA, but not the production of IL-12p40 or IL-6, were shown to be dependent on type I IFN signaling: DCs from mice genetically deficient for the type I IFN receptor or human DCs treated with a neutralizing antibody to the type I IFN receptor produced very low levels of IL-12 (62, 78). The role of endogenous type I IFN in IL-12 production still needs to be fully elucidated. At least low levels of type I IFN production were observed in all the conditions in response to single or combinations of TLR ligands in which IL-12 was induced. Thus, type I IFN is necessary for optimal IL-12 production, but it is not responsible for the TLR synergism. These results were surprising, because although type I IFNs have been shown to have some overlapping functions with IL-12 through STAT4 activation in the induction of IFN-γ production and Th1 responses, they have also been shown at high concentration to suppress IL-12 production by decreasing IL-12p40 expression (79). However, the IL-12p35 gene promoter is responsive, in part, to IFN signaling, suggesting that type I IFN may have a direct transcriptional effect on IL-12, for example by inducing the expression of the IFN regulatory factor (IRF) family members IRF1 and IRF8 that are involved in p35 transcription (80). It is also possible that type 1 IFN removes negative feedback mechanisms for IL-12 production, for example the activation of the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by TLR3 or TLR4 ligands (81, 82) that we have recently reported to be inhibited by endogenously produced type I IFN in both fibroblasts and DCs (83).
In addition to positive regulatory mechanisms, there are several negative control mechanisms that avoid excessive production of the pro-inflammatory IL-12 and IL-23 and may also regulate the relative production of the two cytokines and the balance between Th1 and Th17 responses. Anti-inflammatory cytokines play an important role in regulating the production of IL-12 and IL-23 and often act as negative feedback mechanisms for avoiding excessive inflammation. IL-10, an important cytokine that maintains the balance between effective resistance against pathogens and detrimental systemic inflammation, is a potent inhibitor of IL-12 production by blocking transcription of both its genes through induction of the synthesis of repressor proteins and possibly acting by preventing the transcription of nuclear factor-κB (NF-κB)-induced genes (84–87). The essential regulatory role of IL-10 is demonstrated by the uncontrolled lethal systemic inflammatory response to various pathogens in IL-10-deficient mice (88–90). In addition to IL-12, IL-10 is also an important inhibitor of IL-23 production (63). Transforming growth factor-β (TGF-β) is also an inhibitor of IL-12 production that, unlike IL-10, also reduces the stability of the IL-12 p40 mRNA (91). Surprising was the finding that TNF, another cytokine strictly intertwined with IL-12 in the regulation of the inflammatory response and IFN-γ production, also suppresses IL-12 production (56, 92).
IL-12 production is severely inhibited by ligand binding to Gαs-linked G-protein coupled receptors (GPCRs), mostly through induction of cyclic adenosine monophosphate (cAMP) (93). This was initially reported for IL-12 suppression by prostaglandin E2 (PGE2) (94) and then extended to other Gαs-linked receptors such as α2 adrenergic receptors, adenosine receptor A2a, histamine receptor H2, and vasoactive intestinal peptide receptor (93). Interestingly, PGE2 has been shown to have the opposite effect on IL-23. It increases IL-23 production (95–97), and DCs induced to differentiate in the presence of PGE2 produce more IL-23 than IL-12 (98).
In addition to the differential role of GPCR ligands in regulating either cytokine mentioned above, other conditions that prevalently induce one of the two cytokines have been described. In particular, it has been described that Mycobacterium tuberculosis (63, 99) and zymosan induce preferentially the production of IL-23 versus IL-12 (63, 100, 101). This observation has implicated the possible important role of TLR2, a receptor involved in the recognition of both organisms, and the β-glucan receptor dectin-1 (101) in the regulation of IL-23 production. Indeed, TLR2, unlike most TLRs, has been previously shown to be a relatively poor inducer of IL-12 production (102). Below we discuss our recent studies (63) analyzing the interaction of the receptors triggered by either M. tuberculosis or zymosan in the regulation of IL-12 and IL-23.
Transcriptional regulation of the genes encoding IL-12 and IL-23 chains
An analysis of nucleosome positioning, chromatin remodeling, and transcription factor binding in the regulation of the murine IL-12 p40 gene transcription (103) revealed that the p40 gene promoter in macrophages is normally configured in a nucleosome array. Upon activation of macrophages with TLR ligands such as lipopolysaccharide (LPS), nucleosome 1 is selectively remodeled such that it allows the accessibility of this region by the transcription factor CCAAT/enhancer binding protein (C/EBP) (103). The remodeling of nucleosome 1 is not sufficient, however, for p40 gene transcription, and the p40 promoter contains several elements that are functionally important for its inducible expression (56). An NF-κB element was found to be important for promoter activation in response to LPS (104). A downstream C/EBP site cooperates with Rel proteins binding to the NF-κB site, and overexpression of C/EBPβ was found to be sufficient for p40 promoter activity (105, 106). However, in macrophages from C/EBPβ-deficient mice, p40 transcription was enhanced, while the transcription of the p35 gene and the production of heterodimers were severely impaired (107). An ETS consensus element upstream of the NF-κB binds two members of the ETS family of transcriptional factors, ETS-2 and PU.1, that form a large supramolecular complex with c-Rel and several members of the IRF family, including IRF-1, IRF-2, and IFN-consensus sequence-binding protein (ICSB) or IRF-8 (106, 108, 109). IRF-1 and IRF-8 appear to be positive regulatory factors and IRF-2 a negative one, although p40 expression is dysregulated in mice deficient in any of the three IRFs (108–112). The transcriptional complex binding to the ETS site is particularly important for the enhanced p40 transcription induced by IFN-γ and LPS (109). Several of the inhibitors of IL-12 production have been shown to affect one or more of the transcriptional factors mentioned above. For example, 1,25-dihydroyvitamin D3 reduces binding to the NF-κB site, Fc receptor ligation prevents binding of PU.1 and the transcription complex to the ETS site, whereas the vasoactive intestinal peptide affects binding to both sites (108, 113, 114). A repressor site termed GA-12 (GATA sequence in the IL-12 promoter) was identified between the ETS and the NF-κB sites, which is occupied by a GA-12 binding protein (GAP-12) in unstimulated cells (106). GAP-12 binding was enhanced by treatment with IL-4 and PGE2, two inhibitors of IL-12 p40 expression, and IL-4-mediated early suppression of the IL-12 p40 promoter was shown to be critically dependent on an intact GA-12 sequence (106). Although IL-10 is perhaps the most potent inhibitor of IL-12 p40 transcription, its activity could not be reproduced on the transfected p40 promoter (84).
Analysis of IL-12 p35 gene expression was complicated not only by its ubiquitous expression but also by its low expression, until the priming effect of IFN-γ on its expression was discovered (115). The promoter region of both the mouse and human p35 genes has been cloned (65, 116, 117). It contains putative transcriptional motifs such as Sp1, IFN-γ-response element (γ-IRE), PU.1, and C/EBP. The human p35 gene appears to initiate its transcription from at least two sites: one active in B lymphoblastoid cells, and one active in monocytes (65). The latter follows a ‘TATA’ box-like sequence, suggested in the mouse to be part of an ancestral p35 promoter (117), and it generates a shorter mRNA. Multiple transcription start sites for the murine p35 gene were identified resulting in four p35 mRNA isoforms (118) that are differentially represented in non-stimulated and stimulated cells. Only the isoforms that accumulate after stimulation readily support translation of the p35 chain, indicating that p35 gene expression is regulated by both transcriptional and translational mechanisms (118). The presence of multiple transcription initiation sites in the human and murine IL-12 p35 promoters and the possibility that p35 combined with different chains to form IL-12 and IL-35 in different cell types raises the interesting prospect of cell type specific usage of the promoter.
Like the IL-12p40 and p35 promoter, the activity of the IL-23p19 promoter requires c-Rel (119, 120). c-Rel binds to two proximal NF-κB binding sites in the p19 promoter, and it is absolutely necessary for p19 transcription induced by different TLR triggering (119, 120). The synergy between MyD88 and TRIF-associated TLRs in the transcription of both IL-23 and IL-12-encoding genes is mediated at least in part by the activation of IRF5 (121). Interesting, extracellular signal-regulated kinase (ERK)-mitogen-activated protein kinase (MAPK) has been shown to upregulate expression of IL-23 p19 while it inhibits the expression of IL-12 p40 and has limited effect on IL-12 p35 (122, 123). The ERK dependence of IL-23p19 expression may explain the ability of signaling through TLR2 and dectin-1 receptor to induce both IL-23 and IL-10 production (124).
Post-translational processing of IL-12
IL-12p40 and IL-12p35 chains are processed intracellularly by different mechanisms (58, 125). Whereas processing of p40 conforms to the cotranslational model of signal peptide removal concomitant with translocation into the endoplasmic reticulum (ER), translocation of the p35 pre-protein into the ER is not accompanied by cleavage of the signal peptide; rather, removal of the p35 signal peptide occurs via two sequential cleavages. The first cleavage takes place within the ER at a site localized in the hydrophobic region of the signal peptide. Although the pre-protein is glycosylated upon entry into the ER, the primary cleavage is not affected by glycosylation. Subsequently, the remaining portion of the p35 signal peptide is removed by a second cleavage, possibly involving a metalloprotease, concomitant with additional glycosylation and secretion. Secretion is inhibited by mutation of the second cleavage site or by inhibition of glycosylation with tunicamycin. In contrast, p40 secretion is not affected by inhibition of glycosylation (58, 125). Together, these findings suggest diverse mechanisms for regulation of IL-12 production with an important role for p35 processing in the control of the output of biologically active IL-12 heterodimer. Unlike p35, the p19 chain of IL-23 is not glycosylated (63) (Fig. 1), and the treatment of the producer cells with tunicamycin that prevent the secretion of IL-12 does not affect the production of IL-23 (63). Whether other post-translation mechanisms are important for IL-12 and IL-23 production remains to be investigated.
Induction of IL-23 by M. tuberculosis
Early in our studies, we observed that activation of human DCs with two complex microorganisms, heat-killed M. tuberculosis H37Rv strain and the yeast zymosan, efficiently induced IL-23 but little or no IL-12 (63). This finding was surprising, because these microorganisms contain ligands for different TLRs as well as other PRRs, and the association of TLR agonists such as LPS and the TLR7/8-ligand R848 were able to efficiently induce the production of both IL-23 and IL-12. We observed, however, that the addition of TLR7/8 ligand R848 or IFN-γ priming enabled both H37Rv and zymosan to efficiently induce IL-12 production in DCs with no major effect on IL-23 production (63). Thus, the relative pattern of expression of the two cytokines appears to be strictly dependent on the combination of PRRs engaged by different microorganisms.
M. tuberculosis H37Rv induced IL-12 in DCs only after IFN-γ-priming and/or costimulation with R848, whereas IL-23 was induced by H37Rv alone and modestly affected by costimulation. Mycobacteria have been described to activate DCs through at least four different PRRs (TLR2, TLR4, TLR9, and the cytoplasmic receptor NOD2) (126–130). TLR2 and NOD2 in combination are required for the production of TNF (128). Our results showed that the association of the NOD2 ligand muramyl dipeptide (MDP) with the TLR2 ligands Pam3C or Pam2C or with the TLR7/8 ligand R848 induced both IL-12 and IL-23 in IFN-γ-primed monocyte-derived DCs. Interestingly, however, IL-12 was induced at much higher level by the combination of ligands for NOD2 and TLR7/8, whereas IL-23 was induced by the combination of ligands for NOD2 and TLR2. The relative production of the two cytokines induced by the combinations MDP and Pam3C associated or not with R848 was similar to the pattern induced by H37Rv associated or not with R848, suggesting that NOD2 and TLR2 acting together are crucial receptors for IL-12 and IL-23 expression in response to M. tuberculosis (Fig. 3). We have also shown that DCs from Crohn’s disease patients with a loss-of-function homozygous mutation in the NOD2 gene fail to produce significant levels of IL-12, not only in response to MDP and R848, as expected, but also in response to H37Rv with or without R848 (63).
Fig. 3. Model for the differential regulation of IL-12 and IL-23 production in human dendritic cells by M. tuberculosis or zymosan.

M. tuberculosis induces DCs to produce IL-23, and this production is further increased by IFN-γ. NOD2 and TLR2 are among the main PRRs that recognize M. tuberculosis, and the combination of NOD2 and TLR2 ligands mimics the ability of M. tuberculosis to induce IL-23. Unlike IL-23, M. tuberculosis or the combination of NOD2 and TLR2 ligands induces IL-12 production only if DCs are also stimulated by IFN-γ. Ligands for other TLRs (TLR8 in our model) enhance IL-12 production induced by M. tuberculosis or NOD2 ligands. Dectin-1 and TLR2 are the main PRRs that recognize zymosan. Stimulation of DCs with the dectin-1 ligand β-glucan strongly induces IL-23 production that is increased by costimulation by TLR8 or TLR2 ligands. IFN-γ inhibits the IL-23 production induced by dectin-1 ligands; the association of TLR2 (or TLR8; not depicted) ligands with dectin-1 ligands partially overcomes the inhibition by IFN-γ Dectin-1 ligands induce IL-12 production only when associated with ligands for TLRs such as TLR8. IFN-γ cooperates with the combination of dectin-1 and TLR8 ligands to greatly enhance IL-12 production. TLR2 ligands drastically inhibit IL-12 production induced by β-glucan and TLR8 ligands. The ability of zymosan to induce IL-23 production in a dose-dependent manner can be attributed to the simultaneous engagement of dectin-1 and TLR2, two PRRs that have an activating role for production of this cytokine. The differential production of IL-12 and IL-23 from DCs determines the polarization of Th1 or Th17 T-cell differentiation. IL-12 is essential for the induction of Th1 cells from naive CD4+ T lymphocytes. IL-23, together with TGF-β and IL-6 and with the essential contribution of IL-1β, directs the differentiation of naive CD4+ T cells in Th17 cells. Reproduced from Ref. (63).
The striking ability of NOD2 and TLR signaling to synergize in the differential production of IL-12 or IL-23 as well as previous studies indicating the synergy between NOD2 and TLR for the production of other pro-inflammatory cytokines (128, 131, 132) were at odds with the elegant studies using NOD2−/− mice and other experimental approaches. Such studies and experimental approaches clearly indicated that NOD2 inhibits TLR signaling, and in particular, the TLR2 signaling (133, 134) that we observed to strongly synergize with NOD2 for the induction of IL-23 (63). However, more recent studies appear to have explained this discrepancy (135). When NOD2 is stimulated at the same time as TLR agonists are added, its ability to inhibit TLR signaling is minimal, and, indeed, synergy for the induction of cytokines such as TNF and IL-12 p40 with LPS (TLR4) and CpG (TLR9) is observed (135). However, when human DCs were pretreated with the NOD2 agonist MDP, a profound inhibition of pro-inflammatory cytokine induction by all TLR agonists tested was observed. This inhibition was mediated by the ability of MDP to enhance the activity of IRF4 (135), resulting in inhibition of NFκB activation by TLR signaling but not by TNF signaling (136, 137). These data put forward the interesting concept that when tissues like the gut are exposed to NOD2 ligands, they are desensitized to subsequent induction of inflammation by TLR agonists. This explains the ability of NOD2 agonists, such as MDP, to protect mice and patients from inflammatory bowel disease (135). However, when the NOD2 and TLR ligands are simultaneously expressed on the same pathogen, a synergistic induction of pro-inflammatory cytokines is observed, which, depending on the TLR involved, may preferentially activate the IL-12/Th1 or the IL-23/Th17 axis. It remains to be tested how the NOD2/IRF4 pathway affects the production of IL-23 induced by TLR2 in association with NOD2. This association is particularly of interest, because although the IL-23R polymorphism is associated with both Crohn’s disease and ulcerative colitis, only the former condition shows a strong association with NOD2 polymorphism (138).
In addition to NOD2 and TLR2, M. tuberculosis expresses heat-labile TLR4 ligands that might cooperate with other PRRs for cytokine induction (130). In the mouse, TLR9 plays an important role in IL-12 p40 production, whereas TLR2 is mostly responsible for production of TNF (126). The relative inefficiency of M. tuberculosis to induce IL-12 in human DC cultures may be explained by the fact that human DCs do not express TLR9 (61). However, in vitro costimulation with R848, a ligand for the well-expressed TLR8 on human DCs, may mimic the effect of the mycobacterial TLR9 ligands on mouse DCs.
IL-23 may participate in the protection against M. tuberculosis infection by inducing a Th17 response and facilitating a memory T-cell response (139–143). The emerging role of IL-23, together with the well known and possibly predominant role of IL-12 and IFN-γ in the protection against M. tuberculosis, suggests that a balanced production of IL-23 and IL-12 might be crucial in the defense against this intracellular pathogen (140). Coordinated expression of IL-12 and IL-23 is indeed induced by H37Rv, when IFN-γ is also present.
Although our data and a large body of literature on IL-12 and other pro-inflammatory cytokines suggest that TLRs and NOD2 play the major role in the differential regulation of IL-12/IL-23, recent evidence has suggested the involvement of β-glucan receptor/dectin-1 in M. tuberculosis recognition by human and mouse DCs and in the induction of cytokines (144, 145).
We tested the dectin-1 competitive antagonist laminarin (146) at concentrations that effectively blocked DC activation by zymosan, and we did not find it able to block IL-12/IL-23 induction by M. tuberculosis H37Rv (authors’ unpublished data), arguing against a major role for dectin-1 but consistent with recent findings that recognition of non-virulent M. tuberculosis strains, not the virulent H37Rv, by human cells involves this receptor (144).
Induction of IL-23 by zymosan
We observed that like M. tuberculosis, the yeast zymosan induced IL-23 but not IL-12 production (63). Similar to M. tuberculosis, zymosan also required costimulation of DCs with R848, IFN-γ, or both to efficiently induce IL-12 production. The combination of a TLR2 ligand (Pam2C) and the β-glucan receptor/dectin-1 ligand, β-glucan, which are agonists for the major PRR present in zymosan, allowed us to identify TLR2 as a key PRR that differentially regulates the expression of IL-12 and IL-23 not only in association with NOD2 stimulation but also β-glucan (Figs 3 and 4). Insoluble β-glucan alone induced very high level IL-23 production in human DCs (63), consistent with results recently reported for mouse DCs (100), and production was increased 2–3-fold by costimulation with TLR2 or TLR7/8 agonists. However, IL-12 was only induced by β-glucan and R848 together and not by β-glucan alone or in combination with TLR2 agonists. Most striking, the addition of TLR2 ligands drastically decreased the IL-12 production induced by β-glucan and R848, without affecting the production of IL-23. IL-23 was preferentially induced by high-dose zymosan, whereas IL-12 was more efficiently induced by low-dose zymosan in combination with R848 and/or IFN-γ (63). This paradoxical inverse dose-dependence is likely explained by the differential contribution of dectin-1 and TLR2 ligands in stimulating DCs when zymosan was utilized at low or high doses.
Fig. 4. Role of dectin-1, TLR2, and TLR8 in IL-12 and IL-23 production in DC.

DC treated with IFN-γ (red columns) or left untreated (blue columns) were stimulated with β-glucan or Pam2C (TLR2-ligand), or with a combination of β-glucan plus Pam2C, with or without R848 (TLR7/8-ligand). Cell extracts after 12 h culture were tested for mRNA accumulation using the QuantiGene multiplex assay (left panels). Supernatants after 18 h culture were tested for IL-23 and IL-12 protein production by ELISA (Right panels). Modified from Ref. (63).
IL-10, a potent inhibitor of IL-12 production (85), was induced by zymosan (147, 148), and its production was enhanced by TLR2 ligands, consistent with previous reports (149–151). However, TLR2 ligands continued to exert their inhibitory activity in the presence of a neutralizing anti-IL-10 monoclonal antibody, excluding that IL-10 mediates the inhibition of IL-12 (63). Previous reports described the promotion of Th2 and Treg cell responses by TLR2 ligands via IL-10 production (151, 152). IL-12 promotes Th1 responses and prevents the differentiation of other Th subsets, including Th17, facilitated by IL-23. Thus, the IL-10-independent inhibition of IL-12 and the enhancement of IL-23 production by TLR2 are novel mechanisms by which TLR2 triggering may modulate the Th responses.
Because IFN-γ enhances transcription of both p40 and p35 and as it has a particularly marked effect on production of the IL-12 heterodimer (153), it was reasonable to hypothesize that IFN-γ had a similar effect on IL-23. We (63) showed that IFN-γ was required for optimal IL-12 and IL-23 production in human monocyte-derived DCs stimulated by LPS and in both freshly isolated DCs and monocyte-derived DC stimulated by H37Rv alone or in association with other TLR ligands. However, the effect of IFN-γ on IL-23 production was dependent on the PRR triggered, since IFN-γ effectively inhibited, at both protein and mRNA levels, β-glucan-induced IL-23 production. IFN-γ also inhibited the production of IL-10, as previously reported by others using different inducers (154–156). The mechanism by which IFN-γ inhibits IL-10 reportedly involves the GSK3 and CREB/AP1 transcription factors (156). The finding that both IL-23 and IL-10 were inhibited by IFN-γ raises the possibility of similarities in the pathways by which dectin-1 induces the two genes.
Previous studies have shown that Candida yeasts induced both IL-12 and IL-23, whereas the hyphal form induced only IL-23 (157). Those data are consistent with previous data in mice (158) but not with others reported for human DCs that indicate an inability of the hyphal form to induce IL-12 (159). Because in Candida β-glucan is exposed only in the yeast form and not in the filamentous form (146), it is possible that Candida may induce IL-23 using receptors other than dectin-1. However, laminarin blocked both IL-12 and IL-23 production induced by zymosan (authors’ unpublished results), providing evidence that the β-glucan receptor is involved in the induction of both cytokines by the yeast zymosan. Although the Th1 immune response is still considered the most effective in providing resistance to fungal infection (14), evidence suggesting an important role for the IL-23/IL-17 axis as well as for dectin-1 in anti-fungal resistance is rapidly emerging (157, 160–163).
Differentiation of human Th17 cells
Since their formal identification as a separate T-cell subset (13), there has been much interest in the biology and differentiation of Th17 cells because of the obviously important role that the cytokines produced by these cells play in anti-microbial immunity at mucosal surfaces, autoimmunity, and possibly cancer immunity. IL-23 was originally described as sharing functions with IL-12, particularly as an inducer of IFN-γ production and as a T-cell mitogen, however with a preferential effect on antigen-experienced T cells, unlike IL-12 that is a better inducer of proliferation for naive T cells (28). Although genetically deficient mice and effective neutralizing IL-23 antibodies were not available until recently, the likely involvement of IL-23 in autoimmunity was suggested by results indicating that mice deficient for the p40 chain shared by IL-12 and IL-23 were resistant to models of experimental autoimmunity, such as experimental allergic encephalitis (EAE), whereas mice deficient for the IL-12-specific p35 chain were susceptible, often more so than wildtype mice (25, 164). It was then directly established that IL-23 has an important and sometimes essential role in autoimmune models, such as EAE, colitis, and arthritis (25, 27, 34, 35). The mechanism of induction of autoimmune pathogenesis by IL-23 was connected to its ability to induce production of cytokines of the IL-17 family and others such as IL-6 (37). IL-23 p19 genetically deficient mice and the use of neutralizing antibodies to IL-23 (165) has confirmed this role of IL-23 in the mouse and has opened the possibility that IL-23 might be a good target for the treatment of autoimmunity. The findings discussed above of the association of polymorphisms in the IL23R gene as well and more often in the IL12B gene resulting in several human autoimmunity syndromes and the very promising clinical trials showing a good therapeutic effect of antibodies against the IL-12/IL-23 common chain p40 in psoriasis and in Crohn’s disease (166, 167) clearly supported that possibility.
Because of the almost absolute absence of a Th17 response and IL-17 production in IL-23 p19-deficient mice in the studied models of autoimmunity, it was assumed that IL-23 is necessary and sufficient for the differentiation of the Th17 cells. However, as for the role of IL-12 in the differentiation of Th1 cells, it was found that naive T cells do not express the receptor complex for IL-23 and that the differentiation of Th17 cells and the upregulation of the IL-23 receptors requires TGF-β in the context of an inflammatory environment (168) in which cytokines such as IL-6, but possibly also IL-1 (169, 170), play a major role. The transcription factor RORγT was determined to be necessary for Th17 cytokine production and the expression of the IL-23 receptor complex (171). IL-23 then becomes important for the expansion of the Th17 cells and for increasing their ability to produce IL-17 and other cytokines. IL-21 is also produced by Th17 cells and contributes to their differentiation and expansion with a positive feedback mechanism (172). There has been some controversy whether IL-23 is at all required for Th17 cell differentiation, but it appears that although Th17 cells can differentiate in the absence of IL-23, in most experimental models IL-23 is required for the optimal expansion and activation of Th17 cells. Also, it has been shown that Th17 differentiated in the presence of TGF-β and IL-6 only are not encephalitogenic, but they are endowed with that ability by exposure to IL-23 (173).
The mechanisms of human Th17 cell differentiation were not clear, and a few recently published reports indicate that TGF-β not only was not required for differentiation of human Th17 cells but actually inhibited it (75, 174–176). Most human studies have put more emphasis on the requirement for IL-1 and IL-23 in the differentiation of human Th17 with a role for IL-6 but not for TGF-β. One of the difficulties of the studies with human T cells is that, although naive T cells can be purified to homogeneity, most studies have been performed with partially purified naive T cells, raising the doubt whether the effect of IL-23 observed in some of the published experiments was due to contamination with memory T cells expressing the IL-23 receptor. Cord blood T cells, used in some studies, are always a more reliable source of naive T cells.
It was also recently suggested that human DCs stimulated with different bacteria and bacterial products induce the production of IL-17 by memory T cells but not differentiation of Th17 cells from naive T cells (177). The factors inducing IL-17 production were identified as IL-23 and both IL-1α and IL-1β. In that study, IL-6 and TGF-β were shown to increase IL-17 production and not to inhibit it. In particular, the combination of the NOD2 agonist MDP and TLR2 agonists, mimicking exposure to mycobacteria, induced DCs to produce IL-23 and IL-1 that elicited the production of IL-17 by human CD4+ T cells (177). These results probably need to be reevaluated on the light of the present knowledge that MDP in addition to being a NOD2 agonist can also activate the inflammasome and induce IL-1β processing independently of NOD2 (178–180). However, the use of NOD2-deficient Crohn’s disease patients (177) confirmed that NOD2 was necessary even if possibly not sufficient.
We (63) tested whether the IL-23-containing supernatants from human DCs stimulated by zymosan or β-glucan were able to induce production of IL-17 from naive human CD4+ T cells stimulated polyclonally (Fig. 5). We observed a robust IL-17 production that was not affected or was slightly enhanced by an antibody specifically neutralizing IL-12, whereas it was drastically diminished by an antibody to IL-12 p40, neutralizing both IL-12 and IL-23. IL-1 was required for IL-17 induction in human CD4+ cells as shown by the ability of IL-1RA to completely abolish IL-17 production. Neutralization of IL-23, TGF-β, or IL-6 resulted in a profound but not complete inhibition of IL-17 production (63). These neutralization studies in combination with studies in which we added combinations of recombinant cytokines to the cultured human CD4+ cells indicated that IL-1 together with IL-23, IL-6, and TGF-β were necessary for IL-17 production (63) (Fig. 5). However, even if our studies clearly showed that TGF-β has an important and necessary role for the optimal human Th17 cell differentiation, adding an excessive amount of exogenous TGF-β to the cultures results in inhibition of both IL-17 and IFN-γ production, whereas the addition of a neutralizing anti-TGF-β antibody to CD4+ T cells cultured in the presence of the supernatants from activated DCs inhibited IL-17 production but increased IFN-γ production. In our studies, we used preparations of human peripheral blood naive CD4+ T cells that were approximately 98–99% pure. We found that human T cells with a memory phenotype were not more responsive than the naive T cells to the stimulation with IL-23 and IL-1, excluding that our results could be due to a contamination with memory T cells. Our observation that IL-23 induces human naive T cells to produce IL-17 is consistent with others that used cord blood CD4+ T cells (19, 181). It is possible that at least a subset of naive T cells already express small levels of IL-23 receptor or that the other cytokines necessary for differentiation of Th17 T cells, e.g. IL-1, induce the expression of the receptor on the naive T cells and then IL-23 can act on them amplifying IL-17 production. It is apparent, however, that in cultures of human naive or memory T cells, IL-23 is very important for the induction of cells producing IL-17, although not as essential as IL-1.
Fig. 5. Induction of IL-17, IL-22, and IFN-γ in human CD4+ T cells by supernatants of stimulated monocyte-derived DCs.

Supernatants from differently stimulated DCs were used to induce IL-17, IL-22, and IFN-γ production from human CD4+CD45RO− T lymphocytes stimulated with anti-CD3 and anti-CD28. (A) Supernatants from IFN-γ-primed DCs stimulated with 200 yg/ml zymosan or 1 yg/ml LPS+R848 or from unprimed DCs stimulated with 10 yg/ml β-glucan were evaluated by ELISA for IL-12, IL23, IL-6, and IL-1β production and for the capacity to induce IL-17 and IFN-γ in naive CD4+ T cells (mean ± SE from 10 independent experiments). (B) IL-17 and IFN-γ production was determined by ELISA in naive T-cell cultures in the presence of supernatants from zymosan- or β-glucan–stimulated DCs and in the presence or absence of neutralizing anti–IL-12 p70 (20C2) or anti-p40 (C8.6) antibodies (mean ± SE from seven independent experiments). (C) Effect of neutralizing anti–TGF-β or anti–IL-6 or a mixture of both antibodies on IL-17, IL-22, and IFN-γ production measured by ELISA in naive T cells cultured in the presence of supernatants from zymosan-stimulated DCs. (D) Effect of 10 ng/ml IL-1β on IL-17 and IFN-γ production, as determined by ELISA in naive T cells cultured in the presence of IL-23 or an IL-12–depleted supernatant from LPS+R848–stimulated monocyte-derived DCs. Supernatants were diluted to contain IL-23 at a final concentration of 15 ng/ml. Recombinant IL-23 was used at 15 ng/ml. (C) and (D) show the mean ± SD from triplicate cultures. Similar results were obtained in three independent experiments. Modified from Ref. (63).
We were surprised that the supernatant fluid from DCs stimulated with LPS+R848, which contained equivalent amounts of IL-23 and IL-6 as the supernatant fluids from DCs stimulated with β-glucan or zymosan, failed to induce IL-17 production. We found that the inability of the LPS+R848-induced supernatant to induce IL-17 production was due to the absence of high IL-1β concentrations that were present in the zymosan- and β-glucan-induced supernatants (63). Because the secretion of bioactive IL-1β depends on processing through caspases in the inflammasome, we considered whether the dectin-1 receptor was able to activate the inflammasome, and we confirmed that induction of bioactive IL-1β by β-glucan was blocked by caspase-1 inhibitor, whereas LPS did not induce IL-1β unless adenosine triphosphate (ATP) was added to activate the inflammasome (authors’ unpublished results). Indeed, a recent report (182) conclusively showed that a number of PRR ligands, including zymosan but not LPS, induced cells to excrete ATP that then activated the inflammasome inducing pro-IL-1β processing. Interestingly, it has also been reported that IL-1 may be important in the production of IL-23 by synoviocytes (183), as it was previously shown for IL-12 production by DCs (184). We also observed that caspase-1 inhibitors or IL-1 receptor antagonist (IL-1RA) significantly decreased the production of IL-23 by DCs stimulated with β-glucan (authors’ unpublished observation). Thus, these results strongly suggest that activation of the inflammasome is necessary both for the production of IL-23 and for the development of Th-17 responses.
IL-22 is a cytokine belonging to the IL-10 family originally named IL-10-related T-cell-derived inducible factor (IL-TIF) and thought to be produced specifically by Th1 cells (185, 186). IL-22 shares 22% homology with IL-10, and its receptors are expressed on a variety of epithelial cells on which the cytokine has been shown to exert both damaging and protective effect (185, 186). It was then observed that in human T cells the expression of IL-22 overlaps partially with that of Th17 cells rather than with Th1 cells (187). In vitro IL-22 production by human CD4+ T cells was shown to be induced both in Th1 and Th17 conditions, but IL-6 and TGF-β were shown to inhibit rather than enhance IL-22 production in Th17 co0nditions (19, 188). We observed (Fig. 5) that the supernatant of zymosan-stimulated DCs induces human naive CD4+ T cells to produce both IL-17 and IL-22. Unlike IL-17, the production of IL-22 was enhanced when endogenous TGF-β or IL-6 were neutralized, and it was not significantly affected by inhibition of IL-1 action by IL-1RA. Thus, in human naive T cells, IL-17, IL-22, and IFN-γ are regulated differently.
A series of recent papers (19, 181, 189, 190) using naive peripheral and cord blood naive CD4+ T cells have now clearly established that as in mice, TGF-β plays an important role in the differentiation of human Th17 cells together with IL-1, IL-6, IL-23, and IL-21. Much obviously remains to be studied regarding the exact molecular mechanisms of Th17 cell differentiation in humans compared to mice. Clearly it is prudent to avoid easy extrapolations between the two species, but it is also important not to rush in seeing diversity when it is not there and thus deny the enormous value of mouse immunology, which if intelligently used, can serve as a beacon to orient us in the technically and organizationally challenging human studies. However, it is time to devote more attention to human studies, in parallel to those in experimental animals, and to apply to them the same advanced technology and scientific scrutiny that are state of the art in the mouse studies.
References
- 1.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Creagh EM, O’Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006;27:352–357. doi: 10.1016/j.it.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 4.Janeway CA., Jr Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54:1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Trinchieri G. Biology of natural killer cells. Adv Immunol. 1989;47:187–376. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trinchieri G, et al. Natural killer cell stimulatory factor (NKSF) or interleukin-12 is a key regulator of immune response and inflammation. Prog Growth Factor Res. 1992;4:355–368. doi: 10.1016/0955-2235(92)90016-b. [DOI] [PubMed] [Google Scholar]
- 7.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 8.Rouvier E, Luciani MF, Mattei MG, Denizot F, Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J Immunol. 1993;150:5445–5456. [PubMed] [Google Scholar]
- 9.Yao Z, et al. Human IL-17: a novel cytokine derived from T cells. J Immunol. 1995;155:5483–5486. [PubMed] [Google Scholar]
- 10.Yao Z, Timour M, Painter S, Fanslow W, Spriggs M. Complete nucleotide sequence of the mouse CTLA8 gene. Gene. 1996;168:223–225. doi: 10.1016/0378-1119(95)00778-4. [DOI] [PubMed] [Google Scholar]
- 11.Fossiez F, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Broxmeyer HE. Is interleukin 17, an inducible cytokine that stimulates production of other cytokines, merely a redundant player in a sea of other biomolecules? J Exp Med. 1996;183:2411–2415. doi: 10.1084/jem.183.6.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrington LE, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 14.Romani L. Cell mediated immunity to fungi: a reassessment. Med Mycol. 2008:1–15. doi: 10.1080/13693780801971450. [DOI] [PubMed] [Google Scholar]
- 15.Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13:139–145. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- 16.Bronte V. Th17 and cancer: friends or foes? Blood. 2008;112:214. doi: 10.1182/blood-2008-04-149260. [DOI] [PubMed] [Google Scholar]
- 17.Langowski JL, Kastelein RA, Oft M. Swords into plowshares: IL-23 repurposes tumor immune surveillance. Trends Immunol. 2007;28:207–212. doi: 10.1016/j.it.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Trinchieri G. Interleukin-10 production by effector T cells: Th1 cells show self control. J Exp Med. 2007;204:239–243. doi: 10.1084/jem.20070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Volpe E, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650–657. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 20.Ben-Sasson SZ, Le Gros G, Conrad DH, Finkelman FD, Paul WE. Cross-linking Fc receptors stimulate splenic non-B, non-T cells to secrete interleukin 4 and other lymphokines. Proc Natl Acad Sci USA. 1990;87:1421–1425. doi: 10.1073/pnas.87.4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Min B, et al. Basophils produce IL-4 and accumulate in tissues after infection with a Th2-inducing parasite. J Exp Med. 2004;200:507–517. doi: 10.1084/jem.20040590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 23.Boonstra A, et al. Flexibility of mouse classical and plasmacytoid-derived dendritic cells in directing T helper type 1 and 2 cell development: dependency on antigen dose and differential toll-like receptor ligation. J Exp Med. 2003;197:101–109. doi: 10.1084/jem.20021908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kobayashi M, et al. Identification and purification of Natural Killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med. 1989;170:827–846. doi: 10.1084/jem.170.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cua DJ, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 26.Langrish CL, McKenzie BS, Wilson NJ, de Waal Malefyt R, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev. 2004;202:96–105. doi: 10.1111/j.0105-2896.2004.00214.x. [DOI] [PubMed] [Google Scholar]
- 27.Murphy CA, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oppmann B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 29.Cooper AM, Khader SA. IL-12p40: an inherently agonistic cytokine. Trends Immunol. 2007;28:33–38. doi: 10.1016/j.it.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 30.D’Andrea A, et al. Production of natural killer cell stimulatory factor (interleukin 12) by peripheral blood mononuclear cells. J Exp Med. 1992;176:1387–1398. doi: 10.1084/jem.176.5.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parham C, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 32.Filipe-Santos O, et al. Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol. 2006;18:347–361. doi: 10.1016/j.smim.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 33.Langowski JL, et al. IL-23 promotes tumour incidence and growth. Nature. 2006;442:461–465. doi: 10.1038/nature04808. [DOI] [PubMed] [Google Scholar]
- 34.Hue S, et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–2483. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kullberg MC, et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med. 2006;203:2485–2494. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uhlig HH, et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25:309–318. doi: 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 37.Yen D, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duerr RH, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Begovich AB, et al. The autoimmune disease-associated IL12B and IL23R polymorphisms in multiple sclerosis. Hum Immunol. 2007;68:934–937. doi: 10.1016/j.humimm.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 40.Brionez TF, Reveille JD. The contribution of genes outside the major histocompatibility complex to susceptibility to ankylosing spondylitis. Curr Opin Rheumatol. 2008;20:384–391. doi: 10.1097/BOR.0b013e32830460fe. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y, et al. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet. 2008;4:e1000041. doi: 10.1371/journal.pgen.1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nunez C, et al. IL23R: a susceptibility locus for celiac disease and multiple sclerosis? Genes Immun. 2008;9:289–293. doi: 10.1038/gene.2008.16. [DOI] [PubMed] [Google Scholar]
- 43.Rueda B, et al. The IL23R Arg381Gln non-synonymous polymorphism confers susceptibility to ankylosing spondylitis. Ann Rheum Dis. 2008 doi: 10.1136/ard.2007.080283. in press. [DOI] [PubMed] [Google Scholar]
- 44.Fisher SA, et al. Genetic determinants of ulcerative colitis include the ECM1 locus and five loci implicated in Crohn’s disease. Nat Genet. 2008;40:710–712. doi: 10.1038/ng.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang M, et al. The inflammatory disease-associated variants in IL12B and IL23R are not associated with rheumatoid arthritis. Arthritis Rheum. 2008;58:1877–1881. doi: 10.1002/art.23492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pflanz S, et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4(+) T cells. Immunity. 2002;16:779–790. doi: 10.1016/s1074-7613(02)00324-2. [DOI] [PubMed] [Google Scholar]
- 47.Jankovic D, Trinchieri G. IL-10 or not IL-10: that is the question. Nat Immunol. 2007;8:1281–1283. doi: 10.1038/ni1207-1281. [DOI] [PubMed] [Google Scholar]
- 48.Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol. 2007;25:221–242. doi: 10.1146/annurev.immunol.22.012703.104758. [DOI] [PubMed] [Google Scholar]
- 49.Devergne O, Birkenbach M, Kieff E. Epstein-Barr virus-induced gene 3 and the p35 subunit of interleukin 12 form a novel heterodimeric hematopoietin. Proc Natl Acad Sci USA. 1997;94:12041–12046. doi: 10.1073/pnas.94.22.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Devergne O, et al. A novel interleukin-12 p40-related protein induced by latent Epstein-Barr virus infection in B lymphocytes. J Virol. 1996;70:1143–1153. doi: 10.1128/jvi.70.2.1143-1153.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Collison LW, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 52.Niedbala W, et al. IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. Eur J Immunol. 2007;37:3021–3029. doi: 10.1002/eji.200737810. [DOI] [PubMed] [Google Scholar]
- 53.Lelievre E, et al. Signaling pathways recruited by the cardiotrophin-like cytokine/cytokine-like factor-1 composite cytokine: specific requirement of the membrane-bound form of ciliary neurotrophic factor receptor alpha component. J Biol Chem. 2001;276:22476–22484. doi: 10.1074/jbc.M101681200. [DOI] [PubMed] [Google Scholar]
- 54.Manetti R, et al. Interleukin 12 induces stable priming for interferon gamma (IFN-gamma) production during differentiation of human T helper (Th) cells and transient IFN-gamma production in established Th2 cell clones. J Exp Med. 1994;179:1273–1283. doi: 10.1084/jem.179.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wolf SF, et al. Cloning of cDNA for natural killer cell stimulatory factor, a heterodimeric cytokine with multiple biologic effects on T and natural killer cells. J Immunol. 1991;146:3074–3081. [PubMed] [Google Scholar]
- 56.Ma X, Trinchieri G. Regulation of interleukin-12 production in antigen-presenting cells. Adv Immunol. 2001;79:55–92. doi: 10.1016/s0065-2776(01)79002-5. [DOI] [PubMed] [Google Scholar]
- 57.Snijders A, Hilkens CM, van der Pouw Kraan TC, Engel M, Aarden LA, Kapsenberg ML. Regulation of bioactive IL-12 production in lipopolysaccharide- stimulated human monocytes is determined by the expression of the p35 subunit. J Immunol. 1996;156:1207–1212. [PubMed] [Google Scholar]
- 58.Carra G, Gerosa F, Trinchieri G. Biosynthesis and posttranslational regulation of human IL-12. J Immunol. 2000;164:4752–4761. doi: 10.4049/jimmunol.164.9.4752. [DOI] [PubMed] [Google Scholar]
- 59.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 60.Jarrossay D, Napolitani G, Colonna M, Sallusto F, Lanzavecchia A. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur J Immunol. 2001;31:3388–3393. doi: 10.1002/1521-4141(200111)31:11<3388::aid-immu3388>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 61.Kadowaki N, et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863–869. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gautier G, et al. A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med. 2005;201:1435–1446. doi: 10.1084/jem.20041964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gerosa F, et al. Differential regulation of interleukin 12 and interleukin 23 production in human dendritic cells. J Exp Med. 2008;205:1447–1461. doi: 10.1084/jem.20071450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hayes MP, Murphy FJ, Burd PR. Interferon-gamma-dependent inducible expression of the human interleukin-12 p35 gene in monocytes initiates from a TATA-containing promoter distinct from the CpG-rich promoter active in Epstein-Barr virus-transformed lymphoblastoid cells. Blood. 1998;91:4645–4651. [PubMed] [Google Scholar]
- 66.Ma X, et al. The interleukin 12 p40 gene promoter is primed by interferon gamma in monocytic cells. J Exp Med. 1996;183:147–157. doi: 10.1084/jem.183.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Verreck FA, et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci USA. 2004;101:4560–4565. doi: 10.1073/pnas.0400983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.D’Andrea A, Ma X, Aste-Amezaga M, Paganin C, Trinchieri G. Stimulatory and inhibitory effects of interleukin (IL)-4 and IL-13 on the production of cytokines by human peripheral blood mononuclear cells: priming for IL-12 and tumor necrosis factor alpha production. J Exp Med. 1995;181:537–546. doi: 10.1084/jem.181.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marshall JD, Robertson SE, Trinchieri G, Chehimi J. Priming with IL-4 and IL-13 during HIV-1 infection restores in vitro IL- 12 production by mononuclear cells of HIV-infected patients. J Immunol. 1997;159:5705–5714. [PubMed] [Google Scholar]
- 70.Cella M, Scheidegger D, Plamer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med. 1996;184:747–752. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kalinski P, et al. IL-4 is a mediator of IL-12p70 induction by human Th2 cells: reversal of polarized Th2 phenotype by dendritic cells. J Immunol. 2000;165:1877–1881. doi: 10.4049/jimmunol.165.4.1877. [DOI] [PubMed] [Google Scholar]
- 72.Macatonia SE, et al. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J Immunol. 1995;154:5071–5079. [PubMed] [Google Scholar]
- 73.Schulz O, et al. CD40 triggering of heterodimeric IL-12 p70 production by dendritic cells in vivo requires a microbial priming signal. Immunity. 2000;13:453–462. doi: 10.1016/s1074-7613(00)00045-5. [DOI] [PubMed] [Google Scholar]
- 74.Abdi K, Singh N, Matzinger P. T-cell control of IL-12p75 production. Scand J Immunol. 2006;64:83–92. doi: 10.1111/j.1365-3083.2006.01767.x. [DOI] [PubMed] [Google Scholar]
- 75.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 76.Gerosa F, Baldani-Guerra B, Nisii C, Marchesini V, Carra G, Trinchieri G. Reciprocal activating interaction between natural killer cells and dendritic cells. J Exp Med. 2002;195:327–333. doi: 10.1084/jem.20010938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Edwards AD, et al. Microbial recognition via Toll-like receptor-dependent and -independent pathways determines the cytokine response of murine dendritic cell subsets to CD40 triggering. J Immunol. 2002;169:3652–3660. doi: 10.4049/jimmunol.169.7.3652. [DOI] [PubMed] [Google Scholar]
- 78.Bekeredjian-Ding I, et al. T cell-independent, TLR-induced IL-12p70 production in primary human monocytes. J Immunol. 2006;176:7438–7446. doi: 10.4049/jimmunol.176.12.7438. [DOI] [PubMed] [Google Scholar]
- 79.Cousens LP, et al. Two roads diverged: interferon alpha/beta- and interleukin 12-mediated pathways in promoting T cell interferon gamma responses during viral infection. J Exp Med. 1999;189:1315–1328. doi: 10.1084/jem.189.8.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu J, Guan X, Tamura T, Ozato K, Ma X. Synergistic activation of interleukin-12 p35 gene transcription by interferon regulatory factor-1 and interferon consensus sequence-binding protein. J Biol Chem. 2004;279:55609–55617. doi: 10.1074/jbc.M406565200. [DOI] [PubMed] [Google Scholar]
- 81.Polumuri SK, Toshchakov VY, Vogel SN. Role of phosphatidylinositol-3 kinase in transcriptional regulation of TLR-induced IL-12 and IL-10 by Fc gamma receptor ligation in murine macrophages. J Immunol. 2007;179:236–246. doi: 10.4049/jimmunol.179.1.236. [DOI] [PubMed] [Google Scholar]
- 82.Martin M, Schifferle RE, Cuesta N, Vogel SN, Katz J, Michalek SM. Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. J Immunol. 2003;171:717–725. doi: 10.4049/jimmunol.171.2.717. [DOI] [PubMed] [Google Scholar]
- 83.Hasan UA, Trinchieri G, Vlach J. Toll-like receptor signaling stimulates cell cycle entry and progression in fibroblasts. J Biol Chem. 2005;280:20620–20627. doi: 10.1074/jbc.M500877200. [DOI] [PubMed] [Google Scholar]
- 84.Aste-Amezaga M, Ma X, Sartori A, Trinchieri G. Molecular mechanisms of the induction of IL-12 and its inhibition by IL- 10. J Immunol. 1998;160:5936–5944. [PubMed] [Google Scholar]
- 85.D’Andrea A, Aste-Amezaga M, Valiante NM, Ma X, Kubin M, Trinchieri G. Interleukin-10 inhibits human lymphocyte IFN-γ production by suppressing natural killer cell stimulatory factor/interleukin-12 synthesis in accessory cells. J Exp Med. 1993;178:1041–1048. doi: 10.1084/jem.178.3.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Murray PJ. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci USA. 2005;102:8686–8691. doi: 10.1073/pnas.0500419102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.El Kasmi KC, et al. Cutting edge: A transcriptional repressor and corepressor induced by the STAT3-regulated anti-inflammatory signaling pathway. J Immunol. 2007;179:7215–7219. doi: 10.4049/jimmunol.179.11.7215. [DOI] [PubMed] [Google Scholar]
- 88.Jankovic D, Kullberg MC, Hieny S, Caspar P, Collazo CM, Sher A. In the absence of IL-12, CD4(+) T cell responses to intracellular pathogens fail to default to a Th2 pattern and are host protective in an IL-10(−/−) setting. Immunity. 2002;16:429–439. doi: 10.1016/s1074-7613(02)00278-9. [DOI] [PubMed] [Google Scholar]
- 89.Gazzinelli RT, et al. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF- alpha. J Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- 90.Belkaid Y, et al. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J Exp Med. 2001;194:1497–1506. doi: 10.1084/jem.194.10.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Du C, Sriram S. Mechanism of inhibition of LPS-induced IL-12p40 production by IL-10 and TGF-beta in ANA-1 cells. J Leukoc Biol. 1998;64:92–97. doi: 10.1002/jlb.64.1.92. [DOI] [PubMed] [Google Scholar]
- 92.Ma X, et al. Inhibition of IL-12 production in human monocyte-derived macrophages by TNF. J Immunol. 2000;164:1722–1729. doi: 10.4049/jimmunol.164.4.1722. [DOI] [PubMed] [Google Scholar]
- 93.Braun MC, Kelsall BL. Regulation of interleukin-12 production by G-protein-coupled receptors. Microbes Infect. 2001;3:99–107. doi: 10.1016/s1286-4579(00)01357-5. [DOI] [PubMed] [Google Scholar]
- 94.van der Pouw Kraan TC, Boeije LC, Smeenk RJ, Wijdenes J, Aarden LA. Prostaglandin-E2 is a potent inhibitor of human interleukin 12 production. J Exp Med. 1995;181:775–779. doi: 10.1084/jem.181.2.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sheibanie AF, Khayrullina T, Safadi FF, Ganea D. Prostaglandin E2 exacerbates collagen-induced arthritis in mice through the inflammatory interleukin-23/interleukin-17 axis. Arthritis Rheum. 2007;56:2608–2619. doi: 10.1002/art.22794. [DOI] [PubMed] [Google Scholar]
- 96.Sheibanie AF, et al. The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23-->IL-17 axis. J Immunol. 2007;178:8138–8147. doi: 10.4049/jimmunol.178.12.8138. [DOI] [PubMed] [Google Scholar]
- 97.Sheibanie AF, Tadmori I, Jing H, Vassiliou E, Ganea D. Prostaglandin E2 induces IL-23 production in bone marrow-derived dendritic cells. FASEB J. 2004;18:1318–1320. doi: 10.1096/fj.03-1367fje. [DOI] [PubMed] [Google Scholar]
- 98.Khayrullina T, Yen JH, Jing H, Ganea D. In vitro differentiation of dendritic cells in the presence of prostaglandin E2 alters the IL-12/IL-23 balance and promotes differentiation of Th17 cells. J Immunol. 2008;181:721–735. doi: 10.4049/jimmunol.181.1.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Khader SA, Cooper AM. IL-23 and IL-17 in tuberculosis. Cytokine. 2008 Feb;41:79–83. doi: 10.1016/j.cyto.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.LeibundGut-Landmann S, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 101.Robinson MJ, Sancho D, Slack EC, LeibundGut-Landmann S, Reis e Sousa C. Myeloid C-type lectins in innate immunity. Nat Immunol. 2006;7:1258–1265. doi: 10.1038/ni1417. [DOI] [PubMed] [Google Scholar]
- 102.Agrawal S, et al. Cutting edge: different Toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J Immunol. 2003;171:4984–4989. doi: 10.4049/jimmunol.171.10.4984. [DOI] [PubMed] [Google Scholar]
- 103.Weinmann AS, et al. Nucleosome remodeling at the IL-12 p40 promoter is a TLR-dependent, Rel-independent event. Nat Immunol. 2001;2:51–57. doi: 10.1038/83168. [DOI] [PubMed] [Google Scholar]
- 104.Murphy TL, Cleveland MG, Kulesza P, Magram J, Murphy KM. Regulation of interleukin 12 p40 expression through an NF-kappa B half- site. Mol Cell Biol. 1995;15:5258–5267. doi: 10.1128/mcb.15.10.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Plevy SE, Gemberling JHM, Hsu S, Dorner AJ, Smale ST. Multiple control elements mediate activation of the murine and human interleukin 12 p40 promoters: evidence of functional synergy between C/EBP and Rel proteins. Mol Cell Biol. 1997;17:4572–4588. doi: 10.1128/mcb.17.8.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Becker C, Wirtz S, Ma X, Blessing M, Galle PR, Neurath MF. Regulation of IL-12 p40 promoter activity in primary human monocytes: roles of NF-kappaB, CCAAT/enhancer-binding protein beta, and PU.1 and identification of a novel repressor element (GA-12) that responds to IL-4 and prostaglandin E(2) J Immunol. 2001;167:2608–2618. doi: 10.4049/jimmunol.167.5.2608. [DOI] [PubMed] [Google Scholar]
- 107.Gorgoni B, Maritano D, Marthyn P, Righi M, Poli V. C/EBPbeta Gene Inactivation Causes Both Impaired and Enhanced Gene Expression and Inverse Regulation of IL-12 p40 and p35 mRNAs in Macrophages. J Immunol. 2002;168:4055–4062. doi: 10.4049/jimmunol.168.8.4055. [DOI] [PubMed] [Google Scholar]
- 108.Cappiello MG, Sutterwala FS, Trinchieri G, Mosser DM, Ma X. Suppression of Il-12 transcription in macrophages following Fc gamma receptor ligation. J Immunol. 2001;166:4498–4506. doi: 10.4049/jimmunol.166.7.4498. [DOI] [PubMed] [Google Scholar]
- 109.Ma X, Neurath M, Gri G, Trinchieri G. Identification and characterization of a novel Ets-2-related nuclear complex implicated in the activation of the human interleukin-12 p40 gene promoter. J Biol Chem. 1997;272:10389–10395. doi: 10.1074/jbc.272.16.10389. [DOI] [PubMed] [Google Scholar]
- 110.Gri G, Savio D, Trinchieri G, Ma X. Synergistic regulation of the human interleukin-12 p40 promoter by NFkappaB and Ets transcription factors in Epstein-Barr virus- transformed B cells and macrophages. J Biol Chem. 1998;273:6431–6438. doi: 10.1074/jbc.273.11.6431. [DOI] [PubMed] [Google Scholar]
- 111.Wang IM, Contursi C, Masumi A, Ma X, Trinchieri G, Ozato K. An IFN-gamma-inducible transcription factor, IFN consensus sequence binding protein (ICSBP), stimulates IL-12 p40 expression in macrophages. J Immunol. 2000;165:271–279. doi: 10.4049/jimmunol.165.1.271. [DOI] [PubMed] [Google Scholar]
- 112.Salkowski CA, Kopydlowski K, Blanco J, Cody MJ, McNally R, Vogel SN. IL-12 is dysregulated in macrophages from IRF-1 and IRF-2 knockout mice. J Immunol. 1999;163:1529–1536. [PubMed] [Google Scholar]
- 113.D’Ambrosio D, et al. Inhibition of IL-12 production by 1,25-dihydroxyvitamin D3. Involvement of NF-kappaB downregulation in transcriptional repression of the p40 gene. J Clin Invest. 1998;101:252–262. doi: 10.1172/JCI1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Delgado M, Ganea D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit interleukin-12 transcription by regulating nuclear factor kappaB and Ets activation. J Biol Chem. 1999;274:31930–31940. doi: 10.1074/jbc.274.45.31930. [DOI] [PubMed] [Google Scholar]
- 115.Hayes MP, Wang J, Norcross MA. Regulation of interleukin-12 expression in human monocytes: selective priming by interferon-gamma of lipopolysaccharide-inducible p35 and p40 genes. Blood. 1995;86:646–650. [PubMed] [Google Scholar]
- 116.Tone Y, et al. Structure and chromosomal location of the mouse interleukin-12 p35 and p40 subunit genes. Eur J Immunol. 1996;26:1222–1227. doi: 10.1002/eji.1830260606. [DOI] [PubMed] [Google Scholar]
- 117.Yoshimoto T, Kojima K, Funakoshi T, Endo Y, Fujita T, Nariuchi H. Molecular cloning and characterization of murine IL-12 genes. J Immunol. 1996;156:1082–1088. [PubMed] [Google Scholar]
- 118.Babik JM, Adams E, Tone Y, Fairchild PJ, Tone M, Waldmann H. Expression of murine IL-12 is regulated by translational control of the p35 subunit. J Immunol. 1999;162:4069–4078. [PubMed] [Google Scholar]
- 119.Carmody RJ, Ruan Q, Liou HC, Chen YH. Essential roles of c-Rel in TLR-induced IL-23 p19 gene expression in dendritic cells. J Immunol. 2007;178:186–191. doi: 10.4049/jimmunol.178.1.186. [DOI] [PubMed] [Google Scholar]
- 120.Mise-Omata S, Kuroda E, Niikura J, Yamashita U, Obata Y, Doi TS. A proximal kappaB site in the IL-23 p19 promoter is responsible for RelA- and c-Rel-dependent transcription. J Immunol. 2007;179:6596–6603. doi: 10.4049/jimmunol.179.10.6596. [DOI] [PubMed] [Google Scholar]
- 121.Ouyang X, Negishi H, Takeda R, Fujita Y, Taniguchi T, Honda K. Cooperation between MyD88 and TRIF pathways in TLR synergy via IRF5 activation. Biochem Biophys Res Commun. 2007;354:1045–1051. doi: 10.1016/j.bbrc.2007.01.090. [DOI] [PubMed] [Google Scholar]
- 122.Goodridge HS, Harnett W, Liew FY, Harnett MM. Differential regulation of interleukin-12 p40 and p35 induction via Erk mitogen-activated protein kinase-dependent and -independent mechanisms and the implications for bioactive IL-12 and IL-23 responses. Immunology. 2003;109:415–425. doi: 10.1046/j.1365-2567.2003.01689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Petro TM. ERK-MAP-kinases differentially regulate expression of IL-23 p19 compared with p40 and IFN-beta in Theiler’s virus-infected RAW264.7 cells. Immunol Lett. 2005;97:47–53. doi: 10.1016/j.imlet.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 124.Slack EC, et al. Syk-dependent ERK activation regulates IL-2 and IL-10 production by DC stimulated with zymosan. Eur J Immunol. 2007;37:1600–1612. doi: 10.1002/eji.200636830. [DOI] [PubMed] [Google Scholar]
- 125.Murphy FJ, Hayes MP, Burd PR. Disparate intracellular processing of human IL-12 preprotein subunits: atypical processing of the P35 signal peptide. J Immunol. 2000;164:839–847. doi: 10.4049/jimmunol.164.2.839. [DOI] [PubMed] [Google Scholar]
- 126.Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med. 2005;202:1715–1724. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brightbill HD, et al. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 1999;285:732–736. doi: 10.1126/science.285.5428.732. [DOI] [PubMed] [Google Scholar]
- 128.Ferwerda G, et al. NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog. 2005;1:279–285. doi: 10.1371/journal.ppat.0010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jang S, Uematsu S, Akira S, Salgame P. IL-6 and IL-10 induction from dendritic cells in response to Mycobacterium tuberculosis is predominantly dependent on TLR2-mediated recognition. J Immunol. 2004;173:3392–3397. doi: 10.4049/jimmunol.173.5.3392. [DOI] [PubMed] [Google Scholar]
- 130.Means RT, Jr, Dessypris EN, Krantz SB. Inhibition of human colony-forming-unit erythroid by tumor necrosis factor requires accessory cells. J Clin Invest. 1990;86:538–541. doi: 10.1172/JCI114741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.van Heel DA, et al. Synergistic enhancement of Toll-like receptor responses by NOD1 activation. Eur J Immunol. 2005;35:2471–2476. doi: 10.1002/eji.200526296. [DOI] [PubMed] [Google Scholar]
- 132.van Heel DA, et al. Synergy between TLR9 and NOD2 innate immune responses is lost in genetic Crohn’s disease. Gut. 2005;54:1553–1557. doi: 10.1136/gut.2005.065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–808. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- 134.Yang Z, et al. NOD2 transgenic mice exhibit enhanced MDP-mediated down-regulation of TLR2 responses and resistance to colitis induction. Gastroenterology. 2007;133:1510–1521. doi: 10.1053/j.gastro.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Watanabe T, et al. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest. 2008;118:545–559. doi: 10.1172/JCI33145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Honma K, et al. Interferon regulatory factor 4 negatively regulates the production of proinflammatory cytokines by macrophages in response to LPS. Proc Natl Acad Sci USA. 2005;102:16001–16006. doi: 10.1073/pnas.0504226102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Negishi H, et al. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci USA. 2005;102:15989–15994. doi: 10.1073/pnas.0508327102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol. 2008;8:458–466. doi: 10.1038/nri2340. [DOI] [PubMed] [Google Scholar]
- 139.Fletcher HA. Correlates of immune protection from tuberculosis. Curr Mol Med. 2007;7:319–325. doi: 10.2174/156652407780598520. [DOI] [PubMed] [Google Scholar]
- 140.Khader SA, et al. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J Immunol. 2005;175:788–795. doi: 10.4049/jimmunol.175.2.788. [DOI] [PubMed] [Google Scholar]
- 141.Khader SA, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 142.Umemura M, et al. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin infection. J Immunol. 2007;178:3786–3796. doi: 10.4049/jimmunol.178.6.3786. [DOI] [PubMed] [Google Scholar]
- 143.Wozniak TM, Ryan AA, Britton WJ. Interleukin-23 restores immunity to Mycobacterium tuberculosis infection in IL-12p40-deficient mice and is not required for the development of IL-17-secreting T cell responses. J Immunol. 2006;177:8684–8692. doi: 10.4049/jimmunol.177.12.8684. [DOI] [PubMed] [Google Scholar]
- 144.Yadav M, Schorey JS. The {beta}-glucan receptor Dectin-1 functions together with TLR2 to mediated macrophage activation by mycobacteria. Blood. 2006;108:3168–3175. doi: 10.1182/blood-2006-05-024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rothfuchs AG, et al. Dectin-1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J Immunol. 2007;179:3463–3471. doi: 10.4049/jimmunol.179.6.3463. [DOI] [PubMed] [Google Scholar]
- 146.Gantner BN, Simmons RM, Underhill DM. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J. 2005;24:1277–1286. doi: 10.1038/sj.emboj.7600594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Qin BY, et al. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol. 2003;10:913–921. doi: 10.1038/nsb1002. [DOI] [PubMed] [Google Scholar]
- 148.Rogers NC, et al. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity. 2005;22:507–517. doi: 10.1016/j.immuni.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 149.Re F, Strominger JL. IL-10 released by concomitant TLR2 stimulation blocks the induction of a subset of Th1 cytokines that are specifically induced by TLR4 or TLR3 in human dendritic cells. J Immunol. 2004;173:7548–7555. doi: 10.4049/jimmunol.173.12.7548. [DOI] [PubMed] [Google Scholar]
- 150.Netea MG, et al. Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J Immunol. 2004;172:3712–3718. doi: 10.4049/jimmunol.172.6.3712. [DOI] [PubMed] [Google Scholar]
- 151.Dillon S, et al. A Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J Immunol. 2004;172:4733–4743. doi: 10.4049/jimmunol.172.8.4733. [DOI] [PubMed] [Google Scholar]
- 152.Sutmuller RP, et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485–494. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Liu J, Cao S, Herman LM, Ma X. Differential regulation of interleukin (IL)-12 p35 and p40 gene expression and interferon (IFN)-gamma-primed IL-12 production by IFN regulatory factor 1. J Exp Med. 2003;198:1265–1276. doi: 10.1084/jem.20030026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Donnelly RP, Freeman SL, Hayes MP. Inhibition of IL-10 expression by IFN-g upregulates transcription of TNF-a in human monocytes. J Immunol. 1995;155:1420–1427. [PubMed] [Google Scholar]
- 155.Flores RR, Diggs KA, Tait LM, Morel PA. IFN-gamma negatively regulates CpG-induced IL-10 in bone marrow-derived dendritic cells. J Immunol. 2007;178:211–218. doi: 10.4049/jimmunol.178.1.211. [DOI] [PubMed] [Google Scholar]
- 156.Hu X, et al. IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–574. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 157.Acosta-Rodriguez EV, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 158.d’Ostiani CF, et al. Dendritic cells discriminate between yeasts and hyphae of the fungus Candida albicans. Implications for initiation of T helper cell immunity in vitro and in vivo. J Exp Med. 2000;191:1661–1674. doi: 10.1084/jem.191.10.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Romagnoli G, et al. The interaction of human dendritic cells with yeast and germ-tube forms of Candida albicans leads to efficient fungal processing, dendritic cell maturation, and acquisition of a Th1 response-promoting function. J Leukoc Biol. 2004;75:117–126. doi: 10.1189/jlb.0503226. [DOI] [PubMed] [Google Scholar]
- 160.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis. 2004;190:624–631. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 161.Kleinschek MA, et al. IL-23 enhances the inflammatory cell response in Cryptococcus neoformans infection and induces a cytokine pattern distinct from IL-12. J Immunol. 2006;176:1098–1106. doi: 10.4049/jimmunol.176.2.1098. [DOI] [PubMed] [Google Scholar]
- 162.Saijo S, et al. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat Immunol. 2007;8:39–46. doi: 10.1038/ni1425. [DOI] [PubMed] [Google Scholar]
- 163.Taylor PR, et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol. 2007;8:31–38. doi: 10.1038/ni1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gran B, et al. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol. 2002;169:7104–7110. doi: 10.4049/jimmunol.169.12.7104. [DOI] [PubMed] [Google Scholar]
- 165.Chen Y, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006;116:1317–1326. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ding C, Xu J, Li J. ABT-874, a fully human monoclonal anti-IL-12/IL-23 antibody for the potential treatment of autoimmune diseases. Curr Opin Investig Drugs. 2008;9:515–522. [PubMed] [Google Scholar]
- 167.Wittig BM. Drug evaluation: CNTO-1275, a mAb against IL-12/IL-23p40 for the potential treatment of inflammatory diseases. Curr Opin Investig Drugs. 2007;8:947–954. [PubMed] [Google Scholar]
- 168.Stockinger B, Veldhoen M. Differentiation and function of Th17 T cells. Curr Opin Immunol. 2007;19:281–286. doi: 10.1016/j.coi.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 169.Watanabe H, et al. Danger signaling through the inflammasome acts as a master switch between tolerance and sensitization. J Immunol. 2008;180:5826–5832. doi: 10.4049/jimmunol.180.9.5826. [DOI] [PubMed] [Google Scholar]
- 170.Kryczek I, et al. Cutting edge: opposite effects of IL-1 and IL-2 on the regulation of IL-17+ T cell pool IL-1 subverts IL-2-mediated suppression. J Immunol. 2007;179:1423–1426. doi: 10.4049/jimmunol.179.3.1423. [DOI] [PubMed] [Google Scholar]
- 171.Ivanov II, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 172.Zhou L, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 173.McGeachy MJ, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 174.Chen Z, Tato CM, Muul L, Laurence A, O’Shea JJ. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum. 2007;56:2936–2946. doi: 10.1002/art.22866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Evans HG, Suddason T, Jackson I, Taams LS, Lord GM. Optimal induction of T helper 17 cells in humans requires T cell receptor ligation in the context of Toll-like receptor-activated monocytes. Proc Natl Acad Sci USA. 2007;104:17034–17039. doi: 10.1073/pnas.0708426104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wilson NJ, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 177.van Beelen AJ, et al. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity. 2007;27:660–669. doi: 10.1016/j.immuni.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 178.Hsu LC, et al. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci USA. 2008;105:7803–7808. doi: 10.1073/pnas.0802726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pan Q, et al. MDP-induced interleukin-1beta processing requires Nod2 and CIAS1/NALP3. J Leukoc Biol. 2007;82:177–183. doi: 10.1189/jlb.1006627. [DOI] [PubMed] [Google Scholar]
- 180.Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14:1929–1934. doi: 10.1016/j.cub.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 181.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci USA. 2008;105:8067–8072. doi: 10.1073/pnas.0709684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Liu FL, et al. Interleukin (IL)-23 p19 expression induced by IL-1beta in human fibroblast-like synoviocytes with rheumatoid arthritis via active nuclear factor-kappaB and AP-1 dependent pathway. Rheumatology (Oxford) 2007;46:1266–1273. doi: 10.1093/rheumatology/kem055. [DOI] [PubMed] [Google Scholar]
- 184.Wesa AK, Galy A. IL-1 beta induces dendritic cells to produce IL-12. Int Immunol. 2001;13:1053–1061. doi: 10.1093/intimm/13.8.1053. [DOI] [PubMed] [Google Scholar]
- 185.Dumoutier L, Louahed J, Renauld JC. Cloning and characterization of IL-10-related T cell-derived inducible factor (IL-TIF), a novel cytokine structurally related to IL-10 and inducible by IL-9. J Immunol. 2000;164:1814–1819. doi: 10.4049/jimmunol.164.4.1814. [DOI] [PubMed] [Google Scholar]
- 186.Gurney AL. IL-22, a Th1 cytokine that targets the pancreas and select other peripheral tissues. Int Immunopharmacol. 2004;4:669–677. doi: 10.1016/j.intimp.2004.01.016. [DOI] [PubMed] [Google Scholar]
- 187.Scriba TJ, et al. Distinct, specific IL-17- and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. J Immunol. 2008;180:1962–1970. doi: 10.4049/jimmunol.180.3.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zheng Y, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 189.Yang L, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.O’Garra A, Stockinger B, Veldhoen M. Differentiation of human T(H)-17 cells does require TGF-beta! Nat Immunol. 2008;9:588–590. doi: 10.1038/ni0608-588. [DOI] [PubMed] [Google Scholar]
- 191.Negishi H, et al. Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proc Natl Acad Sci USA. 2006;103:15136–15141. doi: 10.1073/pnas.0607181103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tailor P, Tamura T, Ozato K. IRF family proteins and type I interferon induction in dendritic cells. Cell Res. 2006;16:134–140. doi: 10.1038/sj.cr.7310018. [DOI] [PubMed] [Google Scholar]
- 193.Asselin-Paturel C, et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol. 2001;2:1144–1150. doi: 10.1038/ni736. [DOI] [PubMed] [Google Scholar]
- 194.Byrnes AA, et al. Type I interferons and IL-12: convergence and cross-regulation among mediators of cellular immunity. Eur J Immunol. 2001;31:2026–2034. doi: 10.1002/1521-4141(200107)31:7<2026::aid-immu2026>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 195.Gerosa F, et al. The reciprocal interaction of NK cells with plasmacytoid or myeloid dendritic cells profoundly affects innate resistance functions. J Immunol. 2005;174:727–734. doi: 10.4049/jimmunol.174.2.727. [DOI] [PubMed] [Google Scholar]