

Abstract
Understanding the ways in which pathogens invade and neutralize their hosts is of great interest from both an academic and clinical perspective. However, in many cases genetic tools are unavailable or insufficient to fully characterize the detailed mechanisms of pathogenesis. Small molecule approaches are particularly powerful due to their ability to modulate specific biological functions in a highly controlled manner and their potential to broadly target conserved processes across species. Recently, two approaches that make use of small molecules, activity-based protein profiling (ABPP) and high-throughput phenotypic screening, have begun to find applications in the study of pathways involved in pathogenesis. In this review we highlight ways in which these techniques have been applied to examine bacterial and parasitic pathogenesis as well as discuss possible ways in which these efforts can be expanded in the near future.
Keywords: Chemical genetics, microbial pathogenesis, bacteria, parasites, activity-based probes, small molecule screening
Introduction
Microbial pathogenesis, the collection of mechanisms through which an infectious agent causes disease, is a dynamic process involving concerted interactions between the etiological organism and host. Due to the highly regulated nature of the infection cycle, small molecules that modulate or monitor enzyme activity levels are ideal tools for studying pathogenesis.
In microbial pathogenesis distinct environmental cues often trigger key events such as host cell invasion that are required for disease progression. Small molecules often act rapidly with remarkable spatial and temporal control, allowing these events to be dissected in real-time. Genetic manipulation of targets, on the other hand, often is not possible on a timescale that is sufficient to monitor distinct processes with a high degree of temporal resolution. Even conditional knock down techniques such as RNA interference are limited by the speed of protein translation and degradation before activity levels can be sufficiently changed. In addition, many pathogens lack the machinery necessary for such methods to be used. Together, these limitations underscore the benefits of using pharmacological approaches to study pathogenesis.
In addition to being used to perturb protein function, small molecule probes can be used to profile the activity levels of enzymes involved in different stages of virulence processes. These compounds, known as activity-based probes (ABPs), give an indirect readout of enzymatic activity within a complex system without the need for biochemical purification of individual components. Because many enzymes involved in processes such as host-pathogen interactions are difficult to purify or express recombinantly, this method is extremely powerful for interrogating the precise function of a given factor in pathogenesis. Additionally, many proteins are post-translationally regulated, making it impossible to monitor their activity by examining transcript or protein expression levels alone. Therefore, ABPs are capable of providing a more complete view of a pathogenic mechanism.
Although small molecules have proven to be valuable for studies of a wide range of basic biological processes, they have primarily been used in eukaryotic systems. This is partially because genetic tools are particularly robust in many prokaryotes systems. However, some clinically relevant bacteria are either difficult to genetically manipulate or are genetically intractable. One such pathogen, the anaerobic, Gram-positive bacterium Clostridium difficile, is highly antibiotic-resistant and frequently causes a severe infection of the colon in hospitalized patients. Another is the obligate intracellular bacterium Chlamydia trachomatis, a causative agent of the sexually transmitted disease that bears its name. Similarly, protozoan parasites are often obligate intracellular pathogens and are not easily manipulated by genetic means. Some species have poor DNA transfection efficiencies and homologous recombination rates and/or are not amenable to RNA interference for gene knockdown. Therefore, there is a clear need for alternative methods in a number of biologically and medically relevant organisms.
While small molecules have been used in the past to study microbes, they primarily have been used to kill the organism. However, with the development of more sophisticated screening methods, it is now clear that specific small molecules can be used to target discrete steps in pathogenic mechanisms of interest. Thus, small molecules must also be viewed as tools that can provide new information regarding the biochemical aspects of virulence. Small molecules therefore have great potential to shed light on the details of pathogenesis that will likely help guide our efforts to treat infectious diseases.
Using chemical genetic screens to study components of pathogenesis
In order to functionally dissect biological systems, small molecules must be able to selectively modulate the activities of specific pathway components. This is the principle behind chemical genetics, wherein chemical agents perturb the function of a given gene product with the same degree of specificity as traditional genetic manipulation. To discover suitable small molecules, high-throughput screening methods are traditionally employed. These screens typically can be divided into two classes: forward and reverse chemical genetic screens (Figure 1) (for review see (1)).
Figure 1.

Chemical genetic screens for identifying small molecules that modulate pathogenesis. a) Forward chemical genetic screen in which a high-throughput phenotypic assay is used to find small molecules that cause a desired effect on a pathogenesis phenotype (e.g. host cell invasion, death, rupture, etc.). Subsequently the protein target of a small molecule “hit” is identified. b) Reverse chemical genetic screen in which a high-throughput activity assay for a purified protein of interest is used to find compounds that alter the protein's activity in vitro. Subsequently the identified small molecules are used in a phenotypic assay to determine the role of the targeted protein.
In a forward chemical genetic screen, libraries of small molecules are added to a relevant biological system to search for a desired change in phenotype (Figure 1a). In the case of pathogenesis, examples include changes in the kinetics of invasion, evasion, or host cell death. Once a desired compound is identified based on its activity, the target of this small molecule is then identified. Forward chemical genetic screens, also known as phenotypic screens, have some notable advantages. First, no assumptions are made regarding the importance of individual elements in a pathway; the screen identifies the critical components. Second, if whole organisms are used in the screen, the “hits” are pre-selected to be cell permeable and nontoxic, thereby facilitating their use for subsequent biological assays. However, the most significant downside of phenotypic screens is that identifying and validating the target of the small molecule is often an arduous task. In an effort to address this pitfall, several techniques have been developed to aide in target identification. One recent example combines the mass spectrometry approach SILAC (stable isotope labeling with amino acids in cell culture) with binding assays (2), while another uses massively parallel sequencing to identify causative mutations in bacteria resistant to compounds of interest (3).
Reverse chemical genetic screens begin with a protein of interest and involve screening for compounds that modulate its function (Figure 1b). This often requires the target proteins to be recombinantly expressed in relatively large quantities and the development of a suitable in vitro assay to monitor its activity. Lead compounds identified in these screens are then applied to whole organisms to assess whether alterations of the protein function produce specific phenotypes. If the small molecule inactivates the protein, this technique is analogous to creating a genetic knockout. One of the primary advantages of reverse chemical genetic screens is that the target of the small molecule is already known and therefore a direct link between that protein and a cellular phenotype can be made. However, some of the most significant drawbacks of this type of screen are that lead compounds may lack cell permeability and/or specificity for the target protein, thus making in vivo phenotypic studies difficult.
Bacteria
Pathogenic mechanisms are often conserved in many related bacterial species. Therefore, compounds discovered using chemical genetic screens in microbes can often be used to identify and study basic virulence mechanisms in related organisms with minimal effort. One such mechanism that has been studied using small molecules is the type III secretion system (T3SS), which is used by many Gram-negative species of bacteria to inject pathogenic effectors into a host cell.
To identify compounds that could be used to study T3SS function, a luciferase-based high-throughput phenotypic screen was developed in Yersinia pseudotuberculosis. This screen identified a group of salicylidene acylhydrazides that block the expression and secretion of specific T3SS effector proteins at different phases of the pathway (see Table 1) (4). These compounds were subsequently used to study T3SS in Yersinia, Chlamydia, Salmonella, and Shigella spp., illustrating how the small molecule approach can be applied to study conserved mechanisms in related organisms (4-8). In contrast, a similar approach using genetic techniques would have required the creation of mutants in each organism of interest.
Table 1.
Selected compounds identified using high-throughput screens that have provided insight into mechanisms of microbial pathogenesis.
| Bacteria | |||
|---|---|---|---|
| Compound | Target/Effect | Species | Reference |
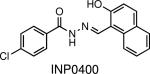 |
Abolishes in vitro virulence by inhibiting the type 3 secretion system (T3SS). | Yersinia, Salmonella, Chlamydia, and Shigella | Negrea et al., 2007; Kauppi et al., 2003; Nordfelth et al., 2005; Veenendaal et al., 2009; Muschiol et al., 2006 |
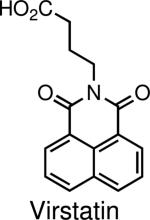 |
Inhibits the transcription factor ToxT, which ablates expression of the virulence factors cholera toxin (CT) and toxin coregulated pilus (TCP). | V. cholerae | Hung et al., 2005 |
 |
Blocks the inhibition of protein synthesis by Shiga toxin and diptheria toxin by interfering with intracellular toxin transport | C. diphtheriae and S. dysenteriae | Saenz et al., 2007 |
 |
Antagonizes or superagonizes quorum sensing. | A. tumefaciens and P. aeruginosa | Geske et al., 2007 |
| Parasites | |||
|---|---|---|---|
| Compound | Target/Effect | Species | Reference |
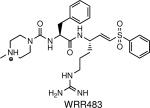 |
Inhibits cysteine protease EhCP1, which was shown to be essential for amebic invasion. | E. histolytica | Melendez-Lopez et al., 2007 |
 |
Blocks egress from red blood cells by inhibiting the serine protease PfSUB1 | P. falciparum | Yeoh et al., 2007 |
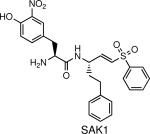 |
Blocks erythrocyte rupture by inhibiting dipeptidyl peptidase 3 (DPAP3) | P. falciparum | Arastu-Kapur et al., 2008 |
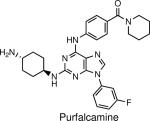 |
Blocks parasite proliferation and possibly motility and invasion by inhibiting calcium-dependant protein kinase 1 (PfCDPK1) | P. falciparum and T. gondii | Kato et al., 2008 |
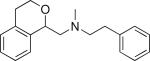 |
Inhibits parasite invasion, motility, and adhesion protein secretion | T. gondii | Carey et al., 2004 |
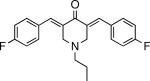 |
Inhibits parasite motility and adhesion protein secretion | T. gondii | Carey et al., 2004 |
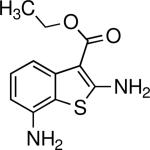 |
Enhances parasite invasion, motility, and constitutive adhesion protein secretion | T. gondii | Carey et al., 2004 |
The salicylidene acylhydrazides identified in this chemical screen have helped elucidate specific functions of the T3SS beyond its known general role in pathogenesis. In the obligate intracellular bacterium Chlamydia, inhibiting the T3SS at different stages of the infection cycle partially blocked host invasion, stopped effector secretion, and also slowed intracellular chlamydial development and replication (5). Host invasion was also inhibited in Salmonella and Shigella (6, 8). These findings were made possible by the high degree of temporal control afforded by small molecule techniques. Additionally, in Salmonella and Yersinia the inhibitors disrupt motility, reaffirming that the T3SS is related to conserved flagellar export (6, 8). However, it is unclear if the T3SS is directly involved in flagellar processing or if the compounds are also targeting the highly related flagellar secretion apparatus. For all three bacterial species, determining if and how these diverse phenotypic effects are interconnected through the T3SS will require deciphering the exact mechanisms by which each compound acts. To date some of the molecules have been shown to inhibit the T3SS by disrupting needle assembly (8). This explains the observation that, in some cases, these compounds are incapable of inhibiting pathogenesis unless they are preincubated with the bacteria during growth.
Another common pathogenic mechanism used by many bacterial species is the deployment of toxins to disrupt host processes. Toxin pathogenesis is an intricate process involving toxin expression, secretion, host uptake and trafficking, and ultimately toxin activity leading to alterations in host cell function. High-throughput chemical genetic screens can be used to discover compounds capable of disrupting this regulated process at different stages. In one such example, a phenotypic screen was used to find small molecules that attenuate toxin expression in Vibrio cholerae, a Gram-negative bacterium that results in severely infectious gastroenteritis (9). The authors found a compound (subsequently named Virstatin; see table 1) that post-translationally inhibits the activity of the virulence regulator ToxT. Because ToxT controls the transcription of the genes encoding cholera toxin (CT) and toxin coregulated pilus (TCP), Virstatin blocks the production of both proteins. Interestingly, Virstatin is capable of preventing V. cholerae from effectively colonizing mice even though the compound fails to kill bacteria in culture. This emphasizes that Virstatin targets a pathogenic mechanism that is distinct from processes required for general viability of the bacterium. More recently a luciferase-based high-throughput screen that assayed toxin-mediated inhibition of protein synthesis was used to discover compounds that inhibit the trafficking of CT and related toxins in host cells (10). These compounds modify the trafficking of the toxins in several ways, and have highlighted the distinct retrograde flow of toxins through the Golgi in the host.
In some cases, pathogenic mechanisms used by bacteria are particularly amenable to manipulation by synthetic compounds because the pathogen itself uses small molecules as key pathway regulators. This is the case for quorum sensing (QS), a method of communication used by many Gram-negative proteobacteria to sense their population density. Several species use mechanisms to trigger virulence only when appropriate colony numbers have been reached in order to maximize the chance of overwhelming the host. This communication is accomplished using N-acylated-L-homoserine lactones, and there has been a significant body of research attempting to manipulate and understand QS using chemical biology. For example, high-throughput screening was used to discover a structurally-unrelated agonist of quorum sensing in the pathogenic strain Pseudomonas aeruginosa that acts through one of the endogenous QS receptors, LasR (11). In another example, focused libraries of synthetic structural analogues of these compounds were screened to discover modulators of quorum sensing in both Pseudomonas aeruginosa and Agrobacterium tumefaciens (12). Interestingly, only minor changes to these molecules can result in both potent inhibitors as well as compounds that act as superagonists of quorum sensing (see table 1). Additionally, some compounds behave as antagonists at low concentrations and agonists at higher concentrations. Several of the identified inhibitors were found to block the production of an essential virulence factor in P. aeruginosa (12). More recently inhibitors of a QS transcription factor have also been discovered, adding to the arsenal of compounds that can be used to dissect this pathogenic mechanism (13).
Parasites
Diseases caused by obligate intracellular protozoan parasites are often the result of multiple rounds of host cell invasion, parasite replication, and host cell lysis. Understanding the mechanisms underlying each of these events is therefore paramount for dissecting pathogenesis. High throughput screens have been utilized successfully to this end. A forward chemical genetic screen involving a microscopy-based assay was used to identify compounds that modulate host invasion of Toxoplasma gondii (14). T. gondii is an highly prevalent protozoan parasite that can cause neurological damage in neonates and immunocompromised individuals.
In order to identify tools that could be used to dissect the process of T. gondii invasion, Carey and co-workers developed a screen in which fluorescent parasites were added to host cells and surface labeling of extracellular parasites was used to quantify efficiency of host cell invasion using microscopy (14). Compounds were selected for their ability to alter the ratio of extracellular to intracellular parasites. Interestingly, this high-throughput phenotypic screen not only identified compounds that inhibited host cell invasion but also compounds that enhanced the process (see table 1). Further mechanistic studies demonstrated that the compounds inhibited or activated distinct aspects of invasion and that the vast majority had some effect on parasite gliding motility. The compounds also were able to uncouple the regulatory mechanisms behind constitutive and pathogenesis-associated secretion of adhesion proteins involved in host recognition and attachment.
Although the compounds identified in this screen will be valuable new tools, the identification of the molecular targets has proven difficult. Recent efforts have included modification of one of the inhibitors in an effort to create a biotinylated version suitable for target isolation by affinity purification (15). As was the case for several of the screens discussed in bacterial systems, the small molecules identified in this study were also broadly applicable, with several of the compounds inhibiting invasion by the related apicomplexan parasite, Plasmodium knowlesi.
Small molecules have also been used to study the pathogenesis of Plasmodium falciparum, the causative agent of the most deadly form of malaria. P. falciparum multiplies within red blood cells in a compartment called the parasitophorous vacuole. In order to escape and continue the infectious cycle, P. falciparum must lyse the vacuole and subsequently the host cell. In an effort to characterize host cell rupture by this pathogen, two groups conducted forward and reverse chemical genetic screens independently. In the forward chemical genetic screen, a fluorescence-activated cell scanning (FACS) method was used to identify compounds that prevented parasites from escaping the host red blood cell (16). The authors identified a chloroisocoumarin and two peptide vinyl sulfones that were capable of inhibiting this key process (see table 1). Biotin-conjugated versions of these covalent inhibitors were used to identify the subtilisin-family serine protease PfSUB1 and dipeptidyl peptidase 3 (DPAP3), an ortholog of the cysteine protease cathepsin C as the relevant targets of the compounds. In the reverse chemical genetic screen, inhibitors of PfSUB1 were identified and used to demonstrate that inhibition of PfSUB1 activity attenuated the parasite's ability to rupture erythrocytes (17). Using their respective inhibitors, both groups identified the serine repeat antigen SERA5 as a downstream target of PfSUB1. Based on these findings the mechanisms leading to SERA5 activation and subsequent parasite egress are beginning to take shape and both proteins may represent valid therapeutic targets.
Reverse chemical genetic screens are often useful for studying the function of essential genes that cannot be knocked out. This was the case for an enzyme known as calcium-dependant protein kinase 1 (PfCDPK1) that is critical for Plasmodium falciparum pathogenesis (18). The authors attempted to knock out the kinase to determine its biological function, but it was found to be essential for viability. Gene expression patterns linked PfCDPK1 to a cluster of motility genes, and subsequently a high-throughput assay was used to identify inhibitors of the protein. A 2,6,9-trisubstituted purine named Purfalcamine (see table 1) was found to potently inhibit both recombinant PfCDPK1 activity and parasite proliferation. Cultures of P. falciparum treated with Purfalcamine arrested in the late schizont stage, and the authors speculate that the compound is most likely affecting motor processes. Similarly, incubation of the compound with T. gondii cultures resulted in blockade of parasite invasion, once again demonstrating the cross-species applicability of these compounds. Thus, using a combination of genomic analysis and chemical perturbation PfCDPK1 was found to be associated with parasite motility, which is essential for host cell invasion.
In a slightly different take on a reverse chemical genetic screen, high-throughput screens have been used to determine the substrate specificity of a target enzyme. This information can then be used to rationally design a specific inhibitor for the target of interest. This technique was used in Entamoeba histolytica, a virulent protozoan parasite that causes liver abscesses and intestinal lesions. The parasite secretes a cysteine protease termed EhCP1 that is capable of cleaving extracellular matrix in host tissues. EhCP1 is also important for host immune system evasion, as it can cleave IgG and the complement protein C3. Using recombinantly expressed EhCP1 Melendez-Lopez and co-workers used a combinatorial library of fluorogenic peptide substrates to determine the substrate specificity of this protease (19). Based on the results of this screen, a selective peptidic vinyl sulfone inhibitor named WRR483 (see table 1) was synthesized and used to monitor the activity of EhCP1 in a human colon xenograft model. Blockade of EhCP1 resulted in a complete inhibition of amebic invasion, demonstrating the necessity of this cysteine protease for parasite virulence.
Using activity-based probes to survey components of pathogenesis
Activity-based probes (ABPs) are a rapidly emerging tool for profiling the regulation of enzyme activity levels in the context of a native cellular environment (for review see (20)). ABPs make use of chemically reactive functional groups to covalently modify the active site of a target enzyme (Figure 2). They are composed of three components: an electrophilic warhead, a region that determines specificity, and a tag for visualization and/or target pulldown. Alternatively, a small surrogate tag containing a bioorthogonal reactive group can be used that allows the bulky visualization or affinity moiety to be attached in a secondary step following target labeling by the probe (21). Depending on the tag employed, enzyme activity can be profiled using a wide variety of methodologies including autoradiography, gel electrophoresis, fluorescent scanning, microarrays, LCMS, etc. (21-23). Thus far ABPs have primarily been developed for several subclasses of hydrolases, although some ligases and transferases that rely on nucleophilic residues in catalysis have also been targeted with ABPs. Because there are numerous proteases involved in both parasitic (24, 25) and bacterial (26, 27) virulence, ABPs have a great deal of potential for profiling microbial pathogenesis.
Figure 2.

a) General schematic of an activity-based probe. b) General mechanism of labeling by an activity-based probe following post-translational enzyme activation.
Several of the properties of ABPs make them ideal tools for studying pathogenesis. General ABPs can be used to simultaneously monitor changes in the activity of entire superfamilies of enzymes during different stages of pathogenesis, such as during microbial growth and interaction with the host. This can lead to the identification of new protein targets that show regulatory patterns that correlate with virulence (Figure 3a). ABPs are also useful for target validation, including identification of the targets of small molecules discovered in forward chemical genetic screens such as those discussed above. Fluorescently quenched versions of probes can also be used to visualize the activation of enzymes in real-time, making them extremely valuable for examining the spatially- and temporally-regulated process of pathogenesis (28, 29).
Figure 3.

Uses of activity-based probes in studying pathogenesis. a) Activity-based protein profiling (ABPP) can be used to identify proteins with regulatory patterns associated with microbial pathogenesis. b) Scheme of competitive ABPP, in which a general activity-based probe that labels an entire family of enzymes can be used to optimize selective inhibitors that only bind a single target protein.
In addition to assaying activity directly, ABPs can be used to develop selective inhibitors for specific enzymes using a method known as competitive activity-based protein profiling (ABPP), in which an ABP with broad specificity is added after inhibitor treatment to show residual activity of all labeled proteins (Figure 3b) (20, 30). In this way, one can determine the selectivity of an inhibitor by monitoring the disappearance of a targeted protein band after pre-treatment with the compound of interest.
Bacteria
There are currently relatively few examples of the use of activity-based probes to study bacterial virulence. This is likely to change as microbiologists and chemists realize the value of these tools to elucidate the roles of different enzymes in pathogenic mechanisms. For example, one highly relevant target that has not yet been explored in this way is the multifunctional autoprocessing repeats-in-toxin (MARTX) family of toxins (27). All MARTX toxins contain a cysteine protease domain (CPD) involved in autoproteolytic activation. These CPDs are activated after binding the small molecule inositol hexakisphosphate (InsP6) in the cytosol of eukaryotic host cells. One could therefore imagine using ABPs to monitor this interesting mode of activation more closely (31). Recently, inhibitors of the MARTX CPD have been identified that may provide a starting point for ABP development (31). Notably, the large glucosylating toxins of Clostridium difficile also contain this CPD and may therefore be able to be studied using the same or similar probes.
One example of the use of an ABP to monitor aspects of bacterial pathogenesis involved following the activity of the host cysteine protease cathepsin B (CatB) in macrophages infected with Salmonella typhimurium (32). For this study the authors synthesized an ABP with an epoxide warhead, azido-E-64 (see table 2), based on a previously constructed probe for cysteine proteases, DCG-04 (33). Azido-E-64 was used to examine the localization of active CatB in endocytic compartments following infection with Salmonella. The authors discovered that active CatB was absent from vacuoles containing Salmonella, suggesting that the bacterium is capable of inhibiting CatB activation. The authors surmise that this could be part of Salmonella's method evading host defense mechanisms (32).
Table 2.
Selected activity-based probes used to dissect mechanisms of microbial pathogenesis.
| Bacteria | |||
|---|---|---|---|
| Compound (TAG, SPECIFICITY, WARHEAD) | Target Enzyme Class | Application | Reference |
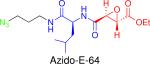 |
Cysteine proteases | Monitoring Cathepsin B activity in host macrophages during Salmonella infection. | Hang et al., 2006 |
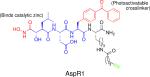 |
Metalloproteases | Identifying a role for peptide deformylase in C. trachomatis growth within host cells. | Balakrishnan et al., 2006 |
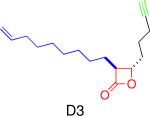 |
Various bacterial enzymes (ligases, transferases, hydrolases, oxidoreductases) | Development of a selective inhibitor for the virulence-associated bacterial protease ClpP | Böttcher et al., 2008; Böttcher et al., 2009 |
| Parasites | |||
|---|---|---|---|
| Compound (TAG, SPECIFICITY, WARHEAD) | Target Enzyme Class | Application | Reference |
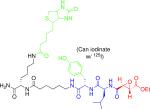 |
Cysteine proteases (clan CA) | Identifying a role for falcipain 1 in host cell invasion by P. falciparum. | Greenbaum et al., 2002 |
| Identifying a role for calpains in host cell rupture by P. falciparum and T. gondii. | Chandramohanad as et al., 2009 | ||
| Identifying enzymes in the pathogenic secretome of Shistosoma larvae. | Dvorak et al., 2008 | ||
| Development a selective inhibitors for P. Falciparum proteases DPAP1 and 3, which are involved in parasite egress. | Arastu-Kapur et al., 2008 | ||
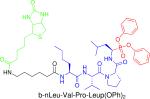 |
Serine proteases | Identifying enzymes in the pathogenic secretome of Shistosoma larvae including cercarial elastase. | Dvorak et al., 2008 |
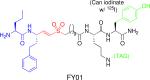 |
Specific probe for CatC and DPAPs | Development a selective inhibitors for P. Falciparum proteases DPAP1 and 3, which are involved in parasite egress. | Arastu-Kapur et al., 2008 |
An ABP was also used to help identify a factor critical for the growth of Chlamydia trachomatis within host cells (34). Two matrix metalloprotease (MMP) inhibitors were found to block chlamydial growth, and subsequent gene sequencing of resistant mutants revealed a point mutation in the promoter of peptide deformylase (PDF). PDF is an enzyme that uses zinc to remove an N-terminal formyl group from bacterial proteins after they are synthesized. A hydroxamate-based ABP termed AspR1 was used to confirm that these inhibitors were indeed targeting PDF, as incubation with the inhibitors competed with activity-based labeling of the enzyme. This study identifies a new target with clinical potential for battling C. trachomatis infection.
In another application, β-lactones and common antibiotic β-lactam scaffolds were used to create ABPs to label different bacterial enzymes in vivo (see table 2) (35-37). Competitive ABPP was subsequently employed to create selective inhibitors for one of these enzymes, ClpP (caseinolytic protein protease), a widely conserved serine protease associated with virulence in bacterial strains such as Staphylococcus aureus (35, 37). S. aureus is a widespread Gram-positive bacterium that can result in food poisoning or “toxic shock syndrome,” which is potentially fatal. A refined ClpP inhibitor was able to block ClpP activity in S. aureus in cell culture. Using the inhibitor, invasive proteolytic and hemolytic activities were confirmed to be regulated directly by ClpP in wild-type and methicillin-resistant (MRSA) strains (35). In addition, these techniques were used to demonstrate that ClpP is upstream of several critical virulence factors in pyrogenic toxin superantigen (PTSA)-producing strains of S. aureus. These included the enterotoxins SEB and SEC3 as well as toxic shock syndrome toxin (TSST-1) (37). Because this data was collected using actual clinical isolates, these studies are highly relevant to the development new therapeutic strategies.
Parasites
Several insights regarding parasitic pathogenesis have been made in the last decade using the broad-spectrum cysteine protease probe DCG-04 (33). These studies are therefore a testament to the value of activity-based protein profiling in studying microbial pathogenesis when the proper tools are available. DCG-04 has an epoxide warhead for covalently labeling the active site cysteines of papain fold proteases. In addition, the probe has a biotin affinity-tag for affinity purification and a tyrosine residue that can be iodinated for visualization (see table 2).
DCG-04 was first used in parasites to discover a role for the cysteine protease falcipain 1 in host cell invasion by P. falciparum (38). This enzyme is a good candidate for profiling with an ABP, as it cannot easily be recombinantly expressed and its transcription, and translation levels do not correlate with its activity. The authors used DCG-04 to profile the regulation of different cysteine proteases throughout the P. falciparum lifecycle. The highest levels of falcipain 1 activity were found in the merezoite stage, which is responsible for host invasion by the parasite. Subsequent competitive ABPP lead to the development of selective inhibitors for falcipain 1. When synchronized parasites were treated with the resulting falcipain 1 inhibitor, there was a decrease in new ring stage parasites but no effect on host cell rupture, implying falcipain 1 has a role in the invasion of new host cells (38).
Competitive ABPP was also used to develop selective inhibitors for DPAP1 and DPAP3 in P. falciparum (16). The authors used the broad-spectrum probes 125I-DCG-04 and FY01 to screen a small library of inhibitors (see table 2) (30, 39). One of the resulting compounds, SAK1 (see table 1), was then used to show DPAP3 likely plays a role in regulation of PfSUB1 maturation.
Recently DCG-04 was used to demonstrate a role for the host calcium-dependent proteases calpain-1 and -2 in host cell rupture by the apicomplexan parasites P. falciparum and T. gondii (40). Synchronized P. falciparum cultures arrest in the late schizont lifecycle stage immediately before rupture when incubated with DCG-04, and the authors used the probe to identify calpain-1 and show that it is active and membrane-associated with the correct timing for egress. Subsequently they used a variety of techniques to confirm that both P. falciparum and T. gondii cannot rupture and escape infected host cells that do not have active calpains. The authors suggest a model wherein these parasites co-opt the host proteases by triggering a calcium cascade that activates the calpains and causes them to remodel host cellular components that enable egress.
It is also possible to use multiple ABPs simultaneously to classify enzymes in complex proteomes involved in pathogenic processes. For example, such a strategy was used to identify secreted proteases involved in host skin penetration by Schistosoma spp. larvae, known as cercariae (41). Shistosoma, or blood flukes, cause Shistosomiasis, which is second only to malaria among globally relevant parasitic diseases. The authors used the probe b-nLeu-Val-Pro-Leu-P(OPh)2 (see table 2) to profile serine protease activity and 125I-DCG04 to profile cysteine protease activity in the secretomes of cercariae from different Schistosoma species. S. mansoni cercariae were found to primarily secrete serine proteases such as cercarial elastase. In contrast, S. japonicum cercariae secrete clan CA cysteine proteases with cathepsin B-like activity. This information is highly relevant for developing new classes of anti-parasitic agents for each of these pathogens.
Clinical benefits of using small molecules to study microbial pathogenesis
Employing small molecules to dissect mechanisms of pathogenesis gives rise to the potential for additional clinical benefits beyond providing new insights in this medically relevant field of study. It is possible that using therapeutics that target virulence pathways instead of microbial viability will put less selective pressure on a pathogen and therefore reduce the chance of resistance developing to a treatment (42-44). This approach should still be efficacious in patients with active immune systems, as virulence factors have been shown to be necessary for productive colonization of the host. It should be noted, however, that this strategy requires regulators of pathogenesis that are not directly involved in cell viability. For many bacteria, pathogenicity is encoded by mobile genetic elements and is independent from essential pathways (45). However, the benefits of targeting virulence may not apply to obligate intracellular organisms such as T. gondii and P. falciparum where pathogenesis is an innate part of the lifecycle and would likely be subject to as much selective pressure as other essential survival pathways. Nonetheless, there are potential clinical benefits to using small molecules to study pathogenesis. By using approaches such as high-throughput phenotypic screens, efforts are necessarily directed towards “druggable” targets that can modulate pathogenic phenotypes (46, 47). This head start may shorten the timeline between laboratory research and the development of successful therapeutics.
Conclusion
Many aspects of chemical manipulation are ideally suited for examining the highly spatially- and temporally-regulated processes involved in microbial pathogenesis. Small molecules are therefore the perfect complement to genetic techniques in these studies. Although currently there are relatively few examples of compounds being used to functionally dissect mechanisms of microbial virulence, this is an area poised for rapid growth. This will undoubtedly occur as more chemists and microbiologists realize the potential value of high-throughput chemical genetic screening and activity-based protein profiling in microbial systems, and the potential value of the results.
Acknowledgements
The authors thank A. Shen for critical evaluation of the manuscript and assistance with editing. This work was supported by NIH grants U54 RR020843, R01 EB005011 and R01 AI078947.
References
- 1.Smukste I, Stockwell BR. Advances in chemical genetics. Annu Rev Genomics Hum Genet. 2005;6:261–286. doi: 10.1146/annurev.genom.6.080604.162136. [DOI] [PubMed] [Google Scholar]
- 2.Ong SE, Schenone M, Margolin AA, Li X, Do K, Doud MK, Mani DR, Kuai L, Wang X, Wood JL, Tolliday NJ, Koehler AN, Marcaurelle LA, Golub TR, Gould RJ, Schreiber SL, Carr SA. Identifying the proteins to which small-molecule probes and drugs bind in cells. Proc Natl Acad Sci U S A. 2009;106:4617–4622. doi: 10.1073/pnas.0900191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nusbaum C, Ohsumi TK, Gomez J, Aquadro J, Victor TC, Warren RM, Hung DT, Birren BW, Lander ES, Jaffe DB. Sensitive, specific polymorphism discovery in bacteria using massively parallel sequencing. Nat Methods. 2009;6:67–69. doi: 10.1038/nmeth.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kauppi AM, Nordfelth R, Uvell H, Wolf-Watz H, Elofsson M. Targeting bacterial virulence: inhibitors of type III secretion in Yersinia. Chem Biol. 2003;10:241–249. doi: 10.1016/s1074-5521(03)00046-2. [DOI] [PubMed] [Google Scholar]
- 5.Muschiol S, Bailey L, Gylfe A, Sundin C, Hultenby K, Bergstrom S, Elofsson M, Wolf-Watz H, Normark S, Henriques-Normark B. A small-molecule inhibitor of type III secretion inhibits different stages of the infectious cycle of Chlamydia trachomatis. Proc Natl Acad Sci U S A. 2006;103:14566–14571. doi: 10.1073/pnas.0606412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Negrea A, Bjur E, Ygberg SE, Elofsson M, Wolf-Watz H, Rhen M. Salicylidene acylhydrazides that affect type III protein secretion in Salmonella enterica serovar typhimurium. Antimicrob Agents Chemother. 2007;51:2867–2876. doi: 10.1128/AAC.00223-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nordfelth R, Kauppi AM, Norberg HA, Wolf-Watz H, Elofsson M. Small-molecule inhibitors specifically targeting type III secretion. Infect Immun. 2005;73:3104–3114. doi: 10.1128/IAI.73.5.3104-3114.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Veenendaal AK, Sundin C, Blocker AJ. Small-molecule type III secretion system inhibitors block assembly of the Shigella type III secreton. J Bacteriol. 2009;191:563–570. doi: 10.1128/JB.01004-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hung DT, Shakhnovich EA, Pierson E, Mekalanos JJ. Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science. 2005;310:670–674. doi: 10.1126/science.1116739. [DOI] [PubMed] [Google Scholar]
- 10.Saenz JB, Doggett TA, Haslam DB. Identification and characterization of small molecules that inhibit intracellular toxin transport. Infect Immun. 2007;75:4552–4561. doi: 10.1128/IAI.00442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muh U, Hare BJ, Duerkop BA, Schuster M, Hanzelka BL, Heim R, Olson ER, Greenberg EP. A structurally unrelated mimic of a Pseudomonas aeruginosa acyl-homoserine lactone quorum-sensing signal. Proc Natl Acad Sci U S A. 2006;103:16948–16952. doi: 10.1073/pnas.0608348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geske GD, O'Neill JC, Miller DM, Mattmann ME, Blackwell HE. Modulation of bacterial quorum sensing with synthetic ligands: systematic evaluation of N-acylated homoserine lactones in multiple species and new insights into their mechanisms of action. J Am Chem Soc. 2007;129:13613–13625. doi: 10.1021/ja074135h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mattmann ME, Geske GD, Worzalla GA, Chandler JR, Sappington KJ, Greenberg EP, Blackwell HE. Synthetic ligands that activate and inhibit a quorum-sensing regulator in Pseudomonas aeruginosa. Bioorg Med Chem Lett. 2008;18:3072–3075. doi: 10.1016/j.bmcl.2007.11.095. [DOI] [PubMed] [Google Scholar]
- 14.Carey KL, Westwood NJ, Mitchison TJ, Ward GE. A small-molecule approach to studying invasive mechanisms of Toxoplasma gondii. Proc Natl Acad Sci U S A. 2004;101:7433–7438. doi: 10.1073/pnas.0307769101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans KM, Haraldsen JD, Pearson RJ, Slawin AM, Ward GE, Westwood NJ. Synthesis and chemical characterisation of target identification reagents based on an inhibitor of human cell invasion by the parasite Toxoplasma gondii. Org Biomol Chem. 2007;5:2063–2069. doi: 10.1039/b704685e. [DOI] [PubMed] [Google Scholar]
- 16.Arastu-Kapur S, Ponder EL, Fonovic UP, Yeoh S, Yuan F, Fonovic M, Grainger M, Phillips CI, Powers JC, Bogyo M. Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nat Chem Biol. 2008;4:203–213. doi: 10.1038/nchembio.70. [DOI] [PubMed] [Google Scholar]
- 17.Yeoh S, O'Donnell RA, Koussis K, Dluzewski AR, Ansell KH, Osborne SA, Hackett F, Withers-Martinez C, Mitchell GH, Bannister LH, Bryans JS, Kettleborough CA, Blackman MJ. Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell. 2007;131:1072–1083. doi: 10.1016/j.cell.2007.10.049. [DOI] [PubMed] [Google Scholar]
- 18.Kato N, Sakata T, Breton G, Le Roch KG, Nagle A, Andersen C, Bursulaya B, Henson K, Johnson J, Kumar KA, Marr F, Mason D, McNamara C, Plouffe D, Ramachandran V, Spooner M, Tuntland T, Zhou Y, Peters EC, Chatterjee A, Schultz PG, Ward GE, Gray N, Harper J, Winzeler EA. Gene expression signatures and small-molecule compounds link a protein kinase to Plasmodium falciparum motility. Nat Chem Biol. 2008;4:347–356. doi: 10.1038/nchembio.87. [DOI] [PubMed] [Google Scholar]
- 19.Melendez-Lopez SG, Herdman S, Hirata K, Choi MH, Choe Y, Craik C, Caffrey CR, Hansell E, Chavez-Munguia B, Chen YT, Roush WR, McKerrow J, Eckmann L, Guo J, Stanley SL, Jr., Reed SL. Use of recombinant Entamoeba histolytica cysteine proteinase 1 to identify a potent inhibitor of amebic invasion in a human colonic model. Eukaryot Cell. 2007;6:1130–1136. doi: 10.1128/EC.00094-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barglow KT, Cravatt BF. Activity-based protein profiling for the functional annotation of enzymes. Nat Methods. 2007;4:822–827. doi: 10.1038/nmeth1092. [DOI] [PubMed] [Google Scholar]
- 21.Sadaghiani AM, Verhelst SH, Bogyo M. Tagging and detection strategies for activity-based proteomics. Curr Opin Chem Biol. 2007;11:20–28. doi: 10.1016/j.cbpa.2006.11.030. [DOI] [PubMed] [Google Scholar]
- 22.Sieber SA, Mondala TS, Head SR, Cravatt BF. Microarray platform for profiling enzyme activities in complex proteomes. J Am Chem Soc. 2004;126:15640–15641. doi: 10.1021/ja044286+. [DOI] [PubMed] [Google Scholar]
- 23.Okerberg ES, Wu J, Zhang B, Samii B, Blackford K, Winn DT, Shreder KR, Burbaum JJ, Patricelli MP. High-resolution functional proteomics by active-site peptide profiling. Proc Natl Acad Sci U S A. 2005;102:4996–5001. doi: 10.1073/pnas.0501205102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carruthers VB, Blackman MJ. A new release on life: emerging concepts in proteolysis and parasite invasion. Mol Microbiol. 2005;55:1617–1630. doi: 10.1111/j.1365-2958.2005.04483.x. [DOI] [PubMed] [Google Scholar]
- 25.McKerrow JH, Caffrey C, Kelly B, Loke P, Sajid M. Proteases in parasitic diseases. Annu Rev Pathol. 2006;1:497–536. doi: 10.1146/annurev.pathol.1.110304.100151. [DOI] [PubMed] [Google Scholar]
- 26.Butler SM, Festa RA, Pearce MJ, Darwin KH. Self-compartmentalized bacterial proteases and pathogenesis. Mol Microbiol. 2006;60:553–562. doi: 10.1111/j.1365-2958.2006.05128.x. [DOI] [PubMed] [Google Scholar]
- 27.Satchell KJ. MARTX, multifunctional autoprocessing repeats-in-toxin toxins. Infect Immun. 2007;75:5079–5084. doi: 10.1128/IAI.00525-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blum G. Use of fluorescent imaging to investigate pathological protease activity. Curr Opin Drug Discov Devel. 2008;11:708–716. [PubMed] [Google Scholar]
- 29.Blum G, Mullins SR, Keren K, Fonovic M, Jedeszko C, Rice MJ, Sloane BF, Bogyo M. Dynamic imaging of protease activity with fluorescently quenched activity-based probes. Nat Chem Biol. 2005;1:203–209. doi: 10.1038/nchembio728. [DOI] [PubMed] [Google Scholar]
- 30.Greenbaum D, Baruch A, Hayrapetian L, Darula Z, Burlingame A, Medzihradszky KF, Bogyo M. Chemical approaches for functionally probing the proteome. Mol Cell Proteomics. 2002;1:60–68. doi: 10.1074/mcp.t100003-mcp200. [DOI] [PubMed] [Google Scholar]
- 31.Lupardus PJ, Shen A, Bogyo M, Garcia KC. Small molecule-induced allosteric activation of the Vibrio cholerae RTX cysteine protease domain. Science. 2008;322:265–268. doi: 10.1126/science.1162403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hang HC, Loureiro J, Spooner E, van der Velden AW, Kim YM, Pollington AM, Maehr R, Starnbach MN, Ploegh HL. Mechanism-based probe for the analysis of cathepsin cysteine proteases in living cells. ACS Chem Biol. 2006;1:713–723. doi: 10.1021/cb600431a. [DOI] [PubMed] [Google Scholar]
- 33.Greenbaum D, Medzihradszky KF, Burlingame A, Bogyo M. Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chem Biol. 2000;7:569–581. doi: 10.1016/s1074-5521(00)00014-4. [DOI] [PubMed] [Google Scholar]
- 34.Balakrishnan A, Patel B, Sieber SA, Chen D, Pachikara N, Zhong G, Cravatt BF, Fan H. Metalloprotease inhibitors GM6001 and TAPI-0 inhibit the obligate intracellular human pathogen Chlamydia trachomatis by targeting peptide deformylase of the bacterium. J Biol Chem. 2006;281:16691–16699. doi: 10.1074/jbc.M513648200. [DOI] [PubMed] [Google Scholar]
- 35.Bottcher T, Sieber SA. Beta-lactones as privileged structures for the active-site labeling of versatile bacterial enzyme classes. Angew Chem Int Ed Engl. 2008;47:4600–4603. doi: 10.1002/anie.200705768. [DOI] [PubMed] [Google Scholar]
- 36.Staub I, Sieber SA. Beta-lactams as selective chemical probes for the in vivo labeling of bacterial enzymes involved in cell wall biosynthesis, antibiotic resistance, and virulence. J Am Chem Soc. 2008;130:13400–13409. doi: 10.1021/ja803349j. [DOI] [PubMed] [Google Scholar]
- 37.Bottcher T, Sieber SA. Structurally refined beta-lactones as potent inhibitors of devastating bacterial virulence factors. Chembiochem. 2009;10:663–666. doi: 10.1002/cbic.200800743. [DOI] [PubMed] [Google Scholar]
- 38.Greenbaum DC, Baruch A, Grainger M, Bozdech Z, Medzihradszky KF, Engel J, DeRisi J, Holder AA, Bogyo M. A role for the protease falcipain 1 in host cell invasion by the human malaria parasite. Science. 2002;298:2002–2006. doi: 10.1126/science.1077426. [DOI] [PubMed] [Google Scholar]
- 39.Yuan F, Verhelst SH, Blum G, Coussens LM, Bogyo M. A selective activity-based probe for the papain family cysteine protease dipeptidyl peptidase I/cathepsin C. J Am Chem Soc. 2006;128:5616–5617. doi: 10.1021/ja060835v. [DOI] [PubMed] [Google Scholar]
- 40.Chandramohanadas R, Davis PH, Beiting DP, Harbut MB, Darling C, Velmourougane G, Lee MY, Greer PA, Roos DS, Greenbaum DC. Apicomplexan parasites co-opt host calpains to facilitate their escape from infected cells. Science. 2009;324:794–797. doi: 10.1126/science.1171085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dvorak J, Mashiyama ST, Braschi S, Sajid M, Knudsen GM, Hansell E, Lim KC, Hsieh I, Bahgat M, Mackenzie B, Medzihradszky KF, Babbitt PC, Caffrey CR, McKerrow JH. Differential use of protease families for invasion by schistosome cercariae. Biochimie. 2008;90:345–358. doi: 10.1016/j.biochi.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 42.Escaich S. Antivirulence as a new antibacterial approach for chemotherapy. Curr Opin Chem Biol. 2008;12:400–408. doi: 10.1016/j.cbpa.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 43.Clatworthy AE, Pierson E, Hung DT. Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol. 2007;3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 44.Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. The biology and future prospects of antivirulence therapies. Nat Rev Microbiol. 2008;6:17–27. doi: 10.1038/nrmicro1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finlay BB, Falkow S. Common themes in microbial pathogenicity revisited. Microbiol Mol Biol Rev. 1997;61:136–169. doi: 10.1128/mmbr.61.2.136-169.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hung DT, Rubin EJ. Chemical biology and bacteria: not simply a matter of life or death. Curr Opin Chem Biol. 2006;10:321–326. doi: 10.1016/j.cbpa.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 47.Morgan RE, Evans KM, Patterson S, Catti F, Ward GE, Westwood NJ. Targeting invasion and egress: from tools to drugs? Curr Drug Targets. 2007;8:61–74. doi: 10.2174/138945007779315678. [DOI] [PubMed] [Google Scholar]