

Abstract
Infections with the diarrheagenic pathogen, Giardia lamblia, are commonly treated with the 5-nitroimidazole (5-NI) metronidazole (Mz), and yet treatment failures and Mz resistance occur. Using a panel of new 2-ethenyl and 2-ethanyl 5-NI derivatives, we found that compounds with a saturated bridge between the 5-NI core and a pendant ring system exhibited only modestly increased antigiardial activity and could not overcome Mz resistance. By contrast, olefins with a conjugated bridge connecting the core and a substituted phenyl or heterocyclic ring showed greatly increased antigiardial activity without toxicity, and several overcame Mz resistance and were more effective than Mz in a murine giardiasis model. Determination of the half-wave potential of the initial one-electron transfer by cyclic voltammetry revealed that easier redox activation correlated with greater antigiardial activity and capacity to overcome Mz resistance. These studies show the potential of combining systematic synthetic approaches with biological and electrochemical evaluations in developing improved 5-NI drugs.
Introduction
The anaerobic protozoan parasite, Giardia lamblia, is a major cause of human diarrheal disease, infecting an estimated 10% of the world’s population in both endemic and epidemic fashion.1 In the U.S., Giardia is the most common cause of waterborne diarrheal disease, with international travelers, hikers, and children in day care centers at particular risk.2 Infection is by fecal–oral transmission and is initiated by ingestion of infectious cysts in contaminated water or through person-to-person contact. After excystation, flagellated trophozoites colonize the upper small intestine where they attach to the epithelial lining but do not invade the mucosa. The duration of giardial infection is variable, but chronic infection and reinfection occur commonly.3 Although ~50% of Giardia infections are asymptomatic, major symptoms include diarrhea, often severe and protracted, with malabsorption, dehydration, weight loss, failure to thrive, cognitive impairment in children, and chronic fatigue in adults.4,5 These symptoms result from a combination of intestinal villus atrophy, loss of brush border microvilli, digestive enzyme deficiencies, and epithelial barrier dysfunction.6,7
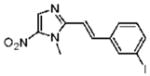
The leading antigiardial agent, metronidazole (Mz, Figure 1), is a synthetic 5-nitroimidazole (NI) derivative introduced in 1959 by Rhône-Poulenc. In addition to Giardia, Mz is active against Entamoeba histolytica and Trichomonas vaginalis, as well as several clinically important anaerobic bacteria (including Clostridium difficile, Helicobacter pylori, and Bacteroides fragilis), making it a highly versatile antibiotic. Mz has a long record of efficacy and safety.8,9 On the basis of the therapeutic utility of Mz, several other 5-NIsa have been developed more recently. For example, tinidazole (1, Figure 1) is a N1-position modified 5-NI, which has been approved by the FDA for the treatment of giardiasis. Not all antigiardial nitro drugs are based on the 5-NI scaffold. Nitazoxanide (2, Figure 1) belongs to an emerging class of 5-nitrothiazole compounds with potent antigiardial activity,10 although Giardia lines resistant to Mz are cross-resistant to nitazoxanide.9 Overall, Mz remains the standard treatment for giardiasis to date, making the discovery and development of new therapeutics an important goal in expanding the arsenal of antibiotics for controlling the infection.
Figure 1.

5-Nitro heterocyclic compounds active against Giardia.
The high specificity of the 5-NIs is due to the requirement for the prodrug to be reduced to toxic free radical intermediates by low redox potential reactions present only in anaerobic microbes.11 These short-lived free radicals cause lethal damage to the microbe, although their cellular targets are not well-defined in either eukaryotes or prokaryotes. Energy metabolism in Giardia, which does not have mitochondria, is fermentative, and electron transport proceeds in the absence of oxidative phosphorylation present in organisms with mitochondria. However, Giardia is microaerotolerant and can reduce O2 and thus protect the highly oxygen-sensitive, key metabolic enzyme pyruvate–ferredoxin oxidoreductase and the iron-containing ferredoxins that activate Mz.12 The oxidoreductase carboxylates pyruvate and donates electrons to ferredoxin, which in turn reduces other components in the electron transport chain. ATP production is linked to pyruvate decarboxylation.13 Reduced ferredoxin activates the critical nitro group of the prodrug Mz, leading to formation of toxic free radicals that kill the parasite. Selective reductive activation in the target microbes, but not mammalian cells, is generally thought to be critical for the potency and selectivity of 5-NI drugs.14 However, relatively little is known about the electrochemical properties of most 5-NIs, making it difficult to draw detailed conclusions about the importance of low redox potential for their antimicrobial activity, selectivity, and mechanisms of action.
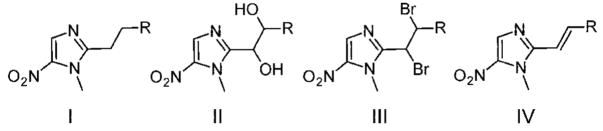
In spite of the efficacy of 5-NI drugs, treatment failures in giardiasis occur in up to 20% of cases.14 Clinical resistance of G. lamblia to Mz and other 5-NIs is proven, and in vitro resistance can be induced so that parasites grow in physiologically relevant concentrations of Mz.15 In addition, resistance has been induced in vitro against all commonly used antigiardial drugs, including different 5-NIs, furazolidone, albendazole, and quinacrine,9,14 further demonstrating the need for new compounds to preempt resistance development. Prior studies suggested that modifications in the 2-position of the imidazole ring can lead to effective 5-NI drugs.16,17 For example, the 2-position modified 5-NI compound 3 (Figure 1, compound 17 in ref 16) is very active against G. lamblia and can overcome Mz resistance.16 Although these 5-NI derivatives were not examined for their toxicity, in vivo efficacy, or mechanisms of action, they raised the possibility that 2-position derivatives of 5-NI may be promising for developing new, potent, and safe drugs against giardiasis.
In the present study, we synthesized a library of substituted 2-styryl 5-NI compounds and evaluated the significance of the olefin bridge directly attached to the NI ring by chemically modifying it by dibromination, dihydroxylation, and reduction. These new compounds were assessed for their potency against G. lamblia in vitro and in vivo, their cytotoxicity in human cells, and their electrochemical properties.
Results and Discussion
Chemistry
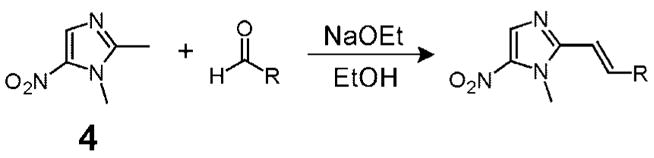
We synthesized 2-styryl 5-NI derivatives using the aldol condensation18 between 1,2-dimethyl-5-nitroimidazole (dimetridazole, 4, Table 1) and a panel of aryl aldehydes in the presence of sodium ethoxide in ethanol as shown in Table 1. By use of this protocol, a diverse array of functionality was incorporated into the 2-position of the NI core ranging from electron-rich aromatic systems (Table 1, entries 2–5, 16, and 18) and electron-poor ones (entries 6–15 and 17) to different heterocyclic scaffolds (entries 23–27). Because certain halogen substitutions in the aromatic ring can yield potent antimicrobial compounds,16,17 we systematically explored most of the possible halogen monosubstitutions around the phenyl ring (entries 6–15). While the yields of these reactions varied widely (13–77%), sufficient material could be produced for biological testing after purification by flash chromatography or recrystallization.
Table 1.
Synthesis of 2-Ethenyl Derivatives of 5-NI by Aldol Condensation
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | aldehyde | product | yield (%) | entry | Aldehyde | product | yield (%) |
| 1 | Benzaldehyde |
 5 |
20 | 15 | 3-fluoro-benzaldehyde |
 19 |
13 |
| 2 | 2-methyl-benzaldehyde |
 6 |
23 | 16 | 3-methoxy-benzaldehyde |
 20 |
32 |
| 3 | 3-methyl-benzaldehyde |
 7 |
42 | 17 | 4-trifluoro-methoxy-benzaldehyde |
21 |
14 |
| 4 | 4-methyl-benzaldehyde |
 8 |
22 | 18 | 2,3,4-trimethoxy-benzaldehyde |
 22 |
16 |
| 5 | 2,4,6-trimethyl-benzaldehyde |
 9 |
29 | 19 | 4-diethoxy-methyl-benzaldehyde |
23 |
24 |
| 6 | 2-bromo-benzaldehyde |
 10 |
20 | 20 | 2-biphenyl-carbaldehyde |
 24 |
21 |
| 7 | 3-bromo-benzaldehyde |
 11 |
19 | 21 | 1-napthyl-aldehyde |
 25 |
28 |
| 8 | 4-bromo-benzaldehyde |
 12 |
72 | 22 | 2-napthyl-aldehyde |
 26 |
19 |
| 9 | 2-chloro-benzaldehyde |
 13 |
24 | 23 | piperonal |
 27 |
31 |
| 10 | 3-chloro-benzaldehyde |
 14 |
19 | 24 | 1-methyl-imidazole-2-carbaldehyde |
 28 |
77 |
| 11 | 4-chloro-benzaldehyde |
15 |
15 | 25 | furan-2-carbaldehyde |
 29 |
53 |
| 12 | 2,4-dichloro-benzaldehyde |
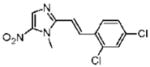 16 |
15 | 26 | 2-thiophene-carbaldehyde |
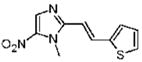 30 |
23 |
| 13 | 2-iodo-benzaldehyde |
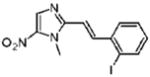 17 |
20 | 27 | 3-thiophene-carbaldehyde |
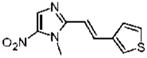 31 |
22 |
| 14 | 3-iodo-benzaldehyde |
 18 |
19 | ||||
In another round of derivatizations, we functionalized the double bond present in the aldol condensation products. This allowed us to probe the biological importance of an unsaturated bridge connecting the NI and the phenyl ring in the new compounds. To this end, we initially attempted to oxidize the double bond in 5 to the corresponding epoxide 32 using m-CPBA (Scheme 1a). Although the epoxide was observed by LCMS, it proved to be difficult to isolate or even capture by treatment of the crude reaction mixture with a suitable nucleophile. A more productive functionalization was achieved by treating olefin 12 with bromine in chloroform to produce tribromide 33 in 88% yield (Scheme 1b). Additionally, the double bonds of nitroimidazoles 8 and 12 were dihydroxylated using the acidic Upjohn procedure19 to furnish diol 34 in 28% and diol 35 in 14% yield (Scheme 1c). Subsequent acetylation of diols 34 and 35 using acetic anhydride produced compounds 36 and 37 in 66% and 92% yields, respectively (Scheme 1c).
Scheme 1.

Last, we evaluated the complete reduction of the C2-olefin in the aldol adducts to their saturated counterparts for direct structure–activity comparisons. Reducing the double bond in the presence of the nitro group on the imidazole ring proved difficult. Thus, as expected, hydrogenation of 5 over palladium or platinum catalysts efficiently hydrogenated the double bond but also reduced the nitro group to the corresponding amine 38, as outlined in Scheme 2a. We also attempted an alkylation strategy where dimetridazole 4 was treated with NaOEt and benzyl bromide but observed no conversion to the desired product (Scheme 2b). A NiCl2/NaBH4 reducing agent system,20 which is a mild source of hydride, was also tried with olefin 5 but with unsatisfactory results, as only the fully reduced product 38 was again observed (Scheme 2c). We further explored constructing the imidazole ring independently followed by a nitration to give the desired product.21 However, the nitration of known imidazole 39 occurred on the undesired aromatic ring, forcing us to abandon this approach (Scheme 2d). Ultimately, we evaluated diimide as the reducing agent. While a more conventional Cu(II)/hydrazine system22 for generating the diimide failed to effect the reaction, tosyl hydrazine in pyridine23 selectively reduced the double bond while leaving the nitro functionality intact (Scheme 2e). This procedure allowed us to convert the aldol adducts 5, 8, and 12 to their fully saturated counterparts 40, 41, and 42 in 32%, 18%, and 17% yields, respectively (Scheme 2e).
Scheme 2.

Improved Antigiardial Activity and Selectivity of 2-Styryl-5-NI Derivatives
Prior work had demonstrated that dibrominated 5-NI compound 3 (Figure 1), a C2-modified 5-NI, had increased potency against G. lamblia relative to Mz.16 In the present study, utilizing a panel of isolates representing assemblages A and B, the two major genetic groupings that contain all human-infectious Giardia, we confirmed that compound 3 (Table 2, entry 1) possessed a 40-fold increased antigiardial activity over Mz (Table 2, entry 10). However, toxicity analysis in the human cell line, HeLa, indicated that dibromide 3 was > 500-fold more toxic than Mz. To consider both the potency and toxicity of the new compounds in comparison to Mz, the in vitro selectivity was determined by calculating the ratio of the IC50 in HeLa cells to EC50 in Giardia as an estimate for the therapeutic index. The most promising candidate compounds for further evaluation will be ones with an equal or higher in vitro selectivity ratio than Mz. In the case of dibromide 3, the selectivity ratio was > 10-fold lower than that of Mz (Table 2). Similarly, a close analogue of 3 bearing a bromine instead of a methyl at the phenyl ring para to the saturated bridge, compound 33 (Table 2, Entry 2), did not overcome the toxic effects of 3 and had even lower antimicrobial activity, further reducing the selectivity ratio. To evaluate whether the toxicity of 3 could be attenuated without affecting its potent antigiardial activity, we synthesized compounds related to dibromide 3 in which the bromine atoms in the bridge were replaced systematically with hydroxyl groups (34 and 35, Table 2, entries 3 and 4) or acetoxy groups (36 and 37, Table 2, entries 5 and 6). These compounds also had selectivity ratios markedly below Mz, rendering them significantly less effective than Mz. Finally, we prepared more direct analogues of dibromide 3, in which the bromine atoms were replaced with hydrogen atoms (40–42, Table 2, entries 7–9). Although elimination of the two bromine atoms in 3 decreased toxicity, it also compromised antigiardial activity and the selectivity ratio. Together, these results suggest that modifications in the saturated bridge at C2 of the nitroimidazole core could not overcome toxicity without markedly reducing antimicrobial activity.
Table 2.
Antigiardial Potency and Toxicity of New 5-NI Alkanes
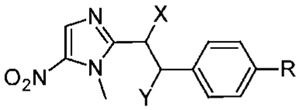 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Activity against G. lambliaa |
||||||||
| entry | name | X | Y | R | EC50 (nM) | pEC50 (Mean ± SE) | toxicity in HeLa cells,b pIC50 (mean ± SE) | selectivity ratio,c IC50/EC50 |
| 1 | 3 | Br | Br | Me | 65 | 7.19 ± 0.15 | 4.95 ± 0.06 | 174 |
| 2 | 33 | Br | Br | Br | 347 | 6.46 ± 0.15 | 5.10 ± 0.15 | 23 |
| 3 | 34 | OH | OH | Me | 832 | 6.08 ± 0.15 | <4.00 | > 120 |
| 4 | 35 | OH | OH | Br | 447 | 6.35 ± 0.19 | 4.04 ± 0.04 | 208 |
| 5 | 36 | OAc | OAc | Me | 316 | 6.50 ± 0.19 | <4.00 | > 319 |
| 6 | 37 | OAc | OAc | Br | 224 | 6.65 ± 0.16 | 4.01 ± 0.01 | 436 |
| 7 | 40 | H | H | H | 209 | 6.68 ± 0.18 | <4.00 | > 474 |
| 8 | 41 | H | H | Me | 282 | 6.55 ± 0.25 | <4.00 | > 357 |
| 9 | 42 | H | H | Br | 1148 | 5.94 ± 0.33 | 4.01 ± 0.01 | 85 |
| 10 | Mz | 2630 | 5.58 ± 0.09 | 2.22 ± 0.08 | 2291 | |||
Activity against G. lamblia was determined as EC50, the compound concentration that inhibited growth of G. lamblia by 50% over the assay period, and is expressed as negative log10 value of the EC50 (pEC50). Higher pEC50 values reflect greater potency. Data are mean ± SE of the average pEC50 of four different G. lamblia isolates (WB/C6, GS/M, 106, and 713). The average pEC50 for each individual isolate is based on the results of three to four separate experiments. Mean EC50 was calculated from the mean combined pEC50 for all four isolates.
Toxicity against HeLa cells was determined as IC50, the compound concentration that killed 50% of HeLa cells in the cytotoxicity assays, and is expressed as negative log10 value of the IC50 (pIC50), with higher values reflecting greater toxicity. Data are the mean ± SE of the results of three separate experiments.
Selectivity ratio, the ratio of IC50 to EC50, reflects the relative selectivity of compound potency against Giardia compared to toxicity in HeLa cells, with higher ratios indicating greater selectivity for the parasite.
On the basis of the disappointing results obtained with the C2-saturated bridge analogues, we reasoned that an unsaturated bridge joining the phenyl ring to the 5-NI core might exert more direct electronic influence on the 5-NI ring and improve antimicrobial activity. To explore this idea, we synthesized a series of 5-NI compounds bearing a substituted styryl unit directly attached to the C2 position as presented in Table 1. The simplest compound of this styryl series with a bare phenyl ring, 5 (Table 3, entry 1), displayed strong antigiardial activity without measurable toxicity, leading to a high selectivity ratio. Substitutions at the phenyl with one or several methyl moieties (6–9, Table 3, entries 2–5) did not improve the activity or selectivity ratio of compound 5. Similarly, halogen substitutions on the phenyl ring (10–19, Table 3, entries 6–15) also did not significantly improve antigiardial activity. Toxicity was only very slightly increased by most of these modifications, with the exceptions of the methyl, chloro, and iodo substitutions at the meta position of the phenyl ring, which increased toxicity moderately (7, 14, 18, Table 3, entries 3, 10, and 14, respectively) but not to the levels of the dibrominated compounds 3 and 33 (Table 2, entries 1 and 2). Addition of larger side chains at the phenyl moiety, including OMe (20 and 22, Table 3, entries 16 and 18), OCF3 (21, Table 3, entry 17), and phenyl (24, Table 3, entry 20), markedly reduced activity against Giardia, while addition of diethoxymethyl (23, Table 3, entry 19) did not affect antigiardial activity or toxicity. Beyond substituted styryl compounds, we performed the aldol condensation reaction between dimetridazole 4 and a panel of heterocyclic and other aromatic aldehydes to furnish compounds 25–31 (Table 1, entries 21–27). Even though these compounds exhibited promisingly low cytotoxic profiles, their antigiardial activity and selectivity ratio (Table 4, entries 1–7) did not surpass that exhibited by the simple phenyl compound 5 (Table 3, entry 1).
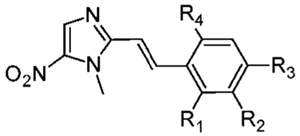
Table 3.
Antigiardial Potency and Toxicity of New 5-NI Alkenes
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| activity against G. lambliaa |
|||||||||
| entry | name | R1 | R2 | R3 | R4 | EC50 (nM) | pEC50 (mean ± SE) | toxicity in HeLa cells,a pIC50 (mean ± SE) | selectivity ratio,a IC50/EC50 |
| 1 | 5 | H | H | H | H | 39 | 7.41 ± 0.12 | <4.00 | >2570 |
| 2 | 6 | Me | H | H | H | 69 | 7.16 ± 0.22 | 4.03 ± 0.03 | 1349 |
| 3 | 7 | H | Me | H | H | 40 | 7.40 ± 0.16 | 4.51 ± 0.24 | 776 |
| 4 | 8 | H | H | Me | H | 162 | 6.79 ± 0.17 | <4.00 | >617 |
| 5 | 9 | Me | H | Me | Me | 339 | 6.47 ± 0.15 | 4.96 ± 0.12 | 32 |
| 6 | 10 | Br | H | H | H | 49 | 7.31 ± 0.20 | 4.14 ± 0.13 | 1479 |
| 7 | 11 | H | Br | H | H | 47 | 7.33 ± 0.20 | 4.06 ± 0.06 | 1862 |
| 8 | 12 | H | H | Br | H | 49 | 7.31 ± 0.27 | 4.14 ± 0.10 | 1479 |
| 9 | 13 | Cl | H | H | H | 72 | 7.14 ± 0.15 | <4.00 | >1380 |
| 10 | 14 | H | Cl | H | H | 22 | 7.66 ± 0.18 | 4.52 ± 0.14 | 1380 |
| 11 | 15 | H | H | Cl | H | 52 | 7.28 ± 0.10 | 4.04 ± 0.05 | 1738 |
| 12 | 16 | Cl | H | Cl | H | 234 | 6.63 ± 0.22 | <4.00 | >427 |
| 13 | 17 | I | H | H | H | 60 | 7.22 ± 0.17 | <4.00 | >1660 |
| 14 | 18 | H | I | H | H | 93 | 7.03 ± 0.16 | 4.50 ± 0.23 | 339 |
| 15 | 19 | H | F | H | H | 41 | 7.39 ± 0.15 | <4.00 | >2455 |
| 16 | 20 | H | OMe | H | H | 155 | 6.81 ± 0.25 | <4.00 | >646 |
| 17 | 21 | H | H | OCF3 | H | 741 | 6.13 ± 0.21 | 4.08 ± 0.09 | 112 |
| 18 | 22 | OMe | OMe | OMe | H | 617 | 6.21 ± 0.27 | <4.00 | >162 |
| 19 | 23 | H | H | CH(OEt)2 | H | 52 | 7.28 ± 0.18 | 4.01 ± 0.01 | 1862 |
| 20 | 24 | Ph | H | H | H | 851 | 6.07 ± 0.23 | <4.00 | >117 |
Activity against G. lamblia, toxicity in HeLa cells, and selectivity ratios were determined and calculated as described in Table 2.
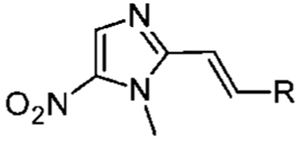
Table 4.
Antigiardial Potency and Toxicity of 5-NI Alkenes with Non-Phenyl Substitutions
 | ||||||
|---|---|---|---|---|---|---|
| activity against g. lambliaa |
||||||
| entry | name | r | EC50 (nM) | pEC50 (Mean ± SE) | toxicity in HeLa cellsa pIC50 (Mean ± SE) | selectivity ratioa (IC50/Ec50) |
| 1 | 25 |  |
100 | 7.00 ± 0.35 | 4.00 ± 0.04 | 1,000 |
| 2 | 26 | 60 | 7.22 ± 0.24 | 4.01 ± 0.01 | 1,622 | |
| 3 | 27 | 69 | 7.16 ± 0.34 | 4.13 ± 0.10 | 1,072 | |
| 4 | 28 | 76 | 7.12 ± 0.12 | 4.04 ± 0.04 | 1,202 | |
| 5 | 29 | 34 | 7.47 ± 0.08 | 4.03 ± 0.03 | 2,754 | |
| 6 | 30 | 81 | 7.09 ± 0.35 | 4.04 ± 0.05 | 1,122 | |
| 7 | 31 | 98 | 7.01 ± 0.27 | <4.00 | >1,023 | |
Activity against G. lamblia, toxicity in HeLa cells, and selectivity ratios were determined and calculated as described in Table 2.
Together, these results show that derivatives of 5-NIs bearing an olefin directly attached to the C2 position of the imidazole ring have potent antigiardial activity. More importantly, most of the analogues possess low cytoxicity and excellent selectivity ratios comparable to or exceeding that of Mz.
Activity of Selected 5-NIs against G. lamblia in a Murine Infection Model
As a next step toward evaluating the ultimate clinical potential of the new 5-NI derivatives, we tested several of the most potent and structurally diverse compounds in a murine model of G. lamblia infection. Normal adult mice were infected orally with trophozoites of the GS/M strain of G. lamblia and left for 2 days to allow full establishment of infection. Mice were then treated with five doses of the compounds over a 3-day period, and infectious load was assessed by counting the number of live trophozoite in the small intestine. Mz, used as reference drug, caused a ~1000-fold reduction in trophozoite numbers, but not parasite eradication, at a dose of 10 mg/kg, while it had no significant effect at the lower dose of 2 mg/kg (Figure 2). Six of the seven tested new 5-NI derivatives were at least as potent as Mz at 10 mg/kg, with four of them (5, 19, 28, and 29) causing complete clearance of the infection. One compound, 12, had no significant activity at either dose. Two of the four compounds, 19 and 28, that caused complete eradication at 10 mg/kg showed significant activity at 2 mg/kg, further demonstrating that they were more potent than Mz (Figure 2). None of the compounds had any clinically observable untoward effects on the mice during the 3-day treatment period at the higher dose (10 mg/kg).
Figure 2.

In vivo potency of new 5-NI compounds against giardiasis. Adult C57BL/6 mice were infected orally with G. lamblia GS/M and given the indicated compounds at 10 or 2 mg/kg or vehicle only by oral gavage five times over a 3-day period starting 2 days after infection. Trophozoite numbers in the small intestine were determined 5 days after infection. Data are mean ± SE of 3–7 animals. Asterisks designate a significant decrease (p < 0.05, t test) relative to infection controls given vehicle only. The gray line depicts the sensitivity of the assay. The in vitro activies of the respective compounds are taken from Tables 2–4.
Further analysis of the in vivo and in vitro data revealed that the in vitro properties of the compounds only partly predicted their in vivo behavior. While a positive correlation existed between in vitro activity and in vivo potency, several notable exceptions were observed (Figure 2). Compound 12 was the fourth most active in vitro (EC50 of 49 nM, Table 3, Entry 8) of the seven new 5-NI derivatives tested in vivo, and yet it showed no activity in vivo. Conversely, compound 28 was only the fifth most active compound in vitro (EC50 of 76 nM, Table 4, entry 4), but it was one of the two most potent compounds in vivo (Figure 2). Thus, compound 12 relatively underperformed, while compound 28 overperformed, in vivo relative to the respective in vitro activity. Nonetheless, these results indicate that several of the new 5-NI compounds have strong in vivo activity and no apparent acute toxicity, supporting the potential utility of these compounds for clinical applications.
Activity of 2-Position Modified 5-NIs against Mz-Resistant G. lamblia
Resistance of G. lamblia to Mz has been documented in the laboratory and threatens to become a serious clinical problem.9,14 We therefore tested the newly synthesized 5-NIs against two independently derived isogenic pairs of Mz-resistant isolates of G. lamblia 713 and G. lamblia 106.24,25 These lines exhibit stable Mz resistance with 30- and 6-fold respective increases in EC50 in the Mz-resistant line relative to the EC50 in the matched parental line, termed here “Mz resistance ratio” or “EC50 (MzR)/EC50 (MzS)”, with a lower resistance ratio indicating a greater ability to overcome Mz resistance. Analysis of the data for the two pairs of G. lamblia lines revealed that the compounds exhibited a range of resistance ratios (Figure 3A and Table 5). Most of the ratios of these compounds were lower than that of Mz for at least one of the two G. lamblia pairs examined, while compounds 5 and 36 were comparable to and compound 40 higher than Mz (Table 5). Importantly, the correlation of resistance ratios between the 713 line and the 106 line was weak, as a substantial number of the compounds we tested overcame Mz resistance in only one line. On the basis of this observation, we divided several of the compounds into three major resistance groups (Figure 3A and Table 5). Group A contained compounds that could substantially overcome Mz resistance in both pairs of G. lamblia lines, while compounds in groups B and C could overcome resistance in one of the two pairs but not (or much less so) in the other.
Figure 3.

Resistance profiles of new 5-NI derivatives. EC50 of all new compounds were determined for MzR and MzS lines of G. lamblia 713 and 106. The ability to overcome Mz resistance is expressed as the ratio of EC50 in the MzR line over EC50 in the MzS line and is plotted for each compound (A, closed circles). Mz (A, open circle) serves as the reference drug. Compounds were categorized arbitrarily into three groups to highlight their functional properties. In (B), resistance ratios for G. lamblia 713 (closed circles) and G. lamblia 106 (open circles) are plotted against the average antimicrobial activity against four Mz-sensitive lines for each drug, using the pEC50 values shown in Tables 2–4.
Table 5.
Activity of New 5-NIs against Mz-Resistant G. lamblia
| activity against G. lamblia 713,a pEC50 (mean ± SE) |
activity against G. lamblia 106,a pEC50 (mean ± SE) |
EC50(MzR)/EC50(MzS)b |
|||||
|---|---|---|---|---|---|---|---|
| groupc | name | MzS | MzR | MzS | MzR | G. lamblia 713 | G. lamblia 106 |
| Mz | 5.83 ± 0.08 | 4.35 ± 0.12 | 5.40 ± 0.10 | 4.59 ± 0.08 | 30.20 | 6.46 | |
| A | 9 | 6.64 ± 0.23 | 5.89 ± 0.02 | 6.69 ± 0.31 | 6.37 ± 0.24 | 5.59 | 2.13 |
| 11 | 7.49 ± 0.09 | 6.90 ± 0.12 | 7.37 ± 0.38 | 6.94 ± 0.11 | 3.95 | 2.73 | |
| 15 | 7.44 ± 0.10 | 6.84 ± 0.10 | 7.14 ± 0.08 | 6.59 ± 0.22 | 4.06 | 3.52 | |
| 19 | 7.59 ± 0.01 | 6.91 ± 0.09 | 7.51 ± 0.27 | 7.03 ± 0.10 | 4.76 | 3.07 | |
| B | 10 | 7.74 ± 0.10 | 6.93 ± 0.14 | 7.38 ± 0.18 | 6.55 ± 0.43 | 6.40 | 6.81 |
| 33 | 6.74 ± 0.09 | 6.32 ± 0.51 | 6.67 ± 0.23 | 5.54 ± 0.23 | 2.62 | 13.67 | |
| C | 12 | 8.09 ± 0.46 | 7.04 ± 0.39 | 7.14 ± 0.17 | 6.70 ± 0.21 | 11.43 | 2.78 |
| 23 | 7.49 ± 0.09 | 6.15 ± 0.12 | 7.19 ± 0.20 | 6.70 ± 0.14 | 22.00 | 3.11 | |
| 24 | 6.46 ± 0.18 | 5.34 ± 0.34 | 6.27 ± 0.27 | 5.91 ± 0.19 | 13.22 | 2.28 | |
| 28 | 7.29 ± 0.21 | 5.93 ± 0.02 | 7.06 ± 0.15 | 6.60 ± 0.14 | 22.96 | 2.84 | |
| other | 5 | 7.69 ± 0.10 | 6.28 ± 0.20 | 7.26 ± 0.24 | 6.40 ± 0.14 | 25.98 | 7.20 |
| 13 | 7.52 ± 0.27 | 6.94 ± 0.16 | 7.22 ± 0.13 | 6.60 ± 0.30 | 3.77 | 4.22 | |
| 29 | 7.69 ± 0.10 | 6.31 ± 0.19 | 7.45 ± 0.28 | 6.44 ± 0.09 | 23.95 | 10.32 | |
| 31 | 7.61 ± 0.61 | 5.98 ± 0.06 | 7.04 ± 0.03 | 6.41 ± 0.06 | 42.93 | 4.25 | |
| 36 | 6.76 ± 0.15 | 5.29 ± 0.03 | 6.29 ± 0.16 | 5.51 ± 0.11 | 29.51 | 6.00 | |
| 40 | 6.99 ± 0.16 | 5.30 ± 0.07 | 6.76 ± 0.13 | 5.59 ± 0.18 | 49.25 | 14.73 | |
pEC50 was determined for the respective Mz-sensitive (MzS) and Mz-resistant (MzR) lines of G. lamblia. Data are mean ± SE of three separate experiments.
The ratio of EC50 in the Mz-resistant line to EC50 in the isogenic Mz-sensitive line (“Mz resistance ratio”) is a measure of the ability of a compound to overcome Mz resistance in the respective G. lamblia lines.
Group refers to the Mz resistance groups depicted in Figure 3A.
These results provide strong pharmacological evidence for more than one mechanism of Mz resistance in G. lamblia. Diversity in antimicrobial resistance mechanism is not surprising, given that the Giardia lines were derived independently,24,25 and is well recognized for other antimicrobials and microbes.26,27 Moreover, different resistance mechanisms have been proposed for G. lamblia, including the down-regulation of the critical pathway required for reductive 5-NI drug activation,13,28,29 although the full extent and relative importance of different resistance mechanisms remain elusive. In any case, the existence of group A compounds suggests that it is possible to develop 5-NIs that can markedly overcome at least two forms of Mz resistance and may be active against other forms of Mz resistance in G. lamblia.
In searching for properties that might explain the resistance grouping of the compounds, we found that the ability to overcome Mz resistance was not correlated with the potency of the respective compounds (Figure 3B). For example, two group A compounds, 9 and 19, exhibited comparable improvements (i.e., decreases) in resistance ratios in both G. lamblia pairs, but compound 9 had ~10-fold lower EC50 against the respective Mz-sensitive parental lines (Table 5). Moreover, compounds 11 and 5 had comparable activities against the Mz-sensitive lines but were markedly different in their resistance profiles (Table 5). Structural analysis also did not reveal any clear explanation for the resistance grouping of the compounds, although some interesting connections could be discerned. For example, among the 2-styryl 5-NIs, two of the four group A compounds (11 and 19) possessed a substitution (i.e., Br and F) at the meta-position in the ring while none of the five group B or C phenyl-carrying styryl compounds did. The importance of the substitution position is illustrated by the set of Br-substituted compounds, 10 (ortho), 11 (meta), and 12 (para). The m-Br containing compound (11, group A) showed excellent improvements in resistance ratios for both G. lamblia pairs, while the o-Br substitution (10, group B) improved the ratio in the 713 line but not the 106 line, and the p-Br substitution (12, group C) improved the ratio in the 106 line but less so in the 713 line (Table 5). On the other hand, these structure–activity relationships did not apply uniformly for other substituents. For example, while the o-Br and p-Br substituted compounds fell distinctly into groups B and C, respectively, the same was not true for the Cl substitutions, where the ortho and para substituted compounds (13 and 15, respectively) had very similar resistance profiles (Table 5). Overall, these results demonstrate that several of the new 5-NIs can markedly, albeit not entirely, overcome resistance to Mz in G. lamblia. However, the ability to do so is not readily predictable on the basis of overall antigiardial activity or structural features.
Electrochemical Properties of 5-NI Alkenes and Alkanes under Anhydrous Conditions
Since structural features of the newly synthesized compounds did not provide consistent explanations for the compounds’ antimicrobial activities, we began to explore their electrochemical properties, since NIs are prodrugs that must be activated by reduction in the target microbes before they exhibit antimicrobial activity. We reasoned that chemical modification of the molecule with groups that perturb the electronic properties of the NI ring would affect the ease with which these molecules undergo reduction. The redox potential of Mz is more negative than the lowest redox couple in mammalian cells, which is believed to be the basis for its high specificity and low toxicity. To determine whether differences in redox activation might explain the ability of the different 5-NIs to overcome Mz resistance, we measured the half-wave potential (E1/2) of the initial one-electron transfer reaction (0/−1 reduction) for groups A, B, and C compounds and selected additional compounds. Measurements were done by cyclic voltammetry in DMSO under anhydrous conditions, with ferrocene as an internal standard and reference point for measuring E1/2.
Representative cyclic voltammograms (CVs) under these conditions for Mz and one of the new 5-NIs, the 3-thiophene compound 31, are shown in Figure 4A. From the similarity of the CVs it can be deduced that both compounds exhibited a similar electrochemical behavior, corresponding to a reversible one-electron reduction to the radical anion. The E1/2 of Mz was −1.561 V under these experimental conditions, which was very close to the E1/2 of −1.557 V for 1-methyl-5-NI, the unsubstituted parent compound of all the new 2-substituted N-methyl-5-NI (Table 6). Most of the new 5-NIs we assayed had more positive E1/2, indicating that they could be reduced more readily, whereas six compounds exhibited more negative E1/2 than Mz and 1-methyl-5-NI (Table 6).
Figure 4.

Electrochemistry of new 5-NI compounds. 5-NIs were subjected to cyclic voltammetry in DMSO (A) or pH 7.0 water (C). Representative scans for Mz and the 3-thiophene compound 31 are shown. Potential is given relative to ferrocene (Fc) in DMSO or a saturated calomel electrode (SCE) in water. Current was normalized against peak cathodic current for each scan. The arrows indicate the scan direction. A summary of the electrochemical data is given in Table 6. E1/2 values in DMSO were plotted against Hammett σ values of the 2-alkenylphenyl 5-NI compounds 5, 7, 8, 11, 12, 14, 15, 18, and 19 (B). E1/2 values in DMSO were also plotted against Ep values in water for selected 5-NIs using the data in Table 6 (D). Data in (B) and (D) are the mean ± SE of three to four experiments.
Table 6.
Electrochemical Properties of Modified 5-NIs
 | |||||
|---|---|---|---|---|---|
| mean ± SE |
|||||
| groupa | name | structure | R | E1/2 in DMSO (V vs Fc)b | Ep in water (V vs SCE)b,c |
| 40 | I | phenyl | −1.593 ± 0.002 | ND | |
| 41 | I | 4-methylphenyl | −1.592 ± 0.003 | ND | |
| 42 | I | 4-bromophenyl | −1.592 ± 0.003 | ND | |
| 22 | IV | 2,3,4-trimethoxyphenyl | −1.584 ± 0.005 | −0.757 ± 0.003 | |
| 27 | IV | 1,3-benzodioxolyl | −1.584 ± 0.001 | −0.778 ± 0.002 | |
| 20 | IV | 3-methoxyphenyl | −1.580 ± 0.007 | −0.763 ± 0.001 | |
| Mz | −1.561 ± 0.001 | −0.751 ± 0.017 | |||
| 1-methyl-5-NI | −1.557 ± 0.001 | ND | |||
| 34 | II | 4-methylphenyl | −1.557 ± 0.001 | ND | |
| 35 | II | 4-bromophenyl | −1.544 ± 0.001 | ND | |
| A | 9 | IV | 2,4,6-trimethylphenyl | −1.529 ± 0.001 | ND |
| 30 | IV | 2-thiopheneyl | −1.528 ± 0.003 | −0.658 ± 0.001 | |
| 8 | IV | 4-methylphenyl | −1.522 ± 0.003 | ND | |
| C | 23 | IV | 4-diethoxymethylphenyl | −1.518 ± 0.003 | ND |
| 5 | IV | phenyl | −1.517 ± 0.001 | −0.634 ± 0.004 | |
| 7 | IV | 3-methylphenyl | −1.516 ± 0.001 | −0.635 ± 0.003 | |
| 6 | IV | 2-methylphenyl | −1.512 ± 0.001 | −0.621 ± 0.001 | |
| C | 24 | IV | 2-biphenyl | −1.512 ± 0.001 | ND |
| 26 | IV | 2-napthyl | −1.509 ± 0.001 | ND | |
| 31 | IV | 3-thiopheneyl | −1.508 ± 0.002 | −0.595 ± 0.001 | |
| C | 12 | IV | 4-bromophenyl | −1.506 ± 0.002 | −0.605 ± 0.007 |
| 29 | IV | 2-furanyl | −1.504 ± 0.003 | ND | |
| A | 15 | IV | 4-chlorophenyl | −1.503 ± 0.001 | −0.584 ± 0.001 |
| 25 | IV | 1-napthyl | −1.502 ± 0.004 | ND | |
| A | 19 | IV | 3-fluorophenyl | −1.502 ± 0.001 | −0.613 ± 0.008 |
| C | 28 | IV | 1-methylimidazolyl | −1.497 ± 0.001 | −0.594 ± 0.014 |
| A | 11 | IV | 3-bromophenyl | −1.497 ± 0.005 | ND |
| 18 | IV | 3-iodophenyl | −1.494 ± 0.004 | ND | |
| B | 33 | III | 4-bromophenyl | −1.493d ± 0.004 | ND |
| 14 | IV | 3-chlorophenyl | −1.492 ± 0.002 | ND | |
| 3 | III | 4-methylphenyl | −1.489d ± 0.001 | ND | |
| 21 | IV | 4-trifluoromethoxyphenyl | −1.487 ± 0.001 | ND | |
| 13 | IV | 2-chlorophenyl | −1.486 ± 0.004 | ND | |
| B | 10 | IV | 2-bromophenyl | −1.481 ± 0.001 | ND |
| 16 | IV | 2,4-dichlorophenyl | −1.469 ± 0.002 | ND | |
Redox activation potential was determined by cyclic voltammetry in DMSO or water (pH 7.0). Half-wave potential (E1/2) of the one-electron transfer reaction (0/−1 reduction) was determined in DMSO, with ferrocene (Fc) as the internal reference. Peak potential, Ep, for the irreversible first electron reduction step was determined in water, using a saturated calomel electrode (SCE) as standard. Data are mean ± SE of three to four separate experiments.
ND, not determined.
The E1/2 values for these compounds are for the reversible wave, but given the different electrochemical response, these values can not be directly compared to the other E1/2 values.
Three of these six compounds had a C2-saturated bridge between the two ring systems (40–42). This is consistent with the electron-donating ability of alkyl groups, which add electron density to the nitroimidazole ring, making reduction more difficult. Since there is no conjugation connecting the imidazole ring to the pendant phenyl ring, substitution on the phenyl ring had no significant effect on the E1/2 (Table 6). The exception to this notion were the two 5-NI alkanes, 3 and 33, both of which showed much more positive E1/2 than Mz (Table 6). However, both compounds had di-Br substitutions in the C2-saturated bridge and showed more complicated electrochemical behavior with an irreversible reduction peak in the CV leading into and overlapping with a reversible wave. The irreversible peak was likely associated with reductive removal of the Br and the reversible wave with reduction of the product NI. Interestingly, both compounds were quite toxic in HeLa cells (Table 2, entries 1 and 2), suggesting that their easier reduction at less negative redox potentials, or their redox-induced structural change and probable side chain release, might contribute to toxicity.
All of the other tested compounds with significantly more positive E1/2 (ranging from 30–90 mV) than Mz and 1-methyl-5-NI were C2-olefinic derivatives of 5-NI, suggesting that the conjugation through the unsaturated link between the imidazole ring and the pendant aromatic ring stabilizes the radical anion through delocalization and thereby favors reduction of the nitro group. The compounds substituted with phenyl (5), methylphenyl (6–8), and trimethylphenyl (9) had 30–50 mV more positive E1/2 than Mz (Table 6). Extended conjugation with larger aromatic groups (24–26) and/or heterocycles with electronegative elements (N, O) (28 and 29) made the compounds easier to reduce (50–65 mV more positive E1/2 than Mz) than the methylphenyl or phenyl derivatives, as did addition of electron-withdrawing halogen groups on the phenyl ring (55–90 mV more positive than Mz). The easiest compound to reduce in this series was 16, which has two electron-withdrawing chlorines on the phenyl ring, giving it a ~90 mV more positive E1/2 than Mz (Table 6).
To gain a better understanding of the observed electrochemical properties of the different 5-NI compounds, we evaluated the substituent effects on the redox potentials using Hammett plots.30–33 In this analysis of aromatic compounds, the E1/2 of a series of meta- and para-substituted derivatives are plotted against the Hammett σ values. The latter are derived from the acid dissociation constants of substituted benzoic acids and provide a quantitative measurement of the electron-donating/accepting character of a particular substituent. Figure 4B shows the Hammett plot for the 2-alkenylphenyl compounds that have simple meta or para substituents for which good σ values are available.34 As expected, we observed an approximately linear correlation with a positive slope, indicating stabilization of negative charge. This correlation suggests that the C2-olefin bridge provides an electronic communication between the pendant phenyl ring and the NI. Dividing the slope of this type of Hammett plot by the Nernst equation coefficient of 0.0592 V yields a dimensionless parameter that is directly comparable to the reaction parameter, ρ, obtained from the slopes of Hammett plots of other types of reactions.32 This calculation gives a ρ of 0.84 for the data in Figure 4B, indicating that the reduction of these compounds is slightly less sensitive to the substituent on the phenyl group than the acid dissociation of benzoic acid with a ρ value of 1.0. Hammett plots for many aromatic redox couples give much larger ρ values, often > 2.30–33 The modest value observed in our experiments is consistent with the substituents being located at some distance from the primary reduction site, the nitro group of the imidazole core.
Electrochemical Properties of 5-NI Alkenes under Aqueous Conditions
We conducted the initial electrochemical measurements in DMSO because the reduction of the 5-NIs is reversible under these conditions, as shown by the oxidation peak observed on the return scan in the CVs (Figure 4A). The resultant E1/2 value (the potential midway between the two peak values) is close to the thermodynamic redox potential for reduction to the radical anion and thus provides a fundamental measure of the ease of adding the first electron to the different 5-NIs. While it is possible that the reduction of the 5-NIs occurs in a nonaqueous environment inside the microbial target cells (e.g., in the active site of a redox protein), it is likely that the reduction occurs in a more aqueous environment. Determination of the thermodynamic potential for reduction of 5-NIs in water is confounded by the fact that the electron transfers in aqueous conditions are irreversible reactions. This is shown by the lack of an oxidation peak on the return scan in the CVs in aqueous solution (Figure 4C). The irreversibility is due to the instability of the radical anion in aqueous solution, which upon protonation is further reduced at the same potential. Because of this, only the peak potential (Ep) for the reduction can be derived from the CVs (Table 6). In contrast to the E1/2, the Ep is strongly affected by the kinetics of the electron transfer reactions and any coupled chemical reactions and may not be directly correlated with the thermodynamic redox potential for the overall reaction. Furthermore, the kinetic influence means that variations in the experimental conditions (e.g., differences in the electrode surface) will affect the Ep, possibly leading to greater scatter in the data.
For all of these reasons, we chose to rely on the E1/2 values in DMSO rather than the aqueous Ep values as our primary indicator of the relative ease of reduction for the different 5-NIs. However, given that the first step in the reduction in aqueous solution at physiological pH is likely the same as that of the overall reaction in DMSO, i.e., one-electron reduction to the radical anion, we reasoned that the E1/2 values in DMSO should give us an indication of the relative ease of reduction of the 5-NIs in aqueous solution. This is particularly likely among compounds of similar structure, for which the difference in solvation energy between DMSO and water would be expected to remain fairly constant across the series. In order to test this hypothesis, we determined the CVs of selected C2-olefinic 5-NI derivatives in neutral (pH 7.0) aqueous solution and correlated the data with the E1/2 values obtained under anhydrous conditions (Table 6). A plot of Ep of the aqueous CVs vs E1/2 for the same compounds in DMSO showed a strong linear correlation (Figure 4D), supporting the notion that the E1/2 values obtained in DMSO give a reliable measure of the relative ease of reduction of the different 5-NIs independent of whether the reduction occurs in an aqueous or nonaqueous environment in the cell. It must be noted that the analysis of the true one-electron redox potential of the new 5-NI compounds in water would likely require other assay approaches, such as pulse radiolysis, although the utility of true aqueous one-electron potentials may be limited if reduction occurs in a purely aqueous environment in the cells conducive to rapid multielectron transfer.
Importance of Redox Potential of Modified 5-NI for Antimicrobial Potency and Selectivity
We next examined the relationship between E1/2 and the ability to overcome Mz resistance (Figure 5A). In both pairs of Mz-sensitive and Mz-resistant G. lamblia lines, compounds with improved (i.e., decreased) resistance ratios generally had more positive E1/2, although the importance of this relationship appeared to differ between the 713 and 106 pairs. In G. lamblia 713, almost all of the tested compounds showed some ability to overcome resistance, with the majority exhibiting more positive E1/2 but a few compounds also showing more negative E1/2 (Figure 5A). By comparison, in G. lamblia 106, only compounds with more positive E1/2 than Mz could overcome Mz resistance. This was not sufficient, however, since only about half of the compounds with more positive E1/2 had a better (i.e., decreased) resistance ratio than Mz while the other half exhibited reduced antigiardial activity compared to Mz in the Mz-resistant line (Figure 5A). Thus, a decrease in the energy requirements for redox activation, as reflected in a more positive E1/2, is generally correlated with the ability of 5-NI to overcome Mz resistance, but such a decrease is neither necessary nor sufficient.
Figure 5.

Relationship of redox potential and antigiardial activity and selectivity for new 5-NI compounds. The redox potential in DMSO (E1/2) of each new 5-NI was plotted against resistance ratios (A), overall antigiardial activity (B), and selectivity ratio relative to HeLa cells (C). For compounds with limited aqueous solubility, the data points represent the ratio of EC50 and the highest test concentration (100 μM) and thus the minimum rather than exact selectivity ratio (closed triangles in C). Mz (open circles) is shown in all panels as the reference drug.
A similar conclusion also holds true for the overall antigiardial activity of the nitro compounds, since we found a positive correlation between E1/2 and activity (Figure 5B). However, several compounds (e.g., 20 and 27) with more negative E1/2 than Mz also exhibited increased antigiardial activity compared to Mz, while others (e.g., 21 and 24) had markedly more positive E1/2 than Mz but only a very modest increase in activity (Tables 2, 3, 4, and 6). Rather surprisingly, comparison of E1/2 and selectivity ratios revealed little correlation between these parameters (Figure 5C). Thus, many of the compounds with more negative E1/2 than Mz showed low selectivity ratios, while compounds with markedly more positive E1/2 had ratios comparable to or exceeding that of Mz. These results indicate that the ease of redox activation is not an important determinant of the selectivity of 5-NIs in Giardia compared to mammalian cells, at least as applied to the compounds tested in this study with their relatively limited E1/2 range.
Conclusions
This study expands on prior work16,17 to demonstrate clearly that modification of the 2-position of the 5-NI core provides a promising avenue for improving its antimicrobial potency and selectivity. All of the new 5-NI derivatives were more potent than Mz against diverse isolates of G. lamblia, and almost a third (10 of 35) of these compounds showed 50- to 100-fold improvement in antigiardial activity. Perhaps more importantly, a number of the new compounds markedly overcame resistance to Mz. With the current series of 5-NI derivatives and quantitative approaches, we did not identify compounds that could overcome Mz resistance completely, as no compound had identical activity against the Mz-resistant and Mz-sensitive pairs of isogenic Giardia lines. However, the quantitative analysis of antigiardial activity revealed that the new compounds displayed a wide spectrum of activities in the face of Mz resistance, suggesting that a larger 5-NI library may well contain compounds whose activity is not compromised by prior Mz resistance.
Antimicrobial potency and ability to overcome Mz resistance were poorly if at all correlated. In fact, the compounds with the most improved Mz resistance ratios had only modest overall activity against the Mz-sensitive G. lamblia isolates, with pEC50 values in the mid-range of those observed in the entire library. These results imply that the molecular properties that determine overall potency are not those that overcome Mz resistance, although detailed molecular conclusions are impossible to draw at present, given the lack of mechanistic insights into the relevant targets of 5-NIs in G. lamblia.
Analysis of the electrochemical properties of the new series of 5-NI compounds revealed fresh insights into the determinants of antigiardial activity. Compounds with more positive redox activation potential, which makes them more readily reducible, generally had greater antimicrobial activity and an improved capacity to overcome Mz resistance. However, this relationship was not universal, as several compounds with more negative redox potentials also had markedly improved antigiardial profiles, while other compounds with more positive redox potential did not. Thus, although easier redox activation can probably explain the improvement in antigiardial activity profile for many of the compounds, it is neither necessary nor sufficient. Other yet to be defined molecular properties must therefore also be important. To our surprise, we did not find a correlation between ease of redox activation and selectivity, as one might have expected from the general recognition that the excellent selectivity of 5-NI compounds for microbial targets over mammalian cells is based on the differences in the lowest redox couples present in these systems.11 Our data suggest that, at least within the relatively negative and narrow E1/2 range of compounds tested in this study, other determinants beyond ease of reduction affect toxicity and selectivity of 5-NIs, thus casting some doubts on the preeminence of the selective redox activation paradigm in explaining the clinical utility of 5-NI drugs.
The 5-NI compounds with a conjugated olefin bridge between the imidazole core and the pendant ring system generally had higher antigiardial activity and better selectivity than those with a saturated alkane connection. In parallel, the energy requirements for nitro reduction were lower (i.e., more positive) in the conjugated compounds and were markedly impacted by the nature of the functional groups on the distant ring, whereas the same was not true in the alkanes. The most straightforward explanation may be that the olefin bridge allows effective electronic communication between the pendant ring system and the NI and thereby helps to fine-tune the nitro reducibility by distant functional groups. It needs to be borne in mind, however, that the electrochemical behavior of 5-NIs within the microbial target cells may be more complex than what can be modeled readily under cyclic voltammetry conditions in DMSO or water. Inside the target cells, the initial, rate-limiting reductive step presumably occurs in the presence of electrolytes, proteins, lipids, and other molecules with varying charges that may affect the charge distribution and, hence, reducibility and stability of the 5-NI radicals. In addition, the initial step is followed by a sequence of multielectron transfers, whose products may interact differently with microbial target molecules than the radical formed in the initial one-electron transfer reaction.35
The study shows that 5-NI derivatives can be developed that are more potent than Mz in an animal model of giardiasis and have no apparent toxic side effects, making them good candidates for further preclinical development. The data caution, however, that in vivo activity can differ markedly from what might be expected on the basis of the observed in vitro activity. Such differences are most likely accounted for by differential pharmacokinetics of the respective compounds, which may be fruitful ground for future studies to develop the clinical potential of the new 5-NI compounds. Our results also stress that new compounds need to be tested rapidly in in vivo models to find truly effective drugs for treating giardiasis. In doing so, it should be possible to minimize the negative impact on development time and resource consumption that follows from the unavoidable attrition of compounds that are effective in vitro but exhibit poor in vivo activity.
Experimental Section
Chemistry
Materials and General Methods
Reagents and solvents were purchased from commercial sources and were used as received. Reaction progress was monitored by thin-layer chromatography on Merck silica gel 60 F-254 with detection by UV. Silica gel 60 (Merck 40–63 μm) was used for flash column chromatography. Melting points are uncorrected and were determined by use of a Thomas-Hoover Uni-Melt capillary melting point apparatus. 1H NMR and 13C NMR spectra were recorded with Bruker DRX-600, Bruker DRX-500, Varian 400, or Bruker AMX-400 spectrometers using DMSO-d6 or CDCl3. Proton magnetic resonance (1H NMR) spectra were recorded at 600, 500, or 400 MHz. Data are presented as follows: chemical shift (parts per million, ppm), multiplicity s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, sep = septet, m = multiplet, br = broad, coupling constant, J (in hertz), and integration. Carbon magnetic resonance (13C NMR) spectra were recorded at 150, 125, or 100 MHz. Data for 13C NMR are reported in terms of chemical shifts (ppm) and coupling constant J (in hertz) for fluorine-containing compounds. High-resolution mass spectra (HRMS) were recorded at the mass spectrometry facility at The Scripps Research Institute, La Jolla, CA. Elemental analyses were performed by Midwest Microlab, LLC, Indianapolis, IN.
General Procedure for the Aldol Condensation
For the synthesis of 2-vinyl-5-nitroimidazoles by aldol condensation, dimetridazole (4, Table 1) was dissolved in EtOH (5.5 mL/mmol of dimetridazole) and treated with sodium ethoxide (NaOEt, 2.0–2.5 equiv). The resulting amber suspension was vigorously stirred at ambient temperature for 5–10 min before the addition of the aryl aldehydes (1.1–1.3 equiv) in small portions or dropwise using a syringe. The reaction mixture was then heated to 65 °C overnight, after which the brown reaction mixture was cooled to ambient temperature, transferred to a separatory funnel, and partitioned (CH2Cl2//H2O). The organic phase was washed with brine (NaCl/H2O), dried over anhydrous Na2SO4, and evaporated in vacuo to generally give a brown residue which was purified by column chromatography over silica gel.
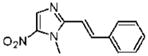
1-Methyl-5-nitro-2-[(E)-2-phenylvinyl]-1H-imidazole (5)
Dimetridazole (5.0 g, 35.4 mmol) was reacted with benzaldehyde (4.5 g, 42.5 mmol, 1.2 equiv) in the presence of NaOEt (6.03 g, 88.6 mmol, 2.5 equiv). The mixture was purified by column chromatography (14% → 33% EtOAc/hexanes) to give 5 as a bright-yellow solid (1.6 g, 20%). 1H NMR (500 MHz, CDCl3) δ 8.10 (s, 1H), 7.89 (d, J = 15.8, 1H), 7.59–7.57 (m, 2H), 7.44–7.37 (m, 3H), 6.90 (d, J = 15.7, 1H), 4.06 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 149.8, 139.8, 135.2, 134.2, 129.8, 129.0, 127.5, 111.2, 32.8; HRMS (ESI+) calcd for C12H12N3O2 [M + H]+ 230.0924, found 230.0931. Anal. (C12H11N3O2) C, H, N.
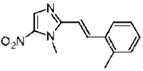
1-Methyl-2-[(E)-2-(2-methylphenyl)vinyl]-5-nitro-1H-imidazole (6)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with 2-methylbenzaldehyde (1.02 g, 8.5 mmol, 1.2 equiv) in the presence of NaOEt (1.2 g, 17.7 mmol, 2.5 equiv). The mixture was purified by column chromatography (85% CH2Cl2/hexanes → CH2Cl2). The material obtained in this manner was further purified by a recrystallization from CH3OH (25 mL) to give 6 as a bright-yellow solid (0.40 g, 23%). 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 15.6, 1H), 8.09 (s, 1H), 7.61 (dd, J = 7.2, 1.6, 1H), 7.30–7.22 (m, 3H), 6.81 (d, J = 15.6, 1H), 4.04 (s, 3H), 2.48 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 149.9, 137.4, 137.4, 137.3, 134.2, 134.1, 130.8, 129.5, 126.3, 125.5, 112.3, 32.7, 19.8; HRMS (ESI+) calcd for C13H14N3O2 [M + H]+ 244.1080, found 244.1076. Anal. (C13H13N3O2) C, H, N.
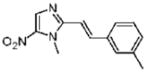
1-Methyl-2-[(E)-2-(3-methylphenyl)vinyl]-5-nitro-1H-imidazole (7)
Dimetridazole (3.5 g, 24.8 mmol) was reacted with 3-methylbenzaldehyde (3.53 mL, 29.8 mmol, 1.2 equiv) in the presence of NaOEt (4.2 g, 62 mmol, 2.5 equiv). The mixture was purified by column chromatography (CH2Cl2 → 10% CH3OH/CH2Cl2) to give 7 as a yellow solid (2.53 g, 42%). 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.80 (d, J = 15.6, 1H), 7.33 (s, 1H), 7.32 (d, J = 7.2, 1H), 7.27–7.21 (m, 1H), 7.14 (d, J = 7.6, 1H), 6.82 (d, J = 15.6, 1H), 3.99 (s, 3H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 149.9, 139.9, 138.6, 135.1, 134.2, 130.7, 128.8, 128.0, 124.7, 110.9, 32.7, 21.3; HRMS (ESI+) calcd for C13H14N3O2 [M+ H]+ 244.1080, found 244.1083. Anal. (C13H13N3O2): C, 64.19; H, 5.39; N, 17.27. Found: C, 64.36; H, 5.50; N, 16.84.
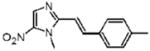
1-Methyl-2-[(E)-2-(4-methylphenyl)vinyl]-5-nitro-1H-imidazole (8)
Dimetridazole (7.0 g, 49.6 mmol) was reacted with 4-methylbenzaldehyde (7.0 mL, 59.5 mmol, 1.2 equiv) in the presence of NaOEt (8.4 g, 124 mmol, 2.5 equiv). The mixture was purified by column chromatography (CH2Cl2 → 10% CH3OH/CH2Cl2) to give 8 as a yellow solid (2.65 g, 22%). 1H NMR (400 MHz, DMSO-d6) δ 8.18 (s, 1H), 7.74 (d, J = 16.0, 1H), 7.68 (d, J = 8.0, 2H), 7.33 (d, J = 16.0, 1H), 7.24 (d, J = 8.0, 2H), 4.02 (s, 3H), 2.34 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 149.9, 139.2, 137.9, 134.2, 132.7, 129.4, 127.7, 127.4, 112.0, 32.7, 20.9; HRMS (ESI+) calcd for C13H14N3O2 [M + H]+ 244.1080, found 244.1074. Anal. (C13H13N3O2): C, 64.19; H, 5.39; N, 17.27. Found: C, 64.32; H, 4.92; N, 16.85.
1-Methyl-2-[(E)-2-(2,4,6-trimethylphenyl)vinyl]-5-nitro-1H-imidazole (9)
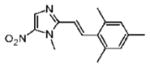
Dimetridazole (1.0 g, 7.1 mmol) was reacted with 2,4,6-trimethylbenzaldehyde (1.36 g, 9.23 mmol, 1.3 equiv) in the presence of NaOEt (966 mg, 14.2 mmol, 2.0 equiv). The mixture was purified by column chromatography (CH2Cl2 → 5% CH3OH/CH2Cl2) to give 9 as a yellow solid (559 mg, 29%). 1H NMR (400 MHz, CDCl3) δ 8.10 (s, 1H), 7.97 (d, J = 16.0, 1H), 6.93 (s, 2H), 6.51 (d, J = 16.0, 1H), 3.99 (s, 3H), 2.39 (s, 6H), 2.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 149.8, 139.5, 138.1, 136.5, 134.0, 131.8, 129.9, 129.2, 116.5, 32.7, 21.2, 21.0; HRMS (ESI+) calcd for C15H18N3O2 [M + H]+ 272.1393, found 272.1397. Anal. (C15H17N3O2): C, 66.40; H, 6.32; N, 15.49. Found: C, 66.87; H, 6.40; N, 15.60.
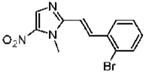
2-[(E)-2-(2-Bromophenyl)vinyl]-1-methyl-5-nitro-1H-imidazole (10)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with 2-bromobenzaldehyde (1.6 g, 8.5 mmol, 1.2 equiv) in the presence of NaOEt (1.2 g, 17.7 mmol, 2.5 equiv). The mixture was purified by crystallization from CH3OH/CH2Cl2 (1/1) to give 10 as a bright-yellow solid (0.44 g, 20%). 1H NMR (600 MHz, CDCl3) δ 8.22 (d, J = 15.7, 1H), 8.12 (s, 1H), 7.67 (dd, J = 7.9, 1.0, 1H), 7.65 (dd, J = 8.1, 0.8, 1H), 7.37 (t, J = 7.4, 1H), 7.24 (td, J = 7.7, 0.7, 1H), 6.87 (d, J = 15.6, 1H), 4.08 (s, 3H); 13C NMR (100 MHz, DMSO-d6 + CD2Cl2) δ 149.2, 139.0, 135.6, 134.8, 133.8, 132.9, 130.6, 127.7, 127.6, 124.2, 115.6, 32.7; HRMS (ESI+) calcd for C12H11BrN3O2 [M + H]+ 308.0029, found 308.0034. Anal. (C12H10BrN3O2) C, H, N.
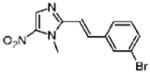
2-[(E)-2-(3-Bromophenyl)vinyl]-1-methyl-5-nitro-1H-imidazole (11)
Dimetridazole (1.5 g, 10.6 mmol) was reacted with 3-bromobenzaldehyde (2.3 g, 12.7 mmol, 1.2 equiv) in the presence of NaOEt (1.8 g, 26.6 mmol, 2.5 equiv). The mixture was purified by column chromatography (20% EtOAc/hexanes) to give 11 as a bright-yellow solid (0.62 g, 19%). 1H NMR (600 MHz, CDCl3) δ 8.04 (s, 1H), 7.75 (d, J = 15.8, 1H), 7.52 (s, 1H), 7.47 (d, J = 7.7, 1H), 7.42 (d, J = 7.7, 2H), 6.88 (d, J = 15.7, 1H), 4.06 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 140.0, 143.1, 138.0, 133.8, 132.5, 130.4, 130.0, 129.7, 126.4, 125.2, 112.3, 32.8; HRMS (ESI+) calcd for C12H11BrN3O2 [M + H]+ 308.0029, found 308.0034. Anal. (C12H10BrN3O2): C, 46.78; H, 3.27; N, 13.64. Found: C, 47.65; H, 3.54; N, 13.08.
2-[(E)-2-(4-Bromophenyl)vinyl]-1-methyl-5-nitro-1H-imidazole (12)
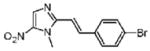
Dimetridazole (5.0 g, 35.4 mmol) was reacted with 4-bromobenzaldehyde (7.8, 42.5 mmol, 1.2 equiv) in the presence of NaOEt (6.0 g, 88.5 mmol, 2.5 equiv). The mixture was purified by column chromatography (CH2Cl2 → 10% CH3OH/CH2Cl2) to give 12 as a yellow solid (7.8 g, 72%). 1H NMR (400 MHz, DMSO-d6) δ 8.06 (s, 1H), 7.81 (d, J = 16.0, 1H), 7.72 (d, J = 8.4, 2H), 7.61 (d, J = 8.4, 2H), 7.42 (d, J = 15.6, 1H), 4.13 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 149.4, 134.7, 134.0, 131.7, 130.8, 129.6, 128.4, 122.5, 114.0, 32.7; HRMS (ESI+) calcd for C12H11BrN3O2 [M + H]+ 308.0029, found 308.0026. Anal. (C12H10BrN3O2) C, H, N.
2-[(E)-2-(2-Chlorophenyl)vinyl]-1-methyl-5-nitro-1H-imidazole (13)
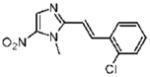
Dimetridazole (1.0 g, 7.1 mmol) was reacted with 2-chlorobenzaldehyde (1.2 g, 8.5 mmol, 1.2 equiv) in the presence of NaOEt (1.2 g, 17.7 mmol, 2.5 equiv). The mixture was purified by crystallization from CH3OH/CH2Cl2 (1/2) to give 13 as a bright-yellow solid (0.45 g, 24%). 1H NMR (500 MHz, CDCl3) δ 8.25 (d, J = 15.8, 1H), 8.12 (s, 1H), 7.69–7.67 (m, 1H), 7.46–7.43 (m, 1H), 7.34–7.30 (m, 2H), 6.92 (d, J = 15.7, 1H), 4.07 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 135.7, 134.6, 134.6, 134.9, 133.6, 130.5, 130.3, 127.1, 127.1, 114.1, 32.9; HRMS (ESI+) calcd for C12H11ClN3O2 [M + H]+ 264.0534, found 264.0539. Anal. (C12H10ClN3O2) C, H, N.
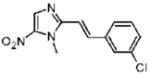
2-[(E)-2-(3-Chlorophenyl)vinyl]-1-methyl-5-nitro-1H-imidazole (14)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with 3-chlorobenzaldehyde (1.2 g, 8.5 mmol, 1.2 equiv) in the presence of NaOEt (1.2 g, 17.7 mmol, 2.5 equiv). The mixture was purified by crystallization from CH3OH to give 14 as a bright-yellow solid (0.36 g, 19%). 1H NMR (600 MHz, CDCl3) δ 8.09 (s, 1H), δ 7.82 (d, J = 15.7, 1H), 7.57 (s, 1H) 7.43 (td, J = 4.6, 1.5, 1H), 7.36–7.35 (m, 2H), 6.90 (d, J = 15.7, 1H), 4.07 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 149.2, 139.1, 138.1, 137.0, 135.0, 134.1, 130.2, 129.6, 126.8, 126.0, 112.6, 32.8; HRMS (ESI+) calcd for C12H11ClN3O2 [M + H]+264.0534, found 264.0536. Anal. (C12H10ClN3O2) C, H, N.
2-[(E)-2-(4-Chlorophenyl)vinyl]-1-methyl-5-nitro-1H-imidazole (15)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with 4-chlorobenzaldehyde (1.2 g, 8.5 mmol, 1.2 equiv) in the presence of NaOEt (1.2 g, 17.7 mmol, 2.5 equiv). The mixture was purified by crystallization from CH3OH/CH2Cl2 (3/1) to give 15 as a bright-yellow solid (0.27 g, 15%). 1H NMR (500 MHz, CDCl3) δ 8.10 (s, 1H), 7.85 (d, J = 15.7, 1H), 7.52 (d, J = 8.5, 2H), 7.40 (d, J = 8.5, 2H), 6.88 (d, J = 15.8, 1H), 4.07 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 149.5, 138.4, 135.7, 134.2, 133.7, 133.4, 129.3, 128.6, 111.7, 32.8; HRMS (ESI+) calcd for C12H11ClN3O2 [M + H]+ 264.0534, found 264.0535. Anal. (C12H10ClN3O2) C, H, N.
2-[(E)-2-(2,4-Dichlorophenyl)vinyl]-1-methyl-5-nitro-1H-imidazole (16)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with 2,4-dichlorobenzaldehyde (1.49 g, 9.23 mmol, 1.3 equiv) in the presence of NaOEt (966 mg, 14.2 mmol, 2.0 equiv). The mixture was purified by column chromatography (CH2Cl2 → 10% CH3OH/CH2Cl2) to give 16 as a yellow solid (316 mg, 15%). 1H NMR (DMSO-d6) δ 8.20 (s, 1H), 8.19 (d, J = 8.8, 1H), 8.02 (d, J = 16.0, 1H), 7.73 (d, J = 2.0, 1H), 7.54 (d, J = 8.8, 1H), 7.53 (d, J = 16.0, 1H), 4.05 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 148.9, 134.3, 134.0, 133.7, 132.0, 131.0, 129.3, 129.2, 128.9, 127.8, 116.6, 32.8; HRMS (ESI+) calcd for C12H10Cl2N3O2 [M + H]+ 298.0145, found 298.0135. Anal. (C12H9Cl2N3O2): C, 48.34; H, 3.04; N, 14.09. Found: C, 48.50; H, 3.75; N, 13.87.
2-[(E)-2-(2-Iodophenyl)vinyl]-1-methyl-5-nitro-1H-imidazole (17)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with 2-iodobenzaldehyde (2.0 g, 8.5 mmol, 1.2 equiv) in the presence of NaOEt (1.2 g, 17.7 mmol, 2.5 equiv). The mixture was purified by crystallization from CH3OH/CH2Cl2 (1/2) to give 17 as a bright-orange solid (0.44 g, 20%). 1H NMR (500 MHz, CDCl3) δ 8.14 (s, 1H), 8.08 (d, J = 15.6, 1H), 7.94 (dd, J = 7.9, 1.2, 1H), 7.63 (dd, J = 7.9, 1.5, 1H), 7.41 (dt, J = 7.4, 0.6, 1H), 7.07 (dt, J = 7.7, 1.5, 1H), 6.79 (d, J = 15.5, 1H), 4.08 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 149.1, 143.1, 140.1, 138.7, 134,3, 130.8, 128.6, 126.8, 114.5, 100.9, 33.0; HRMS (ESI+) calcd for C12H11IN3O2 [M + H]+ 355.9891, found 355.9892. Anal. (C12H10IN3O2) C, H, N.
2-[(E)-2-(3-Iodophenyl)vinyl]-1-methyl-5-nitro-1H-imidazole (18)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with 3-iodobenzaldehyde (2.0 g, 8.5 mmol, 1.2 equiv) in the presence of NaOEt (1.2 g, 17.7 mmol, 2.5 equiv). The mixture was purified by column chromatography (50% CH2Cl2/hexanes → 100% CH2Cl2) to give 18 as a dark-orange solid (0.50 g, 19%). 1H NMR (600 MHz, CDCl3) δ 8.09 (s, 1H), 7.93 (s, 1H), 7.77 (d, J = 15.7, 1H), 7.70 (d, J = 7.9, 1H), 7.50 (d, J = 7.74, 1H), 7.15 (t, J = 7.8, 1H), 6.87 (d, J = 15.7, 1H), 4.07 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 149.2, 139.1, 138.4, 137.8, 137.4, 135.7, 134.1, 130.6, 127.0, 124.5, 94.8, 32.8; HRMS (ESI+) calcd for C12H11IN3O2 [M + H]+ 355.9891, found 355.9902. Anal. (C12H10IN3O2): C, 40.58; H, 2.84; N, 11.83. Found: C, 41.07; H, 2.91; N, 11.12.
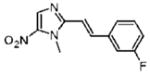
2-[(E)-2-(3-Fluorophenyl)vinyl]-1-methyl-5-nitro-1H-imidazole (19)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with 3-fluorobenzaldehyde (1.1 g, 8.5 mmol, 1.2 equiv) in the presence of NaOEt (1.2 g, 17.7 mmol, 2.5 equiv). The mixture was purified by column chromatography (50% CH2Cl2/hexanes → 100% CH2Cl2) to give 19 as a bright-yellow solid (0.23 g, 13%). 1H NMR (500 MHz, CDCl3) δ 8.10 (s, 1H), 7.86 (d, J = 15.7, 1H), 7.40–7.37 (m, 1H), 7.35–7.34 (m, 1H), 7.29 (dt, J = 10, 2.2, 1H), 7.09 (tdd, J = 8.1, 2.6, 1.0, 1H), 6.90 (d, J = 15.7, 1H), 4.08 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 163.1 (d, J = 245.2), 149.2, 139.1, 138.3 (d, J = 2.9), 137.5 (d, J = 7.3), 134.1, 130.5 (d, J = 8.1), 123.6 (d, J = 2.9), 116.6 (d, J = 21.5), 113.5 (d, J = 21.8), 112.5, 32.8; HRMS (ESI+) calcd for C12H11FN3O2 [M + H]+ 248.0830, found 248.0820. Anal. (C12H10FN3O2) C, H, N.
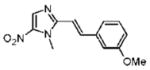
2-[(E)-2-(3-Methoxyphenyl)vinyl]-1-methyl-5-nitro-1H-imidazole (20)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with 3-methoxybenzaldehyde (1.13 mL, 9.23 mmol, 1.3 equiv) in the presence of NaOEt (966 mg, 14.2 mmol, 2.0 equiv). The mixture was purified by column chromatography (CH2Cl2 → 5% CH3OH/CH2Cl2) to give 20 as a yellow solid (588 mg, 32%). 1H NMR (400 MHz, DMSO-d6) δ 8.14 (s, 1H), 7.70 (d, J = 16.0, 1H), 7.35 (d, J = 16.0, 1H), 7.34 (br s, 1H), 7.29 (d, J = 5.2, 1H), 6.93–6.90 (m, 1H), 3.99 (s, 3H), 3.77 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 159.6, 149.7, 139.0, 137.9, 136.8, 134.1, 129.8, 120.5, 115.3, 113.3, 112.4, 55.2, 32.8; HRMS (ESI+) calcd for C13H14N3O3 [M+H]+ 260.1030, found 260.1029. Anal. (C13H13N3O3) C, H, N.
1-Methyl-5-nitro-2-{(E)-2-[4-(trifluoromethoxy)phenyl]vinyl}-1H-imidazole (21)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with 4-(trifluoromethoxy)benzaldehyde (1.33 mL, 9.23 mmol, 1.3 equiv) in the presence of NaOEt (966 mg, 14.2 mmol, 2.0 equiv). The mixture was purified by column chromatography (CH2Cl2 → 10% CH3OH/CH2Cl2) to give 21 as a yellow solid (311 mg, 14%). 1H NMR (400 MHz, DMSO-d6) δ 8.13 (s, 1H), 7.88 (d, J = 8.8, 2H), 7.73 (d, J = 15.6, 1H), 7.38 (d, J = 15.6, 2H), 7.36 (d, J = 8.8, 1H), 3.98 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 149.3, 148.6, 148.5, 148.6, 148.6, 142.0, 139.1, 136.1, 134.8, 134.0, 129.5, 128.0, 121.2, 120.6, 118.7, 114.3, 32.8; HRMS (ESI+) calcd for C13H11F3N3O3 [M + H]+ 314.0747, found 314.0752. Anal. (C13H10F3N3O3): C, 49.85; H, 3.22; N, 13.42. Found: C, 49.92; H, 3.60; N, 13.88.
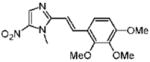
1-Methyl-5-nitro-2-[(E)-2-(2,3,4-trimethoxyphenyl)vinyl]-1H-imidazole (22)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with 2,3,4-trimethoxybenzaldehyde (1.67 g, 8.52 mmol, 1.2 equiv) in the presence of NaOEt (1.2 g, 17.8 mmol, 2.5 equiv). The mixture was purified by column chromatography (CH2Cl2 → 10% CH3OH/CH2Cl2) to give 22 as a yellow solid (362 mg, 16%). 1H NMR (400 MHz, CDCl3) δ 8.06 (s, 1H), 7.94 (d, J = 16.0, 1H), 7.23 (d, J = 8.8, 1H), 6.96 (d, J = 16.0, 1H), 6.68 (d, J = 8.8, 1H), 4.00 (s, 3H), 3.93 (s, 3H), 3.88 (s, 3H), 3.87 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 155.1, 152.9, 150.7, 142.4, 138.7, 135.2, 134.4, 123.4, 122.2, 110.6, 107.5, 61.1, 60.8, 56.0, 32.6; HRMS (ESI+) calcd for C15H18N3O5 [M + H]+ 320.1241, found 320.1240. Anal. (C15H17N3O5) C, H, N.
2-{(E)-2-[4-(Diethoxymethyl)phenyl]vinyl}-1-methyl-5-nitro-1H-imidazole (23)
Dimetridazole (20 g, 142 mmol) was reacted with 4-(diethoxymethyl)benzaldehyde (34.5 mL, 170 mmol, 1.2 equiv) in the presence of NaOEt (24 g, 354 mmol, 2.5 equiv). The mixture was purified by column chromatography (CH2Cl2 → 10% CH3OH/CH2Cl2) to give 23 as a yellow solid (5.67 g, 24%). 1H NMR (400 MHz, CDCl3) δ 8.09 (s, 1H), 7.87 (d, J = 16.0, 1H), 7.56 (d, J = 8.0, 2H), 7.51 (d, J = 8.0, 2H), 6.89 (d, J = 16.0, 1H), 5.52 (s, 1H), 4.05 (s, 3H), 3.65–3.53 (m, 4H, 2 × OCH2CH3), 1.25 (t, J = 7.2, 6H, 2 × OCH2CH3); 13C NMR (100 MHz, CDCl3) δ 149.8, 140.9, 139.4, 135.2, 134.2, 127.3, 111.3, 101.0, 89.8, 61.1, 32.8, 15.2; HRMS (ESI+) calcd for C17H22N3O4 [M + H]+ 332.1605, found 332.1608. Anal. (C17H21N3O4) C, H, N.
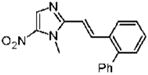
2-[(E)-2-Biphenyl-2-ylvinyl]-1-methyl-5-nitro-1H-imidazole (24)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with biphenyl-2-carbaldehyde (1.67 g, 9.23 mmol, 1.3 equiv) in the presence of NaOEt (966 mg, 14.2 mmol, 2.0 equiv). The mixture was purified by column chromatography (CH2Cl2 → 5% CH3OH/CH2Cl2) to give 24 as a yellow solid (455 mg, 21%). 1H NMR (400 MHz, DMSO-d6) δ 8.13–8.12 (m, 1H), 8.08 (s, 1H), 7.70 (d, J = 15.6, 1H), 7.51 –7.43 (m, 5H), 7.39–7.32 (m, 4H), 4.02 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 149.7, 141.7, 139.8, 138.9, 135.9, 134.0, 132.9, 130.3, 129.5, 129.2, 128.3, 127.7, 127.4, 126.5, 113.7, 32.7; HRMS (ESI+) calcd for C18H16N3O2 [M + H]+ 306.1237, found 306.1225. Anal. (C18H15N3O2) C, H, N.
1-Methyl-2-[(E)-2-(1-naphthyl)vinyl]-5-nitro-1H-imidazole (25)
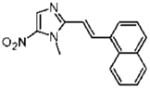
Dimetridazole (1.0 g, 7.1 mmol) was reacted with 1-naphthaldehyde (1.25 mL, 9.23 mmol, 1.3 equiv) in the presence of NaOEt (966 mg, 14.2 mmol, 2.0 equiv). The mixture was purified by column chromatography (CH2Cl2 → 10% CH3OH/CH2Cl2) to give 25 as a yellow solid (554 mg, 28%). 1H NMR (400 MHz, DMSO-d6) δ 8.59 (d, J = 15.6, 1H), 8.26 (d, J = 8.4, 1H), 8.25 (s, 1H), 8.16 (d, J = 8.0, 1H), 8.01–7.99 (m, 2H), 7.66–7.59 (m, 3H), 7.49 (d, J = 15.6, 1H), 4.08 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 149.7, 134.1, 133.7, 133.3, 132.0, 130.6, 129.7, 128.7, 127.0, 126.1, 125.8, 125.6, 124.6, 128.8, 115.5, 32.8; HRMS (ESI+) calcd for C16H14N3O2 [M + H]+ 280.1080, found 280.1085. Anal. (C16H13N3O2) C, H, N.
1-Methyl-2-[(E)-2-(2-naphthyl)vinyl]-5-nitro-1H-imidazole (26)
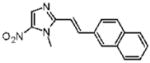
Dimetridazole (1.0 g, 7.1 mmol) was reacted with 2-naphthaldehyde (1.44 g, 9.23 mmol, 1.3 equiv) in the presence of NaOEt (966 g, 14.2 mmol, 2.0 equiv). The mixture was purified by column chromatography (CH2Cl2 → 10% CH3OH/CH2Cl2) to give 26 as a yellow solid (385 mg, 19%). 1H NMR (400 MHz, DMSO-d6) δ 8.22 (s, 1H), 8.21 (br s, 1H), 8.07 (dd, J = 8.8, 1.6, 1H), 7.98–7.92 (m, 4H), 7.56–7.52 (m, 3H), 4.08 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 149.8, 137.8, 134.2, 133.3, 133.1, 133.0, 128.6, 128.3, 128.2, 127.6, 126.9, 126.8, 126.6, 123.9, 113.5, 32.8; HRMS (ESI+) calcd for C16H14N3O2 [M + H]+ 280.1080, found 280.1081. Anal. (C16H13N3O2): C, 68.81; H, 4.69; N, 15.05. Found: C, 68.21; H, 4.78; N, 14.95.
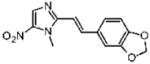
2-[(E)-2-(3a,7a-Dihydro-1,3-benzodioxol-5-yl)vinyl]-1-methyl-5-nitro-1H-imidazole (27)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with piperonal (1.39 g, 9.23 mmol, 1.3 equiv) in the presence of NaOEt (966 mg, 14.2 mmol, 2.0 equiv). The mixture was purified by column chromatography (CH2Cl2 → 10% CH3OH/CH2Cl2) to give 27 as a yellow solid (600 mg, 31%). 1H NMR (400 MHz, DMSO-d6) δ 8.17 (s, 1H), 7.70 (d, J = 15.6, 1H), 7.57 (d, J = 1.6, 1H), 7.25 (d, J = 15.6, 1H), 7.21 (dd, J = 8.0, 1.6z, 1H), 6.96 (d, J = 8.0, 1H), 6.09 (s, 2H), 4.01 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 150.1, 148.4, 148.0, 137.9, 134.3, 130.0, 124.0, 111.0, 108.4, 106.0, 101.4, 89.3, 32.7; HRMS (ESI+) calcd for C13H12N3O4 [M + H]+ 274.0822, found 274.0823. Anal. (C13H11N3O4) C, H, N.
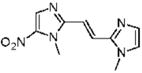
1-Methyl-2-[(E)-2-(1-methyl-1H-imidazol-2-yl)vinyl]-5-nitro-1H-imidazole (28)
Dimetridazole (1.06 g, 7.5 mmol) was reacted with 1-methyl-1H-imidazole-2-carbaldehyde (1.0 g, 9.1 mmol, 1.2 equiv) in the presence of NaOEt (1.27 g, 18.8 mmol, 2.5 equiv). The mixture was purified by column chromatography (CH2Cl2 → 10% CH3OH/CH2Cl2) to give 28 as a yellow solid (1.36 g, 77%). 1H NMR (400 MHz, DMSO-d6) δ 8.20 (s, 1H), 7.64 (d, J = 15.2, 1H), 7.33 (d, J = 15.2, 1H), 7.31 (s, 1H), 7.07 (s, 1H), 3.99 (s, 3H), 3.78 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 149.3, 143.4, 134.1, 129.2, 124.0, 123.0, 113.6, 32.7, 32.4; HRMS (ESI+) calcd for C10H12N5O2 [M + H]+ 234.0985, found 234.0986. Anal. (C10H11N5O2) C, H, N.
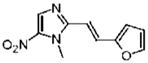
2-[(E)-2-(2-Furyl)vinyl]-1-methyl-5-nitro-1H-imidazole (29)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with furan-2-carbaldehyde (0.65 mL, 7.81 mmol, 1.1 equiv) in the presence of NaOEt (1.2 g, 17.8 mmol, 2.5 equiv). The mixture was purified by column chromatography (CH2Cl2 → 10% CH3OH/CH2Cl2) to give 29 as a yellow solid (825 mg, 53%). 1H NMR (400 MHz, DMSO-d6) δ 8.18 (s, 1H), 7.83 (d, J = 1.6, 1H), 7.58 (d, J = 15.6, 1H), 7.00 (d, J = 15.6, 1H), 6.90 (d, J = 3.2, 1H), 6.64 (dd, J = 3.2, 2.0, 1H), 3.98 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 151.4, 149.5, 144.9, 139.0, 134.2, 124.8, 113.6, 112.7, 110.3, 32.6; HRMS (ESI+) calcd for C10H10N3O3 [M + H]+ 220.0717, found 220.0720. Anal. (C10H9N3O3) C, H, N.
1-Methyl-5-nitro-2-[(E)-2-(2-thienyl)vinyl]-1H-imidazole (30)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with thiophene-2-carbaldehyde (0.86 mL, 9.23 mmol, 1.3 equiv) in the presence of NaOEt (966 mg, 14.2 mmol, 2.0 equiv). The mixture was purified by column chromatography (CH2Cl2 → 10% CH3OH/CH2Cl2) to give 30 as a yellow solid (384 mg, 23%). 1H NMR (400 MHz, DMSO-d6) δ 8.18 (s, 1H), 7.90 (d, J = 1.6, 1H), 7.78 (d, J = 15.6, 1H), 7.70 (dd, J = 5.2, 1.2, 1H), 7.63 (dd, J = 4.8, 2.8, 1H), 7.21 (d, J = 15.6, 1H), 4.00 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 150.0, 138.9, 138.7, 134.2, 132.1, 127.4, 125.6, 112.6, 32.7; HRMS (ESI+) calcd for C10H10N3O2S [M + H]+ 236.0488, found 236.0491. Anal. (C10H9N3O2S): C, 51.05; H, 3.86; N, 17.86. Found: C, 51.48; H, 3.35; N, 17.57.
1-Methyl-5-nitro-2-[(E)-2-(3-thienyl)vinyl]-1H-imidazole (31)
Dimetridazole (1.0 g, 7.1 mmol) was reacted with thiophene-3-carbaldehyde (0.81 mL, 9.23 mmol, 1.3 equiv) in the presence of NaOEt (966 mg, 14.2 mmol, 2.0 equiv). The mixture was purified by column chromatography (CH2Cl2 → 10% CH3OH/CH2Cl2) to give 31 as a yellow solid (367 mg, 22%). 1H NMR (400 MHz, DMSO-d6) δ 8.18 (s, 1H), 7.91 (d, 15.6, 1H), 7.67 (d, J = 5.2, 1H), 7.52 (d, J = 3.6, 1H), 7.15 (dd, J = 5.2, 3.6, 1H), 7.03 (d, J = 15.6, 1H), 3.99 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 149.5, 140.3, 138.9, 134.2, 130.8, 130.1, 128.5, 128.3, 111.6, 32.7; HRMS (ESI+) calcd for C10H10N3O2S [M + H]+ 236.0488, found 236.0493. Anal. (C10H9N3O2S): C, 51.05; H, 3.86; N, 17.86. Found: C, 50.90; H, 3.54; N, 17.67.
2-[1,2-Dibromo-2-(4-bromophenyl)ethyl]-1-methyl-5-nitro-1H-imidazole (33)
Compound 12 (2.05 g, 6.7 mmol) was dissolved in anhydrous CHCl3 (120 mL), and the pale-yellow solution was cooled to 0 °C. Bromine (0.37 mL, 1.12 g, 7.03 mmol, 1.05 equiv) was added via syringe at 0 °C, and the resulting orange solution was stirred at ambient temperature for 4 h. The mixture was transferred to a separatory funnel and partitioned (CH2Cl2//H2O). The organic phase was washed with Na2S2O3 (3 × 100 mL), brine (NaCl/H2O, 2 × 50 mL), dried over Na2SO4, and evaporated in vacuo to give a pale-yellow solid. The residue was purified by column chromatography (hexanes → 30% EtOAc/hexanes) to give 33 as an off-white solid (2.73 g, 88%). 1H NMR (400 MHz, DMSO-d6) δ 8.20 (s, 1H), 7.74 (d, J = 8.4, 2H), 7.61 (d, J = 8.4, 2H), 6.49 (d, J = 11.2, 1H), 6.03 (d, J = 11.6, 1H), 4.01 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 149.1, 139.1, 138.4, 132.3, 131.5, 130.7, 122.3, 51.7, 42.0, 33.3; HRMS (ESI+) calcd for C12H11Br3N3O2 [M + H]+ 465.8296, found 465.8394. Anal. (C12H10Br3N3O2) C, H, N.
1-(1-Methyl-5-nitro-1H-imidazol-2-yl)-2-(4-methylphenyl)-ethane-1,2-diol (34)
Compound 8 (0.69 g, 2.85 mmol) was made into a suspension in t-BuOH/H2O (1:1, 37 mL). Citric acid (1.10 g, 5.7 mmol, 2.0 equiv) was added in one portion followed by potassium osmate dihydrate (2.1 mg, 0.0057 mmol, 0.2 mol %). To the black suspension was added N-methylmorpholine N-oxide (NMO, 0.32 mL, 1.1 equiv) dropwise at ambient temperature, and the resulting suspension was stirred vigorously overnight. The mixture was filtered through Celite, and the brown filtrate was transferred to a separatory funnel and partitioned (CH2Cl2/H2O). The organic phase was washed with brine (2 × 100 mL), dried over Na2SO4, and evaporated in vacuo to give a brown residue. Crude product was purified by column chromatography (EtOAc) to give 34 as a yellowish solid (220 mg, 28%). 1H NMR (400 MHz, CDCl3 + CD3OD) δ 7.86 (s, 1H), 7.01 (s, 4H), 4.92 (d, J = 6.9, 1H), 4.64 (d, J = 6.9, 1H), 3.49 (s, 3H), 3.35 (br s, 2H), 2.23 (s, 3H); 13C NMR (100 MHz, CDCl3 + CD3OD) δ 151.4, 138.0, 135.8, 131.3, 129.0, 129.0, 126.2, 75.6, 70.9, 32.8, 20.9; HRMS (ESI+) calcd for C13H16N3O4 [M + H]+ 278.1135, found 278.1137. Anal. (C13H15N3O4): C, 56.31; H, 5.45; N, 15.15. Found: C, 56.41; H, 5.44; N, 15.11.
1-(4-Bromophenyl)-2-(1-methyl-5-nitro-1H-imidazol-2-yl)-ethane-1,2-diol (35)
Compound 12 (1.42 g, 4.62 mmol) was made into a suspension in 1:1 t-BuOH/H2O (60 mL). Citric acid (1.78 g, 9.25 mmol, 2.0 equiv) was added in one portion followed by potassium osmate dihydrate (3.0 mg, 0.0092 mmol, 0.2 mol %). To the black suspension was added N-methylmorpholine N-oxide (NMO, 0.52 mL, 5.08 mmol, 1.1 equiv) dropwise at ambient temperature, and the resulting suspension was stirred vigorously overnight. The mixture was filtered through Celite, and the brown filtrate was transferred to a separatory funnel and partitioned (CH2Cl2/H2O). The organic phase was washed with brine (2 × 100 mL), dried over Na2SO4, and evaporated in vacuo to give a brown residue. The residue was purified by column chromatography (CH2Cl2 → 5% MeOH/CH2Cl2) to give diol 35 as a white foam (225 mg, 14%). 1H NMR (400 MHz, DMSO-d6) δ 7.96 (s, 1H), 7.43 (d, J = 8.4, 2H), 7.21 (d, J = 8.4, 2H), 6.07 (d, J = 4.8, 1H), 5.81 (d, J = 4.8, 1H), 4.93–4.91 (m, 1H), 4.80–4.78 (m, 1H), 3.85 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 152.1, 140.9, 138.9, 131.6, 130.5, 129.1, 120.2, 74.1, 71.5, 33.4; HRMS (ESI+) calcd for C12H13BrN3O4 [M + H]+ 342.0084, found 342.0086. Anal. (C12H12BrN3O4): C, 42.12; H, 3.54; N, 12.28. Found: C, 41.84; H, 3.77; N, 12.47.
1-(1-Methyl-5-nitro-1H-imidazol-2-yl)-2-(4-methylphenyl)-ethane-1,2-diyl Diacetate (36)
Diol 34 (90 mg, 0.325 mmol) was dissolved in anhydrous pyridine (0.5 mL), and the solution was cooled to 0 °C. Acetic anhydride (132.6 mg, 122 μL, 1.3 mmol, 4.0 equiv) was added dropwise via syringe, and the resulting solution was stirred at ambient temperature for 3 h. The mixture was transferred to a separatory funnel and partitioned (CH2Cl2/H2O). The organic phase was washed with 1.0 N HCl (2 × 50 mL), brine (2 × 50 mL), dried over Na2SO4, and evaporated in vacuo to give a yellow oil. The residue was purified by column chromatography (hexanes → 40% EtOAc/hexanes) to provide acetylated adduct 36 as a yellowish wax (74 mg, 66%). 1H NMR (400 MHz, CDCl3) δ 7.93 (s, 1H), 7.11–7.04 (m, 4H), 6.36 (d, J = 9.4, 1H), 5.93 (d, J = 9.4, 1H), 3.62 (s, 3H), 2.28 (s, 3H), 2.13 (s, 3H), 2.11 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.4, 169.2, 147.0, 139.3, 138.5, 132.5, 131.7, 129.4, 127.0, 74.7, 68.6, 32.7, 21.6, 21.0, 20.6; HRMS (ESI+) calcd for C17H19N3O6Na [M + Na]+ 384.1166, found 384.1172. Anal. (C17H19N3O6): C, 56.51; H, 5.30; N, 11.63. Found: C, 53.32; H, 5.13; N, 10.22.
1-(1-Methyl-5-nitro-1H-imidazol-2-yl)-2-(4-bromophenyl)-ethane-1,2-diyl Diacetate (37)
Diol 35 (200 mg, 0.58 mmol) was dissolved in anhydrous pyridine (10 mL), and the solution was cooled to 0 °C. Acetic anhydride (0.14 mL, 1.45 mmol, 2.5 equiv) was added dropwise via syringe, and the resulting solution was stirred at ambient temperature for 3 h. The mixture was transferred to a separatory funnel and partitioned (CH2Cl2//H2O). The organic phase was washed with 1.0 N HCl (2 × 50 mL), brine (2 × 50 mL), dried over Na2SO4, and evaporated in vacuo to give a yellow oil. The residue was purified by column chromatography (hexanes → 40% EtOAc/hexanes) to provide acetylated adduct 37 as a colorless oil (227 mg, 92%). 1H NMR (400 MHz, CDCl3) δ 7.93 (s, 1H), 7.41 (d, J = 8.4, 2H), 7.13 (d, J = 8.4, 2H), 6.39 (d, J = 9.2, 1H), 5.96 (d, J = 9.2, 1H), 3.73 (s, 3H), 2.13 (s, 3H), 2.11 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.3, 169.1, 146.4, 143.9, 133.9, 132.4, 132.0, 129.0, 123.5, 74.1, 68.0, 32.9, 20.9, 20.6; HRMS (ESI+) calcd for C16H17BrN3O6 [M + H]+ 426.0295, found 426.0300. Anal. (C16H16BrN3O6) C, H, N.
1-Methyl-5-nitro-2-(2-phenylethyl)-1H-imidazole (40)
Compound 5 (1.0 g, 4.37 mmol), p-toluenesulfonhydrazine (4.06 g, 21.82 mmol, 5.0 equiv), and K2CO3 (3.02 g, 21.82 mmol, 5.0 equiv) in pyridine (30 mL) were stirred at 120 °C for 14 h. Purification of the mixture was carried out by preparatory thin layer chromatography on silica gel, eluting with 70% EtOAc/hexanes to furnish 40 as a white solid (0.324 g, 32%). 1H NMR (600 MHz, CDCl3) δ 8.00 (s, 1H), 7.35–7.29 (m, 2H), 7.25 (tt, J = 7.2, 1.7, 1H), 7.14 (dd, J = 7.0, 1.3, 2H), 3.26 (s, 3H), 3.13 (t, J = 7.3, 2H), 3.04 (t, J = 8.0, 2H); 13C NMR (150 MHz, CDCl3) δ 152.6, 139.8, 138.8, 132.6, 128.8, 128.3, 126.8, 33.6, 32.7, 29.7; HRMS (ESI+) calcd for C12H14N3O2 [M + H]+ 232.1080, found 232.1083. Anal. (C12H13N3O2): C, 62.33; H, 5.67; N, 18.17. Found: C, 62.71; H, 5.77; N, 17.57.
1-Methyl-2-[2-(4-methylphenyl)ethyl]-5-nitro-1H-imidazole (41)
Compound 8 (0.9 g, 3.75 mmol), p-toluenesulfonhydrazine (3.49 g, 18.8 mmol, 5.0 equiv), and K2CO3 (2.59 g, 18.8 mmol, 5.0 equiv) in anhydrous pyridine (30 mL) were stirred at 120 °C for 14 h. The mixture was cooled to ambient temperature, transferred to a separatory funnel, and partitioned (CH2Cl2/H2O). The organic phase was washed with brine (NaCl/H2O, 2 × 50 mL), NaHCO3/H2O (2 × 50 mL), dried over Na2SO4, and evaporated in vacuo to give a brown residue. The residue was purified by flash chromatography (CH2Cl2 → 5% MeOH/CH2Cl2) to give 41 as a pale-yellow solid (166 mg, 18%). 1H NMR (400 MHz, CDCl3) δ 7.98 (s, 1H), 7.10 (d, J = 8.0, 2H), 7.03 (d, J = 8.0, 2H), 3.66 (s, 3H), 3.10–3.06 (m, 2H), 3.04–2.99 (m, 2H), 2.33 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 152.7, 136.6, 136.2, 132.5, 129.3, 128.1, 127.9, 33.0, 32.7, 29.6, 20.9; HRMS (ESI+) calcd for C13H16N3O2 [M + H]+ 246.1237, found 246.1239. Anal. (C13H15N3O2): C, 63.66; H, 6.16; N, 17.13. Found: C, 63.15; H, 6.54; N, 17.65.
2-[2-(4-Bromophenyl)ethyl]-1-methyl-5-nitro-1H-imidazole (42)
Compound 12 (0.9 g, 2.95 mmol)), p-toluenesulfonhydrazine (2.73 g, 14.7 mmol, 5.0 equiv), and K2CO3 (2.03 g, 14.7 mmol, 5.0 equiv) in anhydrous pyridine (30 mL) were stirred at 120 °C for 14 h. The mixture was transferred to a separatory funnel and partitioned (CH2Cl2/H2O). The organic phase was washed with 1.0 N HCl (3 × 100 mL), dried over Na2SO4, and evaporated in vacuo to give a brown residue which was purified by flash chromatography (hexanes → 40% EtOAc/hexanes) to provide 42 as an off-white solid (151 mg, 17%). 1H NMR (400 MHz, DMSO-d6) δ 8.03 (s, 1H), 7.47 (d, J = 8.4, 2H), 7.23 (d, J = 8.4, 2H), 3.78 (s, 3H), 3.07–3.04 (m, 2H), 3.03–2.98 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.8, 139.8, 138.7, 132.2, 131.0, 130.7, 119.1, 32.7, 31.3, 28.0; HRMS (ESI+) calcd for C12H13BrN3O2 [M + H]+ 310.0186, found 310.0189. Anal. (C12H12BrN3O2) C, H, N.
Antimicrobial Assays
G. lamblia C6/WB (ATCC 50803), GS/M (ATCC 50580), BRIS/87/HEPU/713 (713),24 BRIS/83/HEPU/106 (106)25 were used as Mz-sensitive lines. The respective isogenic lines, 713-M3 and 106-21D10, were employed as Mz- resistant lines.24,25 C6/WB, 713, and 106 belong to G. lamblia assemblage A, while GS/M belongs to assemblage B. All lines were grown in TYI-S-33 medium supplemented with 10% bovine serum and 1 mg/mL bovine bile (Sigma) under anaerobic conditions.36 The Mz-resistant lines were routinely cultured with 50 μM Mz but were grown without Mz for 2–3 days before experiments. Stocks of the test compounds (10 mM in DMSO) were diluted in PBS to 75 μM, and 1:3 serial dilutions were made in 40 μL TYI-S-33 medium in 96-well plates. Trophozoites were harvested in the mid-logarithmic growth phase and were added to the wells in a 10 μL volume at 1000–2000 cells/well (total volume 50 μL/well). Final compound concentrations ranged from 20 μM to 0.4 nM, covering a 5 log10 range. Appropriately diluted solvent alone was used as a control. Cultures were grown for 2 days at 37 °C under anaerobic conditions (AnaeroPack-Anaero system, Remel). After incubation, cell growth and viability were determined with an ATP assay by adding 50 μL/well BacTiter-Glo microbial cell viability assay reagent (Promega) and measuring the luminescent signal in a microplate reader. The 50% effective concentration (EC50), the drug concentration that inhibits G. lamblia growth by 50% compared to parallel cultures without added drugs, was determined by graphic extrapolation of the concentration–response curves. Antigiardial potency is expressed as negative log10 value of the EC50 (pEC50), with results calculated as mean ± SE of three to four independent experiments. Initial studies showed that the results of the ATP assays correlated well with direct microscopic evaluation.
Cytotoxicity Assays
The human epithelial cell line HeLa (ATCC CCL-2) was maintained in DMEM, supplemented with 10% FBS, 5 mM HEPES, 50 U/mL penicillin, and 50 μg/mL streptomycin, at 37 °C in 5% CO2/95% air. Compound stocks in DMSO were diluted in DMEM to a concentration of 375 μM. Serial 1:3 dilutions were made in DMEM in 96-well plates (40 μ L/well), and 2000 HeLa cells were added per well in a 10 μL volume. Final compound concentrations ranged from 100 μM to 2 nM. Cells were cultured for 2 days, and viable cell numbers were determined with a dye reduction assay. For this, Alamar-Blue reagent (AbD Serotec) was added at 5 μL/well, and cultures were incubated for 4 h, after which fluorescence was assayed in a microplate reader. The 50% inhibitory concentration (IC50), the drug concentration that caused 50% loss of viability in the HeLa cultures compared to parallel cultures without added drugs, was determined by graphic extrapolation of the concentration–response curves. Toxicity is expressed as negative log10 value of the IC50 (pIC50), with results calculated as the mean ± SE of three to four independent experiments.
Murine Giardia Infections
Adult C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME). For infections, trophozoites of G. lamblia GS/M were grown to mid-logarithmic growth phase and given by oral gavage at 107/mouse in a 0.2 mL volume in TYI-S-33 growth medium. After 2 days (to allow establishment of infection), mice were given a test compound in 1% methylcellulose in PBS by oral gavage five times over a 3-day period. Controls received only methylcellulose/PBS. At day 5, the entire small intestine was removed and opened longitudinally, placed into 5 mL PBS, and cooled on ice for 10 min. After vigorous shaking to detach trophozoites, live G. lamblia were counted in a hemocytometer with a phase-contrast microscope. All animal studies were reviewed and approved by the UCSD Institutional Animal Care and Use Committee.
Electrochemical Measurements
E1/2 values were measured by cyclic voltammetry, using a CH Instruments model 600c digital potentiostat with its standard software package. All measurements were conducted under Ar gas in a jacketed, one-compartment cell with a Pt wire counter electrode. For the nonaqueous CVs, a Au disk working electrode (~1 mm diameter) was used along with a Ag wire pseudoreference electrode. For the aqueous CVs, a glass carbon working electrode (~2 mm diameter) was used with a SCE reference electrode. Immediately before use, the working electrode was polished with 0.25 μm diamond polishing paste, rinsed thoroughly with deionized water, polished with 0.05 μm alumina paste, rinsed again with deionized water, then rinsed with acetone and dried with a tissue. The electrolyte solution used for the nonaqueous CVs was 0.10 M NBu4PF6 in DMSO. Prior to use, the solid electrolyte was recrystallized three times from 95% ethanol, dried overnight in vacuum at 100 °C, and stored in a desiccator. The solvent, anhydrous grade DMSO (Aldrich, sure-seal), was passed through a short column of activated alumina directly into the cell. Ferrocene was added as an internal potential reference. For the aqueous CVs, the electrolyte solution was 0.1 M KCl in pH 7.0 phosphate buffer (0.1 M ionic strength). For all experiments, oxygen was removed by bubbling Ar gas through the solution prior to making measurements. The temperature was controlled by using a circulating water bath to run 25 °C water through the outer cell jacket. In most experiments, 5 μL of a 100 mM stock solution of the 5-NI compounds in DMSO was added to 3 mL of electrolyte solution in the cell to give a measurement concentration of 0.17 mM. CVs were recorded at 100 mV/s.
Acknowledgments
This work was supported by NIH U01 Cooperative Research Agreement AI75527 and the UCSD Digestive Diseases Research Development Center (DK80506). J.C.T. was supported by an NIH postdoctoral fellowship under the Ruth L. Kirschstein National Research Service Award program. P.H. was supported by a fellowship from the Swiss National Science Foundation (SSMBS; PASMA 114623). K.K. and L.V.A. received financial support from the CSU-LSAMP program funded by National Science Foundation, Grant HRD-0331537. We thank Taryn K. Fernandes, Cameron A. Collins, and M. Naomi King for help with the cyclic voltammetry studies.
Footnotes
Abbreviations used: CV, cyclic voltammogram; E1/2, half-wave potential; Ep, peak potential; Mz, metronidazole; NI, nitroimidazole.
References
- 1.Huang DB, White AC. An updated review on Cryptosporidium and Giardia. Gastroenterol Clin North Am. 2006;35:291–314. viii. doi: 10.1016/j.gtc.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Thompson RC. Giardiasis as a re-emerging infectious disease and its zoonotic potential. Int J Parasitol. 2000;30:1259–1267. doi: 10.1016/s0020-7519(00)00127-2. [DOI] [PubMed] [Google Scholar]
- 3.Nash TE. Surface antigenic variation in Giardia lamblia. Mol Microbiol. 2002;45:585–590. doi: 10.1046/j.1365-2958.2002.03029.x. [DOI] [PubMed] [Google Scholar]
- 4.Berkman DS, Lescano AG, Gilman RH, Lopez SL, Black MM. Effects of stunting, diarrhoeal disease, and parasitic infection during infancy on cognition in late childhood: a follow-up study. Lancet. 2002;359:564–571. doi: 10.1016/S0140-6736(02)07744-9. [DOI] [PubMed] [Google Scholar]
- 5.Hanevik K, Hausken T, Morken MH, Strand EA, Morch K, et al. Persisting symptoms and duodenal inflammation related to Giardia duodenalis infection. J Infect. 2007;55:524–530. doi: 10.1016/j.jinf.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Buret AG. Pathophysiology of enteric infections with Giardia duodenalis. Parasite. 2008;15:261–265. doi: 10.1051/parasite/2008153261. [DOI] [PubMed] [Google Scholar]
- 7.Troeger H, Epple HJ, Schneider T, Wahnschaffe U, Ullrich R, et al. Effect of chronic Giardia lamblia infection on epithelial transport and barrier function in human duodenum. Gut. 2007;56:328–335. doi: 10.1136/gut.2006.100198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Falagas ME, Walker AM, Jick H, Ruthazer R, Griffith J, et al. Late incidence of cancer after metronidazole use: a matched metronidazole user/nonuser study. Clin Infect Dis. 1998;26:384–388. doi: 10.1086/516306. [DOI] [PubMed] [Google Scholar]
- 9.Wright JM, Dunn LA, Upcroft P, Upcroft JA. Efficacy of antigiardial drugs. Expert Opin Drug Saf. 2003;2:529–541. doi: 10.1517/14740338.2.6.529. [DOI] [PubMed] [Google Scholar]
- 10.Anderson VR, Curran MP. Nitazoxanide: a review of its use in the treatment of gastrointestinal infections. Drugs. 2007;67:1947–1967. doi: 10.2165/00003495-200767130-00015. [DOI] [PubMed] [Google Scholar]
- 11.Edwards DI. Nitroimidazole drugs–action and resistance mechanisms. I. Mechanisms of action. J Antimicrob Chemother. 1993;31:9–20. doi: 10.1093/jac/31.1.9. [DOI] [PubMed] [Google Scholar]
- 12.Dan M, Wang AL, Wang CC. Inhibition of pyruvate–ferredoxin oxidoreductase gene expression in Giardia lamblia by a virus-mediated hammerhead ribozyme. Mol Microbiol. 2000;36:447–456. doi: 10.1046/j.1365-2958.2000.01863.x. [DOI] [PubMed] [Google Scholar]
- 13.Townson SM, Upcroft JA, Upcroft P. Characterisation and purification of pyruvate:ferredoxin oxidoreductase from Giardia duodenalis. Mol Biochem Parasitol. 1996;79:183–193. doi: 10.1016/0166-6851(96)02661-8. [DOI] [PubMed] [Google Scholar]
- 14.Upcroft P, Upcroft JA. Drug targets and mechanisms of resistance in the anaerobic protozoa. Clin Microbiol Rev. 2001;14:150–164. doi: 10.1128/CMR.14.1.150-164.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salas-Herrera IG, Pearson RM, Johnston A, Turner P. Concentration of metronidazole in cervical mucus and serum after single and repeated oral doses. J Antimicrob Chemother. 1991;28:283–289. doi: 10.1093/jac/28.2.283. [DOI] [PubMed] [Google Scholar]
- 16.Upcroft JA, Dunn LA, Wright JM, Benakli K, Upcroft P, et al. 5-Nitroimidazole drugs effective against metronidazole-resistant Trichomonas vaginalis and Giardia duodenalis. Antimicrob Agents Chemother. 2006;50:344–347. doi: 10.1128/AAC.50.1.344-347.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Upcroft JA, Campbell RW, Benakli K, Upcroft P, Vanelle P. Efficacy of new 5-nitroimidazoles against metronidazole-susceptible and -resistant Giardia, Trichomonas, and Entamoeba spp. Antimicrob Agents Chemother. 1999;43:73–76. doi: 10.1128/aac.43.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benakli K, Kaafarani M, Crozet MP, Vanelle P. Competition between C- and O-alkylation reactions in 5-nitroimidazole series: influence of nucleophile. Heterocycles. 1999;51:557–565. [Google Scholar]
- 19.Dupau P, Epple R, Thomas AA, Fokin VV, Sharpless KB. Osmium-catalyzed dihydroxylation of olefins in acidic media: old process, new tricks. Adv Synth Catal. 2002;344:421–433. [Google Scholar]
- 20.Narisada M, Horibe I, Watanabe F, Takeda K. Selective reduction of aryl halides and alpha, beta-unsaturated esters with sodium-borohydride cuprous chloride in methanol and its application to deuterium labeling. J Org Chem. 1989;54:5308–5313. [Google Scholar]
- 21.Ishihara M, Togo H. An efficient preparation of 2-imidazolines and imidazoles from aldehydes with molecular iodine and (diacetoxyiodo) benzene. Synlett. 2006:227–230. [Google Scholar]
- 22.Dandapani S, Jeske M, Curran DP. Synthesis of all 16 stereoisomers of pinesaw fly sex pheromones. Tools and tactics for solving problems in fluorous mixture synthesis. J Org Chem. 2005;70:9447–9462. doi: 10.1021/jo051526a. [DOI] [PubMed] [Google Scholar]
- 23.Kawasaki S, Fujita T, Toyota K, Yoshifuji M. Preparation and properties of fluorenylidene(phenyl)phosphines bearing bulky groups with electron-donating substituents in the ortho position on the phenyl group. Bull Chem Soc Jpn. 2005;78:1082–1090. [Google Scholar]
- 24.Townson SM, Laqua H, Upcroft P, Boreham PF, Upcroft JA. Induction of metronidazole and furazolidone resistance in Giardia. Trans R Soc Trop Med Hyg. 1992;86:521–522. doi: 10.1016/0035-9203(92)90095-t. [DOI] [PubMed] [Google Scholar]
- 25.Boreham PF, Phillips RE, Shepherd RW. Altered uptake of metronidazole in vitro by stocks of Giardia intestinalis with different drug sensitivities. Trans R Soc Trop Med Hyg. 1988;82:104–106. doi: 10.1016/0035-9203(88)90278-7. [DOI] [PubMed] [Google Scholar]
- 26.Rouveix B. Clinical implications of multiple drug resistance efflux pumps of pathogenic bacteria. J Antimicrob Chemother. 2007;59:1208–1209. doi: 10.1093/jac/dkl564. [DOI] [PubMed] [Google Scholar]
- 27.Wright GD. Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv Drug Delivery Rev. 2005;57:1451–1470. doi: 10.1016/j.addr.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 28.Liu SM, Brown DM, O’Donoghue P, Upcroft P, Upcroft JA. Ferredoxin involvement in metronidazole resistance of Giardia duodenalis. Mol Biochem Parasitol. 2000;108:137–140. doi: 10.1016/s0166-6851(00)00194-8. [DOI] [PubMed] [Google Scholar]
- 29.Muller J, Ley S, Felger I, Hemphill A, Muller N. Identification of differentially expressed genes in a Giardia lamblia WB C6 clone resistant to nitazoxanide and metronidazole. J Antimicrob Chemother. 2008;62:72–82. doi: 10.1093/jac/dkn142. [DOI] [PubMed] [Google Scholar]
- 30.Frontana C, Vazquez-Mayagoitia A, Garza J, Vargas R, Gonzalez I. Substituent effect on a family of quinones in aprotic solvents: an experimental and theoretical approach. J Phys Chem A. 2006;110:9411–9419. doi: 10.1021/jp060836+. [DOI] [PubMed] [Google Scholar]
- 31.Aguilar-Martinez M, Cuevas G, Jimenez-Estrada M, Gonzalez I, Lotina-Hennsen B, et al. An experimental and theoretical study of the substituent effects on the redox properties of 2-[(R-phenyl) amine]-1,4-naphthalenediones in acetonitrile. J Org Chem. 1999;64:3684–3694. doi: 10.1021/jo990186o. [DOI] [PubMed] [Google Scholar]
- 32.Leventis N, Rawaswdeh AMM, Zhang GH, Elder IA, Sotiriou-Leventis C. Tuning the redox chemistry of 4-benzoyl-N-methylpyridinium cations through para substitution. Hammett linear free energy relationships and the relative aptitude of the two-electron reduced forms for H-bonding. J Org Chem. 2002;67:7501–7510. doi: 10.1021/jo020489+. [DOI] [PubMed] [Google Scholar]
- 33.Legrand YM, Gray M, Cooke G, Rotello VM. Model systems for flavoenzyme activity: relationships between cofactor structure, binding and redox properties. J Am Chem Soc. 2003;125:15789–15795. doi: 10.1021/ja036940b. [DOI] [PubMed] [Google Scholar]
- 34.Mcdaniel DH, Brown HC. An extended table of Hammett substituent constants based on the ionization of substituted benzoic acids. J Org Chem. 1958;23:420–427. [Google Scholar]
- 35.La-Scalea MA, Serrano SHP, Gutz IGR. Voltammetric behaviour of metronidazole at mercury electrodes. J Braz Chem Soc. 1999;10:127–135. [Google Scholar]
- 36.Clark CG, Diamond LS. Methods for cultivation of luminal parasitic protists of clinical importance. Clin Microbiol Rev. 2002;15:329–341. doi: 10.1128/CMR.15.3.329-341.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]