

Abstract
Pathogenesis and growth of three common women’s cancers (breast, endometrium and ovary) are linked to estrogen. A single gene encodes the key enzyme for estrogen biosynthesis named aromatase, inhibition of which effectively eliminates estrogen production in the entire body. Aromatase inhibitors successfully treat breast cancer, whereas their roles in endometrial and ovarian cancers are less clear. Ovary, testis, adipose tissue, skin, hypothalamus and placenta express aromatase normally, whereas breast, endometrial and ovarian cancers overexpress aromatase and produce local estrogen exerting paracrine and intracrine effects. Tissue-specific promoters distributed over a 93-kilobase regulatory region upstream of a common coding region alternatively control aromatase expression. A distinct set of transcription factors regulates each promoter in a signaling pathway- and tissue-specific manner. In cancers of breast, endometrium and ovary, aromatase expression is primarly regulated by increased activity of the proximally located promoter I.3/II region. Promoters I.3 and II lie 215 bp from each other and are coordinately stimulated by PGE2 via a cAMP-PKA-dependent pathway. In breast adipose fibroblasts exposed to PGE2 secreted by malignant epithelial cells, activation of PKC potentiates cAMP-PKA-dependent induction of aromatase. Thus, inflammatory substances such as PGE2 may play important roles in inducing local production of estrogen that promotes tumor growth.
Keywords: Aromatase, aromatase inhibitor, breast cancer, endometrial cancer, endometriosis, uterine leiomyomata, fibroids, aromatase overexpression, gain-of-function mutations, aromatase excess syndrome, gynecomastia, estrogen, estrogen biosynthesis, endometrium, uterus
I. PHYSIOLOGICAL REGULATION OF AROMATASE EXPRESSION IN HUMAN TISSUES
A. The aromatase enzyme
The aromatase enzyme is localized in the endoplasmic reticulum of estrogen-producing cells [1,2]. Aromatase enzyme complex is comprised of two polypeptides. The first of these is a specific cytochrome P450, namely aromatase cytochrome P450 (the product of the CYP19 gene) [1]. The second is a flavoprotein, NADPH-cytochrome P450 reductase and is ubiquitously distributed in most cells. Thus, cell-specific expression of aromatase P450 (P450arom) determines the presence or absence of aromatase activity. For practical purposes, we will refer to “P450arom” as “aromatase” throughout this text. Since only a single gene (CYP19) encodes aromatase in mice and humans, targeted disruption of this gene or inhibition of its product effectively eliminates estrogen biosynthesis in these species [1].
In the human, aromatase is expressed in a number of cells including the ovarian granulosa cell, the placental syncytiotrophoblast, the testicular Leydig cell, as well as various extraglandular sites including the brain and skin fibroblasts [3]. The principal product of the ovarian granulosa cells during the follicular phase is estradiol. Additionally, aromatase is expressed in human adipose tissue. Whereas the highest levels of aromatase are in the ovarian granulosa cells in premenopausal women, the adipose tissue becomes the major aromatase-expressing body site after menopause (Fig 1) [4,5]. Although aromatase level per adipose tissue fibroblast may be small, the sum of estrogen arising from billions of adipose tissue fibroblasts in the entire body makes a physiologic impact. The principal product of the ovary is the potent estrogen, estradiol. In adipose tissue, estrogenically weak estrone is produced from androstenedione of adrenal origin in relatively large quantities. However, at least half of this peripherally produced estrone is eventually converted to estradiol in extraovarian tissues (Fig 1) [6].
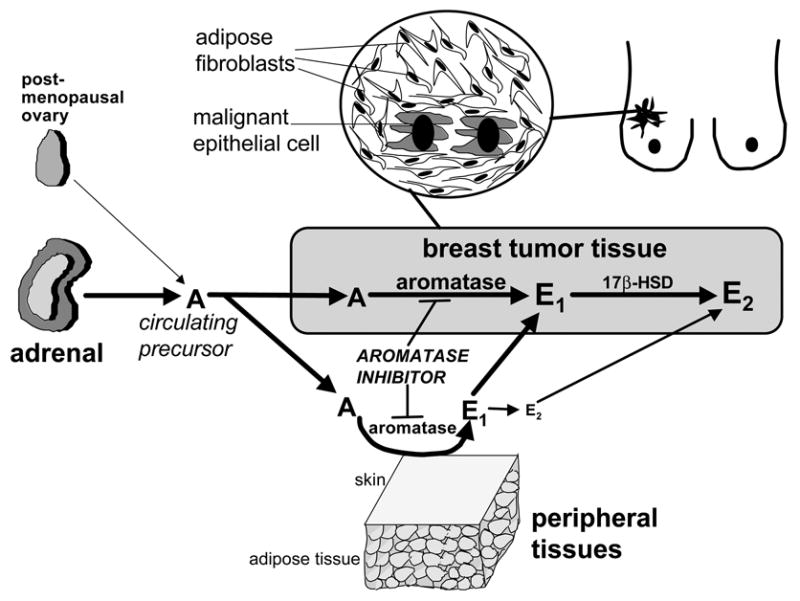
Figure 1. Tissue sites of estrogen production in women.
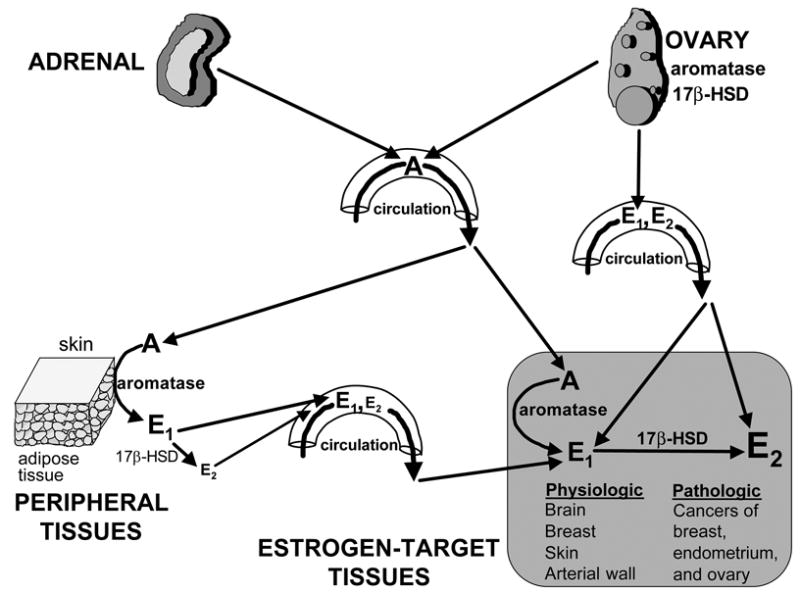
The biologically active estrogen, estradiol (E2) is produced at least in three major sites: 1- direct secretion from the ovary in reproductive-age women; 2- by conversion of circulating androstenedione (A) of adrenal and/or ovarian origins to estrone (E1) in peripheral tissues; and 3- by conversion of A to E1 in estrogen-target tissues. In the latter two instances, estrogenically weak E1 is further converted to E2 within the same tissue. The presence of the enzyme aromatase and 17β-hydroxysteroid dehydrogenase (17β-HSD) is critical for E2 formation at these sites. E2 formation by peripheral and local conversion is particularly important for postmenopausal women and for estrogen-dependent diseases such as breast cancer, endometriosis and endometrial cancer.
B. The CYP19 (aromatase) gene
The aromatase gene is transcribed from the telomere to the centromere, and the region encoding the aromatase protein spans 30 kb of the 3′-end and contains 9 exons (II-X) [7]. The ATG translation start site is located in coding exon II. The upstream (telomeric) 93 kb of the gene contains a number of promoters [2,3]. The most proximal gonad-specific promoter II and two other proximal promoters, I.3 (expressed in adipose tissue and breast cancer) and I.6 (expressed in bone) are found to be located within the 1-kb region upstream of the ATG translation start site in exon II, as expected (Fig 2). Promoter I.2, the minor placenta-specific promoter, is located approximately 13 kb upstream of the ATG site in exon II. The promoters specific for the brain (I.f), endothelial cells (I.7), fetal tissues (I.5), adipose tissue (I.4) and placenta (2a and I.1) are localized in tandem order at ~ 33, 36, 43, 73, 78 and 93- kb, respectively, upstream of the first coding exon, the exon II (Fig 2) [2,8]. In addition to promoter II-specific sequences, transcripts containing two other unique sequences, untranslated exons I.3 and I.4, are present in adipose tissue and in adipose tissue fibroblasts maintained in culture [8]. Transcription initiated by use of each promoter gives rise to a transcript with a unique 5′-untranslated end that contains the sequence encoded in the first exon immediately downstream of this particular promoter (Fig 2). Therefore, the 5′-untranslated region of aromatase mRNA is promoter-specific and may be viewed as a signature of the particular promoter used. It should be emphasized again that all of these 5′-ends are spliced onto a common junction 38 bp upstream of the ATG translation start site [8]. Consequently, the sequence encoding the open reading frame is identical in each case. Thus, the expressed protein is the same regardless of the splicing pattern (Fig 2).
Figure 2. CYP19 (aromatase) gene.
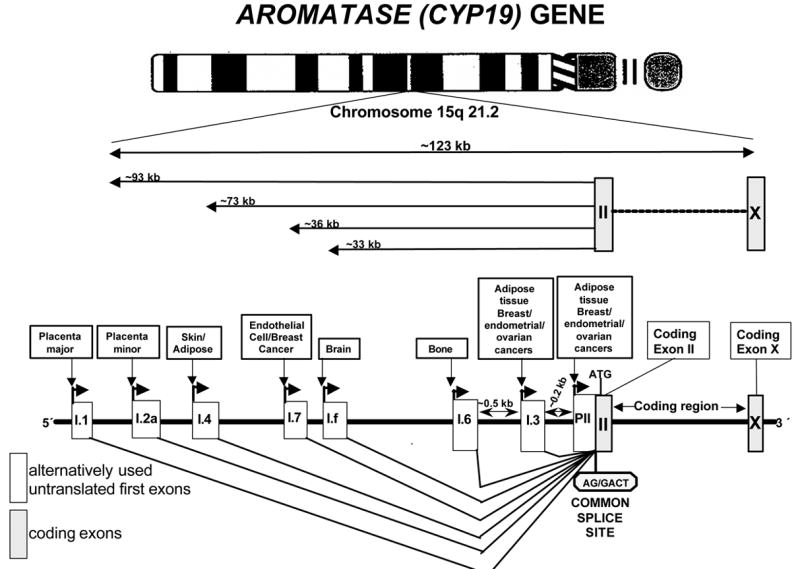
Expression of the aromatase gene is regulated by the tissue-specific activation of a number of promoters via alternative splicing.
C. Normal hormonal pathways that regulate aromatase expression
The primary site of aromatase expression in premenopausal women is the ovarian follicle, where FSH induces aromatase and thus estradiol production in a cyclic fashion [3]. Ovarian aromatase expression is mediated primarily by FSH receptors, cAMP production and activation of the proximal promoter II [3] (Fig 3). Men and postmenopausal also produce estrogen by aromatase that resides in extragonadal tissues such as adipose tissue and skin [3] (Fig 3). Estrogen produced in these extragonadal tissues are of paramount importance for the closure of bone plates and bone mineralization in both men and postmenopausal women, since the phenotype of men with defective genes of aromatase or estrogen receptor-α include severe osteoporosis and extremely tall stature with growth into adulthood [9]. A distal promoter (I.4) located 73 kilobases upstream of the coding region directs aromatase expression in adipose tissue and skin fibroblasts. Promoter I.4 in these tissues is regulated by combined action of a glucocorticoid and a member of the class I cytokine family [e.g., interleukin (IL)-6, IL-11, leukemia inhibitory factor (LIF), oncostatin-M] (Fig 3) [10].
Figure 3. Physiological regulation of aromatase expression.
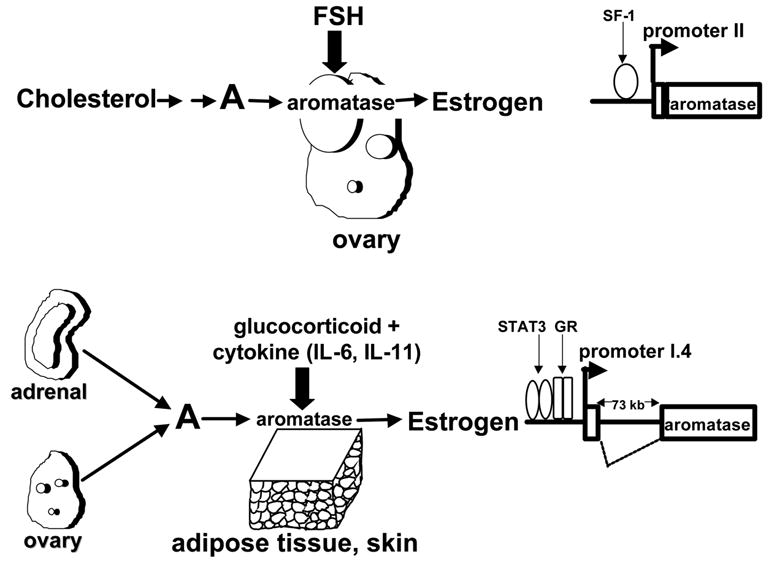
FSH induces aromatase expression via a cAMP-dependent pathway in ovarian granulosa cells via promoter II. Steroidogenic factor-I (SF-1) mediates this action of FSH. On the other hand, a combination of a glucocorticoid and a member of the class I cytokine family induces aromatase expression in skin and adipose tissue fibroblasts via promoter I.4 located 70 kb upstream of the coding region. Binding of signal transducers and activators of transcription (STAT)-3 and glucocorticoid receptor (GR) upstream of promoter I.4 mediate regulation of aromatase expression in these fibroblasts.
The alternative use of promoters comprises the basis for differential regulation of aromatase expression by various hormones, growth factors and cytokines in a tissue-specific manner. For example, extremely high baseline levels of the placental promoter I.1 activity are maintained constitutively in the syncytiotrophoblast and a consequence of decreasing levels of inhibitory transcription factors as cytotrophoblasts differentiate to a syncytiotrophoblast [11,12]. On the other hand, extremely low baseline levels of promoter II in the ovary are stimulated strikingly by FSH via a cAMP-dependent pathway in the developing follicle [3] (Fig 3). Serum, cytokines and growth factors are inhibitory to promoter II. In case of adipose and skin fibroblasts, promoter I.4 is used in vivo and activated coordinately by a glucocorticoid in the presence of a cytokine (IL-6, IL-11, LIF, oncostatin M). Glucocorticoid receptors and the Jak-1/STAT-3 pathway mediate this induction [10].
Promoter use in cultured adipose tissue fibroblasts is a function of hormonal treatments. For example, in vitro studies showed that PGE2 or cAMP analogs stimulate aromatase expression strikingly via proximally located promoters II and I.3, whereas treatment with a glucocorticoid plus a member of the class I cytokine family switches promoter use to I.4 [10,13].
II. PATHOLOGICAL EXPRESSION OF AROMATASE IN WOMEN’S CANCERS
Breast and endometrial cancers are highly responsive to estrogen for growth evident by high concentrations of estrogen receptors in these tissues [14]. Malignant breast and endometrial tumors also produce large amounts of estrogen locally via overexpressing aromatase compared to their normal counterparts [15]. In particular, aromatase overexpression in breast cancer tissue has been shown to be critical, since the use of aromatase inhibitors is clearly therapeutic in breast cancer. Aromatase is also overexpressed in endometrial cancer [16]. Although preliminary trials showed promising results, the therapeutic role of aromatase inhibitors in endometrial cancer is not as clear yet [17,18].
Experimental and epidemiological evidence suggest that estrogen and progesterone are implicated in ovarian carcinogenesis. New data have indicated that estrogen favors neoplastic transformation of the ovarian surface epithelium while progesterone offers protection against ovarian cancer development [19–23]. Since a subset of ovarian cancers was linked to endometriosis and, aromatase is a key molecular target in endometriosis, aromatase expression in ovarian cancer may also be targeted for treatment in selected patients [15]. In fact, recent pilot studies employing aromatase inhibitors have shown various degrees of clinical benefit for patients with advanced stages of ovarian cancer [24–27].
A. AROMATASE AND BREAST CANCER
Paracrine interactions between malignant breast epithelial cells, proximal adipose fibroblasts and vascular endothelial cells are responsible for estrogen biosynthesis and lack of adipogenic differentiation in breast cancer tissue. It appears that malignant epithelial cells secrete factors that inhibit the differentiation of surrounding adipose fibroblasts to mature adipocytes and also stimulate aromatase expression in these undifferentiated adipose fibroblasts [28]. The in vivo presence of malignant epithelial cells also enhances aromatase expression in endothelial cells in breast tissue [29]. We developed a model in breast cancer, which reconciles the inhibition of adipogenic differentiation and estrogen biosynthesis in a positive feedback cycle.
The desmoplastic reaction (formation of the dense fibroblast layer surrounding malignant epithelial cells) is essential for structural and biochemical support for tumor growth. In fact, the pathologists refer to 70% of breast carcinomas as “scirrhous” type indicating the rock-like consistency of these tumors [30]. This consistency comes from the tightly packed undifferentiated adipose fibroblasts around malignant epithelial cells. Malignant epithelial cells achieve this by secreting large quantities of TNF and IL-11 that inhibit the differentiation of fibroblasts to mature adipocytes. Thus, large numbers of these estrogen-producing cells are maintained proximal to malignant cells [28,29]. At the same time, a separate set of factors secreted by malignant epithelial cells activates aromatase expression in surrounding adipose fibroblasts [28,29].
Malignant epithelial cells induce aromatase via activation of aberrant promoters in breast cancer tissue and adipose fibroblasts proximal to tumor (Fig 4). The breast adipose tissue in disease-free women maintains low levels of aromatase expression primarily via promoter I.4 that lies 73 kb upstream of the common coding region [8] (Fig 4). The proximal promoters I.3 and II are used only minimally in normal breast adipose tissue [8]. Transcription via activity of promoters II and I.3 in the breast tumor fibroblasts and malignant epithelial cells, however, is strikingly increased [31–34] (Fig 4). Additionally, the endothelial-type promoter I.7 is also upregulated in breast tumor tissue [35] (Fig 4). Thus, it appears that the prototype estrogen-dependent malignancy breast cancer takes advantage of four promoters utilized in various cell types for aromatase expression (Fig 4). The sum of aromatase mRNA species arising from these four promoters markedly increase total aromatase mRNA levels in breast cancer compared with the normal breast that uses almost exclusively promoter I.4 (Fig 4).
Figure 4. Promoter use for aromatase expression in normal and malignant breast tissues.
The levels of total aromatase mRNA levels in breast cancer tissue are strikingly higher than normal breast tissue. The normal breast adipose tissue maintains low levels of aromatase expression primarily via promoter I.4. Promoters I.3 and II are used only minimally in normal breast adipose tissue. Promoter II and I.3 activities in the breast cancer, however, are strikingly increased. Additionally, the endothelial-type promoter I.7 is also upregulated in breast cancer. Thus, it appears that the prototype estrogen-dependent malignancy breast cancer takes advantage of four promoters (II, I.3, I.7 and I.4) for aromatase expression. The sum of aromatase mRNA species arising from these four promoters markedly increase total aromatase mRNA levels in breast cancer compared with the normal breast.
1. Tissue origins of estrogen in postmenopausal women with breast cancer
Estrogen arises from two sources in a postmenopausal woman with breast cancer. First, estrogen that arises from extraovarian body sites such as subcutaneous adipose tissue and skin reaches breast cancer by way of circulation in an endocrine manner. Second, estrogen locally produced in breast cancer tissue makes an impact via paracrine or intracrine mechanisms.
a) Endocrine effect: aromatase in adipose tissue and skin
The potential roles of extraovarian aromatase in human physiology and pathology were also recognized initially in the 1960s [36]. These studies demonstrated that the conversion rate of plasma androstenedione to estrone in humans increased as a function of obesity and aging [5,37]. These studies also revealed the importance of extraovarian tissues (primarily adipose tissue and skin) as the origin of estrogen in postmenopausal women (see Fig 1). Extraovarian estrogen formation was shown to be correlated positively with excess body weight in both pre- and postmenopausal women, and may be increased as much as ten-fold in morbidly obese postmenopausal women [5,37]. This elevation in association with both obesity and aging bears a striking relationship to the incidence of endometrial hyperplasia and cancer, which are more commonly observed in elderly obese women [38]. It is now recognized that the continuous production of estrogen by the adipose tissue in these women is one of the risk factors of endometrial hyperplasia and cancer.
Evidence is also suggestive of a role of estrogens produced by adipose tissue in the pathogenesis of the breast cancer. For example, breast cancer incidence correlates positively with the body fat content or serum estradiol levels in postmenopausal women suggesting that estrogen collectively produced in all extraovarian sites reach the breast tissue by circulation in an endocrine fashion and stimulate tumor growth [39,40]. A role for adipose tissue estrogen biosynthesis in promoting the growth of breast cancer is implied because of the palliative effects of adrenalectomy in the past. As estrone production by adipose tissue is dependent on plasma androstenedione secreted by the adrenal cortex as substrate, the role of adrenalectomy is explicable in terms of the denial of substrate precursor for adipose tissue estrogen biosynthesis. Today, reduction of estrogen biosynthesis in the adipose tissue is accomplished by the use of aromatase inhibitors in the treatment of breast cancer [41,42] (Fig 5).
Figure 5. Origins of estrogen in postmenopausal breast cancer.
This figure exemplifies the important pathologic roles of extraovarian (peripheral) and local estrogen biosynthesis in an estrogen-dependent disease in postmenopausal women. The estrogen precursor androstenedione (A) originates primarily from the adrenal in the postmenopausal woman. Aromatase expression and enzyme activity in extraovarian tissues such as fat increases with advancing age. The aromatase activity in skin and subcutaneous adipose fibroblasts gives rise to formation of systemically available estrone (E1) and to a smaller extent estradiol (E2). The conversion of circulating A to E1 in undifferentiated breast adipose fibroblasts compacted around malignant epithelial cells and subsequent conversion of E1 to E2 in malignant epithelial cells provide high tissue concentrations of E2 for tumor growth. The clinical relevance of these findings is exemplified by the successful use of aromatase inhibitors to treat breast cancer.
b) Paracrine/intracrine effect local aromatase in breast cancer tissue
Mechanisms giving rise to increased local concentrations of estrogen in breast cancer via aromatase overexpression within the tumor tissue has been demonstrated by a number investigators [43–45]. These studies showed strikingly increased local levels of estrone, estrone sulfate and eastradiol in breast tumor tissue compared with circulating estrogen levels [43–45].
A series of paracrine interactions between malignant breast epithelial cells and surrounding adipose stroma were uncovered and explained increased local estrogen levels in the breast bearing a cancer. For example, independent studies from at least six different laboratories were indicative of striking increases in aromatase enzyme activity and mRNA levels in breast fat adjacent to cancer compared with those in distal fat or disease-free breast adipose tissue [32–34,46–49] (Fig 5). We also found that an elevation in aromatase expression in adipose stroma surrounding malignant epithelial cells is regulated by complex cellular, molecular and genomic mechanisms [29,31,47]. Interestingly, the overall aromatase expression in breast adipose tissue in mastectomy specimens bearing a breast tumor was significantly higher than that in benign breast tissue removed for reduction mammoplasty [31,50,51].
Estrogens can act both directly or indirectly on human breast cancer cells to promote proliferation. Breast cancer cells in culture elaborate a number of growth stimulants in response to estrogen, which can act in an autocrine and paracrine manner to stimulate their growth. However, there is also evidence that estrogens can directly induce proliferation of breast cancer cells. The pathologic significance of local aromatase activity in breast cancer was recognized based on the following in vitro data. MCF7 breast cancer cells, which were stably transfected to express an MMTV-promoter-driven human aromatase cDNA and inoculated into oophorectomized nude mice, remained dependent on circulating androstenedione for their rapid growth [52]. Another evidence for the importance of local aromatase expression in the breast tissue came from an in vivo mouse model demonstrating that aromatase overexpression in breast tissue is sufficient for maintaining hyperplasia in the absence of circulating estrogen and that aromatase inhibitors abrogated hyperplasia [53]. These transgenic mice with MMTV-promoter-driven local aromatase in breast tissue are more prone for breast cancer development [54].
2. Role of adipose tissue in aromatase expression in malignant breast tumor tissue
a) Cell types that express aromatase in breast cancer
Breast adipose tissue is primarily comprised of mature lipid-containing cells and other stromal elements. This latter group of cells in the breast adipose tissue was characterized using immunohistochemical methods [55]. Ninety % of these resident cells of adipose tissue are fibroblasts, i.e., the potential precursors of mature adipocytes and another 7% represented endothelial cells. The majority (80–90%) of aromatase transcripts in adipose tissue was demonstrated to reside in fibroblasts compared with mature adipocytes [55]. Moreover, aromatase enzyme activity was found to reside primarily in the fibroblast component of the adipose tissue in a previous study from the same laboratory [56].
Immunoreactive aromatase was localized to both the malignant epithelial cells and surrounding fibroblasts in breast tumor tissues [57–59]. Different antibodies, however, showed variable affinity to malignant epithelial cells vs fibroblasts [60]. Immunoreactive aromatase was also observed in endothelial cells in normal breast tissue and breast tumors. Recently published data using RNA from laser-captured breast tumor cells showed aromatase mRNA in both stromal and malignant epithelial cells in tumor tissues from 3 patients [60]. Markedly high levels of aromatase enzyme activity have been consistently detected in breast adipose fibroblasts freshly isolated from breast tissue with or without cancer [55,56]. Aromatase enzyme activity in malignant breast epithelial cells, on the other hand, is either undetectable or extremely low [61].
Adjacent adipose tissue including the dense fibroblast layer seems to account for the majority of aromatase expression in breast tumors for the following reasons. First, the quantity of adjacent adipose tissue surrounding a clinically detectable breast tumor is comparatively very large, e.g., the volume of adipose tissue within 1-inch radius of a 1 ml breast tumor is 129 ml. Second, the most intense aromatase immunostaining was observed in the adipose tissue fibroblasts located in and around the fibrous capsule (i.e., desmoplastic reaction) surrounding malignant cells [48]. Third, levels of aromatase expression and activity in fibroblasts isolated from breast adipose tissue or tumor are 10–15 times higher than those found in malignant epithelial cells or cell lines [61].
b) Impaired adipogenic differentiation in breast cancer: link to aromatase overexpression
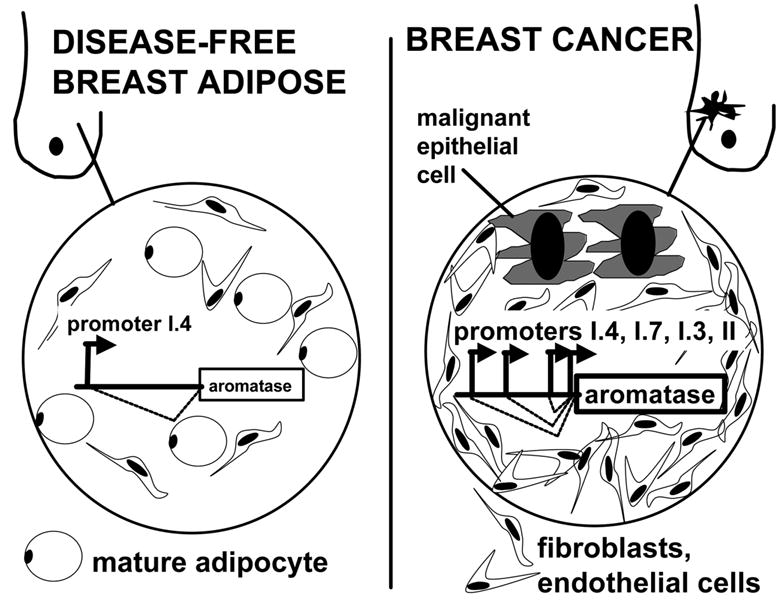
Extraordinarily large quantities of TNF and IL-11 are produced and secreted by malignant breast epithelial cells [28] (Fig 6). These two cytokines mediate one of the most commonly observed biological phenomena in breast tumors, namely the desmoplastic reaction. Desmoplastic reaction or accumulation of fibroblasts around malignant epithelial cells serves to maintain the strikingly hard consistency in many of these tumors (i.e., the traditional macroscopic description of malignant breast tumors as “scirrhous cancer”) and increased local concentrations of estrogen via aromatase overexpression localized to these undifferentiated fibroblasts. The inhibition of differentiation of fibroblasts to mature adipocytes mediated by TNF and IL-11 is the key event responsible for desmoplastic reaction, because neither malignant cell-conditioned media nor these cytokines caused the proliferation of adipose tissue fibroblasts [28]. Moreover, blocking both TNF and IL-11 in cancer cell-CM using neutralizing antibodies is sufficient to reverse this antidifferentiative effect of cancer cells completely (Fig 6) [28]. In summary, desmoplastic reaction primarily occurs via the action of cytokines (TNF and IL-11) secreted by the malignant epithelial cells to inhibit the differentiation of adipose tissue fibroblasts to mature adipocytes. This tumor-induced block in adipocyte differentiation is mediated by the selective inhibition of expression of the essential adipogenic transcription factors, namely, C/EBPα and PPARγ [28] (Fig 6).
Figure 6. Detail of epithelial-stromal interaction via estrogen and cytokines in breast cancer.
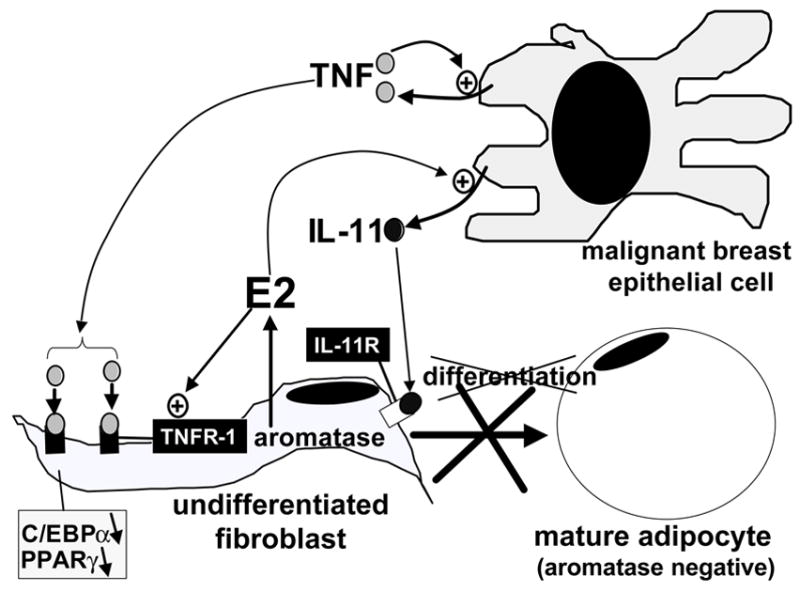
Estradiol (E2) increases secretion of antiadipogenic cytokines (IL-11) from malignant epithelial cells and upregulates their antiadipogenic-type receptors (TNF-receptor type 1, TNFR1) in undifferentiated fibroblasts. These redundant mechanisms give rise to accumulation of undifferentiated fibroblasts around malignant epithelial cells (desmoplastic reaction), which express aromatase and form E2.
Estrogen per se seems to potentiate this anti-adipogenic action via indirect mechanisms. For example, treatment of T47D breast cancer cells with estradiol increased the mRNA levels of IL-11 by 3-fold [62]. Moreover, the cellular actions of TNF are mediated by two distinct receptors TNFR1 (also known as p60 in humans) and TNFR2 (p80). TNFR1 but not TNFR2 was found to be responsible for the inhibition of adipocyte differentiation using mutants of TNF specific for the stimulation of either receptor type [63]. We recently demonstrated that TNFR1 is responsible for inhibition of adipocyte differentiation in breast cancer [64]. Interestingly, estradiol enhances this anti-adipogenic effect by inducing TNFR1 levels in adipose fibroblasts [64] (Fig 6).
Thus, large amounts of antiadipogenic cytokines (e.g., TNF and IL-11) secreted by malignant epithelial cells serve to maintain increased numbers of the aromatase-expressing cell type, i.e., undifferentiated adipose fibroblast, in breast tumor tissue. This is further enhanced by stimulatory effects of estrogen on IL-11 production in cancer cells and on the TNF receptor type that mediates adipogenic inhibition (Fig 6).
2. Transcriptional mechanisms responsible for elevated aromatase expression in breast cancer
Alternative promoter use is a major mechanism that mediates increased aromatase expression in breast cancer. The normal breast adipose tissue maintains low levels of aromatase expression primarily via promoter I.4 that lies 73 kb upstream of the common coding region (see Fig 4). The proximally located promoters I.3 and II are used only minimally in normal breast adipose tissue. Promoter II and I.3 activities in the breast cancer, however, are strikingly increased [31–34]. Additionally, the endothelial-type promoter I.7 is also upregulated in breast cancer [35]. Thus, it appears that the prototype estrogen-dependent malignancy breast cancer takes advantage of four promoters (II, I.3, I.7 and I.4) for aromatase expression (see Fig 4). The sum of aromatase mRNA species arising from these four promoters markedly increase total aromatase mRNA levels in breast cancer compared with the normal breast that uses almost exclusively promoter I.4 (see Fig 4).
a) Upregulation of promoters I.3 and II
Using an in vivo approach, we and two other groups demonstrated by quantitative exon-specific RT-PCR that the use of the proximal promoters II/I.3 are strikingly upregulated in adipose tissue adjacent to breast cancer and in breast cancer tissue per se [31,33,34]. As noted earlier, promoters II and I.3 are located within 215 bp from each other and are coordinately induced by cAMP-dependent or independent mechanisms in adipose fibroblasts in breast tumors. These promoters possibly share common regulatory DNA motifs.
Increased promoter I.3/II activity is in part, the basis for increased aromatase expression in peri- and intratumoral adipose tissue fibroblasts (see Fig 4) [31]. Over the past several years, others and we sought to elucidate the mechanisms underlying this cancer-induced increase in promoters I.3/II activity in adipose tissue fibroblasts.
PGE2 induces aromatase via promoters I.3 and II employing both cAMP/PKA- and PKC-dependent pathways [13]. In this model, PGE2 stimulates binding activity of an orphan nuclear receptor LRH-1 to a nuclear receptor half-site (−136/−124 bp) upstream of promoter II [65,66]. Treatment with PGE2 strikingly increased both LRH-1 expression and its binding activity to the aromatase promoter II in cultured adipose fibroblasts. LRH-1 overexpression significantly increased aromatase promoter II activity and aromatase enzyme activity in cultured adipose fibroblasts (Fig 7). From an in vivo perspective, LRH-1 was upregulated in undifferentiated fibroblasts in breast tumor tissue compared with those in disease-free breast tissue. The recently reported increases in COX-2 expression and beneficial effects of nonsteroidal anti-inflammatory in breast cancer support this model [67,68]. In vivo evidence for increased PGE2 formation in breast cancer, however, is still lacking at this time.
Figure 7. Effects of PGE2, PKA, PKC and sodium butyrate (NaBu) on aromatase expression in breast adipose fibroblasts.
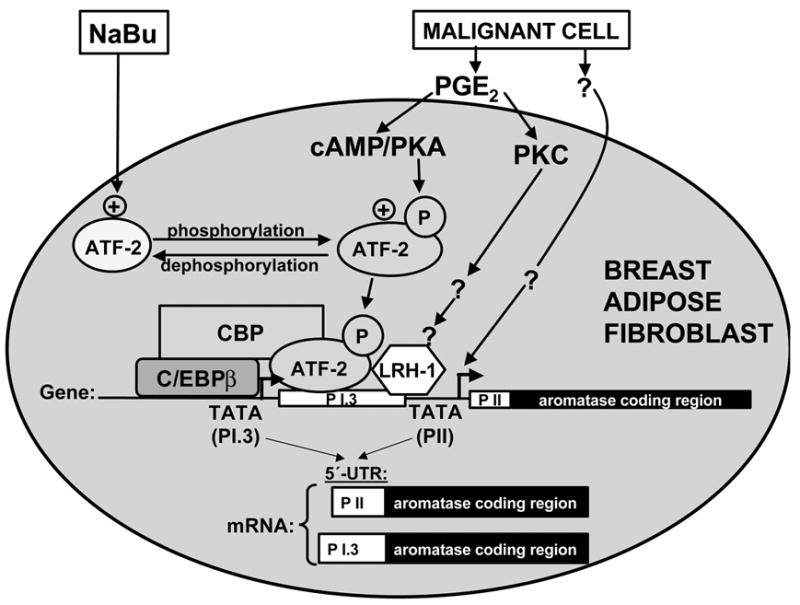
We recently found that sodium-butyrate (NaBu) profoundly decreased promoter I.3/II-specific aromatase mRNA expression induced by PGE2 or a surrogate hormomonal cocktail made of dibutyryl cAMP (Bt2cAMP) plus the PKC activator phorbol diacetae (PDA). MCM, Bt2cAMP+PDA or NaBu regulated aromatase mRNA levels or enzyme activity only specifically via promoters I.3/II but not other promoters. Recruitment of phosphorylated ATF-2 by a CRE (-211/-199) in the promoter I.3/II region conferred the response to malignant epithelial cells conditioned medium, PGE2 or Bt2cAMP+PDA. Malignant cell-conditioned medium, PGE2 or Bt2cAMP+PDA stabilized a complex comprised of phosphorylated ATF-2, C/EBPβ and CBP in the common regulatory region of promoters I.3/II. The inhibitory effect of NaBu on transcription was not accompanied by comparable changes in overall histone acetylation patterns of promoters I.3/II. NaBu treatment, however, consistently decreased ATF-2 phosphorylation and disrupted the activating complex. Taken together, these findings represent a novel mechanism of NaBu action and provide evidence that aromatase activity can be attenuated in a signaling pathway- and tissue-specific fashion. Our data also suggested that malignant cells secreted substances other than PGE2. These unknown substances were associated with signaling pathways other than cAMP-PKA in the activation of aromatase promoters I.3 and II.
In addition to the LRH-1-binding site, we isolated two cis-acting elements that conferred stimulation of promoter I.3/II use in response to cAMP plus or minus PKC activation, the signaling pathways induced by PGE2 [29] [69]. Two critical elements were determined as a C/EBP site (−317/−304) and a CRE (−211/−197), since mutation of either element abolished cAMP-induced promoter activity [69] (Fig 7).
PGE2 or a cAMP analog (plus or minus PKC activator) induced phosphorylation of ATF-2 and its binding to the CRE [69](Fig 7). This CRE is occupied by nonphosphorylated ATF-2 in fibroblasts treated with benign epithelial cell-conditioned medium associated with inactivation of promoters I.3 and II. Moreover, chromatin immunoprecipitation PCR showed that the activator transcriptional complex in a malignant environment contains C/EBPβ, phosphorylated ATF-2 and p300/CBP, whereas the inactivator complex (benign environment) contained nonphosphorylated ATF-2 [69] (Fig 7).
We recently found that sodium-butyrate (NaBu) profoundly decreased cAMP analog (plus or minus PKC activator)-induced promoter I.3/II-specific aromatase mRNA expression. A cAMP analog (plus or minus a PKC activator) or NaBu regulated aromatase mRNA levels or enzyme activity only via promoters I.3/II but not promoters I.1 or I.4 in breast, ovarian, placental and hepatic cells. Mechanistically, recruitment of phosphorylated ATF-2 by a CRE (−211/−199) in the promoter I.3/II region conferred the response to a cAMP analog (plus or minus PKC activator). Treatment with a cAMP analog (plus or minus PKC activator) stabilized a complex comprised of phosphorylated ATF-2, C/EBPβ and CBP in the common regulatory region of promoters I.3/II (Fig 7). The inhibitory effect of NaBu on transcription was not accompanied by comparable changes in overall histone acetylation patterns of promoters I.3/II. NaBu treatment, however, consistently decreased ATF-2 phosphorylation and disrupted the activating complex (Fig 7). Together, these findings represent a novel mechanism of NaBu action and provide evidence that aromatase activity can be attenuated in a signaling pathway- and tissue-specific fashion [69].
In an alternative experimental model, we found that conditioned medium from malignant breast epithelial cells (MCF7 or T47D) markedly induced aromatase expression in adipose tissue fibroblasts via promoters I.3 and II [29] [69](Fig 7). Malignant epithelial cell-conditioned medium also induced phosphorylation of ATF-2 and its binding activity to the promoter I.3/II region. As in the case of treatment with a cAMP analog (plus or minus a PKC activator), incubation of breast adipose fibroblasts with malignant epithelial cell-conditioned medium stabilized a complex made of phosphorylated ATF-2, C/EBPβ and CBP at the aromatase promoter I.3/II region [69]. NaBu also disrupts this transcriptional complex assembled in response to malignant epithelial cell conditioned medium [69]. We hypothesize that a hormonal cocktail secreted from malignant epithelial cells induces aromatase in undifferentiated adipose fibroblasts via redundant pathways. Although PGE2 seems to be an important component of this cocktail, other substances can also induce aromatase expression via promoters I.3/II [29]. For example, the addition of a COX-2 inhibitor or an adenylyl cyclase inhibitor does not reverse induction of aromatase expression by malignant epithelial cell-conditioned medium [29].
A unified model for promoter II/I.3 activation in breast cancer therefore predicts that malignant epithelial cells secrete a number of factors including PGE2 (Fig 7). These factors induce a number of signaling pathways in a redundant fashion to activate the transcription of the aromatase gene via promoter I.3/II in adipose fibroblasts. PGE2 possibly arising from malignant epithelial cells is a candidate factor for activation of promoters I.3 and II in breast cancer. This, however, has not been demonstrated in vivo. Neither PGE2 nor its downstream regulators cAMP or LRH-1-binding site in promoter II were found to be essential for activation of promoters II in adipose fibroblasts treated with malignant cell-conditioned medium [29]. In disease-free breast tissue, incorporation of a number of transcriptional repressors into the multimeric complex that occupies promoter I.3/II region is associated with inhibition of transcription. Malignant epithelial cell conditioned medium, on the other hand, gives rise to replacement of this inhibitory complex by an activator transcriptional complex comprised of distinct factors such as phosphorylated ATF-2, C/EBPβ, p300/CBP and possibly LRH-1 [69] (Fig 7).
In summary, the proximal promoters II and I.3 clustered within a 215 bp region are coordinately regulated. They remain quiescent in fibroblasts of normal breast tissue via redundant binding of multiple transcriptional inhibitors (Fig 7). In a malignant breast environment, however, these promoter regions are occupied by multiple transcriptional enhancers as a result of activation of multiple signaling pathways in a fail-safe fashion to increase aromatase expression in breast fibroblasts (Fig 7).
b) Regulation of promoters II and I.3 in MCF-7 cells
A group of investigators studied the regulation of a number of gene reporter constructs of the promoter II/I.3 region [70]. They found that ERR alpha-1 and CREB1 upregulate and EAR-2, COUP-TFI, RAR gamma, Snail and Slug proteins downregulate this promoter region [70]. The in vivo relevance of these findings will become clearer in future, once the relative significance of aromatase enzyme activity and estrogen biosynthesis is demonstrated in malignant epithelial cells.
c) Upregulation of promoter I.7 in breast cancer
Studies summarized above employed exon-specific RT-PCR analysis of 5′-untranslated ends of aromatase mRNA in breast cancer tissues. This limited strategy permitted only the detection of promoters previously identified from healthy tissues [31,33,34]. Discovery-driven approaches designed to identify novel promoter regions in breast cancer or adjacent adipose tissues, however, have not been published until recently. To identify novel promoter regions in cancer tissues and proximal fat, we employed the 5′-rapid amplification of cDNA ends (RACE) procedure using total RNA isolated from breast cancer and proximal adipose tissue samples. We cloned a novel 101-bp untranslated first exon (I.7) that comprises the 5′-end of 29–54% of aromatase mRNA isolated from breast cancer tissues [35] (Fig 10). The levels Aromatase mRNA with exon I.7 were significantly increased in breast cancer tissues and adipose tissue adjacent to tumors (Fig 10). We identified a promoter immediately upstream of exon I.7 and mapped this to about 36 kb upstream of ATG translation start site of the aromatase gene [35] (Fig 10). Promoter I.7 is a TATA-less promoter containing cis-regulatory elements found in megakaryocytic and endothelial type promoters (Fig 10). Maximal promoter activity could be demonstrated in human microvascular endothelial cells (Fig 10). Binding of the transcription factor GATA-2 to a specific GATA cis-regulatory element in this promoter was critical for its regulation in endothelial cells [35]. In conclusion, promoter I.7 is a GATA-2-regulated endothelial type promoter of the human aromatase gene and may increase estrogen biosynthesis in vascular endothelial cells of breast cancer. The activity of this promoter may also be important for intracrine and paracrine effects of estrogen on blood vessel physiology (Fig 10).
3. Summary of regulation of aromatase expression in breast cancer
Several alternative cellular and molecular mechanisms serve to maintain excessive levels of aromatase activity in breast stroma proximal to malignant epithelial cells. First, malignant epithelial cell-derived factors induce aromatase overexpression via the transcription factors LRH-1, C/EBPβ and phosphorylated ATF-2 (see Fig 5). These factors are incorporated into a multimeric transcriptional complex that occupies the aromatase promoter I.3/II region in adipose tissue fibroblasts adjacent to epithelial cells. Second, aromatase is overexpressed in vascular endothelial cells of tumor tissue via binding of GATA-2 and other endothelial type transcription factors to promoter I.7. These factors may also mediate angiogenesis in tumor tissue. Moreover, estrogen is known to induce the angiogenic factor VEGF in cancer cells [71–74] (see Fig 5). Third, we demonstrated recently that anti-adipogenic cytokines IL-11 and TNF secreted by malignant epithelial cells block the differentiation of the aromatase-expressing cells (fibroblasts) to mature adipocytes that do not express aromatase (see Fig 5). Thereby, these cytokines secreted abundantly by malignant epithelial cells serve to maintain a dense layer of aromatase-expressing fibroblasts proximal to malignant epithelial cells to provide structural and hormonal support. Fourth, the expression of IL-11 in malignant epithelial cells and antiadipogenic type TNF receptors in adjacent adipose tissue fibroblasts are upregulated by estrogen produced as a consequence of elevated aromatase activity in breast tumors. This positive feedback involving complex epithelial-stromal interactions favor higher numbers of undifferentiated fibroblasts, angiogenesis and increased local estrogen concentrations in breast tumors (see Fig 5). These four mechanisms interact to maintain high levels of estrogen production in a breast tumor.
4. Aromatase inhibitors in the treatment of breast cancer
Today, aromatase inhibitors are the most effective endocrine-treatment of estrogen-responsive breast cancer [75] (see Fig 7). Six recent head-to-head randomized clinical trials published since 2000 demonstrated the superiority of aromatase inhibitors to tamoxifen in the treatment of breast cancer [76–83]. Long-term side effect profiles of these agents will determine whether aromatase inhibitors will replace tamoxifen or other selective estrogen receptor modulators in the long run.
There are two intriguing implications of these results. First, it is pharmacologically more efficacious to block estrogen formation rather than its action at least by currently approved estrogen antagonists or SERMs. Second, the local effect of aromatase inhibitors at the target tissue level to block local estrogen formation possibly represents the most critical mechanism for the superior therapeutic potential of aromatase inhibitors (see Fig 7).
Targeting aromatase in breast cancer as a therapeutic strategy was first conceptualized in the 1960s [75]. Aminoglutethimide was the first aromatase inhibitor tested for this purpose. Although the first generation aromatase inhibitor aminoglutethimide was as efficacious as tamoxifen in the treatment of breast cancer, its adverse side effects precluded its widespread use [75]. Tamoxifen was introduced in the 1970s and became the gold standard for hormonal treatment of breast cancer [75]. Second generation aromatase inhibitors were tested in Europe in the 1980s and were found to be as efficacious as tamoxifen [75]. Finally, the third generation aromatase inhibitors were approved in the U.S. to treat postmenopausal breast cancer in the 1990s and proven to be superior to tamoxifen [76–85]. These new inhibitors have a benign side effect profile and suppress estrogen production in extraovarian tissues and within the breast cancer tissue itself. This effectively blocks estrogenic action, reduces recurrences and prolongs disease-free survival in postmenopausal women with breast cancer [76,77]. Aromatase inhibitors are also effective in the treatment of breast cancer that became resistant to treatment with tamoxifen [80].
In these studies, tumors that express estrogen receptor (ER) were more responsive to aromatase inhibitors compared with the tumors with an unknown receptor status [76,77]. Future studies are required to determine whether aromatase inhibitors might be beneficial in ER-negative tumors via ER-independent mechanisms.
B. AROMATASE AND ENDOMETRIAL CANCER
The role of aromatase expression and the therapeutic use of aromatase inhibitors have been well defined for breast cancer endometriosis. Aromatase is also expressed in endometrial cancer tissue, and aromatase inhibitors have been used to treat endometrial cancer [16–18,86]. The pathologic significance of local estrogen biosynthesis via aromatase expression in endometrial cancer tissue or the therapeutic value of aromatase inhibitors in its management, however, is not clear at the moment.
Endometrial cancer is the most common gynecological malignancy found in women [87]. These tumors have high concentrations of estrogen receptors, their growth is clearly enhanced by estrogen, and unopposed estrogen exposure (in the absence of progesterone) predisposes women to development of endometrial cancer [87]. Although there is no consistent evidence of increased concentrations of circulating estrogen in women with endometrial cancer, the local concentration of estradiol in endometrial cancer tissues was reported to be higher than that in blood and in the endometrium of cancer-free women [88–92]. It is therefore conceivable that endometrial cancer itself synthesizes estradiol in situ, which then contributes to growth and possibly carcinogenesis.
A conversion study demonstrated significant conversion of androstenedione to estrone in endometrial cancer tissue [93]. Aromatase protein and mRNA were detected in endometrial cancer using immunohistochemistry and RT-PCR, whereas aromatase expression was low or undetectable in endometrial hyperplasia (a precursor lesion of endometrial cancer) [16,94]. These observations are suggestive that intratumoral aromatase may play a role in the pathology of endometrial cancer. Immunoreactive aromatase was found in malignant epithelial, endometrial stromal and myometrial cells. Aromatase in stromal but not epithelial cells correlated positively with advanced surgical stage and poor survival (Dr. Makio Shozu, personal communication).
Currently available 3rd generation aromatase inhibitors may be used for endocrine treatment of endometrial cancer [95]. Treatment of endometrial cancer tissues in vitro with aromatase inhibitors demonstrated that in situ depletion of estrogen results in decreased cell proliferation of tumor cells [96]. Treatment of women with endometrial cancer with aromatase inhibitors blocked estrogen production in vivo in tumor tissue [97]. Safety data from the clinical trials of postmenopausal women with breast cancer indicated a preventive role of aromatase inhibitors in that an aromatase inhibitor reduced the risk of endometrial cancer [98].
On the other hand, the therapeutic efficacy of aromatase inhibitors in advanced endometrial cancer is not clear. The Gynecologic Oncology Group (GOG) performed a phase II trial of anastrozole in advanced, recurrent, or persistent endometrial cancer [18]. Twenty-three patients were entered, all with grade 2 or 3 cancers. Two partial responses were noted [18]. In a recent report, two cases of reproductive-aged women with grade 1 endometrial cancer who were treated with medroxyprogesterone acetate and anastrozole daily for 3 and 6 months subsequently reverted to normal endometrium. A progestin combined with the elimination of production of estrogen may be an effective therapy in well-differentiated endometrial cancer in the obese premenopausal woman [17]. Thus, there are some early encouraging results. It is, however, too early to predict whether aromatase inhibitors will be used widely in the treatment of endometrial cancer.
III. AROMATASE AND OVARIAN CANCER
1. Estrogen, progesterone and ovarian cancer
Since premenopausal ovary contains large amounts of estrogen and progesterone, and ovarian cancer largely develops in postmenopausal women, it was hypothesized that ovarian cancer was not an ovarian steroid-responsive disease. In view of the protective effect of oral contraceptives and pregnancy, the disruptive and inflammatory effects of incessant ovulation on the surface epithelium were hypothesized to represent the primary mechanism of ovarian carcinogenesis [19,20,23].
Accumulating evidence, however, are suggestive of significant roles of estrogen and progesterone in ovarian cancer. A retrospective 1979–1998 cohort study of 44,241 postmenopausal women revealed that estrogen only hormone replacement therapy (HRT), particularly for 10 or more years, significantly increased risk of ovarian cancer. Relative risks for 10 to 19 years and 20 or more years were 1.8 (95% CI, 1.1–3.0) and 3.2 (95% CI, 1.7–5.7), respectively (p-value for trend <0.001), This study did not show an increased risk in women who used short-term estrogen plus progestin HRT, but authors suggested that risk associated with longer-term estrogen plus progestin HRT warrants further investigation [99]. The estrogen plus progestin WHI HRT study found a trend for increased ovarian cancer risk, which was not significant [100]. These are some of the first pieces of important epidemiological evidence supporting that estrogen plays a critical role in the pathophysiology of ovarian cancer. This has also been supported by other epidemiological and experimental data [19,20,23].
Estrogens appear to favor neoplastic transformation of the ovarian surface epithelium while progesterone offers protection against ovarian cancer development [21,22]. Specifically, estrogens, particularly those present in ovulatory follicles, are both genotoxic and mitogenic to ovarian surface epithelial cells. In contrast, pregnancy-equivalent levels of progesterone are highly effective as apoptosis inducers for ovarian epithelial and ovarian cancer cells. In this regard, high-dose progestin may exert an exfoliation effect and rid an aged ovarian surface epithelium of pre-malignant cells. A limited number of clinical studies have demonstrated efficacies of antiestrogens, aromatase inhibitors, and progestins alone or in combination with chemotherapeutic drugs in the treatment of ovarian cancer. As a result of increased life expectancy in most countries, the number of women taking HRT continues to grow. Therefore, knowledge of the mechanism of action of steroid hormones on the ovarian surface epithelium and ovarian cancer is of paramount significance to HRT risk assessment and to the development of novel therapies for the prevention and treatment of ovarian cancer [19,20,23].
2. A possible link between endometriosis, ovarian cancer, aromatase and prostaglandin formation
Another piece of evidence regarding the role of estrogen in ovarian carcinogenesis comes form the biological and epidemiological links of endometrioid ovarian cancer to endometriosis [19,20,23]. It was proposed that endometrioid and/or undifferentiated ovarian cancer may develop from the endometriotic implants seeded on the ovarian surface epithelium [19,20,23]. Endometriosis is an estrogen-dependent disease that affects 6–10% of U.S. women of reproductive age (approximately 4–7 million) [101]. Endometriosis is a systemic disorder that is characterized by the presence of endometrium-like tissue in ectopic sites outside the uterus, primarily on pelvic peritoneum and ovaries [101]. Local estrogen formation via aromatase expression in endometriosis is extraordinarily important for its pathogenesis and treatment [86,102–106]. In fact, work from our laboratory and other investigators over the past 10 years uncovered a molecular link between inflammation and estrogen production in endometriosis [86]. This is mediated by a positive feedback cycle that favors expression of aromatase and COX-2 and continuous local production of estradiol and PGE2 in endometriotic tissue [107–109]. In this vicious cycle, PGE2 induces aromatase, whereas the product of aromatase enzyme, i.e., estradiol, induces COX-2 and thus PGE2 formation. It is possible that PGE2–induced aromatase expression may also play a biological role and represent a therapeutic target in a subset of ovarian carcinomas related to endometriosis.
3. Aromatase inhibitors for the treatment of ovarian cancer
Ovarian cancer is the fourth most common cause of cancer death in women. Most patients present at an advanced stage, and the disease commonly relapses after primary surgery and chemotherapy. Relapsed ovarian cancer is not curable in the majority of women, and responses to salvage chemotherapy are often sustained for less than a year at the expense of toxicities that may diminish quality of life.
In experimental models of ovarian cancer, it was demonstrated that moderate-high expression of ERα is associated with a growth response to estrogen, and these models are growth-inhibited by antiestrogen strategies both in vitro and in vivo [110–112]. In addition, a number of proteins are estrogen regulated, and these include the PR, the EGF receptor, and HSP27 [113–116].
Clinical studies of tamoxifen in chemoresistant ovarian cancer have suggested that a subset of unselected patients respond to tamoxifen treatment, but the characteristics of responding tumors have not been defined. In one study, 105 patients in first relapse received tamoxifen, and an overall response rate of 17% was reported [117]. This was later reanalyzed to give a 13% response rate in cisplatin-resistant disease and 15% in cisplatin-sensitive disease [118]. Another trial of tamoxifen reported a 17% response rate in chemoresistant disease [119]. ERα has been suggested to be linked with clinical response, but to date no significant association has been demonstrated [117].
The therapeutic role of aromatase inhibitors have recently been explored in advanced ovarian cancer. Phase II studies suggested that aromatase inhibitors are at least as effective as other hormonal treatments in advanced ovarian cancer [24–27]. Because of the significant epidemiological association of estrogen with ovarian cancer, the minimal side effects of aromatase inhibitors, and demonstrated activity of hormonal therapies in other endocrine-associated malignancies, further study is needed.
Acknowledgments
Support: This research work was supported, in part, by the NIH grants CA67167, HD38691 and HD46260, and grants from the AVON Foundation, Northwestern Memorial Foundation, Lynn Sage Cancer Research Foundation and the Friends of Prentice (to SEB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Simpson ER, Clyne C, Rubin G, Boon WC, Robertson K, Britt K, Speed C, Jones M. Aromatase--a brief overview. Annu Rev Physiol. 2002;64:93–127. doi: 10.1146/annurev.physiol.64.081601.142703. [DOI] [PubMed] [Google Scholar]
- 2.Sebastian S, Bulun SE. A highly complex organization of the regulatry region of the human CYP19 (aromatase) gene revealed by the human genome project. J Clin Endocrinol Metab. 2001;86:4600–4602. doi: 10.1210/jcem.86.10.7947. [DOI] [PubMed] [Google Scholar]
- 3.Simpson ER, Mahendroo MS, Means GD, Kilgore MW, Hinshelwood MM, Graham-Lorence S, Amarneh B, Ito Y, Fisher CR, Michael MD, Mendelson CR, Bulun SE. Aromatase cytochrome P450, the enzyme responsible for estrogen biosynthesis. Endocrine Reviews. 1994;15:342–355. doi: 10.1210/edrv-15-3-342. [DOI] [PubMed] [Google Scholar]
- 4.Bulun SE, Simpson ER. Competitive RT-PCR analysis indicates levels of aromatase cytochrome P450 transcripts in adipose tissue of buttocks, thighs, and abdomen of women increase with advancing age. Journal of Clinical Endocrinology and Metabolism. 1994;78:428–432. doi: 10.1210/jcem.78.2.8106632. [DOI] [PubMed] [Google Scholar]
- 5.Grodin JM, Siiteri PK, MacDonald PC. Source of estrogen production in postmenopausal women. Journal of Clinical Endocrinology and Metabolism. 1973;36:207–214. doi: 10.1210/jcem-36-2-207. [DOI] [PubMed] [Google Scholar]
- 6.MacDonald PC, Madden JD, Brenner PF, Wilson JD, Siiteri PK. Origin of estrogen in normal men and in women with testicular feminization. Journal of Clinical Endocrinology and Metabolism. 1979;49:905–916. doi: 10.1210/jcem-49-6-905. [DOI] [PubMed] [Google Scholar]
- 7.Shozu M, Sebastian S, Takayama K, Hsu WT, Schultz RA, Neely K, Bryant M, Bulun SE. Estrogen excess associated with novel gain-of-function mutations affecting the aromatase gene. N Engl J Med. 2003;348:1855–1865. doi: 10.1056/NEJMoa021559. [DOI] [PubMed] [Google Scholar]
- 8.Mahendroo MS, Mendelson CR, Simpson ER. Tissue-specific and hormonally-controlled alternative promoters regulate aromatase cytochrome P450 gene expression in human adipose tissue. Journal of Biological Chemistry. 1993;268:19463–19470. [PubMed] [Google Scholar]
- 9.Bulun S. Aromatase deficiency and estrogen resistance: from molecular genetics to clinic. 2000;2000:31–39. doi: 10.1055/s-2000-13481. [DOI] [PubMed] [Google Scholar]
- 10.Zhao NJY, Bulun SE, Mendelson CR, Simpson ER. Aromatase P450 gene expression in human adipose tissue: Role of a Jak/STAT pathway in regulation of the adipose-specific promoter. Journal of Biological Chemistry. 1995;270:16449–16457. doi: 10.1074/jbc.270.27.16449. [DOI] [PubMed] [Google Scholar]
- 11.Kamat A, Alcorn J, Kunczt C, Mendelson C. Characterization of the regulatory regions of the human aromatase (P450arom) gene involved in placenta-specific expression. Mol Endocrinol. 1998;12:1764–1777. doi: 10.1210/mend.12.11.0190. [DOI] [PubMed] [Google Scholar]
- 12.Jiang B, Kamat A, Mendelson C. Hypoxia prevents induction of aromatase expression in human trophoblast cells in culture: potential inhibitory role of the hypoxia-inducible transcription factor Mash-2 (mammalian achaete-scute homologous protein-2) Mol Endocrinol. 2000;14:1661–1673. doi: 10.1210/mend.14.10.0539. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Y, Agarwal VR, Mendelson CR, Simpson ER. Estrogen biosynthesis proximal to a breast tumor is stimulated by PGE2 via cyclic AMP, leading to activation of promoter II of the CYP19 (aromatase) gene. Endocrinology. 1996;137:5739–5742. doi: 10.1210/endo.137.12.8940410. [DOI] [PubMed] [Google Scholar]
- 14.Bulun SE, Noble LS, Takayama K, Michael MD, Agarwal V, Fisher C, Zhao Y, Hinshelwood MM, Ito Y, Simpson ER. Endocrine disorders associated with inappropriately high aromatase expression. Journal of Steroid Biochemistry and Molecular Biology. 1997;61:133–139. [PubMed] [Google Scholar]
- 15.Bulun SE, Lin Z, Imir G, Amin S, Demura M, Yilmaz B, Martin R, Utsunomiya H, Thung S, Gurates B, Tamura M, Langoi D, Deb S. Regulation of aromatase expression in estrogen-responsive breast and uterine disease: from bench to treatment. Pharmacol Rev. 2005;57:359–383. doi: 10.1124/pr.57.3.6. [DOI] [PubMed] [Google Scholar]
- 16.Bulun SE, Economos K, Miller D, Simpson ER. CYP19 (aromatase cytochrome P450) gene expression in human malignant endometrial tumors. Journal of Clinical Endocrinology and Metabolism. 1994;79:1831–1834. doi: 10.1210/jcem.79.6.7989490. [DOI] [PubMed] [Google Scholar]
- 17.Burnett BAAF, Amezcua C. Anastrozole, an aromatase inhibitor, and medroxyprogesterone acetate therapy in premenopausal obese women with endometrial cancer: a report of two cases successfully treated without hysterectomy. Gynecol Oncol. 2004;94:832–834. doi: 10.1016/j.ygyno.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 18.Rose BVPG, VanLe L, Bell J, Walker JL, Lee RB. A phase II trial of anastrozole in advanced recurrent or persistent endometrial carcinoma: a Gynecologic Oncology Group Study. Gynecol Oncol. 2000;78:212–216. doi: 10.1006/gyno.2000.5865. [DOI] [PubMed] [Google Scholar]
- 19.Somigliana E, Vigano P, Parazzini F, Stoppelli S, Giambattista E, Vercellini P. Association between endometriosis and cancer: a comprehensive review and a critical analysis of clinical and epidemiological evidence. Gynecol Oncol. 2006;101:331–341. doi: 10.1016/j.ygyno.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 20.Vercellini P, Scarfone G, Bolis G, Stellato G, Carinelli S, Crosignani PG. Site of origin of epithelial ovarian cancer: the endometriosis connection. Bjog. 2000;107:1155–1157. doi: 10.1111/j.1471-0528.2000.tb11116.x. [DOI] [PubMed] [Google Scholar]
- 21.Schildkraut JM, Calingaert B, Marchbanks PA, Moorman PG, Rodriguez GC. Impact of progestin and estrogen potency in oral contraceptives on ovarian cancer risk. J Natl Cancer Inst. 2002;94:32–38. doi: 10.1093/jnci/94.1.32. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez GC, Nagarsheth NP, Lee KL, Bentley RC, Walmer DK, Cline M, Whitaker RS, Isner P, Berchuck A, Dodge RK, Hughes CL. Progestin-induced apoptosis in the Macaque ovarian epithelium: differential regulation of transforming growth factor-beta. J Natl Cancer Inst. 2002;94:50–60. doi: 10.1093/jnci/94.1.50. [DOI] [PubMed] [Google Scholar]
- 23.Ho SM. Estrogen, progesterone and epithelial ovarian cancer. Reprod Biol Endocrinol. 2003;1:73. doi: 10.1186/1477-7827-1-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papadimitriou CA, Markaki S, Siapkaras J, Vlachos G, Efstathiou E, Grimani I, Hamilos G, Zorzou M, Dimopoulos MA. Hormonal therapy with letrozole for relapsed epithelial ovarian cancer. Long-term results of a phase II study. Oncology. 2004;66:112–117. doi: 10.1159/000077436. [DOI] [PubMed] [Google Scholar]
- 25.Bowman A, Gabra H, Langdon SP, Lessells A, Stewart M, Young A, Smyth JF. CA125 response is associated with estrogen receptor expression in a phase II trial of letrozole in ovarian cancer: identification of an endocrine-sensitive subgroup. Clin Cancer Res. 2002;8:2233–2239. [PubMed] [Google Scholar]
- 26.del Carmen MG, Fuller AF, Matulonis U, Horick NK, Goodman A, Duska LR, Penson R, Campos S, Roche M, Seiden MV. Phase II trial of anastrozole in women with asymptomatic mullerian cancer. Gynecol Oncol. 2003;91:596–602. doi: 10.1016/j.ygyno.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 27.Lee DMEJ, Hughes JL, Lee JH, Kavanagh JJ. Metastasis to sigmoid colon mucosa and submucosa from serous borderline ovarian tumor: response to hormone therapy. Int J Gynecol Cancer. 2006;16:295–299. doi: 10.1111/j.1525-1438.2006.00206.x. [DOI] [PubMed] [Google Scholar]
- 28.Meng L, Zhou J, Hironobu S, Suzuki T, Zeitoun K, Bulun S. TNFalpha and IL-11 secreted by malignant breast epithelial cells inhibit adipocyte differentiation by selectively downregulating C/EBPalpha and PPARgamma: mechanism of desmoplastic reaction. Cancer Research. 2001;61:2250–2255. [PubMed] [Google Scholar]
- 29.Zhou J, Gurates B, Yang S, Sebastian S, Bulun SE. Malignant breast epithelial cells stimulate aromatase expression via promoter II in human adipose fibroblasts: an epithelial-stromal interaction in breast tumors mediated by C/EBPbeta. Cancer Research. 2001;61:2328–2334. [PubMed] [Google Scholar]
- 30.Haagensen CD. Diseases of the breast. W.B. Saunders Company; Philadelphia: 1986. [Google Scholar]
- 31.Agarwal VR, Bulun SE, Leitch M, Rohrich R, Simpson ER. Use of alternative promoters to express the aromatase cytochrome P450 (CYP19) gene in breast adipose tissues of cancer-free and breast cancer patients. Journal of Clinical Endocrinology and Metabolism. 1996;81:3843–3849. doi: 10.1210/jcem.81.11.8923826. [DOI] [PubMed] [Google Scholar]
- 32.Harada N, Utsumi T, Takagi Y. Tissue-specific expression of the human aromatase cytochrome P450 gene by alternative use of multiple exons I and promoters, and switching of tissue-specific exons I in carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:11312–11316. doi: 10.1073/pnas.90.23.11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Utsumi T, Harada N, Maruta M, Takagi Y. Presence of alternatively spliced transcripts of aromatase gene in human breast cancer. Journal of Clinical Endocrinology and Metabolism. 1996;81:2344–2349. doi: 10.1210/jcem.81.6.8964875. [DOI] [PubMed] [Google Scholar]
- 34.Zhou C, Zhou D, Esteban J, Murai J, Siiteri PK, Wilczynski S, Chen S. Aromatase gene expression and its exon I usage in human breast tumors. Detection of aromatase messenger RNA by reverse transcription-polymerase chain reaction. Journal of Steroid Biochemistry and Molecular Biology. 1996;59:163–171. doi: 10.1016/s0960-0760(96)00100-8. [DOI] [PubMed] [Google Scholar]
- 35.Sebastian S, Takayama K, Shozu M, Bulun S. Cloning and characterization of a novel endothelial promoter of the human CYP19 (aromatase P450) gene that is up-regulated in breast cancer tissue. Mol Endocrinol. 2002;16:2243–2254. doi: 10.1210/me.2002-0123. [DOI] [PubMed] [Google Scholar]
- 36.MacDonald PC, Grodin JM, Siiteri PK. The utilization of plasma androstenedione for estrone production in women. In: Ebling CGc-eFJG., editor. Excerpta Medica Int. Congr. Series No. 184, Progress in Endocrinology. Proceedings of the Third International Congress of Endocrinology June 30-July 5, 1968 (Mexico). Excerpta Medica Foundation; Amsterdam. 1968. pp. 770–776. [Google Scholar]
- 37.Hemsell GJDL, Brenner PF, Siiteri PK, MacDonald PC. Plasma precursors of estrogen. II. Correlation of the extent of conversion of plasma androstenedione to estrone with age. J Clin Endocrin Metab. 1974;38:476–479. doi: 10.1210/jcem-38-3-476. [DOI] [PubMed] [Google Scholar]
- 38.MacDonald PC, Edman CD, Hemsell DL, Porter JC, Siiteri PK. Effect of obesity on conversion of plasma androstenedione to estrone in postmenopausal women with and without endometrial cancer. American Journal of Obstetrics and Gynecology. 1978;130:448–455. doi: 10.1016/0002-9378(78)90287-9. [DOI] [PubMed] [Google Scholar]
- 39.Hankinson S, Willett W, Manson J, Colditz G, Hunter D, Spiegelman D, Barbieri R, Spiezer F. Plasma sex steroid hormone levels and risk of breast cancer in postmenopausal women. J National Cancer Institute. 1999;90:1292–1299. doi: 10.1093/jnci/90.17.1292. [DOI] [PubMed] [Google Scholar]
- 40.Huang Z, Hankinson SE, Colditz GA, Stampfer MJ, Hunter DJ, Manson JE, Hennekens CH, Rosner B, Speizer FE, Willett WC. Dual effects of weight and weight gain on breast cancer risk. JAMA. 1997;278:1407–1411. [PubMed] [Google Scholar]
- 41.Brodie A, Lu Q, Liu Y, Long B. Aromatase inhibitors and their antitumore effects in model systems. Endocrine-Related Cancer. 1999;6:205–210. doi: 10.1677/erc.0.0060205. [DOI] [PubMed] [Google Scholar]
- 42.Brodie A, Lu Q, Long B. Aromatase and its inhibitors. J Steroid Biochem and Mol Biol. 1999;69:205–210. doi: 10.1016/s0960-0760(99)00051-5. [DOI] [PubMed] [Google Scholar]
- 43.Chetrite G, Cortes-Prieto J, Philippe J, Wright F, Pasqualini J. Comparison of estrogen concentrations, estrone sulfatase and aromatase activities in normal, and in cancerous, human breast tissues. J Steroid Biochemistry and Molecular Biology. 2000;72:23–27. doi: 10.1016/s0960-0760(00)00040-6. [DOI] [PubMed] [Google Scholar]
- 44.Geisler J, Berntsen H, Lonning P. A novel HPLC-RIA method for the simultaneous detection of estrone, estradiol and estrone sulphate levels in breast cancear tissue. J Steroid Biochemistry & Molecular Biology. 2000;72:259–264. doi: 10.1016/s0960-0760(00)00036-4. [DOI] [PubMed] [Google Scholar]
- 45.Van Landeghem AAJ, Poortman J, Nabuurs M, Thijssen JHH. Endogenous concentration and subcellular distribution of estrogens in normal and malignant human breast tissue. Cancer Research. 1985;45:2907–2912. [PubMed] [Google Scholar]
- 46.O'Neill JS, Elton RA, Miller WR. Aromatase activity in adipose tissue from breast quadrants: a link with tumor site. British Medical Journal. 1988;296:741–743. doi: 10.1136/bmj.296.6624.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bulun SE, Price TM, Mahendroo MS, Aitken J, Simpson ER. A link between breast cancer and local estrogen biosynthesis suggested by quantification of breast adipose tissue aromatase cytochrome P450 transcripts using competitive polymerase chain reaction after reverse transcription. Journal of Clinical Endocrinology and Metabolism. 1993;77:1622–1628. doi: 10.1210/jcem.77.6.8117355. [DOI] [PubMed] [Google Scholar]
- 48.Sasano H, Nagura H, Harada N, Goukon Y, Kimura M. Immunolocalization of aromatase and other steroidogenic enzymes in human breast disorders. Human Pathology. 1994;25:530–535. doi: 10.1016/0046-8177(94)90127-9. [DOI] [PubMed] [Google Scholar]
- 49.Reed MJ, Topping L, Coldham NG, Purohit A, Ghilchik MW, James VHT. Control of aromatase activity in breast cancer cells: the role of cytokines and growth factors. Journal of Steroid Biochemistry and Molecular Biology. 1993;44:589–596. doi: 10.1016/0960-0760(93)90264-w. [DOI] [PubMed] [Google Scholar]
- 50.Agarwal VR, Ashanullah CI, Simpson ER, Bulun SE. Alternatively spliced transcripts of the aromatase cytochrome P450 (CYP19) gene in adipose tissue of women. Journal of Clinical Endocrinology and Metabolism. 1997;82:70–74. doi: 10.1210/jcem.82.1.3655. [DOI] [PubMed] [Google Scholar]
- 51.Agarwal VR, Bulun SE, Simpson ER. Quantitative detection of alternatively spliced transcripts of the aromatase cytochrome P450 (CYP19) gene in aromatase-expressing human cells by competitive RT-PCR. Mol Cellular Probes. 1995;9:453–464. doi: 10.1006/mcpr.1995.0069. [DOI] [PubMed] [Google Scholar]
- 52.Yue W, Zhou D, Chen S, Brodie A. A new nude mouse model for postmenopausal breast cancer using MCF-7 cells transfected with the human aromatase gene. Cancer Research. 1994;54:5092–5095. [PubMed] [Google Scholar]
- 53.Tekmal R, Gill KNK, Fowler K. Aromatase overexpression and breast hyperplasia, an in vivo model--continued overexpression of aromatase is sufficient to maintain hyperplasia without circulating estrogens, and aromatase inhibitors abrogate these preneoplasatic changes in mammary glands. Endocrine-Related Cancer. 1999;6:307–314. doi: 10.1677/erc.0.0060307. [DOI] [PubMed] [Google Scholar]
- 54.Kovacic SCA, Simpson ER, Clyne CD. Inhibition of aromatase transcription via promoter II by short heterodimer partner in human preadipocytes. Mol Endocrinol. 2004;18:252–259. doi: 10.1210/me.2003-0211. [DOI] [PubMed] [Google Scholar]
- 55.Price T, Aitken J, Head J, Mahendroo MS, Means GD, Simpson ER. Determination of aromatase cytochrome P450 messenger RNA in human breast tissues by competitive polymerase chain reaction (PCR) amplification. Journal of Clinical Endocrinology and Metabolism. 1992;74:1247–1252. doi: 10.1210/jcem.74.6.1592866. [DOI] [PubMed] [Google Scholar]
- 56.Ackerman GE, Smith ME, Mendelson CR, MacDonald PC, Simpson ER. Aromatization of androstenedione by human adipose tissue stromal cells in monolayer culture. Journal of Clinical Endocrinology and Metabolism. 1981;53:412–417. doi: 10.1210/jcem-53-2-412. [DOI] [PubMed] [Google Scholar]
- 57.Santen RJ, Martel J, Hoagland M, Naftolin F, Roa L, Harada N, Hafer L, Zaino R, Santner SJ. Stromal spindle cells contain aromatase in human breast tumors. Journal of Clinical Endocrinology and Metabolism. 1994;79:627–632. doi: 10.1210/jcem.79.2.8045987. [DOI] [PubMed] [Google Scholar]
- 58.Sasano H, Frost A, Saitoh R, Harada N, Poutanen M, Vihko R, Bulun S, Silverberg S, Nagura H. Aromatase and 17beta-hydroxysteroid dehydrogenase type 1 in human breast carcinoma. Journal of Clinical Endocrinology and Metabolism. 1996;81:4042–4046. doi: 10.1210/jcem.81.11.8923858. [DOI] [PubMed] [Google Scholar]
- 59.Brodie A, Long B, Lu Q. Aromatase expression in the human breast. Breast Cancer Res Treat. 1998;49:S85–91. doi: 10.1023/a:1006029612990. [DOI] [PubMed] [Google Scholar]
- 60.Sasano H, Edwards DP, Anderson TJ, Silverberg SG, Evans DB, Santen RJ, Ramage P, Simpson ER, Bhatnagar AS, Miller WR. Validation of new aromatase monoclonal antibodies for immunohistochemistry: progress report. J Steroid Biochem Mol Biol. 2003;86:239–244. doi: 10.1016/s0960-0760(03)00363-7. [DOI] [PubMed] [Google Scholar]
- 61.Pauley R, Santner S, Tait L, Bright R. Regulation of CYP19 aromatase transcription in breast stromal fibroblasts. J Clin Endocrin & Metabo. 2000;85:837–846. doi: 10.1210/jcem.85.2.6345. [DOI] [PubMed] [Google Scholar]
- 62.Crichton MB, Nichols JE, Zhao Y, Bulun SE, Simpson ER. Expression of transcripts of interleukin-6 and related cytokines by human breast tumors, breast cancer cells, and adipose stromal cells. Molecular and Cellular Endocrinology. 1996;118:215–220. doi: 10.1016/0303-7207(96)03761-6. [DOI] [PubMed] [Google Scholar]
- 63.Hube F, Hauner H. The two tumor necrosis factor receptors mediate opposite effects on differentiation and glucose metabolism in human adipocytes in primary culture. Endocrinology. 2000;141:2582–2588. doi: 10.1210/endo.141.7.7561. [DOI] [PubMed] [Google Scholar]
- 64.Deb ASS, Imir AG, Yilmaz MB, Suzuki T, Sasano H, Bulun SE. Estrogen regulates expression of tumor necrosis factor receptors in breast adipose fibroblasts. J Clin Endocrinol Metab. 2004;89:4018–4024. doi: 10.1210/jc.2004-0127. [DOI] [PubMed] [Google Scholar]
- 65.Clyne CD, Speed CJ, Zhou J, Simpson ER. Liver receptor homologue-1 (LRH-1) regulates expression of aromatase in preadipocytes. J Biol Chem. 2002;277:20591–20597. doi: 10.1074/jbc.M201117200. [DOI] [PubMed] [Google Scholar]
- 66.Karuppu D, Kalus A, Simpson ER, Clyne CD. Aromatase and prostaglandin inter-relationsips in breast adipose tissue: significance for breast cancer development. Breast Cancer Research and Treatment. 2002;76:103–109. doi: 10.1023/a:1020531329686. [DOI] [PubMed] [Google Scholar]
- 67.Brueggemeier RW, Quinn AL, Parrett ML, Joarder FS, Harris RE. Correlation of aromatase and cyclooxygenase gene expression in human breast cancer specimens. Cancer Lett. 1999:27–35. doi: 10.1016/s0304-3835(99)00050-6. [DOI] [PubMed] [Google Scholar]
- 68.Richards JA, Petrel TA, Brueggemeier RW. Signaling pathways regulating aromatase and cyclooxygenases in normal and malignant breast cells. J Steroid Biochem. 2002;80:203–212. doi: 10.1016/s0960-0760(01)00187-x. [DOI] [PubMed] [Google Scholar]
- 69.Deb S, Jianfeng Z, Amin SA, Gonca IA, Bertan YM, Zihong L, Bulun SE. A novel role of sodium butyrate in the regulation of cancer-associated aromatase promoters I.3 and II by disrupting a transcriptional complex in breast adipose fibroblasts. J Biol Chem. 2006;281:2585–2597. doi: 10.1074/jbc.M508498200. [DOI] [PubMed] [Google Scholar]
- 70.Chen S, Itoh T, Wu K, Zhou D, Yang C. Transcriptional regulation of aromatase expression in human breast tissue. J Steroid Biochem Mol Biol. 2002;83:93–99. doi: 10.1016/s0960-0760(02)00276-5. [DOI] [PubMed] [Google Scholar]
- 71.Ruohola J, Valve E, Karkkainen M, Joukov V. Vascular endothelial growth factors are differentially regulated by steroid hormones and antiestrogens in breast cancer cells. Mol Cell Endocrinol. 1999;149:29–40. doi: 10.1016/s0303-7207(99)00003-9. [DOI] [PubMed] [Google Scholar]
- 72.Nakamura J, Lu Q, Aberdeen G, Albrecht E, Brodie A. The effect of estrogen on aromatase and vascular endothelial growth factor messenger ribonucleic acid in the normal nonhuman primate mammary gland. J Clin Endocrinol Metab. 1999;84:1432–1437. doi: 10.1210/jcem.84.4.5641. [DOI] [PubMed] [Google Scholar]
- 73.Nakamura J, Savinov A, Lu Q, Brodie A. Estrogen regulates vascular endothelial growth/permeability factor expression in 7,12-dimethylbenz(a)anthracene-induced rat mammary tumors. Endocrinology. 1996;137:5589–5596. doi: 10.1210/endo.137.12.8940388. [DOI] [PubMed] [Google Scholar]
- 74.Shekhar M, Werdell J, Tait L. Interaction with endothelial cells is a prerequisite for branching ductalalveolar morphogenesis and hyperplasia of preneoplastic human breast epithelial cells: regulation by estrogen. Cancer Reserach. 2000;60:439–449. [PubMed] [Google Scholar]
- 75.Santen R. To block estrogen's synthesis or action: that is the question. J Clin Endocrinol Metab. 2002;87:3007–3012. doi: 10.1210/jcem.87.7.8589. [DOI] [PubMed] [Google Scholar]
- 76.Baum M, Budzar AU, Cuzick J, Forbes J, Houghton JH, Klijn JG, Sahmoud T, Group AT. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. Lancet. 2002;359:2131–2139. doi: 10.1016/s0140-6736(02)09088-8. [DOI] [PubMed] [Google Scholar]
- 77.Baum M, Buzdar A, Cuzick J, Forbes J, Houghton J, Howell A, Sahmoud T. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early-stage breast cancer: results of the ATAC (Arimidex, Tamoxifen Alone or in Combination) trial efficacy and safety update analyses. Cancer. 2003;98:1802–1810. doi: 10.1002/cncr.11745. [DOI] [PubMed] [Google Scholar]
- 78.Buzdar VIAU, Sainsbury R. The impact of hormone receptor status on the clinical efficacy of the new-generation aromatase inhibitors: a review of data from first-line metastatic disease trials in postmenopausal women. Breast J. 2004;10:211–217. doi: 10.1111/j.1075-122X.2004.21320.x. [DOI] [PubMed] [Google Scholar]
- 79.Bonneterre J, Thurlimann B, Robertson JF, et al. Anastrozole versus tamoxifen as first-line therapy for advanced breast cancer in 668 postmenopausal women: results of the tamoxifen or Arimidex randomized group efficacy and tolerability study. J Clin Oncol. 2000;18:3748–3757. doi: 10.1200/JCO.2000.18.22.3748. [DOI] [PubMed] [Google Scholar]
- 80.Goss PE, Ingle JN, Martino S, Robert NJ, Muss HB, Piccart MJ, Castiglione M, Tu D, Shepherd LE, Pritchard KI, Livingston RB, Davidson NE, Norton L, Perez EA, Abrams JS, Therasse P, Palmer MJ, Pater JL. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N Engl J Med. 2003;349:1793–1802. doi: 10.1056/NEJMoa032312. [DOI] [PubMed] [Google Scholar]
- 81.Mouridsen H, Gershanovich M, Sun Y, Perez-Carrion R, Boni Monnie C, Apffelstaedt J, Smith R, Sleeboom HP, Janicke F, Pluzanska A, Dank M, Becquart D, Bapsy PP, Salminen E, Snyder R, Lassus M, Verbeek JA, Staffler B, Chaudri-Ross HA, Dugan M. Superior efficacy of letrozole versus tamoxifen as first-line therapy for postmenopausal women with advanced breast cancer: results of a phase III study of the International Letrozole Breast Cancer Group. J Clin Oncol. 2001;19:2596–2606. doi: 10.1200/JCO.2001.19.10.2596. [DOI] [PubMed] [Google Scholar]
- 82.Milla-Santos A, Milla L, Portella J, Rallo L, Pons M, Rodes E, Casanovas J, Puig-Gali M. Anastrozole Versus Tamoxifen as First-Line Therapy in Postmenopausal Patients With Hormone-Dependent Advanced Breast Cancer: A Prospective, Randomized, Phase III Study. Am J Clin Onc. 2003;26:317–322. doi: 10.1097/01.COC.0000047126.10522.F9. [DOI] [PubMed] [Google Scholar]
- 83.Paridaens R, Dirix L, Lohrisch C, Beex L, Nooij M, Cameron D, Biganzoli L, Cufer T, Duchateau L, Hamilton A, Lobelle JP, Piccart M. Mature results of a randomized phase II multicenter study of exemestane versus tamoxifen as first-line hormone therapy for postmenopausal women with metastatic breast cancer. Ann Oncol. 2003;14:1391–1398. doi: 10.1093/annonc/mdg362. [DOI] [PubMed] [Google Scholar]
- 84.Lu Q, Yue W, Wang J, Liu Y, Long B, Brodie A. The effects of aromatase inhibitors and antiestrogens in the nude mouse model. Breast Cancer Research & Treatment. 1998;50:63–71. doi: 10.1023/a:1006004930930. [DOI] [PubMed] [Google Scholar]
- 85.Dixon J, Renshaw L, Bellamy C, Stuart M, Hoctin-Boes G, Miller W. The effects of neoadjuvant anastrozole (Arimidex) on tumor volume in postmenopausal women with breast cancer: a randomized, double-blind, single-center study. Clin Cancer Res. 2000;6:2229–2235. [PubMed] [Google Scholar]
- 86.Bulun SE, Yang S, Fang Z, Gurates B, Tamura M, Zhou J, Sebastian S. Role of aromatase in endometrial disease. J Steroid Biochem Mol Biol. 2001;79:19–25. doi: 10.1016/s0960-0760(01)00134-0. [DOI] [PubMed] [Google Scholar]
- 87.Inoue M, Kyo S, Fujita M, Enomoto T, Kondoh G. Coexpression of the c-kit receptor and the stem cell factor in gynecological tumors. Cancer Res. 1994;54:3049–3053. [PubMed] [Google Scholar]
- 88.Nagasako S, Asanuma N, Nagata Y. [Plasma concentration of estrogens and androgens in postmenopausal women with or without endometrial cancer] Nippon Sanka Fujinka Gakkai Zasshi. 1988;40:707–713. [PubMed] [Google Scholar]
- 89.Potischman N, Swanson CA, Siiteri P, Hoover RN. Reversal of relation between body mass and endogenous estrogen concentrations with menopausal status. J Natl Cancer Inst. 1996;88:756–758. doi: 10.1093/jnci/88.11.756. [DOI] [PubMed] [Google Scholar]
- 90.Sherman ME, Sturgeon S, Brinton LA, Potischman N, Kurman RJ, Berman ML, Mortel R, Twiggs LB, Barrett RJ, Wilbanks GD. Risk factors and hormone levels in patients with serous and endometrioid uterine carcinomas. Mod Pathol. 1997;10:963–968. [PubMed] [Google Scholar]
- 91.Vermeulen-Meiners C, Poortman J, Haspels AA, Thijssen JH. The endogenous concentration of estradiol and estrone in pathological human postmenopausal endometrium. J Steroid Biochem. 1986;24:1073–1078. doi: 10.1016/0022-4731(86)90362-6. [DOI] [PubMed] [Google Scholar]
- 92.Berstein LM, Tchernobrovkina AE, Gamajunova VB, Kovalevskij AJ, Vasilyev DA, Chepik OF, Turkevitch EA, Tsyrlina EV, Maximov SJ, Ashrafian LA, Thijssen JH. Tumor estrogen content and clinico-morphological and endocrine features of endometrial cancer. J Cancer Res Clin Oncol. 2003;129:245–249. doi: 10.1007/s00432-003-0427-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yamamoto T, Kitawaki J, Urabe M, Honjo H, Tamura T, Noguchi T, Okada H, Sasaki H, Tada A, Terashima Y, Nakamura J, Yoshihama M. Estrogen production of endometrium and endometrial cancer tissue; influence of aromatase on proliferation of endometrial cancer cells. Journal of Steroid Biochemistry and Molecular Biology. 1993;44:463–468. doi: 10.1016/0960-0760(93)90251-q. [DOI] [PubMed] [Google Scholar]
- 94.Watanabe K, Sasano H, Harada N, Ozaki M, Niikura H, Sato S, Yajima A. Aromatase in human endometrial carcinoma and hyperplasia. Immunohistochemical, in situ hybridization, and biochemical studies. American Journal of Pathology. 1995;146:491–500. [PMC free article] [PubMed] [Google Scholar]
- 95.Berstein L, Maximov S, Gershfeld E, Meshkova I, Gamajunova V, Tsyrlina E, Larionov A, Kovalevskij A, Vasilyev D. Neoadjuvant therapy of endometrial cancer with the aromatase inhibitor letrozole: endocrine and clinical effects. Eur J Obstet Gynecol Reprod Biol. 2002;105:161–165. doi: 10.1016/s0301-2115(02)00147-1. [DOI] [PubMed] [Google Scholar]
- 96.Sasano H, Sato S, Ito K, Yajima A, Nakamura J, Yoshihama M, Ariga K, Anderson TJ, Miller WR. Effects of aromatase inhibitors on the pathobiology of the human breast, endometrial and ovarian carcinoma. Endocr Relat Cancer. 1999;6:197–204. doi: 10.1677/erc.0.0060197. [DOI] [PubMed] [Google Scholar]
- 97.Yamamoto T, Fukuoka M, Fujimoto Y, Kitawaki J, Nakakoshi M, Yoshihama M, Okada H. Inhibitory effect of a new androstenedione derivative, 14 alpha-hydroxy-4-androstene-3,6,17-trione (14 alpha-OHAT) on aromatase activity of human uterine tumors. J Steroid Biochem. 1990;36:517–521. doi: 10.1016/0022-4731(90)90167-q. [DOI] [PubMed] [Google Scholar]
- 98.Duffy S, Jackson TL, Lansdown M, Philips K, Wells M, Pollard S, Clack G, Cuzick J, Coibion M, Bianco AR. The ATAC adjuvant breast cancer trial in postmenopausal women: baseline endometrial subprotocol data. Bjog. 2003;110:1099–1106. [PubMed] [Google Scholar]
- 99.Lacey JV, Jr, Mink PJ, Lubin JH, Sherman ME, Troisi R, Hartge P, Schatzkin A, Schairer C. Menopausal hormone replacement therapy and risk of ovarian cancer. Jama. 2002;288:334–341. doi: 10.1001/jama.288.3.334. [DOI] [PubMed] [Google Scholar]
- 100.Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, Bonds D, Brunner R, Brzyski R, Caan B, Chlebowski R, Curb D, Gass M, Hays J, Heiss G, Hendrix S, Howard BV, Hsia J, Hubbell A, Jackson R, Johnson KC, Judd H, Kotchen JM, Kuller L, LaCroix AZ, Lane D, Langer RD, Lasser N, Lewis CE, Manson J, Margolis K, Ockene J, O'Sullivan MJ, Phillips L, Prentice RL, Ritenbaugh C, Robbins J, Rossouw JE, Sarto G, Stefanick ML, Van Horn L, Wactawski-Wende J, Wallace R, Wassertheil-Smoller S. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. Jama. 2004;291:1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- 101.Giudice L, Kao L. Endometriosis. Lancet. 2004;364:1789–1799. doi: 10.1016/S0140-6736(04)17403-5. [DOI] [PubMed] [Google Scholar]
- 102.Zeitoun K, Takayama K, Michael MD, Bulun SE. Stimulation of aromatase P450 promoter (II) activity in endometriosis and its inhibition in endometrium are regulated by competitive binding of SF-1 and COUP-TF to the same cis-acting element. Molecular Endocrinology. 1999;13:239–253. doi: 10.1210/mend.13.2.0229. [DOI] [PubMed] [Google Scholar]
- 103.Yang S, Fang Z, Takashi S, Hironobu S, Jianfeng Z, Gurates B, Tamura M, Ferrer K, Bulun SE. Regulationof aromatase P450 expression in endometriotic and endometrial stromal cells by CCAT/enhancer binding proteins: decreased C/EBPbeta in endometriosis is associated with overexpression of aromatase. J Clin Endocrinol Metab. 2002;87:2336–2345. doi: 10.1210/jcem.87.5.8486. [DOI] [PubMed] [Google Scholar]
- 104.Gurates B, Sebastian S, Yang S, Zhou J, Tamura M, Fang Z, Suzuki T, Sasano H, Bulun SE. WT1 and DAX-1 inhibit aromatase P450 expression in human endometrial and endometriotic stromal cells. J Clin Endocrinol Metab. 2002;87:4369–4377. doi: 10.1210/jc.2002-020522. [DOI] [PubMed] [Google Scholar]
- 105.Fang ZJ, Yang S, Gurates G, Tamura M, Simpson ER, Evans D, Bulun SE. Genetic or enzymatic disruption of aromatase inhibits the growth of ectopic uterine tissue. J Clin Endocrinol Metab. 2002;87:3460–3466. doi: 10.1210/jcem.87.7.8683. [DOI] [PubMed] [Google Scholar]
- 106.Kitawaki J, Noguchi T, Amatsu T, Maeda K, Tsukamoto K, Yamamoto T, Fushiki S, Osawa Y, Honjo H. Expression of aromatase cytochrome P450 protein and messenger ribonucleic acid in human endometriotic and adenomyotic tissues but not in normal endometrium. Biology of Reproduction. 1997;57:514–519. doi: 10.1095/biolreprod57.3.514. [DOI] [PubMed] [Google Scholar]
- 107.Noble LS, Takayama K, Putman JM, Johns DA, Hinshelwood MM, Agarwal VR, Zhao Y, Carr BR, Bulun SE. Prostaglandin E2 stimulates aromatase expression in endometriosis-derived stromal cells. Journal of Clinical Endocrinology and Metabolism. 1997;82:600–606. doi: 10.1210/jcem.82.2.3783. [DOI] [PubMed] [Google Scholar]
- 108.Tsai SJ, Wu MH, Lin CC, Sun HS, Chan HM. Regulation of steroidogenic acute regulatory protein expression and progesterone production in endometriotic stromal cells. Journal of Clinical Endocrinology & Metabolism. 2001;86:5765–5773. doi: 10.1210/jcem.86.12.8082. [DOI] [PubMed] [Google Scholar]
- 109.Sun HS, Hsiao KY, Hsu CC, Wu MH, Tsai SJ. Transactivation of steroidogenic acute regulatory protein in human endometriotic stromalcells is mediated by the prostaglandin EP2 receptor. Endocrinology. 2003;144:3934–3942. doi: 10.1210/en.2003-0289. [DOI] [PubMed] [Google Scholar]
- 110.Langdon SP, Hawkes MM, Lawrie SS, Hawkins RA, Tesdale AL, Crew AJ, Miller WR, Smyth JF. Oestrogen receptor expression and the effects of oestrogen and tamoxifen on the growth of human ovarian carcinoma cell lines. Br J Cancer. 1990;62:213–216. doi: 10.1038/bjc.1990.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Langdon SP, Ritchie A, Young K, Crew AJ, Sweeting V, Bramley T, Hillier S, Hawkins RA, Tesdale AL, Smyth JF, et al. Contrasting effects of 17 beta-estradiol on the growth of human ovarian carcinoma cells in vitro and in vivo. Int J Cancer. 1993;55:459–464. doi: 10.1002/ijc.2910550323. [DOI] [PubMed] [Google Scholar]
- 112.Langdon SP, Crew AJ, Ritchie AA, Muir M, Wakeling A, Smyth JF, Miller WR. Growth inhibition of oestrogen receptor-positive human ovarian carcinoma by anti-oestrogens in vitro and in a xenograft model. Eur J Cancer. 1994;30A:682–686. doi: 10.1016/0959-8049(94)90545-2. [DOI] [PubMed] [Google Scholar]
- 113.Langdon SP, Hirst GL, Miller EP, Hawkins RA, Tesdale AL, Smyth JF, Miller WR. The regulation of growth and protein expression by estrogen in vitro: a study of 8 human ovarian carcinoma cell lines. J Steroid Biochem Mol Biol. 1994;50:131–135. doi: 10.1016/0960-0760(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 114.Langdon SP, Gabra H, Bartlett JM, Rabiaz GJ, Hawkins RA, Tesdale AL, Ritchie AA, Miller WR, Smyth JF. Functionality of the progesterone receptor in ovarian cancer and its regulation by estrogen. Clin Cancer Res. 1998;4:2245–2251. [PubMed] [Google Scholar]
- 115.Simpson BJ, Langdon SP, Rabiasz GJ, Macleod KG, Hirst GL, Bartlett JM, Crew AJ, Hawkins RA, Macineira-Perez PP, Smyth JF, Miller WR. Estrogen regulation of transforming growth factor-alpha in ovarian cancer. J Steroid Biochem Mol Biol. 1998;64:137–145. doi: 10.1016/s0960-0760(97)00159-3. [DOI] [PubMed] [Google Scholar]
- 116.Langdon SP, Rabiasz GJ, Hirst GL, King RJ, Hawkins RA, Smyth JF, Miller WR. Expression of the heat shock protein HSP27 in human ovarian cancer. Clin Cancer Res. 1995;1:1603–1609. [PubMed] [Google Scholar]
- 117.Hatch KD, Beecham JB, Blessing JA, Creasman WT. Responsiveness of patients with advanced ovarian carcinoma to tamoxifen. A Gynecologic Oncology Group study of second-line therapy in 105 patients. Cancer. 1991;68:269–271. doi: 10.1002/1097-0142(19910715)68:2<269::aid-cncr2820680209>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 118.Markman M, Iseminger KA, Hatch KD, Creasman WT, Barnes W, Dubeshter B. Tamoxifen in platinum-refractory ovarian cancer: a Gynecologic Oncology Group Ancillary Report. Gynecol Oncol. 1996;62:4–6. doi: 10.1006/gyno.1996.0181. [DOI] [PubMed] [Google Scholar]
- 119.Ahlgren JD, Ellison NM, Gottlieb RJ, Laluna F, Lokich JJ, Sinclair PR, Ueno W, Wampler GL, Yeung KY, Alt D, et al. Hormonal palliation of chemoresistant ovarian cancer: three consecutive phase II trials of the Mid-Atlantic Oncology Program. J Clin Oncol. 1993;11:1957–1968. doi: 10.1200/JCO.1993.11.10.1957. [DOI] [PubMed] [Google Scholar]