

Abstract
Helicobacter pylori infection causes severe dysplasia manifested as gastrointestinal intraepithelial neoplasia (GIN) after 28 weeks post H. pylori infection (WPI) in cancer-prone, hypergastrinemic male INS-GAS mice. We examined the efficacy of the NSAID sulindac (400 ppm in drinking water) alone, the CCK2/gastrin receptor antagonist YM022 (45 mg/kg/week) alone, and sulindac or YM022 combined with H. pylori eradication therapy to prevent H. pylori-associated gastric cancer in male INS-GAS mice. Treatments started at 22 WPI, and mice were euthanized at 28 WPI. In uninfected mice, all treatments significantly delayed development of spontaneous GIN (p<0.05). In H. pylori-infected mice, sulindac alone or YM022 alone had no protective effect on H. pylori-associated GIN. Importantly, sulindac exacerbated the severity of H. pylori-associated gastritis despite decreased gastric PEG2 levels. However, sulindac combined with H. pylori antimicrobial eradication reduced the incidence of GIN (p<0.05), whereas YM022 combined with antimicrobial eradication did not reduce GIN. In infected mice, sulindac or YM022 treatment did not alter gastric expression of the proinflammatory cytokines Ifn-γ and Tnf-α and mucosal cell proliferation. Sulindac or YM022 combined with antimicrobial eradication down-regulated mRNA levels of Ifnγ and Tnfα, and mucosal cell proliferation (p<0.05). We conclude that sulindac enhances H. pylori gastritis and may promote inflammation-mediated gastric carcinogenesis. The combination of sulindac and antimicrobial H. pylori eradication was beneficial for reducing proinflammatory cytokine mRNA in the stomach and preventing progression from severe dysplasia to gastric cancer in H. pylori-infected INS-GAS mice.
Keywords: sunlindac, gastric cancer, antimicrobials, Helicobacter pylori, gastrin receptor antagonist (YM022)
Introduction
Based on experimental animal models and epidemiological evidence, Helicobacter pylori infection causes persistent chronic gastritis, which in susceptible individuals, may progress to atrophy, intestinal metaplasia, dysplasia, and finally intestinal-type gastric cancer (1-4). H. pylori infection resulted in overexpression of type 2 cyclooxygenase (Cox-2) in primary gastric cancer and gastric cancer cell lines of human and mouse gastric epithelial cells (5, 6). Double transgenic mice that constitutively expressed Cox-2 and prostaglandin E synthase-1 (Pges-1) in gastric epithelial cells had higher levels of prostaglandin E2 (PGE2) in the stomach and spontaneously develop macrophage infiltration, metaplasia, and gastric adenocarcinoma. This Cox-2/PGE2-related gastric cancer was suppressed by a Cox-2 inhibitor, NS-398 (7), suggesting that Cox-2/PGE2 pathway may contribute to helicobacter-associated gastric carcinogenesis. Recent studies suggested a positive association between hypergastrinemia, H. pylori infection, and gastric cancer in humans and mice (8-11). Hypergastrinemia and helicobacter infection synergistically promoted gastric cancer in male transgenic INS-GAS mice overexpressing amidated gastrin (8, 9, 11). Because decades of H. pylori infection are a prerequisite for gastric cancer development in susceptible hosts (1), chemoprevention is one of the promising approaches in gastric cancer prevention.
Since overexpression of Cox-2 and production of PGE2 are strongly associated with increased proliferation and reduced apoptosis in gastrointestinal epithelial tumor cells (12), non-steroid anti-inflammatory drugs (NSAIDs) that inhibit cyclooxygenase (Cox) activities and production of PGE2 are among the most widely tested compounds for cancer chemoprevention (13, 14). Inhibition of Cox activities decreased cell proliferation in gastric and intestinal cell lines that constitutively expresses Cox-2 (15, 16). Animal models demonstrated that the non-selective Cox inhibitor, sulindac, prevents oral-esophageal cancer and colon cancer (17, 18). Epidemiological studies also associate consumption of aspirin or other NSAIDs with a reduced risk of colorectal cancer (19) (20) In addition, clinical trials confirmed that sulindac, and the selective Cox-2 inhibitor celecoxib, inhibit the number and growth of adenomatous polyps in patients with familial adenomatous polyposis (21, 22). A lower risk of gastric cancer has been associated with NSAIDs in a dose-dependent manner (23). Considering the association between Cox-2/PGE2 pathway and helicobacter-associated gastric carcinogenesis, NSAIDs have been proposed as candidates for chemoprevention of gastric cancer. However, recent data indicate that suppression of PGE2 by Cox-2 inhibition enhanced Th1 proinflammatory immune responses in H. pylori-infected humans (24, 25); this raises the possibility that NSAIDs may increase H. pylori-associated gastritis and enhance inflammation-mediated gastric carcinogenesis (2).
It also has been demonstrated that long-term treatment of CCK2/gastrin receptor antagonist YF476 prevented the development of helicobacter-associated gastric cancer in INS-GAS mice (26, 27), suggesting that the gastrin signaling pathway provides another potential target for cancer chemoprevention. However, the effect of short-term CCK2/gastrin receptor antagonist treatment has not been analyzed. Long-term treatment of sulindac did not cause tissue injury of gastric mucosa of mice (18) (A. Rustagi personal communication). A recent study demonstrated that gastric PGE2 in H. pylori infection was mainly derived from Cox-1, and the selective Cox-2 inhibitor, rofecoxib, did not suppress PGE2 production or gastric epithelial proliferation, biomarkers of carcinogenesis (28).
We examined the chemopreventive effects of the non-selective Cox inhibitor sulindac and CCK2/gastrin receptor antagonist YM022 (29), an analogue of YF476 (26), alone or in combination with H. pylori antimicrobial eradication during the chronic stage of H. pylori infection in male, INS-GAS mice.
Materials and Methods
Mice
Specific pathogen-free (including Helicobacter spp.) male INS-GAS mice on a FVB/N background were maintained in an AAALAC accredited facility housed in microisolator cages, and given a commercial rodent diet (Prolab 3000, Richmond, Indiana) and water ad libitum. The animal protocol was approved by the MIT Committee on Animal Care.
Experimental design
Fifty-four, 6-8 week old male mice were infected by oral gavage with 0.2 ml of H. pylori (SS1 strain) on alternate days for a total of 3 doses (30). The H. pylori inoculum was adjusted with PBS to OD600=1.0 (30). Helicobacter-uninfected mice were sham-dosed with 0.2 ml of PBS. Antimicrobial therapy used to eradicate H. pylori consisted of omeprazole (400 μmol/kg/day, Sigma-Aldrich, St. Louis, MO), metronidazole (14.2 mg/kg/day, Sigma-Aldrich), and clarithromycin (7.15 mg/kg/day, Abbott Chicago, IL) twice a day for 7 days. This regimen has been used successfully in eradicating H. pylori from experimentally infected mice (31, 32). Antimicrobial H. pylori therapy was administered at 22 weeks post H. pylori infection (WPI). Sulindac was dissolved in buffer (Na2HPO4 4 mM, pH 7.4) at the final concentration of 400 ppm, given ad libitum, and changed daily from 22 WPI (18)YM022 was dissolved in polyethylene glycerol 300 (Sigma) by stirring for 24 hours at room temperature, and 45 mg/kg was injected subcutaneously once a week starting at 22 WPI (27). Mice were euthanized at 28 WPI.
Tissue collection and gastric pH measurement
Mice were fasted overnight (8 - 14 hours) before necropsy. Following CO2 asphyxiation, blood was immediately collected by cardiac puncture. The stomach was clamped at both esophageocardial and pyloroduodenal junctions. Sterile water (1.5 ml) was injected into the stomach, the stomach was massaged gently, and the pH of the aspirated gastric contents were measured using a pH meter (Orion PerpHecT model 320, Thermo Scientific, Waltham, MA).
Histological analysis
The stomach was incised along the greater curvature. Linear gastric strips from the lesser curvature were fixed in 10% neutral-buffered formalin or 70% ethanol, embedded, cut at 4 μm, and stained with hematoxylin and eosin (H&E). Tissue sections were scored for gastric lesions using previously published criteria by two veterinary pathologists (A.B.R and B.R.) blinded to sample identity (33). A dysplasia score of 3.0 was considered low-grade gastrointestinal intraepithelial neoplasia (GIN). Dysplasia scores equal or higher than 3.5 represented high-grade GIN (34). Ki-67 immunostaining (BD Biosciences, San Diego, CA) was used to measure epithelial proliferation of gastric mucosa. The ratio of Ki-67-positive to total epithelial nuclei in all glands of the proximal corpus was quantified manually to ascertain the Ki-67 labeling index (LI), and results were averaged from 3-4 mice per group.
Confirmation of H. pylori eradication by quantitative PCR
A longitudinal strip of gastric tissue from the greater curvature was digested with proteinase K at 55°C overnight followed by DNA extraction with phenol:chloroform:isoamyl alcohol (25:24:1) and ethanol precipitation. H. pylori colonization levels in gastric tissue were quantified using 100 ng of genomic DNA by a fluorogenic quantitative PCR assay with urease B primers as previously describedt (35).
Determination of PGE2 in mouse gastric tissues
Approximately 30 to 50 mg of frozen mouse gastric tissue was ground to a fine powder using a liquid-nitrogen-cooled mortar (Fisher Scientific Co., Fair Lawn, NJ). Samples were then transferred to sealed microcentrifuge tubes, and three times the volume of ice-cold PBS buffer containing 0.1% Butylated hydroxytoluene (BHT, Sigma-Aldrich) and 1 mM EDTA (Sigma-Aldrich) were added. The sample was then homogenized by an Ultrasonic Processor (Misonix, Farmingdale, NJ) at 0 °C for 3 minutes. An aliquot (100μl) of homogenate was transferred to a glass tube (13 × 100 mm) and subjected to extraction of prostaglandins using a method previously described (36). The results were expressed as nanograms of PGE2 / mg protein.
Quantitative analysis of mRNA expression
A longitudinal strip of gastric tissue from the anterior wall was harvested and snap-frozen in liquid nitrogen and then stored at -70°C. Total RNA was extracted with Trizol reagent (Invitrogen, Carlsbad, CA). cDNA was synthesized from 5 μg of total RNA with High Capacity cDNA Archive kit. mRNA levels of Ifn-γ, Tnf-α, Il-4, cycloxygenase 1 (Cox-1 or ptgs1, ABio), cycloxygenase 2 (Cox-2 or Ptgs2), and glyceraldehyde-3-phosphate dehydrogenase (Gapdh) were quantified with TaqMan gene expression assays using an ABI Prism Sequence Detection System 7700 (Applied Biosystems, Foster City, CA). mRNA levels of each gene were normalized to the mRNA level of internal control Gapdh and compared to the data of uninfected mice using the ΔΔCT method (User Bulletin #2, ABio).
Statistical analysis
Gastric lesion scores and Ki-67 LI for proliferation indices were compared using the Mann-Whitney U test. PGE2 levels, expression levels of cytokines, and IgG titers were compared using the student t test. Incidences of low-grade and high-grade GIN in the treatment groups were compared to controls by Chi-square test and Fisher's exact t test using commercial software (Graphpad Prism 4.0, GraphPad Software, Inc., San Diego, CA). Data were presented as mean +/- standard error.
Results
The effect of chemopreventive therapies on gastric pH in uninfected and H. pylori-infected INS-GAS mice
To assess the impact of H. pylori infection with or without treatment on the gastric acidity of INS-GAS mice, the pH of gastric aspirate was measured. Untreated, uninfected INS-GAS mice had a gastric pH=4.76+/-2.2. Among H. pylori-uninfected mice, gastric pH was reduced by sulindac (pH=3.37+/-0.16, p<0.001), and sulindac and antimicrobial therapy also reduced gastric pH (pH=3.85+/-0.38, p=0.07) (Supplemental Fig. 1). Uninfected mice that received sulindac or sulindac and antimicrobial therapy had comparable gastric pH (p=0.21). In contrast, uninfected mice that received gastrin receptor antagonist YM022 or YM022 and antimicrobial therapy did not alter gastric pH (pH=4.72+/-0.25 and 4.59+/-0.2, respectively).
H. pylori infection and antimicrobial eradication were confirmed by quantitative PCR at necropsy. H. pylori was successfully eradicated in all mice (100%) that received antimicrobial therapy at 22 WPI (data not shown). H. pylori infection significantly increased the gastric pH in INS-GAS mice (pH=6.28+/-0.12, p<0.001). Among H. pylori-infected mice, the gastric pH was reduced by sulindac (pH=4.96+/-0.22) and sulindac and antimicrobial therapy (pH=4.59+/-0.22) (both p<0.001). H. pylori-infected mice that received sulindac or sulindac and antimicrobial therapy had a similar gastric pH (p=0.24). Infected mice that received YM022 had a gastric pH (pH=6.35+/-0.05) comparable to that in untreated, infected mice. However, YM022 and antimicrobial therapy reduced the gastric pH (pH=5.55+/-0.25) in the infected mice when compared to untreated infected mice (p=0.07) or infected mice that received YM022 (p<0.01).
Sulindac reduces gastric PGE2 levels in INS-GAS mice
Among H. pylori-uninfected mice, sulindac reduced gastric PGE2 levels (1.09+/-0.27 ng/mg protein) compared to untreated mice (3.03+/-0.59 ng/mg protein, p<0.01) (Fig. 4d). Mice that received sulindac and antimicrobial therapy had lower gastric PGE2 levels (1.62+/-0.34 ng/mg protein, p=0.09) than untreated mice. Compared to untreated mice, gastric PGE2 levels were slightly higher in mice that received YM022 (4.33+/-0.71 ng/mg protein, p=0.17) or YM022 and antimicrobial therapy (5.51+/-0.64 ng/mg protein, p<0.05).
Figure 4.

mRNA expression levels of Th1 cytokines Ifn-γ and Tnf-α (a), Cox-1 and Cox-2 (b), Th2 cytokine Il-4 (c), and gastric PGE2 levels in H. pylori-uninfected mice. Data are presented as fold change relative to untreated, uninfected mice. Sulindac significantly up-regulated gastric Ifn-γ and Tnf-α (a) mRNA levels (p<0.01 and <0.05, respectively). Ifn-γ mRNA levels in mice receiving sulindac and antimicrobial (eradication) were lower than those in mice receiving sulindac (p<0.05), but were higher than untreated mice (p<0.05). Tnf-α mRNA levels in mice receiving sulindac and antimicrobial therapy were comparable to untreated mice or mice receiving sulindac. In contrast, YM022 or the combination of YM022 and antimicrobial therapy had no effect on gastric expression of Ifn-γ and Tnf-α. Gastric expression of Cox-1, Cox-2 (b), or Il-4 (c) was not influenced by sulindac or YM022 alone or in combination with antimicrobial therapy. Gastric PGE2 levels (d) were lower in mice receiving sulindac alone (p<0.01) and a combination of sulindac and antimicrobial therapy (p=0.09), but higher in mice receiving a combination of YM022 and antimicrobial therapy (p<0.05). (*, p<0.05; **, p<0.01 when compared to untreated, uninfected mice or comparison as indicated).
H. pylori-infected INS-GAS mice had significantly higher gastric PGE2 levels (9.73+/-0.81 ng/mg protein, p<0.01) when compared to uninfected mice. H. pylori-induced gastric PGE2 levels were inhibited by sulindac (1.29+/-0.48 ng/mg protein, p<0.01) (Fig. 5d). Infected mice receiving sulindac and antimicrobial therapy had significantly lower gastric PGE2 levels (3.47+/-0.76 ng/mg protein, p<0.01) than untreated, infected mice. YM022 alone reduced gastric PGE2 levels (6.83+/-0.81 ng/mg protein, p<0.05) in H. pylori-infected mice, YM022 and antimicrobial therapy did not alter gastric PGE2 levels (9.49+/-0.93 ng/mg protein) when compared to untreated, infected mice.
Figure 5.
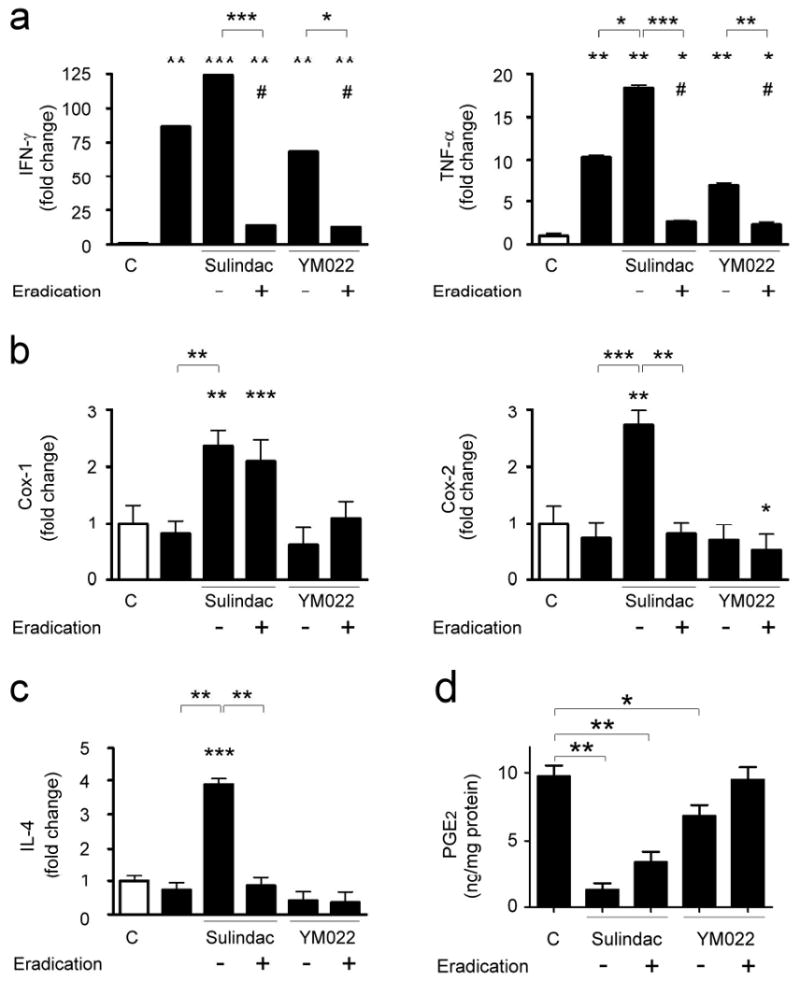
mRNA expression levels of Th1 cytokines Ifn-γ and Tnf-α (a), Cox-1 and Cox-2 (b), Th2 cytokines Il-4 (c), and gastric PGE2 levels in H. pylori-infected mice. Data are presented as fold changes relative to untreated, uninfected mice. White bars, uninfected INS-GAS mice; black bars, H. pylori-infected mice. H. pylori infection significantly up-regulated gastric expression of Ifn-γ and Tnf-α (a) (both p<0.01), but did not alter mRNA levels of Cox-1, Cox-2 (b), and Il-4 (c). mRNA levels of Ifn-γ, Tnf-α, Cox-1, Cox-2, and Il-4 were higher in infected mice that received sulindac than untreated, infected mice (p<0.05), whereas mRNA levels of Ifn-γ, Tnf-α, Cox-2, and Il-4 were suppressed in infected mice receiving a combination of sulindac and H. pylori eradication than those receiving sulindac (p<0.01). YM022 treatment did not alter mRNA levels of Ifn-γ, Tnf-α, Cox-1, Cox-2, and Il-4 in the H. pylori-infected mice. Infected mice receiving a combination of YM022 and H. pylori eradication had down-regulated Ifn-γ and Tnf-α expression compared to those receiving YM022 (p<0.05 or <0.01, respectively). Gastric PGE2 levels (d) were significantly reduced in the mice receiving sulindac, a combination of sulindac and eradication therapy (p<0.01) or YM022 (p<0.05). (Compared to untreated, uninfected mice or comparison between indicated groups: *, p<0.05; **, p<0.01; ***, p<0.001. Compared to untreated, H. pylori-infected mice: #, p<0.05).
Sulindac and YM022 inhibit gastric preneoplasia in uninfected INS-GAS mice
H. pylori-uninfected mice developed mild inflammation, epithelial defects, moderate hyperplasia, and severe oxyntic atrophy, intestinal metaplasia, and dysplasia at 34-36 weeks of age (equivalent to 28 WPI) (Fig. 1a and 2). Of the uninfected mice, 66.67% developed low-grade GIN and none developed high-grade GIN. Sulindac treatment started at 28 to 30 weeks of age (equivalent to 22 WPI) significantly reduced premalignant lesions (p<0.01), and incidence of gastric cancer (p<0.05) in uninfected mice (Fig. 2). Low-grade GIN was observed in 10% of the uninfected INS-GAS mice that received sulindac, whereas high-grade GIN was not diagnosed. Sulindac and antimicrobial therapy started at 28 to 30 weeks of age also significantly reduced the severity of premalignant (p<0.05) and malignant lesions (p<0.05) in the uninfected INS-GAS mice. None of the uninfected mice that received the combination of sulindac and antimicrobial therapy developed GIN.
Figure 1.

Gastric histology in mice. At 32-34 weeks old, uninfected male mice exhibited spontaneous gastric epithelial dysplasia with inflammation (a). H. pylori infection significantly increased inflammation, oxyntic atrophy, foveolar hyperplasia, and dysplasia in the corpus of age-matched male mice at 28 (WPI) (b). Sulindac treatment exacerbated inflammation but not hyperplasia or dysplasia in H. pylori-infected mice (c). H. pylori antimicrobial therapy and sulindac significantly reduced inflammation, oxyntic atrophy, foveolar hyperplasia, and dysplasia (d). Tissues were stained with H&E; bar = 400 μM.
Figure. 2.

Gastric histological scores in sulindac groups; inflammation (a), atrophy (b), dysplasia (c), and GIN (d). Sulindac and a combination of sulindac and antimicrobial therapy reduced incidence of both low-grade and high-grade GIN in H. pylori-uninfected (p<0.05 and p<0.001, respectively), whereas the combination of sulindac and H. pylori eradication reduced gastric cancer incidence in H. pylori-infected mice (p<0.05). (*, p<0.05; **, p<0.01; ***, p<0.001).
YM022 treatment slightly reduced dysplasia (p=0.079), and the incidence of gastric cancer (p=0.087) in uninfected mice (Fig. 3). Low-grade GIN was diagnosed in 25% of the YM022-treated uninfected mice. YM022 and antimicrobial therapy resulted in less severe dysplasia (p<0.01), and incidence gastric cancer (p<0.01) in the uninfected mice. Additionally, uninfected mice that received YM022 or the combination of YM022 and antimicrobial therapy had comparable severity of gastric lesions (p>0.05).
Figure 3.

Gastric histological scores in YM022 groups; inflammation (a), atrophy (b), dysplasia (c), and GIN (d). YM022 or a combination of YM022 and antimicrobial therapy had no effect on the incidence of gastric cancer in H. pylori-uninfected or –infected mice (p<0.05). (*, p<0.05; **, p<0.01; ***, p<0.001).
H. pylori infection promotes gastric carcinogenesis
Helicobacter infection promotes gastric carcinogenesis in mice (9, 11). H. pylori-infected mice developed more severe gastric inflammation (p=0.065) and epithelial defects (p<0.01) than uninfected mice (Fig. 2). H. pylori infection also promoted the development of gastric cancer in all infected mice.
Sulindac increases gastric preneoplasia, while Sulindac and antimicrobial therapy reduces gastric carcinogenesis in H. pylori-infected mice
In H. pylori-infected mice, sulindac exacerbated the severity of inflammation (p<0.01), oxyntic atrophy (p<0.05), and intestinal metaplasia (p<0.05) (Fig. 1c and 2). Epithelial hyperplasia was reduced by sulindac (p<0.01, data not shown). The severity of dysplasia and the incidence of gastric cancer were comparable between untreated infected mice and infected mice that received sulindac. All infected mice that received sulindac developed gastric cancer with both low- and high-grade GIN being observed in a H. pylori-infected INS-GAS mice (80% and 20%, respectively). Compared to untreated H. pylori-infected mice, infected mice that received sulindac and antimicrobial therapy had significantly less severe oxyntic atrophy (p<0.05) and a lower incidence of gastric cancer (p<0.05) (Fig. 1d and 2). Gastric cancer was observed in 64% (38% high and 27% low grade GIN) of the infected mice that received sulindac and antimicrobial therapy. The severity of inflammation, epithelial defects, hyperplasia, intestinal metaplasia, and dysplasia were comparable between the untreated infected mice and infected mice that received sulindac and antimicrobial therapy. Additionally, when compared to H. pylori-infected mice that received sulindac, severity of inflammation (p<0.001), oxyntic atrophy (p<0.001), and intestinal metaplasia (p<0.01), and incidence of gastric cancer (p<0.01) were significantly reduced in the infected mice that received the combination therapy.
Compared to untreated H. pylori-infected mice, infected mice that received YM022 or a combination of YM022 and antimicrobial therapy showed similar pathology scores (Fig. 3). Gastric cancer developed in all the mice that received YM022 and antimicrobial therapy. Low- and high-grade GIN were observed in 75% and 25% of infected mice that received YM022, and in 44% and 56% of infected mice that received YM022 and antimicrobial therapy, respectively. YM022 or YM022 and antimicrobial therapy had no significant effect on gastric cancer incidence in H. pylori-infected mice.
Expression of proinflammatory Ifn-γ and Tnf-α, Cox-1, and Cox-2 in the stomach is up-regulated by sulindac but down-regulated by sulindac and antimicrobial therapy
Among H. pylori-uninfected mice, sulindac significantly up-regulated gastric mRNA levels of Ifn-γ and Tnf-α (p<0.05) (Fig. 4). mRNA levels of Ifn-γ were down-regulated in the mice that received sulindac and antimicrobial therapy compared to uninfected mice that received sulindac (p<0.05), however, the gastric Ifn-γ mRNA levels in the mice that received sulindac and antimicrobial therapy were still higher than those in the untreated mice (p<0.05). H. pylori-uninfected mice that received combination therapy had similar gastric Tnf-α expression, compared to untreated mice or mice that received sulindac. In contrast, Cox-1 and Cox-2 expression levels were not altered by sulindac or the combination therapy. Expression levels of proinflammatory cytokines and Cox enzymes were not changed in uninfected mice by YM022 or the combination of YM022 and antimicrobial therapy. Additionally, gastric Il-4 mRNA levels were not altered by sulindac or YM022, alone or combined with antimicrobial therapy, respectively.
H. pylori infection upregulated gastric mRNA levels of Ifn-γ and Tnf-α in male INS-GAS mice (p<0.01) (Fig. 5). H. pylori infection, however, had no effect on mRNA levels of Cox-1, Cox-2, and Il-4 in the stomach of mice. Compared to untreated H. pylori-infected mice, infected mice that received sulindac had significantly higher mRNA levels of Ifn-γ, Tnf-α, Cox-1, Cox-2, and Il-4 (p<0.05). Infected mice that received sulindac and antimicrobial therapy had reduced mRNA levels of Ifn-γ and Tnf-α than those in untreated infected INS-GAS mice (p<0.05). H. pylori-infected mice that received sulindac and antimicrobial therapy had higher Cox-1 mRNA levels (p<0.001) and comparable Cox-2 and Il-4 mRNA levels when compared to untreated infected mice. Compared to infected mice that received sulindac, infected mice that received sulindac and antimicrobial therapy had down-regulated mRNA levels of Ifn-γ, Tnf-α, Cox-2, and Il-4 (p<0.01). YM022 did not alter gastric mRNA levels of Ifn-γ, Tnf-α, Cox-1, Cox-2, and Il-4 in the infected mice (Fig. 5). H. pylori-infected INS-GAS mice that received YM022 and antimicrobial therapy had comparable mRNA levels of Cox-1, Cox-2, and Il-4, and significantly lower mRNA levels of Ifn-γ and Tnf-α when compared to untreated infected mice and infected mice that received YM022 (p<0.05).
Sulindac and antimibrobial therapy decreased Th1-associated, H. pylori-specific IgG2a antibody responses
H. pylori infection significantly induced H. pylori-specific, Th1-associated IgG2a responses in mice (p<0.001) (Supplemental Fig. 2a). Sulindac treatment did not alter H. pylori-infected IgG2a levels in H. pylori-infected mice (p=0.09). Infected mice that received sulindac and antimicrobials had H. pylori-specific IgG2a levels comparable to untreated infected mice. Additionally, H. pylori-specific IgG2a levels in infected mice that received the sulindac and antimicrobial therapy were significantly lower than those in infected mice that received sulindac (p<0.05). In contrast, YM022 alone or YM022 and antimicrobial therapy did not alter H. pylori-specific IgG2a levels in the infected mice. H. pylori infection also induced H. pylori, Th2-associated IgG1 responses in mice (p<0.001) (Supplemental Fig. 2b). Treatment modalities in H. pylori-infected mice had no significant effect on H. pylori-specific IgG1 levels compared to untreated infected mice.
Gastric mucosal cell proliferation was reduced by a combination of antimicrobial therapy and sulindac or YM022
Proliferating epithelial cells detected by Ki-67 immunohistochemical staining were observed in the isthmus regions of corpus mucosa in uninfected mice (Fig. 6b). In untreated H. pylori-infected mice, proliferating cells in the corpus expanded from the isthmus regions to hypertrophic foveolar regions, and the Ki-67 labeling indices (LI) were higher than in uninfected mice (p<0.01) (Fig. 6a). Among H. pylori-infected mice, proliferating cells were observed in the hypertrophic foveolar regions of the corpus in those mice receiving sulindac or YM022. The distribution of proliferative cells was mainly in the isthmus region of the corpus mucosa in the infected mice receiving combination of sulindac or YM022 and H. pylori eradication. Ki-67 LI positively correlated with severity of gastric pathology and GIN; Ki-67 LI in H. pylori-infected mice receiving sulindac or YM022 was similar to untreated, H. pylori-infected mice. Infected mice receiving the combination of sulindac or YM022 and H. pylori antimicrobial eradication had lower a Ki-67 LI compared to untreated, H. pylori-infected mice and infected mice that received sulindac or YM022 (p<0.05, respectively).
Figure 6.

Cell proliferation in gastric mucosa; Ki-67 labeling indices (LI) (b), and immunohistochemical staining of Ki-67 (c). Ki-67 LI were compared in corpus mucosa (b). H. pylori infection resulted in significantly higher Ki-67 LI in mice (p<0.01). Sulindac or YM022 did not alter H. pylori-associated increase of Ki-67 LI in INS-GAS mice. H. pylori-infected mice receiving the combination of sulindac or YM022 and H. pylori eradication had significantly lower Ki-67 LI compared to untreated, infected mice (both p<0.05) or infected mice that received sulindac or YM022 (p<0.05, respectively). Proliferating cells were positively stained for Ki-67 (c). In helicobacter-uninfected INS-GAS mice, proliferating cells were restricted to the isthmus regions. Zone of proliferating cells expanded from isthmus regions to foveolar regions in the untreated, H. pylori-infected mice and infected mice that received sulindac or YM022. Proliferating cells were restricted to the isthmus regions irrespective of the hyperplastic foveolar glands in the infected mice that received a combination of sulindac or YM022 and H. pylori eradication. White bar, H. pylori-uninfected mice; black bars, H. pylori-infected mice; Ki-67 positive cells were stained in brown color. (Compared to uninfected, untreated mice or comparison between indicated groups: *, p<0.05; **, p<0.01; ***, p<0.001. Compared to infected mice: #, p<0.05).
Discussion
The role of Cox-2 in promoting cancer is mainly attributed to PGE2 production and its ability to promote cell proliferation, migration, and angiogenesis and inhibit apoptosis (37). NSAIDs prevent some types of gastrointestinal cancer and other solid tumors in humans (37). In epidemiological studies, the use of NSAIDs is generally associated with a lower risk gastric cancer. However, a chemopreventive effect of NSAIDs on the development of gastric cancer among H. pylori infected individuals has not been conclusively shown in human clinical trials (36, 38). Patients that underwent H. pylori eradication therapy, chronic use of celecoxib was associated with a higher regression rate of gastric precancerous intestinal metaplasia (36). However, in patients that had received H. pylori eradication therapy, treatment with another selective Cox-2 inhibitor, rofecoxib, for 2 years did not reduce intestinal metaplasia (38). Considering that cancer chemoprevention by NSAIDs is modulated by both Cox-dependent and -independent pathways (39), NSAIDs may have variable efficacy in their abilities to prevent gastric cancer.
Consistent with a role for Cox-2/PGE2 in carcinogenesis, double-transgenic mice expressing Cox-2 and Pges-1 in the gastric mucosa overproduce PGE2 and develop gastric hyperplasia, metaplasia, and cancer in the glandular stomach (7). Since H. pylori infection up-regulates Cox-2 expression and PGE2 production (5, 6), it is reasonable to hypothesize that suppression of PGE2 should prevent H. pylori-associated gastric carcinogenesis. However, H. pylori-associated gastritis and Tnf-α expression are more severe in Cox-1-/- or Cox-2-/- mice (40) and in H. pylori-infected mice receiving NSAIDs (41). Furthermore, Cox-2 inhibition enhances Th1 immune responses against H. pylori (24, 25, 41). CD4+CD25+ regulatory T cell (Treg) function dampens H. pylori-associated gastritis in C56BL/6 mice (30). PGE2 enhances the inhibitory function of human Treg cells in vitro (42). Therefore, Treg cell function in the H. pylori-infected mice is likely to be compromised by reduced PGE2 production in sulindac-treated animals. Suppressed Treg function may have also contributed to enhanced H. pylori-associated inflammation in the infected mice receiving sulindac alone, suggesting that PGE2 induced in response to H. pylori infection plays a role in dampening gastric inflammation and reducing inflammation-mediated gastric carcinogenesis (2). The benefit of PGE2 suppression by NSAIDs in suppressing mediators of cancer progression, however, is not significant to counteract the adverse effect of Cox-2 inhibition associated with Th1-predominant inflammation caused by H. pylori infection.
As previously described, H. pylori infection promoted gastric carcinogenesis in INS-GAS male mice (8, 32). Although treatment with sulindac suppressed H. pylori-induced gastric PGE2 levels, sulindac alone did not prevent gastric cancer in chronically infected INS-GAS mice. Moreover, sulindac exacerbated H. pylori-induced gastritis and up-regulated expression of proinflammatory cytokines. In contrast, a combination of sulindac and antimicrobial therapy reduced gastric PGE2 levels and gastric inflammation, and prevented the development of H. pylori-associated gastric cancer. These findings suggest that in the presence of H. pylori infection, selected NSAIDs enhance Th1-predominant proinflammatory responses to H. pylori antigens (24), and may promote the development of inflammation-mediated gastric cancer in a susceptible host with chronic H. pylori infection (2). In previous studies suppression of PGE2 by Cox-2 inhibitors did not alter Th2-associated Il-4 expression; instead, the treatment up-regulated mRNA levels of Th1-associated Il-12 and Tnf-α in gastric tissue of H. pylori-infected humans (24, 25). Sulindac treatment up-regulated gastric Ifn-γ and Tnf-α as well as IL-4 mRNA in H. pylori-infected mice. Sulindac-mediated up-regulation of Th1 proinflammatory cytokines positively correlated with the severity of H. pylori-associated gastric lesions in infected mice. Sulindac up-regulated gastric Ifn-γ and Tnf-α mRNA levels in H. pylori-uninfected INS-GAS mice, even though gastric lesions and gastric cancer risk were reduced. Sulindac also up-regulated Cox-1 and Cox-2 mRNA levels in H. pylori-infected INS-GAS mice, despite the fact that it reduced gastric PGE2 levels as a result of its known Cox-inhibitory effects (43). Sulindac is a prodrug that is converted to sulfone and sulindac sulfide, and the sulfide form has been shown to activate PPAR-γ and induce Cox-2 expression (44). The significance of up-regulated mRNA expression of Cox-1 and Cox-2 in H. pylori-infected mice that received sulindac is not clear and needs further study. The combination of sulindac and H. pylori eradication reduced the H. pylori-specific IgG2a titers, which were not significantly altered by sulindac treatment.
Sulindac prevented gastrin-driven carcinogenesis in H. pylori-uninfected male mice, despite up-regulation of proinflammatory cytokines. Moreover, the combination of sulindac and antimicrobial therapy were more effective in preventing gastric lesions, reducing cell proliferation in gastric mucosa and incidence of gastric cancer in H. pylori-uninfected mice whose stomachs are known to be colonized with enteric flora(32). Normal microbiota colonize all regions of the bowel, including the upper gastrointestinal tract of mice (45). Antimicrobial therapy with omeprazole, metronidazole and clarithromycin resulted in dynamic changes of the gastric microbiota in humans (46). These findings in humans and mice suggest that gastric flora other then H. pylori contribute to gastric inflammation and cancer (2).
Chemoprevention of helicobacter-associated gastric cancer in INS-GAS mice was previously observed in long-term (6 months) suppression of CCK2/gastrin signaling by treatment with YF476, which was initiated during the acute stage of helicobacter infection (27). We observed that the CCK2/gastrin receptor antagonist, YM022, alone or in combination with antimicrobial therapy reduced gastric cancer risk in H. pylori-uninfected INS-GAS mice. However, the chemopreventive effect of YM022 alone or in combination with antimicrobial therapy was not observed in H. pylori-infected INS-GAS mice when the 6-week treatment was initiated during the chronic stage of H. pylori infection. YM022 blockade of gastrin signaling (27) was not sufficient to counteract YM022-associated gastric pH elevation which may facilitate overgrowth of certain gastric flora following H. pylori eradication therapy (32). These differing results in our current study and the previous report using YF476 might reflect intrinsic differences in the two antagonists, but were more likely due to the different treatment schedules in these two studies. The data, however, are consistent with gastrin playing an important role early, but not later, in the multistep progression to gastric cancer (11).
Gastric proinflammatory cytokines and cell proliferation markers were down-regulated using the combination of YM022 and H. pylori eradication in H. pylori-infected INS-GAS mice, suggesting that this regimen has chemopreventive potential for gastric cancer. Regression of preneoplastic gastric intestinal metaplasia following H. pylori eradication is positively correlated with the interval that patients have been free of H. pylori infection (47). This suggests that regression of H. pylori-associated gastric lesions and gastric cancer risk may not be observed during the first few years after treatment (48). A longer period of follow-up may be necessary to examine the chemopreventive effect of helicobacter-associated gastric cancer in INS-GAS mice using a combination of YM022 and H. pylori eradication.
Our data demonstrate that NSAIDs are an effective treatment modality in preventing of H. pylori-associated gastric cancer provided that H. pylori is eradicated before NSAIDs treatment is initiated. This study highlights the possible adverse effect of selected NSAIDs in promoting H. pylori-associated gastritis and cancer progression in susceptible hosts, despite the fact that PGE2 suppression by NSAIDs has chemopreventive properties in treatment of colon cancer and other solid tumors (19, 20, 37). A probable mechanism by which NSAIDs exacerbates H. pylori-associated gastritis and gastric cancer is via an enhanced proinflammatory Th1 immune response. Our study is consistent with the Maastricht III Consensus Report which recommends H. pylori eradication for the prevention of peptic ulcer and gastric cancer, especially in patients with long-term NSAID usage (49).
Supplementary Material
Acknowledgments
We are grateful to Dr. Keiji Miyata and Dr. Hidenobu Yuki (Astellas Pharma Inc, previously Yamanouchi Pharmaceutical Co., Tokyo, Japan) for providing YM022, Kristen Clapp and Juri Miyamae for technical assistance, and Kathy Cormier for her histology expertise.
Grant Support: NIH grants R01AI37750, P01CA26731, P30ES02109 (J.G. Fox), and R01CA093405-07A1 (T.C. Wang and J.G. Fox).
References
- 1.Correa P. A human model of gastric carcinogenesis. Cancer research. 1988;48:3554–60. [PubMed] [Google Scholar]
- 2.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. (Invited review) J Clin Invest. 2007;117:60–9. doi: 10.1172/JCI30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1:1311–5. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 4.IARC Working Group on the Evaluation of Carcinogenic Risk to Humans. Schistosomes liverlukes and Helicobacter pylori.: International Association for Research against Cancer. 1994:1–241. [Google Scholar]
- 5.Akhtar M, Cheng Y, Magno RM, et al. Promoter methylation regulates Helicobacter pylori-stimulated cyclooxygenase-2 expression in gastric epithelial cells. Cancer research. 2001;61:2399–403. [PubMed] [Google Scholar]
- 6.Kim TI, Lee YC, Lee KH, et al. Effects of nonsteroidal anti-inflammatory drugs on Helicobacter pylori-infected gastric mucosae of mice: apoptosis, cell proliferation, and inflammatory activity. Infection and immunity. 2001;69:5056–63. doi: 10.1128/IAI.69.8.5056-5063.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oshima H, Oshima M, Inaba K, Taketo MM. Hyperplastic gastric tumors induced by activated macrophages in COX-2/mPGES-1 transgenic mice. The EMBO journal. 2004;23:1669–78. doi: 10.1038/sj.emboj.7600170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fox JG, Rogers AB, Ihrig M, et al. Helicobacter pylori-associated gastric cancer in INS-GAS mice is gender specific. Cancer research. 2003;63:942–50. [PubMed] [Google Scholar]
- 9.Fox JG, Wang TC, Rogers AB, et al. Host and microbial constituents influence Helicobacter pylori-induced cancer in a murine model of hypergastrinemia. Gastroenterology. 2003;124:1879–90. doi: 10.1016/s0016-5085(03)00406-2. [DOI] [PubMed] [Google Scholar]
- 10.Mulholland G, Ardill JE, Fillmore D, Chittajallu RS, Fullarton GM, McColl KE. Helicobacter pylori related hypergastrinaemia is the result of a selective increase in gastrin 17. Gut. 1993;34:757–61. doi: 10.1136/gut.34.6.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang TC, Dangler CA, Chen D, et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology. 2000;118:36–47. doi: 10.1016/s0016-5085(00)70412-4. [DOI] [PubMed] [Google Scholar]
- 12.Williams CS, DuBois RN. Prostaglandin endoperoxide synthase: why two isoforms? The American journal of physiology. 1996;270:G393–400. doi: 10.1152/ajpgi.1996.270.3.G393. [DOI] [PubMed] [Google Scholar]
- 13.Chan TA. Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. The lancet oncology. 2002;3:166–74. doi: 10.1016/s1470-2045(02)00680-0. [DOI] [PubMed] [Google Scholar]
- 14.Janne PA, Mayer RJ. Chemoprevention of colorectal cancer. The New England journal of medicine. 2000;342:1960–8. doi: 10.1056/NEJM200006293422606. [DOI] [PubMed] [Google Scholar]
- 15.Sawaoka H, Kawano S, Tsuji S, Tsujii M, Murata H, Hori M. Effects of NSAIDs on proliferation of gastric cancer cells in vitro: possible implication of cyclooxygenase-2 in cancer development. Journal of clinical gastroenterology. 1998;27 1:S47–52. doi: 10.1097/00004836-199800001-00009. [DOI] [PubMed] [Google Scholar]
- 16.Sheng H, Shao J, Kirkland SC, et al. Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J Clin Invest. 1997;99:2254–9. doi: 10.1172/JCI119400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moorghen M, Ince P, Finney KJ, Sunter JP, Appleton DR, Watson AJ. A protective effect of sulindac against chemically-induced primary colonic tumours in mice. The Journal of pathology. 1988;156:341–7. doi: 10.1002/path.1711560411. [DOI] [PubMed] [Google Scholar]
- 18.Opitz OG, Harada H, Suliman Y, et al. A mouse model of human oral-esophageal cancer. J Clin Invest. 2002;110:761–9. doi: 10.1172/JCI15324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kune GA, Kune S, Watson LF. Colorectal cancer risk, chronic illnesses, operations, and medications: case control results from the Melbourne Colorectal Cancer Study. Cancer research. 1988;48:4399–404. [PubMed] [Google Scholar]
- 20.Rostom A, Dube C, Lewin G, et al. Nonsteroidal anti-inflammatory drugs and cyclooxygenase-2 inhibitors for primary prevention of colorectal cancer: a systematic review prepared for the U.S. Preventive Services Task Force. Ann Intern Med. 2007;146:376–89. doi: 10.7326/0003-4819-146-5-200703060-00010. [DOI] [PubMed] [Google Scholar]
- 21.Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. The New England journal of medicine. 2000;342:1946–52. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 22.Labayle D, Fischer D, Vielh P, et al. Sulindac causes regression of rectal polyps in familial adenomatous polyposis. Gastroenterology. 1991;101:635–9. doi: 10.1016/0016-5085(91)90519-q. [DOI] [PubMed] [Google Scholar]
- 23.Wang WH, Huang JQ, Zheng GF, Lam SK, Karlberg J, Wong BC. Non-steroidal anti-inflammatory drug use and the risk of gastric cancer: a systematic review and meta-analysis. Journal of the National Cancer Institute. 2003;95:1784–91. doi: 10.1093/jnci/djg106. [DOI] [PubMed] [Google Scholar]
- 24.Meyer F, Ramanujam KS, Gobert AP, James SP, Wilson KT. Cutting edge: cyclooxygenase-2 activation suppresses Th1 polarization in response to Helicobacter pylori. J Immunol. 2003;171:3913–7. doi: 10.4049/jimmunol.171.8.3913. [DOI] [PubMed] [Google Scholar]
- 25.Pellicano AIM, Leone I, Larussa T, Luzza F. Enhanced activation of cyclooxygenase-2 downregulates Th1 signaling pathway in Helicobacter pylori-infected human gastric mucosa. Helicobacter. 2007:193–9. doi: 10.1111/j.1523-5378.2007.00498.x. [DOI] [PubMed] [Google Scholar]
- 26.Semple G, Ryder H, Rooker DP, et al. (3R)-N-(1-(tert-butylcarbonylmethyl)-2,3-dihydro-2-oxo-5-(2-pyridyl)-1H-1, 4-benzodiazepin-3-yl)-N′-(3-(methylamino)phenyl)urea (YF476): a potent and orally active gastrin/CCK-B antagonist. Journal of medicinal chemistry. 1997;40:331–41. doi: 10.1021/jm960669+. [DOI] [PubMed] [Google Scholar]
- 27.Takaishi S, Cui G, Frederick DM, et al. Synergistic inhibitory effects of gastrin and histamine receptor antagonists on Helicobacter-induced gastric cancer. Gastroenterology. 2005;128:1965–83. doi: 10.1053/j.gastro.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 28.Scheiman JM, Greenson JK, Lee J, Cryer B. Effect of cyclooxygenase-2 inhibition on human Helicobacter pylori gastritis: mechanisms underlying gastrointestinal safety and implications for cancer chemoprevention. Alimentary pharmacology & therapeutics. 2003;17:1535–43. doi: 10.1046/j.1365-2036.2003.01587.x. [DOI] [PubMed] [Google Scholar]
- 29.Nishida A, Miyata K, Tsutsumi R, et al. Pharmacological profile of (R)-1-[2,3-dihydro-1-(2′-methylphenacyl)-2-oxo-5-phenyl-1H-1,4-benzodiazepin-3-yl]-3-(3-methylphenyl)urea (YM022), a new potent and selective gastrin/cholecystokinin-B receptor antagonist, in vitro and in vivo. The Journal of pharmacology and experimental therapeutics. 1994;269:725–31. [PubMed] [Google Scholar]
- 30.Lee CW, Rao VP, Rogers AB, et al. Wild-type and interleukin-10-deficient regulatory T cells reduce effector T-cell-mediated gastroduodenitis in Rag2-/- mice, but only wild-type regulatory T cells suppress Helicobacter pylori gastritis. Infection and immunity. 2007;75:2699–707. doi: 10.1128/IAI.01788-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Zanten SJ, Kolesnikow T, Leung V, O'Rourke JL, Lee A. Gastric transitional zones, areas where Helicobacter treatment fails: results of a treatment trial using the Sydney strain mouse model. Antimicrobial agents and chemotherapy. 2003;47:2249–55. doi: 10.1128/AAC.47.7.2249-2255.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee CW, Rickman B, Rogers AB, Ge Z, Wang TC, Fox JG. Helicobacter pylori eradication prevents progression of gastric cancer in hypergastrinemic INS-GAS mice. Cancer research. 2008;68:3540–8. doi: 10.1158/0008-5472.CAN-07-6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rogers AB, Taylor NS, Whary MT, Stefanich ED, Wang TC, Fox JG. Helicobacter pylori but not high salt induces gastric intraepithelial neoplasia in B6129 mice. Cancer research. 2005;65:10709–15. doi: 10.1158/0008-5472.CAN-05-1846. [DOI] [PubMed] [Google Scholar]
- 34.Boivin GP, Washington K, Yang K, et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology. 2003;124:762–77. doi: 10.1053/gast.2003.50094. [DOI] [PubMed] [Google Scholar]
- 35.Maurer KJ, Rogers AB, Ge Z, Wiese AJ, Carey MC, Fox JG. Helicobacter pylori and cholesterol gallstone formation in C57L/J mice: a prospective study. American journal of physiology. 2006;290:G175–82. doi: 10.1152/ajpgi.00272.2005. [DOI] [PubMed] [Google Scholar]
- 36.Yang HB, Cheng HC, Sheu BS, Hung KH, Liou MF, Wu JJ. Chronic celecoxib users more often show regression of gastric intestinal metaplasia after Helicobacter pylori eradication. Alimentary pharmacology & therapeutics. 2007;25:455–61. doi: 10.1111/j.1365-2036.2006.03224.x. [DOI] [PubMed] [Google Scholar]
- 37.Backlund MG, Mann JR, Dubois RN. Mechanisms for the prevention of gastrointestinal cancer: the role of prostaglandin E2. Oncology. 2005;69 1:28–32. doi: 10.1159/000086629. [DOI] [PubMed] [Google Scholar]
- 38.Leung WK, Ng EK, Chan FK, et al. Effects of long-term rofecoxib on gastric intestinal metaplasia: results of a randomized controlled trial. Clin Cancer Res. 2006;12:4766–72. doi: 10.1158/1078-0432.CCR-06-0693. [DOI] [PubMed] [Google Scholar]
- 39.Grosch S, Maier TJ, Schiffmann S, Geisslinger G. Cyclooxygenase-2 (COX-2)-independent anticarcinogenic effects of selective COX-2 inhibitors. Journal of the National Cancer Institute. 2006;98:736–47. doi: 10.1093/jnci/djj206. [DOI] [PubMed] [Google Scholar]
- 40.Li GQ, Xia HH, Chen MH, et al. Effects of cyclooxygenase-1 and -2 gene disruption on Helicobacter pylori-induced gastric inflammation. The Journal of infectious diseases. 2006;193:1037–46. doi: 10.1086/500984. [DOI] [PubMed] [Google Scholar]
- 41.Tanigawa T, Watanabe T, Hamaguchi M, et al. Anti-inflammatory effect of two isoforms of COX in H. pylori-induced gastritis in mice: possible involvement of PGE2. American journal of physiology. 2004;286:G148–56. doi: 10.1152/ajpgi.00137.2003. [DOI] [PubMed] [Google Scholar]
- 42.Baratelli F, Lin Y, Zhu L, et al. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J Immunol. 2005;175:1483–90. doi: 10.4049/jimmunol.175.3.1483. [DOI] [PubMed] [Google Scholar]
- 43.Felts AS, Ji C, Stafford JB, et al. Desmethyl derivatives of indomethacin and sulindac as probes for cyclooxygenase-dependent biology. ACS chemical biology. 2007;2:479–83. doi: 10.1021/cb700077z. [DOI] [PubMed] [Google Scholar]
- 44.Meade EA, McIntyre TM, Zimmerman GA, Prescott SM. Peroxisome proliferators enhance cyclooxygenase-2 expression in epithelial cells. J Biol Chem. 1999;274:8328–34. doi: 10.1074/jbc.274.12.8328. [DOI] [PubMed] [Google Scholar]
- 45.Sarma-Rupavtarm RB, Ge Z, Schauer DB, Fox JG, Polz MF. Spatial distribution and stability of the eight microbial species of the altered schaedler flora in the mouse gastrointestinal tract. Applied and environmental microbiology. 2004;70:2791–800. doi: 10.1128/AEM.70.5.2791-2800.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adamsson I, Nord CE, Lundquist P, Sjostedt S, Edlund C. Comparative effects of omeprazole, amoxycillin plus metronidazole versus omeprazole, clarithromycin plus metronidazole on the oral, gastric and intestinal microflora in Helicobacter pylori-infected patients. The Journal of antimicrobial chemotherapy. 1999;44:629–40. doi: 10.1093/jac/44.5.629. [DOI] [PubMed] [Google Scholar]
- 47.Mera R, Fontham ET, Bravo LE, et al. Long term follow up of patients treated for Helicobacter pylori infection. Gut. 2005;54:1536–40. doi: 10.1136/gut.2005.072009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mera R, Fontham ET, Bravo LE, et al. Re: Randomized double-blind factorial trial of three treatments to reduce the prevalence of precancerous gastric lesions. Journal of the National Cancer Institute. 2006;98:1426. doi: 10.1093/jnci/djj388. [DOI] [PubMed] [Google Scholar]
- 49.Malfertheiner P, Megraud F, O'Morain C, et al. Current concepts in the management of Helicobacter pylori infection: the Maastricht III Consensus Report. Gut. 2007;56:772–81. doi: 10.1136/gut.2006.101634. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.