

Abstract
Genome-wide pervasive transcription has been reported in many eukaryotic organisms1-7, revealing a highly interleaved transcriptome organization that involves hundreds of novel non-coding RNAs8. These recently identified transcripts either exist stably in cells (Stable Unannotated Transcripts) or are rapidly degraded by the RNA surveillance pathway (Cryptic Unstable Transcripts). One characteristic of pervasive transcription is the extensive overlap of SUTs and CUTs with previously annotated features, which prompts the questions of how these transcripts are generated, and whether they exert function9. Single-gene studies have shown that transcription of SUTs and CUTs can be functional, through mechanisms involving the generated RNAs10,11 or their generation itself12-14. To date, a complete transcriptome architecture including SUTs and CUTs has not been described in any organism. Knowledge about the position and genome-wide arrangement of these transcripts will be instrumental in understanding their function8,15. We provide here a comprehensive analysis of these transcripts in the context of multiple conditions, a mutant of the exosome machinery and different strain backgrounds. We show that both SUTs and CUTs display distinct patterns of distribution at specific locations. Most of the newly identified transcripts initiate from nucleosome-free regions (NFRs) associated with the promoters of other transcripts (mostly protein-coding genes), or from NFRs at the 3’ ends of protein-coding genes. Likewise, about half of all coding transcripts initiate from NFRs associated with promoters of other transcripts. These data change our view of how a genome is transcribed, suggesting that bidirectionality is an inherent feature of promoters. Such an arrangement of divergent and overlapping transcripts may provide a mechanism for local spreading of regulatory signals – that is, coupling the transcriptional regulation of neighbouring genes via transcriptional interference or histone modification.
To obtain a comprehensive survey of the structure and expression level of transcripts across the yeast genome, we used tiling arrays3 to profile wild-type transcriptomes in ethanol (YPE), glucose (YPD, SDC) and galactose (YPGal), which together encompass the main laboratory growth conditions of yeast (Supplementary Table 1 and 2). Transcript start and end positions were mapped to the genome by a segmentation algorithm16 and subsequent manual curation. To identify CUTs, profiles were measured for a deletion mutant of RRP6, coding for an important component of the nuclear exosome, which is involved in the degradation of CUTs17,18. Transcripts specific to the rrp6Δ mutant were designated as CUTs (Methods). Expression profiles are provided in a searchable web-database (http://steinmetzlab.embl.de/NFRsharing).
Altogether, 7,272 transcripts were identified, comprising 5,171 verified or uncharacterized ORF transcripts (ORF-Ts), 847 SUTs and 925 CUTs (Fig. 1, Supplementary Table 3). We took advantage of data from different conditions to disambiguate cases of overlapping or immediately adjacent transcripts (Methods). We only used transcripts with confidently mapped 5’ ends for analyses involving start sites (5,084 ORF-Ts, 823 SUTs and 704 CUTs) (Methods and Supplementary Table 4). For validation, we compared our data to transcript start sites (TSS) mapped by 5’ RACE19. 81% (1,039 of 1,281) of TSSs agreed within 50 bases with the 5’ RACE results (Supplementary Fig. 1); 3% higher than a recent Solexa sequencing approach19. Furthermore, a comparison of our 3’ ends with the Solexa dataset showed agreement of 61% (2,774 of 4,551) within 50 bases. In addition, we tested several CUT boundaries and they agreed well with our RT-PCR and 5’ RACE validations (Supplementary Fig. 2 and Supplementary Table 5). Altogether, 102 SUTs had higher expression level in rrp6Δ compared to wild-type (Supplementary Table 6), suggesting that the distinction between CUTs and SUTs is in some cases condition-dependent, as for example the CUT antisense of PHO84, which is stabilized in old cells11. CUTs were, overall, shorter (median length 440 bases) than SUTs (median length 761 bases; p < 2 × 10-16, Wilcoxon test).
Figure 1. Transcript maps.
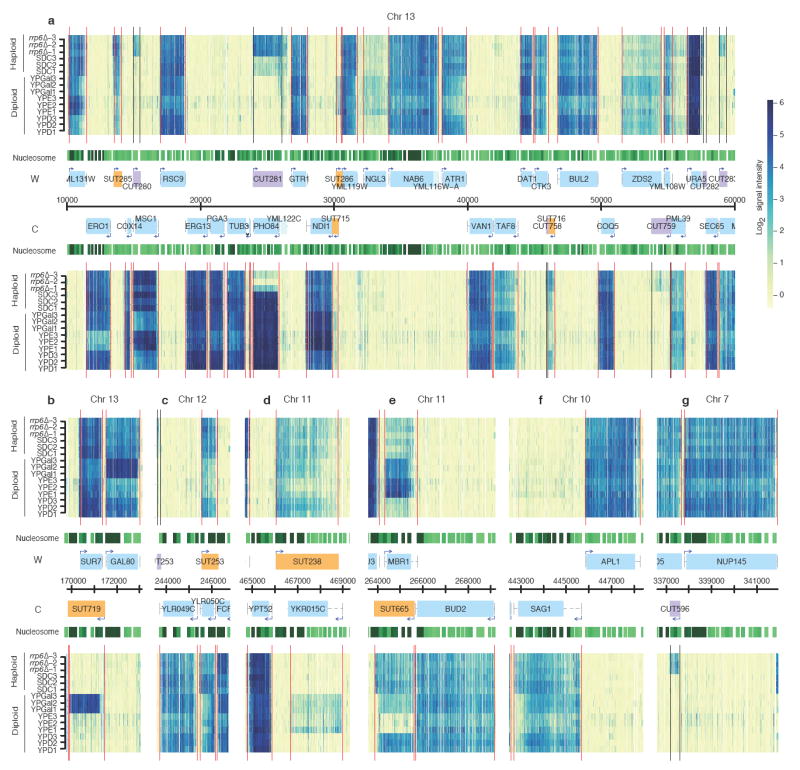
a, Expression data along 50 kb of chromosome XIII (x-axis) for the Watson (W, upper half) and the Crick (C, lower half) strands. Normalized signal intensities are shown for the profiled samples (y-axis): 3 replicates each for rrp6Δ S96 haploid strain, S96 haploid strain in SDC, S1003 diploid strain in YPGal and S1003 diploid strain in YPE; and 3 rows (summarizing 9 replicates) for S1003 diploid strain in YPD. Vertical lines represent inferred transcript boundaries. Nucleosome positions (green tracks, darker for more significant scores22) and genome annotations are shown in the centre: annotated ORFs (blue boxes) and their mapped UTRs (dashed grey lines), SUTs (orange boxes), CUTs (purple boxes) and transcript start sites (arrows).
b ~ g, Examples of transcriptional arrangements; layout as in a. b, tandem gene pair with antisense, GAL80 shares a NFR with SUT719, antisense of SUR7; c, antisense SUT253 originating from both a 5’ NFR (of YLR049C) and a 3’ NFR (of YLR050C); d, antisense SUT238 originating from a 5’ NFR (of YPT52); e, SUT665 originating from a 3’ NFR (of BUD2); f, divergent promoter of two ORF-Ts with a long UTRs; g, CUT596 originating from a 5’ NFR (of NUP145).
Nucleosome-free promoter regions (or 5’ NFRs), which facilitate transcription by allowing RNA polymerase to bind to DNA, have been reported as hallmarks of gene promoters20-24. To test whether unannotated transcripts have such hallmarks, we compared our transcript positions with nucleosome maps22,25. Consistent with promoter activity at NFRs, all classes of transcripts, ORF-Ts, CUTs and SUTs, exhibited depletion of nucleosomes upstream of their TSS (Fig. 2a). Furthermore, no nucleosome was detected between 422 of the 666 (63%) non-overlapping divergent transcript pairs involving at least one unannotated transcript (Methods and Supplementary Table 7). This suggests that these pairs share a single 5’ NFR that may function as a bidirectional promoter.
Figure 2. Properties of divergent transcript pairs.
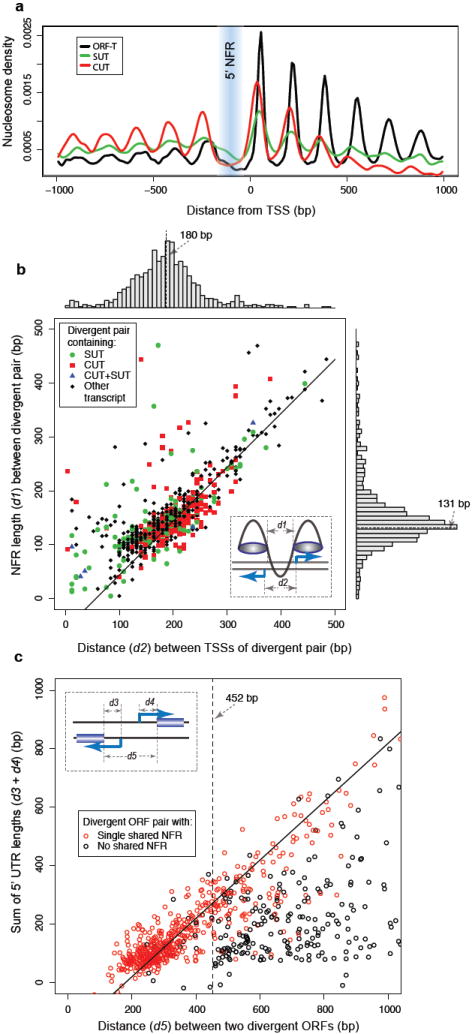
a, Nucleosome density22 relative to TSSs, averaged over ORF-Ts (black line), SUTs (green line) and CUTs (red line).
b, Scatterplot and histograms of shared NFR length (d1) and distance between TSSs (d2) of divergent pairs sharing a 5’ NFR. The line corresponds to the regression d1 = d2 − 2c, where the value c = 22 bases was determined from the mode of the distribution of differences between d1 and d2, and corresponds to a typical distance between NFR and TSS.
c, Scatterplot of the sum of 5’ UTR lengths (d3 + d4) vs. the distance (d5) between coding sequences of divergent ORF-T pairs. The solid line corresponds to the regression d5 = d3 + d4 + b, where the value b = 180 bases for the typical TSS distance between divergent pairs is taken from panel b above. The vertical dotted line at d5 = 452 bases is an estimate of the minimal distance for two ORFs to have separate NFRs.
To further investigate the set of potential bidirectional promoters in the yeast genome, we analyzed all 1,049 non-overlapping divergent transcript pairs that shared a single 5’ NFR. The distribution of distances between their TSSs had an estimated mode at 180 bases, while their shared NFR lengths had a mode at 131 bases (Fig. 2b). The size of the shared 5’ NFRs increased with the inter-transcript distances, in a relationship consistent with a model of a single NFR surrounded by two regions inside the flanking nucleosomes from which transcripts initiate22,25.
In our analysis, 612 of 931 non-overlapping divergent protein-coding transcript pairs were found to share a single 5’ NFR (66%, Supplementary Table 7). This fraction is considerably higher than the 30% of divergent ORF pairs that were previously estimated to share promoters26. Previous studies may have underestimated the number of bidirectional promoters by considering only distances between ORF start codons. Indeed, for divergent ORF-T pairs sharing a 5’ NFR (Fig. 2c, red dots), the total UTR length increased with the distance between the start codons, consistent with a typical size of the inter-transcript distance of a shared promoter being ~180 bases, as evident from Fig. 2b. This relationship holds for a wide range of inter-ORF distances, including cases greater than 1,000 bases, such as SAG1 and APL1 (Fig. 1f). In contrast, divergent ORF-T pairs separated by multiple NFRs showed no correlation between total UTR length and distance separating start codons (Fig. 2c, black dots). Moreover, most of these pairs were separated by more than 452 bases, which is approximately the minimal size of a region spanned by two NFRs (2 × 131 bases), a nucleosome (146 bases) and two intra-nucleosome regions (2 × 22 bases; Supplementary Fig. 3). These results suggest that bidirectional promoter usage is frequent for divergent transcript pairs involving unannotated transcripts and protein-coding genes in any combination.
To determine how many of the 5’ NFRs initiate transcripts bidirectionally, we selected all nucleosome-depleted regions longer than 80 bases immediately upstream of TSSs, defining a set of 3,965 5’ NFRs (Methods and Supplementary Fig. 4). Of these, 1,318 (33%) were bidirectional, involving half of all transcripts with a mapped 5’ NFR (2656 of 5339, Supplementary Tables 8-10). The sequences of NFRs detected as bidirectional promoters did not differ significantly from the other 5’ NFRs by content of palindromic sequences or GC nucleotides. Among transcripts with mapped 5’ NFRs, 61% of unannotated transcripts and 48% of protein-coding transcripts initiated bidirectionally from shared 5’ NFRs rather than initiating from their own promoters (Fig. 3b). Of the unannotated transcripts, 90% shared the 5’ NFR with a protein-coding transcript. These results suggest that bidirectionality is an inherent property of promoters. In addition to bidirectional transcription, a small number of transcripts were found to initiate in tandem orientation from shared 5’ NFRs (Fig. 3b). This number is likely underestimated, however, due to the difficulty of distinguishing immediately adjacent tandem transcripts by microarray hybridization. Altogether, our results suggest that multiple transcripts often initiate from NFRs at promoters in yeast. Additional transcripts will likely be detected by profiling other conditions or mutants other than rrp6Δ.
Figure 3. 5’ and 3’ NFR sharing.
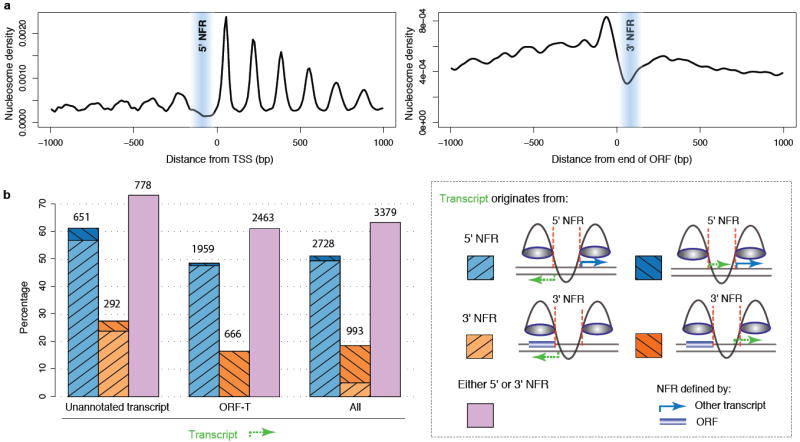
a, Nucleosome density relative to TSSs, averaged over all transcripts (left panel) and relative to translation stop sites, averaged over all ORF-Ts (right panel).
b, Transcripts initiating from 5’ or 3’ NFRs of other transcripts. The first block of bars corresponds to unannotated transcripts (1,063), the second to ORF-Ts (4,039), and the third to all transcripts (5,339) with mapped 5’ NFRs. Within each block, the bars correspond to different orientations of the transcript relative to the 5’ or 3’ NFR it originates from: divergently from a 5’ NFR (light blue), in tandem from a 5’ NFR (dark blue), in antisense to an ORF from a 3’ NFR (light orange), in tandem to an ORF from a 3’ NFR (dark orange), in any orientation from a 5’ or 3’ NFR (pink). See Supplementary Table 11 for a list of these pairs.
In addition to NFRs at promoters, nucleosome-free regions downstream of stop codons have been reported for the vast majority of ORFs and are suspected to play a role in transcription termination as well as in the generation of transcripts antisense to the ORF22. To better characterize such NFRs, we selected all nucleosome-depleted regions longer than 80 bases immediately downstream of stop codons of all verified and uncharacterized ORFs that we detected expressed, defining a set of 2,616 3’ NFRs (Supplementary Table 9). 827 of them initiated a transcript. We observed that 27% of unannotated transcripts with a mapped 5’ NFR initiated from the 3’ NFR of an ORF (Fig. 3b). Together, 3’ and 5’ shared NFRs thus accounted for the majority (73%) of SUT or CUT initiation, and for the majority (61%) of ORF-T initiation (Fig. 3b, Supplementary Table 10 and 11 for a list of all pairs). Altogether, these results show a surprisingly high level of NFR sharing, not only in bidirectional promoters but also in 3’ NFRs.
The high level of NFR sharing may explain a large extent of antisense transcription3, i.e. transcription on opposite strands. 70% of all antisense transcripts with mapped 5’ NFR initiated from a shared nucleosome free region. For example, 269 unannotated transcripts initiating from the 3’ NFR of an ORF were transcribed antisense to the ORF (for example YLR050C and MBR1, Figs. 1c, 1e). Another recurrent configuration is an antisense transcript starting from the 5’ NFR of a downstream tandem transcript. These configurations associate three transcripts; an example is GAL80, whose 5’ NFR initiates a transcript antisense to its upstream gene SUR7 (Fig. 1b). Notably, the level of SUR7 was lowest in YPGal medium, where the antisense and GAL80 had the highest expression (18 further examples are given in Supplementary Table 12). To generalize these observations, we analyzed expression correlations across growth conditions among transcript pairs involving at least one SUT. We observed significant expression anti-correlation between sense-antisense pairs, while bidirectional pairs of transcripts showed a tendency for co-expression (Supplementary Fig. 5 and Supplementary Table 13; p < 10-7, Pearson’s product moment correlation test). These findings fit the patterns displayed by individual cases of transcriptional interference or inhibitory histone modifications10-14,27.
The extent to which the genome-wide set of unannotated transcripts play a biological role, or are merely transcriptional side products (noise) originating from nucleosome depleted regions9, is unknown. The action of transcription itself can be functional even if the transcription product is not. This is the case, for example, with the transcription of the ncRNAs SRG1 and IME4 antisense, which mediate transcriptional silencing13,14. To explore the conservation of transcription initiation from 5’ and 3’ NFRs, we profiled the transcriptome of YJM78928, a highly diverged relative of the laboratory strain S288c. In rich media (YPD), about fifty percent (380/769) of the SUTs expressed in S288c were also found expressed in YJM789 (Methods). The frequencies with which these 380 conserved SUTs were observed sharing NFRs with other transcripts were similar to those in the overall dataset. These results indicate that the interlaced architecture of transcript initiation from 5’ and 3’ NFRs is conserved between these strains of S. cerevisiae. Why some of the unannotated transcripts are stable and others unstable remains to be explored. The parasite Giardia lamblia produces an abundance of antisense transcripts originating bidirectionally from promoters29, and consistent with our rrp6Δ results, its genome lacks orthologs to several nuclear exosome components. Likewise, the function of bidirectional transcription requires further exploration. One hypothesis is that bidirectional transcription has a role in maintaining an open chromatin structure at promoters. In other instances the combined action of bidirectional promoters and transcriptional regulation by these transcripts, or their generation, may provide a mechanism to spread transcriptional regulatory signals locally in the genome.
Methods Summary
cDNA for hybridization was prepared using random- or random plus oligo-dT- priming with the addition of actinomycin D during reverse transcription30. The hybridization data were normalized and segmented using the Bioconductor package ‘tilingArray’16. Segments were then manually curated. Further details can be found in Methods and Supplementary Information.
Supplementary Material
Acknowledgments
We thank Asifa Akhtar, Andreas Ladurner, Stephanie Blandin, Raeka Aiyar, Eugenio Mancera Ramos, Emilie Fritsch for helpful comments on the manuscript, Joern Toedling for helpful discussion and for the template of the website, Charles Girardot for data submission to ArrayExpress, Nick Proudfoot for access to experiment equipment, and the contributors to the Bioconductor (www.bioconductor.org) and R (http://www.r-project.org) projects for their software. This work was supported by grants to L.M.S. from the National Institutes of Health and Deutsche Forschungsgemeinschaft, by a SystemsX fellowship to E.G., a Roche fellowship to J.C. and grants to F.S. from SNF and NCCR Frontiers in Genetics.
Footnotes
Author Contributions L.M.S., Z.X. and W.W. designed the research; Z.X. and W.W. annotated the transcripts with the help of J.G. and F.P.; W.W. and Z.X. performed analysis of the transcripts with the help of J.G.; F.P. and S.C. performed the array hybridizations; J.C. E.G. and F.S. provided samples for the rrp6 mutant, designed and performed validation RT-PCR and 5’ RACE experiments; L.M.S., J.G., F.S. and W.H. supervised the research; L.M.S., Z.X., W.W., J.G. and W.H. wrote the manuscript.
Author Information Raw data are available from ArrayExpress (http://www.ebi.ac.uk/arrayexpress) under accession number E-TABM-590.
References
- 1.Bertone P, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306(5705):2242–2246. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- 2.Carninci P, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309(5740):1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 3.David L, et al. A high-resolution map of transcription in the yeast genome. Proc Natl Acad Sci USA. 2006;103(14):5320–5325. doi: 10.1073/pnas.0601091103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dutrow N, et al. Dynamic transcriptome of Schizosaccharomyces pombe shown by RNA-DNA hybrid mapping. Nat Genet. 2008;40(8):977–986. doi: 10.1038/ng.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li L, et al. Genome-wide transcription analyses in rice using tiling microarrays. Nat Genet. 2006;38(1):124–129. doi: 10.1038/ng1704. [DOI] [PubMed] [Google Scholar]
- 6.Stolc V, et al. A gene expression map for the euchromatic genome of Drosophila melanogaster. Science. 2004;306(5696):655–660. doi: 10.1126/science.1101312. [DOI] [PubMed] [Google Scholar]
- 7.Wilhelm BT, et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453(7199):1239–1243. doi: 10.1038/nature07002. [DOI] [PubMed] [Google Scholar]
- 8.Kapranov P, Willingham AT, Gingeras TR. Genome-wide transcription and the implications for genomic organization. Nat Rev Genet. 2007;8(6):413–423. doi: 10.1038/nrg2083. [DOI] [PubMed] [Google Scholar]
- 9.Struhl K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat Struct Mol Biol. 2007;14(2):103–105. doi: 10.1038/nsmb0207-103. [DOI] [PubMed] [Google Scholar]
- 10.Berretta J, Pinskaya M, Morillon A. A cryptic unstable transcript mediates transcriptional trans-silencing of the Ty1 retrotransposon in S. cerevisiae. Genes Dev. 2008;22(5):615–626. doi: 10.1101/gad.458008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camblong J, et al. Antisense RNA stabilization induces transcriptional gene silencing via histone deacetylation in S. cerevisiae. Cell. 2007;131(4):706–717. doi: 10.1016/j.cell.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 12.Bird AJ, Gordon M, Eide DJ, Winge DR. Repression of ADH1 and ADH3 during zinc deficiency by Zap1-induced intergenic RNA transcripts. EMBO J. 2006;25(24):5726–5734. doi: 10.1038/sj.emboj.7601453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hongay CF, Grisafi PL, Galitski T, Fink GR. Antisense transcription controls cell fate in Saccharomyces cerevisiae. Cell. 2006;127(4):735–745. doi: 10.1016/j.cell.2006.09.038. [DOI] [PubMed] [Google Scholar]
- 14.Martens JA, Laprade L, Winston F. Intergenic transcription is required to repress the Saccharomyces cerevisiae SER3 gene. Nature. 2004;429(6991):571–574. doi: 10.1038/nature02538. [DOI] [PubMed] [Google Scholar]
- 15.Birney E, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huber W, Toedling J, Steinmetz LM. Transcript mapping with high-density oligonucleotide tiling arrays. Bioinformatics. 2006;22(16):1963–1970. doi: 10.1093/bioinformatics/btl289. [DOI] [PubMed] [Google Scholar]
- 17.Davis CA, Ares M. Accumulation of unstable promoter-associated transcripts upon loss of the nuclear exosome subunit Rrp6p in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2006;103(9):3262–3267. doi: 10.1073/pnas.0507783103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wyers F, et al. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell. 2005;121(5):725–737. doi: 10.1016/j.cell.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 19.Nagalakshmi U, et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320(5881):1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee W, et al. A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet. 2007;39(10):1235–1244. doi: 10.1038/ng2117. [DOI] [PubMed] [Google Scholar]
- 21.Shivaswamy S, et al. Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation. PLoS Biol. 2008;6(3):e65. doi: 10.1371/journal.pbio.0060065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mavrich TN, et al. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008 doi: 10.1101/gr.078261.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whitehouse I, Rando OJ, Delrow J, Tsukiyama T. Chromatin remodelling at promoters suppresses antisense transcription. Nature. 2007;450(7172):1031–1035. doi: 10.1038/nature06391. [DOI] [PubMed] [Google Scholar]
- 24.Yuan GC, et al. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309(5734):626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- 25.Albert I, et al. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446(7135):572–576. doi: 10.1038/nature05632. [DOI] [PubMed] [Google Scholar]
- 26.Hermsen R, ten Wolde PR, Teichmann S. Chance and necessity in chromosomal gene distributions. Trends Genet. 2008;24(5):216–219. doi: 10.1016/j.tig.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 27.Uhler JP, Hertel C, Svejstrup JQ. A role for noncoding transcription in activation of the yeast PHO5 gene. Proc Natl Acad Sci USA. 2007;104(19):8011–8016. doi: 10.1073/pnas.0702431104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei W, et al. Genome sequencing and comparative analysis of Saccharomyces cerevisiae strain YJM789. Proc Natl Acad Sci U S A. 2007;104(31):12825–12830. doi: 10.1073/pnas.0701291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teodorovic S, Walls CD, Elmendorf HG. Bidirectional transcription is an inherent feature of Giardia lamblia promoters and contributes to an abundance of sterile antisense transcripts throughout the genome. Nucleic Acids Res. 2007;35(8):2544–2553. doi: 10.1093/nar/gkm105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perocchi F, Xu Z, Clauder-Münster S, Steinmetz LM. Antisense artifacts in transcriptome microarray experiments are resolved by actinomycin D. Nucleic Acids Res. 2007;35(19):e128. doi: 10.1093/nar/gkm683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.