

Abstract
Mutations of HFE are associated with hereditary hemochromatosis, but their influence on host susceptibility to infection is incompletely understood. We report that mice lacking one or both Hfe alleles are protected from septicemia with Salmonella Typhimurium, displaying prolonged survival and improved control of bacterial replication. This increased resistance is paralleled by an enhanced production of the enterochelin-binding peptide lipocalin-2 (Lcn2), which reduces the availability of iron for Salmonella within Hfe-deficient macrophages. Accordingly, Hfe−/−Lcn2−/− macrophages are unable to efficiently control the infection or to withhold iron from intracellular Salmonella. Correspondingly, the protection conferred by the Hfe defect is abolished in Hfe−/− mice infected with enterochelin-deficient Salmonella as well as in Hfe−/−Lcn2−/− mice infected with wild-type bacteria. Thus, by induction of the iron-capturing peptide Lcn2, absence of functional Hfe confers host resistance to systemic infection with Salmonella, thereby providing an evolutionary advantage which may account for the high prevalence of genetic hemochromatosis.
Introduction
HFE, a nonclassical MHC-I molecule, is the protein product of the gene mutated in hereditary hemochromatosis (HH) type I (also termed HFE-related HH), a prevalent autosomal recessive iron overload disorder. Two missense mutations, C282Y and H63D, in the HFE gene account for most HH cases. The C282Y mutation is found with an allelic frequency of 8% to 12% in people of northern European descent, whereas the H63D substitution is also present in other populations.1–4
Despite considerable efforts to elucidate its physiologic function, the precise role of HFE remains incompletely understood. On the one hand, HFE controls cellular iron homeostasis both by lowering the affinity of transferrin receptor-1 (TfR1) for iron-laden transferrin and by influencing cellular iron efflux.5 On the other hand, through its interactions with TfR1 and TfR2 expressed on hepatocytes, HFE modifies the formation of the key iron-regulatory hormone hepcidin antimicrobial peptide (Hamp) and thus affects systemic iron balance.6–11
Although parenchymal organs such as liver, heart, or pancreas progressively accumulate iron in subjects with HFE-related HH, cells of the mononuclear phagocyte system are depleted of this metal.12,13 The mononuclear phagocyte system is centrally involved in the maintenance of body iron homeostasis as it recycles iron from senescent erythrocytes to the circulation by the transmembrane protein ferroportin-1 (Fpn1; also termed Slc40a1).14,15 Importantly, the expression of Fpn1 on cells is controlled by the acute-phase reactant Hamp. Both iron overload and inflammation stimulate the formation of Hamp, which binds to Fpn1, resulting in its internalization and proteolysis with subsequent blockade of iron export.16,17 Macrophages need certain amounts of iron for the generation of ROS, which represents an important mechanism of innate immune defense. However, iron also exerts negative regulatory effects on IFN-γ or LPS-inducible macrophage immune effector pathways, including the formation of nitric oxide, the expression of MHC class II molecules, and the generation of TNF-α.18–20
On infections with intracellular microbes, including Salmonella, host phagocytes and invading pathogens have a shared requirement for iron. Consequently, competitive interactions between macrophage iron transporters and microbial iron acquisition systems form a central battlefield that determines the course of disease.21–23 Salmonella enterica Serovar Typhimurium (S Typhimurium) is a facultative intracellular microbe whose pathogenicity depends on its ability to invade macrophages, thus exploiting these cells as a habitat for multiplication and for spreading within the host.24 To acquire the scarce amounts of free iron present within mononuclear phagocytes, S Typhimurium has evolved siderophore-dependent and -independent mechanisms,25,26 both of which are linked to its virulence.27,28
In response to infections, innate immune cells secrete the antimicrobial peptide lipocalin-2 (Lcn2; also known as neutrophil gelatinase-associated lipocalin, siderocalin, or 24p3), which captures iron-laden bacterial siderophores, such as enterochelin (also known as enterobactin) and carboxymycobactins.29,30 Intriguingly, the interaction of an iron-siderophore-Lcn2 complex with its receptor (LcnR) results in the import of iron into mammalian cells and subsequent iron storage.31 However, an iron-free siderophore can be bound by Lcn2 and taken up into host cells by LcnR. Intracellularly, this imported siderophore can complex iron and export it out of cells, resulting in cellular iron deprivation.31
Given the central importance of iron for the growth and proliferation of intracellular pathogens and the well-known relation between intramacrophage iron availability and immune function, we hypothesized that Hfe deficiency, which results in mononuclear iron depletion, may be advantageous to the host during infections with intraphagocytic microbes such as S Typhimurium.
Methods
Cell culture and Salmonella infection in vitro
Thioglycolate-elicited primary peritoneal macrophages were harvested from C57BL/6 mice of different genotypes (detailed in supplemental Methods, available on the Blood website; see the Supplemental Materials link at the top of the online article), matched for sex and age, and cultured in RPMI (purchased from Biochrom AG) containing 5% heat-inactivated fetal calf serum (FCS; PAA Laboratories GmbH), 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 10 mM HEPES (all from Sigma-Aldrich). Cell preparations used for subsequent experiments had at least 90% to 95% purity, as determined by the expression of F4/80 on the cell surface in fluorescence-activated cell sorting analysis.
Wild-type (WT) S Typhimurium strain ATCC14028 and its isogenic derivatives deficient in iroBC, entC, entC sit feo, ssrA, and SPI-1, respectively, were generated and used as described.25,28,32 Briefly, macrophages were infected with S Typhimurium at a multiplicity of infection of 5 at 37°C for 24 hours, unless otherwise indicated. Thereafter, macrophages were washed 3 times in PBS and subjected to RNA isolation. In certain experiments, macrophages were treated with recombinant murine Lcn2 (apo-Lcn2, free of iron and microbial siderophores; purchased from R&D Systems), a monoclonal anti–mouse Lcn2 antibody (clone 228418; purchased from R&D Systems), a rat IgG2A isotype control antibody (clone 54447; purchased from R&D Systems), or the appropriate solvent. For quantification of intracellular Salmonella by gentamicin protection assay, macrophages were lysed at different time points after infection in 0.5% deoxycholic acid (Sigma-Aldrich) and plated under sterile conditions onto LB agar plates.
To determine the effects of endogenous or recombinant Lcn2 on Salmonella growth, strains were grown in complete RPMI without antibiotics. Bacterial growth was followed by photometric measurement of absorbance at 600 nm. The antibacterial effects of Lcn2 were evaluated in mid-logarithmic cultures.
To test for viability, primary macrophages were isolated, and kits were used to determine cellular lactate dehydrogenase release (Sigma-Aldrich) and caspase-1 activity (Calbiochem) according to the manufacturer's recommendations.
Quantification of iron uptake and release by macrophages
Macrophage iron uptake and release studies were performed as detailed elsewhere32 using 12.5 μg/mL 59Fe-labeled transferrin or 5 μM 59Fe-citrate (DuPont New England Nuclear). For iron release experiments, cells were first incubated with 5 μM 59Fe-citrate for 4 hours to allow iron loading and then washed 4 times. After an additional incubation for 2 hours, cellular iron release was measured by a γ-counter.33 In parallel to each iron release study, a trypan-blue exclusion assay was performed to ensure that neither treatment interfered with the integrity of the macrophage cell-surface membrane.
Determination of iron acquisition by S Typhimurium
Experiments to measure bacterial iron acquisition were performed as described in detail elsewhere.32 In brief, Salmonella-infected macrophages were washed 3 times and repleted with serum-free, HEPES-buffered RPMI. After the addition of 5 μM 59Fe-citrate, cells were incubated for an additional 8 hours. Intracellular bacilli were harvested according to a modified protocol as described.32,34
Determination of cellular iron content
The intracellular iron content of primary macrophages was measured by Graphite furnace atomic absorption spectrometry, carried out exactly as described.35 Values were normalized for the total cellular protein content.
Immunoprecipitation
Acute-phase sera were elicited by intraperitoneal inoculation of 108 WT S Typhimurium that had been heat-inactivated at 70°C for 20 minutes. Endogenous Lcn2 was removed from acute-phase sera by immunoprecipitation with the monoclonal anti-Lcn2 antibody or an isotype control antibody (see “Salmonella infection in vitro”) and protein G Sepharose (Amersham) according to the manufacturer's recommendations.
Salmonella infection in vivo
All animal experiments described were performed in accordance with Austrian legal requirements after approval by the Austrian authorities. Mice were maintained at the central animal facilities of the Medical University of Innsbruck and were given free access to water and food. Hfe−/− and Lcn2−/− mice were generated as described,29,36 crossed back on a C57BL/6 (Nramp1s/s) background for at least 12 generations, and transferred to the specific pathogen-free unit of the local animal facility by embryonic transfer. Double knockout (KO) animals were generated as described in supplemental Methods.
For in vivo infection experiments, male mice were used at 20 to 26 weeks of age and fed a standard diet (180 mg Fe/kg, C1000 from Altromin). Mice were infected intraperitoneally with 500 CFU S Typhimurium suspended in 200 μL PBS. Animals were monitored twice daily for signs of illness, and moribund mice were killed. Forty-eight and 96 hours after infection, mice were randomly selected for the determination of colony counts. Bacterial load in livers and spleens was determined by plating serial dilutions of organ homogenates on LB agar under sterile conditions. In parallel, organs were frozen in liquid nitrogen for subsequent preparation of RNA and protein as detailed in supplemental Methods. Mice selected for the determination of colony counts and gene expression studies were not considered for the recording of survival times.
Blood counts
Blood samples were drawn under anesthesia by retroorbital puncture and collected in heparinized tubes. An aliquot of blood was used for complete blood counts performed on an animal blood counter instrument.
Detection of cytokines, nitrite, and reactive oxygen species
Cytokine concentrations in serum samples and cell culture supernatants were determined by specific enzyme-linked immunoabsorbent assay (ELISA) kits (kits for IFN-γ, TNF-α, IL-1β, IL-6, IL-10, and IL-12p70 obtained from BD PharMingen; kit for Lcn2 obtained from R&D Systems; kit for heterodimeric IL-23 obtained from eBioscience). Determinations of nitrite and total oxidative capacity in cell culture supernatants were performed by commercially available colorimetric assays.
Statistical analysis
Statistical analysis was carried out using a SPSS statistical package (SPSS Inc). Calculations for statistical differences were carried out by analysis of variance test with the use of Bonferroni correction for multiple tests (parametric) or by Kruskal-Wallis test (nonparametric). For comparison of survival experience between subgroups, the Wilcoxon (Gehan) statistic was used.
Results
Cellular iron content of primary WT, Hfe+/−, and Hfe−/− macrophages
We first confirmed that peritoneal macrophages isolated from Hfe+/− and Hfe−/− mice had significantly reduced intracellular iron levels compared with their Hfe+/+ WT counterparts (Figure 1A). To elucidate the putative underlying mechanisms, we studied the expression of iron metabolic genes in macrophages. We found that Hfe+/− and Hfe−/− mononuclear phagocytes expressed significantly higher mRNA levels of Lcn2, which has the capacity to mediate the bidirectional transport of siderophore-bound iron across the cell surface membrane (Figure 1B). In contrast, the transcript levels of other proteins implicated in macrophage iron handling such as divalent metal transporter-1 (Dmt1; Slc11a1), TfR1, Fpn1, Hamp, LcnR, heme oxygenase-1 (Hmox1) and feline leukemia virus subgroup C receptor (Flvcr) were not affected by the Hfe genotype (data not shown). As modulation of Lcn2 expression might also affect the viability of macrophages by induction of apoptosis in these cells, we measured cellular release of lactate dehydrogenase and caspase-1 activity in Hfe+/+ and Hfe−/− macrophages, observing no differences for the Hfe genotype (data not shown).
Figure 1.

Macrophage iron content and Lcn2 expression as a function of Hfe genotype. Thioglycolate-elicited primary peritoneal macrophages were isolated from mice of the indicated genotypes grown under control conditions or infected with WT Salmonella Typhimurium (S Tm). The total cellular iron content was measured by atomic absorption spectrometry (A,C,D) and normalized for protein content. The transcriptional expression of genes contributing to macrophage transmembrane iron fluxes was determined by quantitative RT-PCR and normalized for Hprt expression (B). Iron export from WT and Hfe−/− macrophages was determined in 59Fe-transport studies. Data from 3 to 5 independent experiments were compared by analysis of variance (ANOVA) using Bonferroni correction or Kruskal-Wallis test, with statistical significance as indicated. Values are depicted as lower quartile, median, and upper quartile (boxes); minimum/maximum ranges (A-D); or as mean ± SEM (E), respectively.
To determine the net effect of enhanced Lcn2 expression on the intracellular iron content of mononuclear phagocytes, we measured cellular iron levels of peritoneal macrophages obtained from C57BL/6 Hfe+/+, Hfe−/−, Lcn2−/−, and Hfe−/−Lcn2−/− double KO mice. We observed increased intracellular iron levels in macrophages harvested from Lcn2−/− mice compared with WT controls. Furthermore, Hfe−/−Lcn2−/− macrophages displayed an iron content similar to that observed in Hfe+/+Lcn2−/− phagocytes but significantly higher than in Hfe−/−Lcn2+/+ phagocytes (Figure 1C). Of interest, we measured a substantial reduction in macrophage iron content after Salmonella infection, with the lowest iron levels observed within infected Hfe−/− cells (Figure 1D). In parallel, iron release was stimulated on Salmonella infection as described22 and was significantly higher in Hfe−/− macrophages, both in response to infection or stimulation with IFN-γ, compared with Hfe+/+ macrophages (Figure 1E).
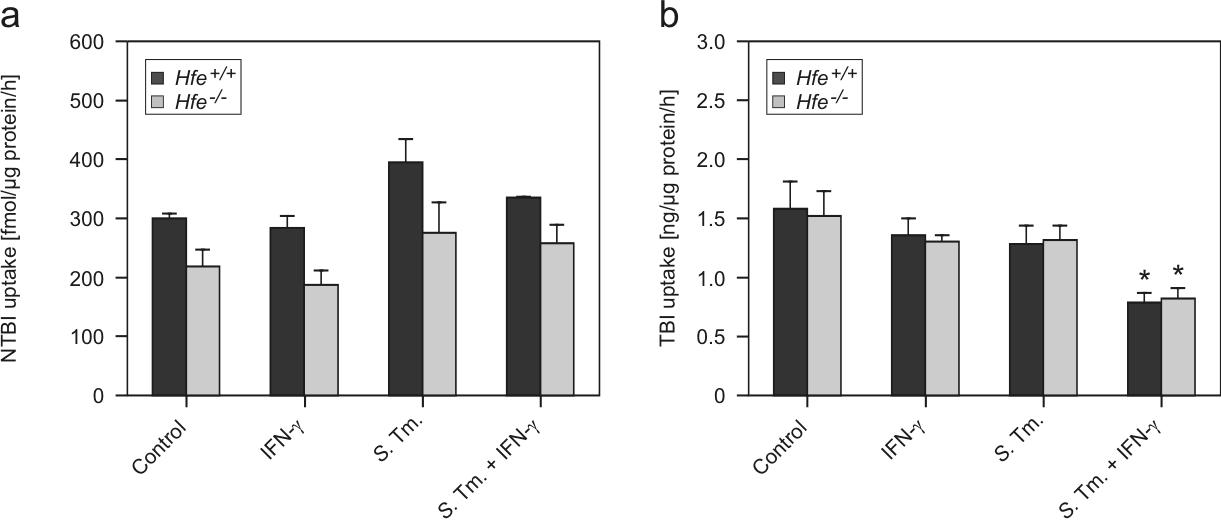
In contrast, with regard to iron uptake we found no significant difference in the acquisition of nontransferrin bound iron or transferrin-bound iron between Hfe+/+ and Hfe−/− macrophages (supplemental Figure 1A-B).
Effects of Hfe genotype and Lcn2 production on intramacrophage Salmonella survival
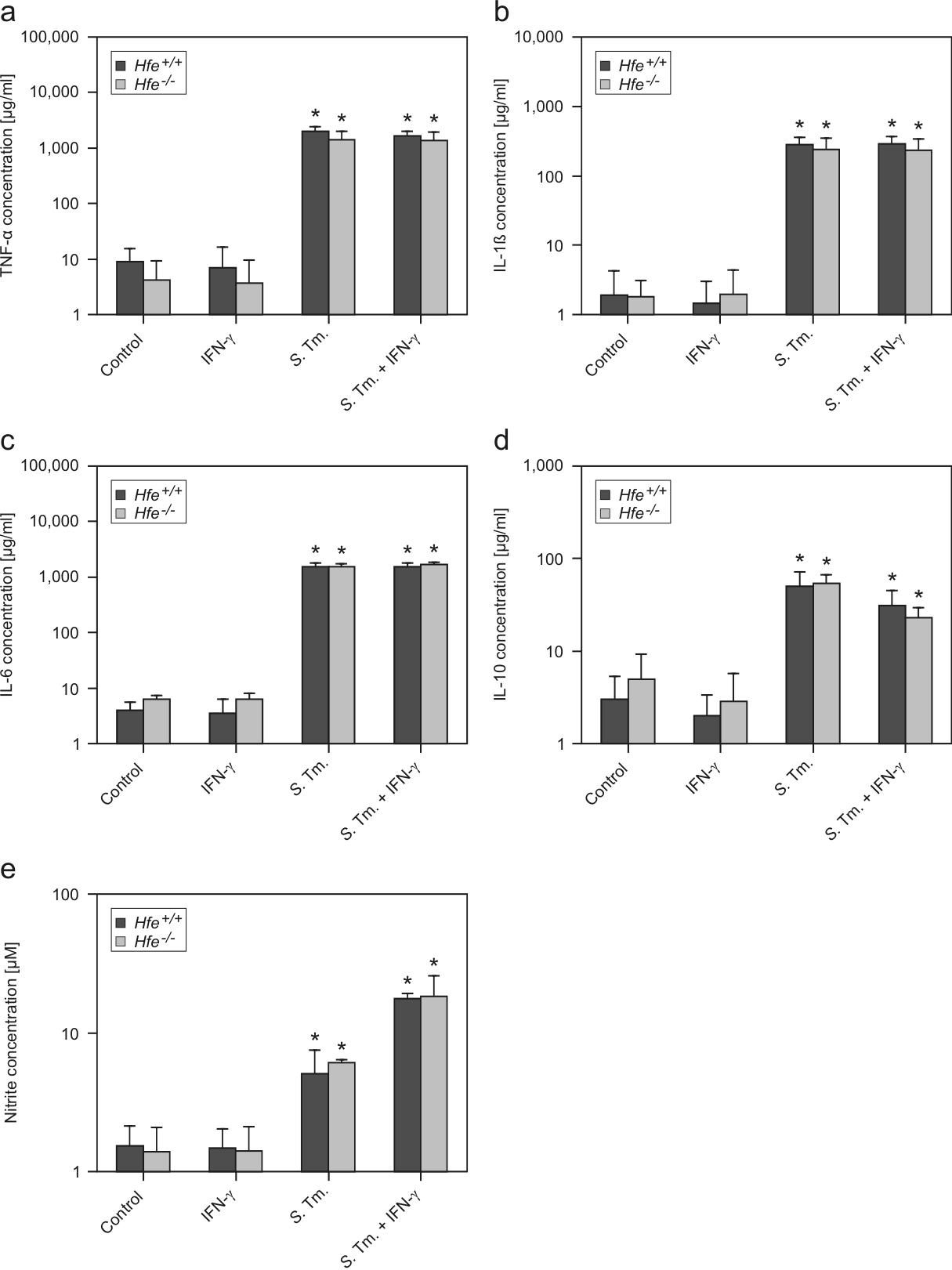
Because iron is essential for the proliferation of intracellular pathogens, we hypothesized that Hfe-deficient phagocytes might better control Salmonella infection. Therefore, we studied the intramacrophage survival of S Typhimurium within WT, Hfe+/−, and Hfe−/− phagocytes. We found that the bacterial burden in isolated primary macrophages was not affected by the Hfe genotype at early time points studied, namely 30 minutes, 1 hour, 2 hours, and 6 hours after infection, suggesting that Hfe may not affect phagocytosis and early elimination of Salmonella (data not shown; Table 1). In contrast, after an infection period of 16 and 24 hours, the microbial burden was significantly lower within Hfe+/− and Hfe−/− phagocytes compared with their WT counterparts (Table 1). When analyzing the expression of several immune genes by quantitative reverse transcription–polymerase chain reaction (RT-PCR), we found that Hfe+/+ and Hfe−/− macrophages expressed comparable mRNA levels of inducible nitric oxide synthase (iNOS) and phagocyte oxidase–p47 (Table 2). Furthermore, the expression levels of mRNA corresponding to inflammatory cytokines/chemokines TNF-α, IL-1β, IL-6, IL-12, IL-23, macrophage inflammatory protein 1α, and monocyte chemoattractant protein 1 in Salmonella-infected or IFN-γ–stimulated macrophages were not affected by their Hfe genotype (Table 2). Accordingly, the concentrations of TNF-α, IL-1β, IL-6, and IL-10 as well as nitrite were similar in culture supernatants of Hfe+/+ and Hfe−/− phagocytes (supplemental Figure 2).
Table 1.
Antimicrobial activity (in ×103 CFU) of WT and Hfe-deficient macrophages
| Bacterial load | Hfe+/+ | Hfe+/− | Hfe−/− |
|---|---|---|---|
| 2 h after infection | 370.67 ± 22.57 | 364.67 ± 40.20 | 367.33 ± 29.37 |
| 6 h after infection | 260.67 ± 15.69 | 245.33 ± 14.32 | 233.33 ± 13.66 |
| 16 h after infection | 86.83 ± 3.59 | 55.50 ± 4.64* | 41.75 ± 3.67† |
| 24 h after infection | 25.36 ± 1.16 | 11.94 ± 0.57‡ | 10.02 ± 0.56§ |
WT, Hfe+/−, and Hfe−/− macrophages were challenged with S Typhimurium WT strain at a MOI of 5, and their antimicrobial activity was evaluated by enumeration of surviving bacteria at indicated times. Data were log-transformed and compared by ANOVA and shown as mean ± SEM for 5 independent experiments.
WT indicates wild-type; MOI, multiplicity of infection; and ANOVA, analysis of variance.
P = .002 for the comparison of WT and Hfe+/− macrophages.
P < .001 for the comparison of WT and Hfe−/− macrophages.
P < .001 for the comparison of WT and Hfe+/− macrophages.
P < .001 for the comparison of WT and Hfe−/− macrophages.
Table 2.
Expression of macrophage immune response genes in WT and Hfe-deficient macrophages
| Hfe genotype/stimulation gene name | Hfe+/+ control | Hfe−/− control | Hfe+/+S Tm | Hfe−/−S Tm |
|---|---|---|---|---|
| iNOS | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.21 ± 0.09* | 0.19 ± 0.06* |
| phox-p47 | 0.68 ± 0.05 | 0.75 ± 0.08 | 1.16 ± 0.25 | 1.25 ± 0.16 |
| TNF-α | 0.03 ± 0.00 | 0.03 ± 0.00 | 2.83 ± 0.35* | 2.21 ± 0.37* |
| IL-1β | 0.01 ± 0.00 | 0.01 ± 0.00 | 175.18 ± 29.84* | 141.71 ± 45.16* |
| IL-6 | 0.31 ± 0.15 | 0.58 ± 0.03 | 296.38 ± 57.90* | 257.65 ± 61.94* |
| IL-10 | 0.24 ± 0.03 | 0.51 ± 0.03 | 3.44 ± 0.64* | 4.59 ± 0.45* |
| IL-12p35 | 0.01 ± 0.00 | 0.02 ± 0.00 | 142.70 ± 22.48* | 148.43 ± 5.82* |
| IL-12/23p40 | 0.15 ± 0.04 | 0.09 ± 0.02 | 377.64 ± 84.73* | 315.80 ± 27.64* |
| IL-18 | 0.39 ± 0.06 | 0.74 ± 0.07 | 3.70 ± 0.86* | 2.86 ± 0.44* |
| IL-23p19 | 0.01 ± 0.00 | 0.00 ± 0.00 | 0.90 ± 0.20* | 1.38 ± 0.05* |
| Mip-1α | 0.01 ± 0.00 | 0.01 ± 0.00 | 1.71 ± 0.27* | 1.13 ± 0.22* |
| Mcp-1 | 0.08 ± 0.02 | 0.20 ± 0.03 | 3.75 ± 0.46* | 4.14 ± 2.04* |
| TGF-β | 0.95 ± 0.22 | 0.71 ± 0.10 | 1.56 ± 0.40 | 1.67 ± 0.49 |
| MHC-II | 2.80 ± 0.34 | 2.16 ± 0.57 | 5.69 ± 0.75* | 4.62 ± 0.15* |
| Lcn2 | 1.9 ± 0.6 | 8.3 ± 1.0† | 4434.1 ± 1695.3* | 12 228.3 ± 1149.6*† |
Primary Hfe+/+ and Hfe−/− macrophages were treated with complete RPMI (control) or infected with WT Salmonella Typhimurium (S Tm) for 24 hours. Expressions of macrophage immune response genes were analyzed by quantitative RT-PCR. Specific values of target genes were normalized to those of Hprt and depicted as means ± SEM of arbitrary expression unit (AU). Values from at least 4 independent experiments were compared by Kruskal-Wallis analysis.
WT indicates wild-type; S Tm, Salmonella Typhimurium; iNOS, inducible nitric oxide synthase; TNF, tumor necrosis factor; IL, interleukin; Mip, macrophage inflammatory protein; Mcp, monocyte chemotactic protein; TGF, transforming growth factor; MHC, major histocompatibility complex; and Lcn, lipocalin.
P < .05 as compared with control cells of the corresponding genotype.
P < .05 for the comparison of Hfe+/+ and Hfe−/− phagocytes subjected to the same stimulation.
Notably, we observed increased Lcn2 protein secretion by Hfe−/− macrophages in response to infection compared with Hfe+/+ cells (Figure 2A), whereas mRNA levels of LcnR, Fpn1, and Hamp were not different between cells of these 2 genotypes (data not shown).
Figure 2.

Macrophage Lcn2 production and antibacterial activity. Macrophages were harvested from WT and Hfe−/− mice and treated with 100 U/mL IFN-γ or infected with WT S Typhimurium (S Tm) for 24 hours. The production of Lcn2 was investigated in culture supernatants by ELISA (A). Data were log-transformed and compared by ANOVA and are shown as mean ± SEM of at least 3 independent experiments. WT, Hfe+/−, and Hfe−/− mice were infected with WT Salmonella. The bacterial burden was determined by gentamicin protection assay (B). In parallel experiments, the acquisition of 59Fe by intramacrophage Salmonella was determined (C). In additional experiments, the survival of S Typhimurium within C57BL/6 WT, Hfe−/−, Lcn2−/−, and Hfe−/−Lcn2−/− macrophages (D) as well as bacterial iron uptake within these cells (E) were determined. These data were analyzed by ANOVA and shown as mean ± SEM of at least 3 independent experiments. *P < .05 compared with the solvent-treated control of the corresponding genotype, #P < .05 for the comparison of cells of different genotype subjected to the same stimulatory regimen.
To determine whether the high levels of Lcn2 produced by Hfe-deficient macrophages directly contribute to the clearance of S Typhimurium, we infected primary peritoneal macrophages with bacteria, added a neutralizing anti–mouse Lcn2 antibody or an isotype control antibody, and determined the intracellular bacterial burden 16 and 24 hours after infection The neutralization of Lcn2 abolished the differences in the bacterial load observed in macrophages of different Hfe genotypes at both time points (Figure 2B; data not shown). Notably, the secretion of macrophage-derived cytokines TNF-α, IL-1β, IL-6, IL-10, IL-12, and IL-23 and the production of ROS and RNS did not differ between macrophages of these Hfe genotypes and was unaffected by the antibody-mediated neutralization of Lcn2 (data not shown).
Importantly, the neutralization of Lcn2 promoted iron acquisition by internalized bacteria and nearly abolished the differences with respect to Salmonella iron acquisition within Hfe WT, Hfe+/−, and Hfe−/− macrophages (Figure 2C). To validate these results, we analyzed C57BL/6 Hfe+/+, Hfe−/−, Lcn2−/−, and Hfe−/−Lcn2−/− macrophages for their capacity to withhold iron from engulfed S Typhimurium and to kill the bacteria after phagocytosis. Although Hfe−/−Lcn2+/+ macrophages inhibited Salmonella replication significantly better than did WT cells, this effect was abrogated in Hfe−/−Lcn2−/− macrophages (Figure 2D). In parallel, the increased ability of Hfe−/−Lcn2+/+ phagocytes to withhold iron from internalized bacteria depended on the presence of Lcn2, because Salmonella iron acquisition was not different between bacteria residing within Hfe+/+Lcn2−/− and Hfe−/−Lcn2−/− cells (Figure 2E).
Course of S Typhimurium infection in Hfe WT, Hfe+/−, and Hfe−/− mice
To investigate the influence of Hfe on the course of invasive Salmonella infection in vivo, Hfe+/−, Hfe−/−, and congenic C57BL/6 WT mice were injected intraperitoneally with a S Typhimurium WT strain. Thirteen (93%) of 14 C57BL/6 control mice died by days 2 to 7 of infection. In comparison, 27% of Hfe+/− and 33% of Hfe−/− mice remained alive at the end of the observation period (Figure 3A). Bacterial loads in livers (Figure 3B) and spleens (Figure 3C) of randomly selected Hfe+/− and Hfe−/− mice were significantly lower compared with their WT littermates, whereas no statistically significant difference in bacterial load was found between Hfe+/− and Hfe−/− animals. Thus, deficiency in 1 or 2 Hfe copies was associated with protection against acute S Typhimurium infection in vivo.
Figure 3.

Effect of Hfe functionality on Salmonella infection in vivo. C57BL/6 WT, Hfe+/−, and Hfe−/− mice were infected intraperitoneally with 500 CFU of WT S Typhimurium. Survival was monitored during an observation period of 10 days and compared between subgroups using the Wilcoxon (Gehan) test (A). P = .007 for Hfe+/+ versus Hfe+/−, P = .049 for Hfe+/+ versus Hfe−/−. Bacterial loads in at least 6 animals per group were determined in livers (B) and spleens (C) of randomly selected animals at days 2 and 4 after infection. Data were log-transformed and compared by ANOVA with Bonferroni correction. Values are depicted as lower quartile, median, and upper quartile (boxes), and minimum/maximum ranges and statistical significance are indicated.
Iron homeostasis, immune response, and peripheral blood count in Hfe WT and Hfe−/− mice in response to Salmonella infection
On examination of tissue expression of selected immune and iron metabolism genes by quantitative RT-PCR, we found that iNOS and phagocyte oxidase–gp47 mRNA expression were not different between spleens of Hfe−/− and C57BL/6 control littermates (supplemental Table 1). However, mRNA levels of Lcn2 were significantly higher in both livers and spleens of Hfe−/− animals compared with their WT counterparts (Figures 4A-B). Similarly, Hfe+/− mice displayed higher Lcn2 transcript expression in both organs compared with C57BL/6 mice (data not shown). In contrast, mRNA levels of Fpn1 and Hamp in both livers and spleens were not significantly different between infected WT and Hfe-deficient animals, although solvent-treated Hfe−/− mice tended to have the lowest hepatic Hamp mRNA expression (details not shown).
Figure 4.

Lcn2 expression in response to Salmonella infection. The mRNA expressions of Lcn2 in livers (A) and spleens (B) of solvent-treated and Salmonella-infected Hfe+/+ and Hfe−/− mice were studied by quantitative RT-PCR. Data from at least 6 mice per group were log-transformed and compared by ANOVA test with Bonferroni correction. Values are depicted as lower quartile, median, and upper quartile (boxes), and minimum/maximum ranges and statistical significances are indicated. Protein levels of Fpn1, Lcn2 iNOS, Hmox1, and actin in livers (C) and spleens (D) of Hfe+/+, Hfe+/−, and Hfe−/− mice were determined by immunoblot analysis. Apparent molecular weights are indicated on the right-hand side of the respective immunoblots. One of at least 3 representative blots is shown. Lcn2 concentrations in serum samples of Hfe+/+, Hfe+/−, and Hfe−/− mice were measured by a specific ELISA (E). The effect of endogenous Lcn2 in acute-phase sera on bacterial growth was measured as optical density of mid-logarithmic liquid cultures (F).
Moreover, Lcn2 protein expression was higher in the livers and spleens of Hfe+/− and Hfe−/− mice compared with WT animals, both under control conditions and after Salmonella infection (Figure 4C-D). In contrast, although Hfe-deficient control mice displayed higher Fpn1 protein levels than did WT controls, no substantial difference in Fpn1 protein expression was found between Salmonella-infected WT and Hfe-deficient animals. Furthermore, the Hfe genotype did not influence iNOS or Hmox1 protein expression in the spleens of either control or infected mice (Figure 4D).
When studying the expression of inflammatory genes in the spleen, we found that mRNA levels of the T cell–derived cytokines IFN-γ, IL-4, IL-13, IL-17A, IL-17F, and TGF-β and of the master switch transcription factors T-bet, GATA-3, RORγt, and Foxp3 were not influenced by the Hfe genotype in solvent-treated or infected animals (supplemental Table 1; data not shown). Moreover, we detected no differences in TNF-α, IL-1β, IL-6, IL-10, IL-12p35, IL-12/23p40, IL-18, and IL23p19 mRNA levels compared with WT and Hfe−/− mice. Correspondingly, serum levels of TNF-α, IL-6, IL-10, IL-12, IL-23, IFN-γ, and nitrite were not different between Hfe+/+ and Hfe−/− animals, whereas IL-1β remained undetectable in these serum samples (supplemental Figure 3; data not shown). In contrast, Lcn2 levels in the sera of Hfe+/− and Hfe−/− mice were significantly higher compared with their WT littermates (Figure 4E).
To determine whether the higher levels of Lcn2 found in the sera of Hfe+/− and Hfe−/− animals contribute to the improved control of Salmonella replication observed in these mice, acute-phase sera were obtained and analyzed for their antibacterial activity in liquid bacterial cultures. Of note, acute-phase sera of mice lacking one or both Hfe alleles limited the growth of WT Salmonella significantly better than did the sera from WT animals (Figure 4F). These differences were abrogated after immunoprecipitation with a neutralizing α-Lcn2 antibody or on the addition of 100 μM ferrous sulfate, implicating an underlying Lcn2-mediated and iron-dependent mechanism.
To rule out an influence of Hfe dysfunction on resistance to S Typhimurium by an effect on hematopoiesis, we analyzed blood samples of solvent-treated and infected WT C57BL/6 and Hfe−/− mice. Blood cell numbers and hemoglobin concentrations were not significantly influenced by the Hfe genotype (supplemental Table 2; data not shown).
Influence of bacterial enterochelin production on the course of Salmonella infection in Hfe WT and Hfe−/− mice and macrophages
Because Lcn2 is known to sequester iron-siderophore complexes, we next investigated the contribution of Salmonella iron acquisition systems to the phenotype of improved bacterial control observed in Hfe−/− macrophages. We found that the protective effect conferred by the absence of Hfe was abrogated on infection with a Salmonella entC mutant, which is unable to produce the bacterial siderophore enterochelin, as well as with an entC sit feo triple mutant. In contrast, the antibacterial effects of Hfe−/− macrophages against WT Salmonella were also observed against ssrA, SPI-I, and iroBC mutant Salmonella, the latter able to produce enterochelin but unable to produce and use salmochelins (Figure 5A). The improved bacterial control exerted by Hfe−/− macrophages thus mimicked the effects of recombinant murine Lcn2 added to liquid Salmonella cultures (supplemental Figure 4).
Figure 5.

Infection with mutant Salmonella strains and infection of Hfe−/−Lcn2−/− double KO mice. Hfe+/+ and Hfe−/− macrophages were infected with WT S Typhimurium and various mutants, and bacterial numbers were enumerated by plating of cell lysates (A). Hfe+/+ and Hfe−/− mice were intraperitoneally infected with 500 CFU entC mutant S Typhimurium. Hfe+/+ mice infected with the identical number of WT S Typhimurium served as a comparison. Survival of subgroups was monitored during an observation period of 10 days and compared using the Wilcoxon (Gehan) test (B). P = .001 for Hfe+/+ WT Salmonella versus Hfe+/+ entC mutant Salmonella, P = .951 for Hfe+/+ entC mutant Salmonella versus Hfe−/− entC mutant Salmonella. C57BL/6 WT, Hfe−/−, Lcn2−/−, and Hfe−/−Lcn2−/− mice were infected with 500 CFU of WT S Typhimurium. At day 2 of infection, animals were killed, and the bacterial load in livers (C) and spleens (D) enumerated by plating appropriated dilutions of organ homogenates. Data are presented as detailed in the legend to Figure 3. Primary peritoneal macrophages of the indicated genotypes were infected with S Typhimurium (S Tm) or left untreated (Ctrl.). After 24 hours, the expression of Fpn1, Lcn2, and Actin was determined by Western blot as described in the “Immunoblotting” section of supplemental Methods. One of 3 representative experiments is shown.
To establish whether bacterial enterochelin production is also linked to the protective effects of Hfe deficiency in vivo, we infected Hfe−/− mice and their WT littermates intraperitoneally with 500 CFU of the entC mutant S Typhimurium strain. Remarkably, 46% of WT animals and 45% of Hfe−/− mice infected with entC-deficient Salmonella survived beyond day 10 of infection. In comparison, the infection of WT mice with WT S Typhimurium caused the rapid death of 11 of 12 animals (∼ 92%) by days 1 to 6 of infection (Figure 5B). Thus, the protective effects of Hfe−/− deficiency may be partially mediated by a mechanism involving impaired bacterial enterochelin utilization, because no difference in the course of Salmonella infection was observed when enterochelin-deficient bacteria were inoculated.
To determine whether the increased Lcn2 production in Hfe−/− mice is directly linked to their increased antibacterial resistance, C57BL/6 Hfe+/+, Hfe−/−, Lcn2−/−, and Hfe−/−Lcn2−/− double KO mice were infected with WT Salmonella. After 2 days, bacterial loads in livers and spleens were significantly lower in Hfe−/− mice compared with Hfe+/+ littermates (Figure 5C-D). Of interest, Hfe+/+Lcn2−/− and Hfe−/−Lcn2−/− animals displayed similar bacterial loads, which were significantly higher compared with Hfe−/−Lcn2+/+ mice, suggesting that Lcn2 is the main mediator of increased resistance to systemic Salmonella infection in Hfe-deficient mice.
Finally, to investigate the possibility that the decreased iron withholding capacity of Lcn2-deficient phagocytes may be related to altered Fpn1 expression, primary peritoneal Hfe+/+, Hfe−/−, Lcn2−/−, and Hfe−/−Lcn2−/− double KO macrophages were prepared and treated with medium or infected with WT Salmonella. Lack of functional Hfe caused an increase in Lcn2 protein expression in infected cells. In contrast, in isolated macrophages, Fpn1 protein levels were increased on Salmonella infection but unaffected by Hfe or Lcn2 genotype (Figure 5E).
Discussion
This study shows that the absence of functional Hfe protects mice against invasive infections with the intracellular pathogen S Typhimurium. This effect can be attributed to the restriction of intracellular iron availability for bacteria, achieved by iron export from infected macrophages and scavenging of the bacterial catecholate siderophore enterochelin.
At least 2 pathways could be responsible for increased iron export from Hfe−/− macrophages; first, mononuclear cells express Fpn1, which pumps ferrous iron out of the cell. Infection with intracellular pathogens results in increased expression of this protein, thus limiting intracellular iron availability.22,37 This pathway has recently been implicated in the resistance of Hfe−/− mice to oral infection with Salmonella in a model of bacterial enterocolitis.38 In our study, Fpn1 mRNA expression did not differ between Hfe+/+ and Hfe−/− peritoneal macrophages and was not differentially modulated on infection with Salmonella. Rather, there was a uniform induction of Fpn1 protein expression in Salmonella-infected primary macrophages compared with control cells, which appeared to be independent of Hfe and/or Lcn2 functionality. It is well established that in addition to cytokine- and inflammation-mediated regulation of Fpn1 mRNA expression,33,37,39 a major regulatory pathway for Fpn1 expression is posttranslational binding of Hamp to Fpn1, which results in Fpn1 internalization and degradation.16,17 Moreover, although splenic expression of Fpn1 protein was higher in uninfected Hfe-deficient mice than in Hfe+/+ littermates, such differences were not observed after infecting Hfe+/+, Hfe+/−, and Hfe−/− mice with Salmonella. This indicates that the differences in iron export observed between infected WT and Hfe-deficient mice are not primarily due to differential expression of Fpn1. Consequently, a second mechanism for cellular iron release and the limitation of iron for intracellular microbes must exist, which according to our studies is due to enhanced expression of Lcn2. Of note, cellular iron release under control conditions was reduced by genetic ablation of Lcn2, because Hfe−/−Lcn2−/− macrophages exported significantly less iron than did Hfe−/−Lcn2+/+ cells (data not shown). Moreover, the abrogation of enhanced iron export as well as resistance to Salmonella infection in Hfe−/−Lcn2−/− macrophages in comparison to Hfe−/−Lcn2+/+ cells suggests that Lcn2 may shuttle siderophore-bound iron out of macrophages and may account for the observed differences between WT and Hfe-deficient phagocytes. Because Lcn2 is unable to bind iron directly, Lcn2-mediated iron export may involve a siderophore. Our data are thus compatible with a model in which a bacterial siderophore such as enterochelin or an as yet not characterized mammalian siderophore31,40 contributes to macrophage iron export. Nevertheless, the existence of mammalian siderophores remains rather speculative.31,40
We found no differences in inflammatory radicals or cytokine production between Salmonella-infected Hfe+/+ and Hfe−/− macrophages which contrasts with a recent report showing reduced proinflammatory cytokine secretion by macrophages from Hfe−/− mice macrophages compared with macrophages obtained from C57BL/6 mice not bred in parallel. This effect was suggested to result from iron-controlled translation of TNF-α and IL-6 mRNAs,38 which disagrees with other observations showing that iron chelation enhances, and iron loading reduces, the formation of TNF-α and other proinflammatory cytokines by macrophages.19,41–43 Moreover, in our mice bred under identical conditions on a completely congenic background, we did not detect Hfe-dependent differences in macrophage Fpn1 levels or cytokine secretion during experimental Salmonella infection. Although the addition of synthetic Hamp to macrophages in vitro resulted in a 24% to 41% decrease of iron release from control and Salmonella-infected macrophages (data not shown), this effect was independent of the Hfe genotype and failed to reduce the described differences in macrophage iron export between WT and Hfe-deficient cells.
Of interest, Hfe-deficient mice displayed reduced bacterial burdens in both liver and spleen, although it is well established that Hfe-deficient hepatocytes accumulate iron,8,44 a fact that is supposed to promote rather than impair bacterial growth and proliferation. We suggest that Kupffer cells, which are thought to be iron deficient in persons with hemochromatosis,12 may represent the primary habitat of Salmonella in the liver and account for the low bacterial load within this organ.45 Although Lcn2 does not appear to account for hepatocellular iron accumulation in Hfe-deficient mice,46 high levels of Lcn2 present within the extracellular space of the liver may contribute to improved intrahepatic bacterial control in Hfe-deficient animals. Alternatively, circulating Lcn2 may impair the spread of S Typhimurium from the primary site of infection to systemic sites, including the liver. This is supported by our data showing that the antibody-mediated removal of Lcn2 from acute-phase sera abolished the differences observed between sera from WT and Hfe-deficient mice.
HFE-associated hereditary hemochromatosis is a prevalent autosomal recessive disorder, and the HFE mutations C282Y and H63D occur with surprisingly high allelic frequencies in populations of Northern and Western European origin.1,3,4,47 It has thus been suggested that HFE defects may be of selective evolutionary advantage. Two major hypotheses have emerged as possible explanations; on one hand, HFE deficiency may enable more effective absorption of dietary iron. This may prevent severe iron deficiency when dietary iron content is low or iron demand is high, for instance during pregnancy or infancy.48 Alternatively, HFE mutations may confer increased resistance against intramacrophage pathogens that have an essential requirement for iron. Our data provide evidence for a protective role of Hfe mutations in a mouse model of typhoid fever. The association of both homozygous and heterozygous Hfe defects with enhanced resistance to S Typhimurium infection implies that mutated Hfe may fulfill the function of a dominant protective allele (P = .001 for 37 Hfe+/+ vs 11 Hfe+/− mice, P = .004 for 37 Hfe+/+ vs 21 Hfe−/− mice, P = .582 for 11 Hfe+/− vs 21 Hfe−/− mice; data combined from all observations on survival using WT S Typhimurium). Although the results of these investigations cannot be directly extrapolated to humans, it is plausible that HFE-related hemochromatosis may be of selective advantage in infections with microbes capable of survival within macrophages.49
It is well established that the growth of S Typhimurium within infected phagocytes is influenced by natural resistance-associated macrophage protein 1 (Nramp1), which acts as a transporter of iron and other divalent metal ions at the phagolysosomal membrane.50 The topology of the Nramp1 protein relative to the phagolysosomal lumen and its exact transport function are still under debate; however, studies on isolated phagosomes have suggested that Nramp1 shifts iron into the phagolysosome to promote the formation of ROS by Fenton chemistry.51 Other data, in contrast, rather support the idea that Nramp1 transports iron out of these pathogen-containing organelles to deprive pathogens from the essential nutrient iron.52,53 Nevertheless, our experiments were performed in mice on a pure C57BL/6 and thus Nramp1-susceptible genetic background (supplemental Methods), ruling out any possible influence of Nramp1 genotype in our model.
Lcn2, whose production is augmented in Hfe-deficient macrophages, has the strongest affinity for enterochelin, a catecholate-type siderophore primarily synthesized by S Typhimurium and Escherichia coli. Because all known enterobacteria are able to use enterochelin for iron uptake,54 it is probable that the increased resistance conferred by Hfe deficiency may be extended to other pathogens that are sensitive to alterations in intracellular iron availability such as Chlamydia spp or Legionella pneumophila.55 In any case, monocytes isolated from otherwise healthy subjects with HH type I have been shown to withhold iron from engulfed Mycobacterium tuberculosis more efficiently than cells from healthy volunteers.56
In summary, the present study highlights the central role of macrophage iron homeostasis for the fate of infections with intracellular microbes. We demonstrate that the absence of functional Hfe is protective against invasive infections with the intracellular pathogen S Typhimurium as a result of enhanced iron limitation mediated by the increased formation of Lcn2.
Supplementary Material
Acknowledgments
We thank Drs Klaus Schümann and Siamak Bahram for providing Hfe−/− mice and Drs Alan Aderem, Niels Borregaard, and Jack B. Cowland for providing Lcn2−/− mice. We thank Dr Nikos Yannoutsos for performing embryonic transfers, Mrs Sabine Engl for outstanding technical support, and Dr Katharina Kurz for helpful advice in statistical analysis.
This work was supported by the Verein zur Förderung von Forschung und Weiterbildung in molekularer Immunologie und Infektiologie an der Medizinischen Universität Innsbruck and by grants from the Austrian Research Fund FWF, Project P-19664, the European Community, Project Euroiron1 (G.W.), and the National Institutes of Health (AI54052 and AI39557; M.-L.V.C. and F.C.F.).
Footnotes
The online version of this article contains a data supplement.
Presented as oral presentation at the 46th Interscience Conference for Antimicrobial Agents and Chemotherapy (ICAAC), Washington, DC, October 24-27, 2008, and at the European Congress for Clinical Microbiology and Infectious Diseases (ECCMID), Helsinki, Finland, May 16-19. 2009.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.N. designed and performed cell culture and animal experiments, analyzed data, generated figures, and wrote the manuscript; I.T. interpreted data and assisted in preparing the manuscript; A.S. performed animal experiments and FACS analyses; M.T., G.F., E.L., and M.S. performed experiments; M.-L.V.C. and K.H. constructed Salmonella mutants and edited the manuscript; S.A. provided Lcn2−/− mice; F.C.F. provided Salmonella mutants, interpreted data, and cowrote the manuscript; and G.W. guided the overall concept of the studies reported, analyzed and interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Günter Weiss, Medical University of Innsbruck, Department of Internal Medicine I, Clinical Immunology and Infectious Diseases, Anichstr 35; A-6020 Innsbruck, Austria; e-mail: guenter.weiss@i-med.ac.at.
References
- 1.Pietrangelo A. Hereditary hemochromatosis–a new look at an old disease. N Engl J Med. 2004;350(23):2383–2397. doi: 10.1056/NEJMra031573. [DOI] [PubMed] [Google Scholar]
- 2.Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13(4):399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 3.Anderson GJ, Powell LW. HFE and non-HFE hemochromatosis. Int J Hematol. 2002;76(3):203–207. doi: 10.1007/BF02982788. [DOI] [PubMed] [Google Scholar]
- 4.Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710–3717. doi: 10.1182/blood-2005-05-1857. [DOI] [PubMed] [Google Scholar]
- 5.Drakesmith H, Sweetland E, Schimanski L, et al. The hemochromatosis protein HFE inhibits iron export from macrophages. Proc Natl Acad Sci U S A. 2002;99(24):15602–15607. doi: 10.1073/pnas.242614699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bridle KR, Frazer DM, Wilkins SJ, et al. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet. 2003;361(9358):669–673. doi: 10.1016/S0140-6736(03)12602-5. [DOI] [PubMed] [Google Scholar]
- 7.Ahmad KA, Ahmann JR, Migas MC, et al. Decreased liver hepcidin expression in the Hfe knockout mouse. Blood Cells Mol Dis. 2002;29(3):361–366. doi: 10.1006/bcmd.2002.0575. [DOI] [PubMed] [Google Scholar]
- 8.Vujić Spasić M, Kiss J, Herrmann T, et al. Hfe acts in hepatocytes to prevent hemochromatosis. Cell Metab. 2008;7(2):173–178. doi: 10.1016/j.cmet.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 9.Nicolas G, Viatte L, Lou DQ, et al. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat Genet. 2003;34(1):97–101. doi: 10.1038/ng1150. [DOI] [PubMed] [Google Scholar]
- 10.Piperno A, Girelli D, Nemeth E, et al. Blunted hepcidin response to oral iron challenge in HFE-related hemochromatosis. Blood. 2007;110(12):4096–4100. doi: 10.1182/blood-2007-06-096503. [DOI] [PubMed] [Google Scholar]
- 11.Gao J, Chen J, Kramer M, Tsukamoto H, Zhang AS, Enns CA. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 2009;9(3):217–227. doi: 10.1016/j.cmet.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cairo G, Recalcati S, Montosi G, Castrusini E, Conte D, Pietrangelo A. Inappropriately high iron regulatory protein activity in monocytes of patients with genetic hemochromatosis. Blood. 1997;89(7):2546–2553. [PubMed] [Google Scholar]
- 13.Moura E, Noordermeer MA, Verhoeven N, Verheul AF, Marx JJ. Iron release from human monocytes after erythrophagocytosis in vitro: an investigation in normal subjects and hereditary hemochromatosis patients. Blood. 1998;92(7):2511–2519. [PubMed] [Google Scholar]
- 14.Knutson MD, Oukka M, Koss LM, Aydemir F, Wessling-Resnick M. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc Natl Acad Sci U S A. 2005;102(3):1324–1328. doi: 10.1073/pnas.0409409102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117(3):285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 16.De Domenico I, Ward DM, Langelier C, et al. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol Biol Cell. 2007;18(7):2569–2578. doi: 10.1091/mbc.E07-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5709):2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 18.Oexle H, Kaser A, Most J, et al. Pathways for the regulation of interferon-gamma-inducible genes by iron in human monocytic cells. J Leukoc Biol. 2003;74(2):287–294. doi: 10.1189/jlb.0802420. [DOI] [PubMed] [Google Scholar]
- 19.Recalcati S, Taramelli D, Conte D, Cairo G. Nitric oxide-mediated induction of ferritin synthesis in J774 macrophages by inflammatory cytokines: role of selective iron regulatory protein-2 downregulation. Blood. 1998;91(3):1059–1066. [PubMed] [Google Scholar]
- 20.Weiss G, Werner-Felmayer G, Werner ER, Grunewald K, Wachter H, Hentze MW. Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J Exp Med. 1994;180(3):969–976. doi: 10.1084/jem.180.3.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collins HL. The role of iron in infections with intracellular bacteria. Immunol Lett. 2003;85(2):193–195. doi: 10.1016/s0165-2478(02)00229-8. [DOI] [PubMed] [Google Scholar]
- 22.Nairz M, Theurl I, Ludwiczek S, et al. The co-ordinated regulation of iron homeostasis in murine macrophages limits the availability of iron for intracellular Salmonella typhimurium. Cell Microbiol. 2007;9(9):2126–2140. doi: 10.1111/j.1462-5822.2007.00942.x. [DOI] [PubMed] [Google Scholar]
- 23.Schaible UE, Kaufmann SH. Iron and microbial infection. Nat Rev Microbiol. 2004;2(12):946–953. doi: 10.1038/nrmicro1046. [DOI] [PubMed] [Google Scholar]
- 24.Vazquez-Torres A, Jones-Carson J, Baumler AJ, et al. Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature. 1999;401(6755):804–808. doi: 10.1038/44593. [DOI] [PubMed] [Google Scholar]
- 25.Hantke K, Nicholson G, Rabsch W, Winkelmann G. Salmochelins, siderophores of Salmonella enterica and uropathogenic Escherichia coli strains, are recognized by the outer membrane receptor IroN. Proc Natl Acad Sci U S A. 2003;100(7):3677–3682. doi: 10.1073/pnas.0737682100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rabsch W, Methner U, Voigt W, Tschape H, Reissbrodt R, Williams PH. Role of receptor proteins for enterobactin and 2,3-dihydroxybenzoylserine in virulence of Salmonella enterica. Infect Immun. 2003;71(12):6953–6961. doi: 10.1128/IAI.71.12.6953-6961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Janakiraman A, Slauch JM. The putative iron transport system SitABCD encoded on SPI1 is required for full virulence of Salmonella typhimurium. Mol Microbiol. 2000;35(5):1146–1155. doi: 10.1046/j.1365-2958.2000.01783.x. [DOI] [PubMed] [Google Scholar]
- 28.Crouch ML, Castor M, Karlinsey JE, Kalhorn T, Fang FC. Biosynthesis and IroC-dependent export of the siderophore salmochelin are essential for virulence of Salmonella enterica Serovar Typhimurium. Mol Microbiol. 2008;67(5):971–983. doi: 10.1111/j.1365-2958.2007.06089.x. [DOI] [PubMed] [Google Scholar]
- 29.Flo TH, Smith KD, Sato S, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432(7019):917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 30.Berger T, Togawa A, Duncan GS, et al. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2006;103(6):1834–1839. doi: 10.1073/pnas.0510847103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell. 2005;123(7):1293–1305. doi: 10.1016/j.cell.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 32.Nairz M, Fritsche G, Brunner P, Talasz H, Hantke K, Weiss G. Interferon-gamma limits the availability of iron for intramacrophage Salmonella typhimurium. Eur J Immunol. 2008;38(7):1923–1936. doi: 10.1002/eji.200738056. [DOI] [PubMed] [Google Scholar]
- 33.Ludwiczek S, Aigner E, Theurl I, Weiss G. Cytokine-mediated regulation of iron transport in human monocytic cells. Blood. 2003;101(10):4148–4154. doi: 10.1182/blood-2002-08-2459. [DOI] [PubMed] [Google Scholar]
- 34.Olakanmi O, Schlesinger LS, Ahmed A, Britigan BE. Intraphagosomal Mycobacterium tuberculosis acquires iron from both extracellular transferrin and intracellular iron pools. Impact of interferon-gamma and hemochromatosis. J Biol Chem. 2002;277(51):49727–49734. doi: 10.1074/jbc.M209768200. [DOI] [PubMed] [Google Scholar]
- 35.Theurl I, Theurl M, Seifert M, et al. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood. 2008;111(4):2392–2399. doi: 10.1182/blood-2007-05-090019. [DOI] [PubMed] [Google Scholar]
- 36.Bahram S, Gilfillan S, Kuhn LC, et al. Experimental hemochromatosis due to MHC class I HFE deficiency: immune status and iron metabolism. Proc Natl Acad Sci U S A. 1999;96(23):13312–13317. doi: 10.1073/pnas.96.23.13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Zandt KE, Sow FB, Florence WC, et al. The iron export protein ferroportin 1 is differentially expressed in mouse macrophage populations and is present in the mycobacterial-containing phagosome. J Leukoc Biol. 2008;84(3):689–700. doi: 10.1189/jlb.1107781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L, Johnson EE, Shi HN, Walker WA, Wessling-Resnick M, Cherayil BJ. Attenuated inflammatory responses in hemochromatosis reveal a role for iron in the regulation of macrophage cytokine translation. J Immunol. 2008;181(42):2723–2731. doi: 10.4049/jimmunol.181.4.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang F, Liu XB, Quinones M, Melby PC, Ghio A, Haile DJ. Regulation of reticuloendothelial iron transporter MTP1 (Slc11a3) by inflammation. J Biol Chem. 2002;277(42):39786–39791. doi: 10.1074/jbc.M201485200. [DOI] [PubMed] [Google Scholar]
- 40.Fernandez-Pol JA. Isolation and characterization of a siderophore-like growth factor from mutants of SV40-transformed cells adapted to picolinic acid. Cell. 1978;14(3):489–499. doi: 10.1016/0092-8674(78)90235-0. [DOI] [PubMed] [Google Scholar]
- 41.Ibrahim AS, Gebermariam T, Fu Y, et al. The iron chelator deferasirox protects mice from mucormycosis through iron starvation. J Clin Invest. 2007;117(9):2649–2657. doi: 10.1172/JCI32338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mencacci A, Cenci E, Boelaert JR, et al. Iron overload alters innate and T helper cell responses to Candida albicans in mice. J Infect Dis. 1997;175(6):1467–1476. doi: 10.1086/516481. [DOI] [PubMed] [Google Scholar]
- 43.Weiss G, Fuchs D, Hausen A, et al. Iron modulates interferon-gamma effects in the human myelomonocytic cell line THP-1. Exp Hematol. 1992;20(5):605–610. [PubMed] [Google Scholar]
- 44.Andrews NC. The iron transporter DMT1. Int J Biochem Cell Biol. 1999;31(10):991–994. doi: 10.1016/s1357-2725(99)00065-5. [DOI] [PubMed] [Google Scholar]
- 45.Vazquez-Torres A, Vallance BA, Bergman MA, et al. Toll-like receptor 4 dependence of innate and adaptive immunity to Salmonella: importance of the Kupffer cell network. J Immunol. 2004;172(10):6202–6208. doi: 10.4049/jimmunol.172.10.6202. [DOI] [PubMed] [Google Scholar]
- 46.Huang H, Akira S, Santos MM. Is the iron donor lipocalin 2 implicated in the pathophysiology of hereditary hemochromatosis? Hepatology. 2009;49(3):1012–1016. doi: 10.1002/hep.22699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andrews NC, Schmidt PJ. Iron homeostasis. Annu Rev Physiol. 2007;69:69–85. doi: 10.1146/annurev.physiol.69.031905.164337. [DOI] [PubMed] [Google Scholar]
- 48.Bulaj ZJ, Griffen LM, Jorde LB, Edwards CQ, Kushner JP. Clinical and biochemical abnormalities in people heterozygous for hemochromatosis. N Engl J Med. 1996;335(24):1799–1805. doi: 10.1056/NEJM199612123352403. [DOI] [PubMed] [Google Scholar]
- 49.Weinberg ED. Survival advantage of the hemochromatosis C282Y mutation. Perspect Biol Med. 2008;51(1):98–102. doi: 10.1353/pbm.2008.0001. [DOI] [PubMed] [Google Scholar]
- 50.Gruenheid S, Pinner E, Desjardins M, Gros P. Natural resistance to infection with intracellular pathogens: the Nramp1 protein is recruited to the membrane of the phagosome. J Exp Med. 1997;185(4):717–730. doi: 10.1084/jem.185.4.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuhn DE, Lafuse WP, Zwilling BS. Iron transport into mycobacterium avium-containing phagosomes from an Nramp1(Gly169)-transfected RAW264.7 macrophage cell line. J Leukoc Biol. 2001;69(1):43–49. [PubMed] [Google Scholar]
- 52.Jabado N, Jankowski A, Dougaparsad S, Picard V, Grinstein S, Gros P. Natural resistance to intracellular infections: natural resistance-associated macrophage protein 1 (Nramp1) functions as a pH-dependent manganese transporter at the phagosomal membrane. J Exp Med. 2000;192(9):1237–1248. doi: 10.1084/jem.192.9.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nairz M, Fritsche G, Crouch ML, Barton HC, Fang FC, Weiss G. Slc11a1 limits intracellular growth of Salmonella enterica sv. Typhimurium by promoting macrophage immune effector functions and impairing bacterial iron acquisition. Cell Microbiol. 2009;11(9):1365–1381. doi: 10.1111/j.1462-5822.2009.01337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Faraldo-Gómez JD, Sansom MS. Acquisition of siderophores in gram-negative bacteria. Nat Rev Mol Cell Biol. 2003;4(2):105–116. doi: 10.1038/nrm1015. [DOI] [PubMed] [Google Scholar]
- 55.Paradkar PN, De Domenico I, Durchfort N, Zohn I, Kaplan J, Ward DM. Iron depletion limits intracellular bacterial growth in macrophages. Blood. 2008;112(3):866–874. doi: 10.1182/blood-2007-12-126854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olakanmi O, Schlesinger LS, Britigan BE. Hereditary hemochromatosis results in decreased iron acquisition and growth by Mycobacterium tuberculosis within human macrophages. J Leukoc Biol. 2007;81(1):195–204. doi: 10.1189/jlb.0606405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}