

Abstract
Use of monolithic supports for enzyme immobilization rapidly expanded since we published the previous part in this series concerned with this topic almost three years ago. Many groups worldwide realized the benefits of applying monolith as a support and used a variety of techniques to immobilize many different enzymes. Although some of these new developments are a refinement of the methods developed previously, some notable new approaches have also been reported. This review summarizes the literature published since 2006 and demonstrates the broad variability of reactive monolith prepared from silica as well as from organic polymers in shapes of disks, columns, and capillaries. All these monoliths were prepared using direct formation from reactive precursors or activation of preformed inactive structures. Interestingly, most of the applications of monolithic enzyme reactors targets proteolytic digestion of proteins for proteomic analysis.
Keywords: Enzyme reactor, Monolith, Support, Immobilization, Review
1 Introduction
Although the first reported bioreactors have been prepared using enzyme immobilized on particulate supports almost 100 years ago [1], the advent of practical enzyme immobilization coincides with the development of a new field – enzyme engineering in the 1970s [2]. Despite the massive publication activity at that time, only a few commercial processes are currently carried out with immobilized enzyme as a catalyst [3,4]. The revival of immobilized enzyme reactors occurred in the past decade and a significant increase in the number of reports describing their preparation and use can be observed [5-12]. Immobilized enzymes are now founding application in a wide variety of areas. One of the most important domains of immobilized enzyme reactors is the protein digestion in proteomic research since proteolysis is a key process in studying the primary structure of proteins and can also be used for the detection of posttranslational modifications such as glycosylation, phosphorylation, methylation, etc. In addition, the application of bioreactors containing immobilized enzymes is also growing in biotechnology, chemical synthesis, and pharmaceutical industry, where these reactors enable rapid screening of enzyme inhibitor candidates as well as detailed characterization of binding interactions and reaction mechanism [13-15].
In the past, particles were dominating supports for immobilization of enzymes [16,17]. However, organic polymer and silica-based monoliths have recently become an attractive alternative for the immobilization. Initially, monolithic materials have been introduced as efficient stationary phases for liquid chromatography characterized by a rapid mass transfer due to the convective flow. Convection consequently becomes a dominant transport mechanism since the diffusion resistance to the mass transfer is almost completely avoided [18].
Supports for immobilization of biomolecules in the format of monolithic disks have emerged in the early 1990s [19] and the monolithic column formats were then used for the first time in 1996 [20]. The latter soon became a support of choice for enzyme immobilization due to their unique properties that include better accessibility of the active site for substrates, very low back pressure, stability in most solvents, and versatility of functional group available on pore surface of the monoliths. One of the most significant advantages of the monolithic enzyme reactor is also the ease of preparation in any size and shape, which only depends on the container where the polymerization process occurs. The burst of reports concerned with monolithic supports also led to publication of several excellent reviews summarizing the achievements [21-26]. In our previous paper in this series [27], we reviewed the progress in field of enzymes immobilized on monolithic supports until 2005. This review article concerns developments in the field since that time until late 2008. In order to simplify orientation in the text and easily compare the new contributions with those published previously, we again divide all the reports according to the monolithic support used for the immobilization.
2 Enzyme immobilization techniques
During pioneering studies in the 1970s, several general strategies enabling enzyme immobilization have been designed and developed. They were divided in four basic categories [28]:
a. Matrix entrapment, which prevents the enzyme from free displacement in a system since the pores of the matrix are mostly smaller that the molecule of enzyme.
b. Encapsulation, representing formation of membrane around the droplet of enzyme, which is typically in solution. The membrane must be permeable to diffusion of small substrate and product molecules.
c. Adsorption leading to formation of physical links between the enzyme molecule and the surface of the support via hydrogen bonds, hydrophobic interactions, ionic bonds, and the like.
d. Covalent bonding consisting in formation of a covalent link between the enzyme and the support carrying reactive functional groups.
Clearly, monolith can be used in all of these approaches except encapsulation.
The advantages of enzymes immobilized on the monolith over their soluble counterparts include the following: (i) one batch of enzyme can be used continuously for an extended period of time; (ii) the enzymatic reaction is readily stopped by replacing the stream of substrate solution with a neat buffer; (iii) the enzyme is stabilized by immobilization; (iv) the product of the enzymatic reaction is not contaminated with free enzyme or peptide fragments from it; (v) reaction rate is enhanced due to the high local concentration of immobilized enzyme. It is also well known that properties of the support such as size, porosity, chemistry, and mechanical strength may affect characteristics of the immobilized enzyme. Therefore, the selection of the support matrix is the key factor controlling the activity and the applicability of the resulting bioreactor.
3 Silica-based monoliths
Two different approaches for immobilization of enzymes using the silica-based monoliths have been reported recently: (i) activation of preformed monoliths followed by covalent immobilization and (ii) entrapment in the matrix.
3.1 Activated silica
The chemical modification leading to activation of monolithic silica support is most often performed in situ using silanes containing a reactive functionality such as 3-mercaptopropyl, 3-aminopropyl, and 3-glycidoxypropyl. The following enzyme immobilization is then carried out in a single reaction step.
Temporini et al. prepared a number of bioreactors based on the commercially available silica-based monolithic 100 × 4.6 mm ID silica column activated with 3-glycidoxypropyltrimethoxysilane [29-32]. The reaction path resulting in enzyme immobilization is shown in Fig. 1. Optimization of this modification reaction that provides the epoxide functionalities and represents the first step in the preparation of immobilized chymotrypsin reactor was studied at room temperature and a significantly elevated temperatures [30]. This variable was found to be critical for preserving stability of the monolithic structure. Use of a temperature exceeding 110 °C during the reaction can increase the internal pressure in the sealed monolithic column and lead to cracks in the its structure.
Fig. 1.

Activation of silica-based monolith using 3-glycidoxypropyltrimethoxy-silane and immobilization of enzyme through its amine functionalities
The activated monolithic supports were used for immobilization of proteolytic enzymes such as trypsin, chymotrypsin, and pronase. The on-line interfacing of bioreactors with HPLC/MS led to an integrated analytical system enabling protein digestion, high resolution separation of formed peptides, and their identification by ESI/MS/MS. Since the support used for immobilization may affect the amount of immobilized enzyme and its activity, the performance of trypsin bioreactors prepared using both conventional particulate and monolithic silica support was compared [29]. For example, trypsin was immobilized in situ on the epoxy-modified monolithic silica 25 × 4.6 mm ID Chromolith® Flash column and on 150 × 4.6 mm ID column packed with microparticulate epoxy-modified silica. Both bioreactors were used for a 20 min long digestion of human serum albumin and human growth hormone. No matter what column dimension and type of the silica support were used, a lower catalytic efficiency was always observed for the bioreactor prepared using silica beads. This was ascribed to the reduced accessibility of immobilized enzyme in the small pores of silica particles. It is also possible that the diffusional mass transport enabling transfer of large substrate molecules to the active site of immobilized enzyme was much slower that convective transfer operatiing in the monolithic reactor. Another important yet not mentioned reason for this lower activity can be the difference in time used for the immobilization reaction that was 24 h for packed reactor but only 4 h for the monolith. Since no inhibitor that would avoid the undesirable autodigestion of trypsin was added to the enzyme solution during the immobilization, the poor performance of the packed reactor could also be related to immobilization of partly digested proteolytic enzyme that is inactive.
The same group also investigated the kinetic characteristics of the immobilized chymotrypsin bioreactor and the effects of various parameters on enzymatic activity using a synthetic chromogenic substrate - N-benzoyl-L-tyrosine-p-nitroanilide [30]. They then used bovine serum albumin as a model protein to demonstrate the applicability of the optimized enzyme reactor to protein analysis. Specifically, albumin was digested on-line, resulting peptides were captured in a C18 trap column to achieve desalting, and then desorbed and separated using a packed C18 analytical column interfaced through an electrospray source with a mass spectrometer for the identification of molecular masses. Complete digestion of serum albumin was achieved in 120 min with a remarkably high sequence coverage of 97.3%.
The proteolytic enzymes also play an important role in the emerging area of glycoproteomics. An automated analytical approach for simultaneous characterization of both glycans and peptide moieties in pronase-generated glycopeptide mixtures has also been reported by Temporini et al. [31]. The integrated analytical system included an immobilized pronase reactor for the on-line digestion of model glycoprotein - ribonuclease B, highly hydrophobic porous graphite carbon trap column for selective enrichment of glycopeptides, normal-phase LC column for their separation, and a MS detector. The experimental setup is shown in Fig. 2.
Fig. 2.

Experimental setup integrating pronase monolithic reactor, Hypercarb trap column and normal phase LC with ESI/MS. Reproduced with permission from [31]. Copyright 2007 American Chemical Society.
Pronase E was immobilized on the epoxy-modified silica-based 50 × 4.6 mm ID Chromolith SpeedRod using the same method developed for attachment of trypsin and chymotrypsin mentioned above. Since the time needed for digestion of ribonuclease B was only 20 min, the proposed system reduces the glycoprotein analysis time from about 3 days typical of most of the traditional off-line methods, to only about 1 h.
The immobilized pronase E and trypsin reactors were also integrated into a chromatographic system used for the analysis of phosphoproteins [32]. The proteolytic digestion was carried out in an on-line mode with a selective enrichment using titanium dioxide trap column followed by the release and separation of phosphopeptides using LC/MS/MS. Since titanium dioxide exhibits a high affinity for phosphorylated peptides under acidic conditions while their release is promoted in the basic eluents, the addition of a hydrophobic trap column was necessary to collect all peptides formed in bioreactor that was run under basic pH to achieve good protein digestion. The hydrophobic trap column enabled change of the mobile phase prior to the selective isolation of phosphopeptides from the mixture. Using a gradient elution, the phosphorylated peptides were then retained in the titanium dioxide column while the non-phosphorylated peptides were separated. After switching the pH of the mobile phase to basic, the phosphorylated peptides were released from the titanium dioxide unit and separated. The feasibility of this approach was demonstrated with ESI/MSn identification of ß-casein used as a model protein and insulin-like grow factor-binding protein 1 (IGFBP-1) isolated from amniotic fluid.
Ma et al. [33] reported immobilized trypsin reactor prepared in a 100 μm ID capillary using the sol-gel method. Tetraethoxysilane and 3-aminopropyltriethoxysilane were utilized as precursors, and cetyltrimethylammonium bromide as a template. The electrostatic interaction played an important role in the formation of a desirable network. The resulting monolithic support bearing amine groups was activated using a common bifunctional reagent, glutaraldehyde, and trypsin was then covalently immobilized as shown in Fig. 3. The solution used in immobilization reaction contained trypsin, benzamidine inhibitor to minimize the autolysis of trypsin, and sodium cyanoborohydride to enhance the stability of immobilized enzyme achieved via reduction of the unstable aldimine functionality to a secondary amine. This approach saved one reaction step – aminolysis - that would be required for immobilization starting from an epoxide modified support.
Fig. 3.

Preparation of an organic-inorganic hybrid silica monolithic matrix bearing amine groups (a) and immobilization of trypsin after activation of the support with glutaraldehyde (b). Reproduced with permission from [33]. Copyright 2008 American Chemical Society.
The digestion of a decapeptide (C-myc, EQKLISEEDL) was found 6,600 fold faster with the immobilized trypsin compared to enzyme in solution. Since digestion of large proteins is more difficult than that of small substrates, the performance of the reactor was also demonstrated with digestion of myoglobin. The sequence coverage for on-column digestion was 92%, the same as in-solution. However, the residence time of myoglobin in the immobilized enzyme reactor was mere 30 s, about 1,500 times shorter than 12 h required to achieve the same degree of digestion in solution. This reactor was also used for digestion of a protein mixture consisting of cytochrome c, myoglobin, carbonic anhydrase, and BSA. The digests were collected and analyzed by LC-MS/MS.
Applicability of the microreactor for real-life proteome analysis was then demonstrated by digestion of 20 μg of proteins extracted from E. coli using a 10 cm long microreactor and LC/MS/MS. A total of 208 proteins were identified after 150 s digestion time in the immobilized enzyme reactor, while only 176 proteins were recognized after the 24 h long digestion in-solution.
3.2 Entrapment via sol-gel
Sol-gel entrapment includes forming an enzyme-doped sol and its infusion into a capillary prior to gelation. This process leads to a monolithic bed that contains enzymes entrapped within the silica matrix. An important aspect of the fabrication of such column is the ability to produce a bimodal pore size distribution consisting of both mesopores that retain the enzyme and macropores that provide channels to allow flow through of liquids at a manageable back pressure.
Kawakami et al. [15] used a two-step sol-gel method to immobilize lipase F-AP15 from Rhizopus oryzae. Lipases are versatile enzymes used for chiral syntheses and highly active when immobilized in alkyl-substituted silicates. For example, monolithic reactor was first prepared in a 50 × 1.6 mm ID poly(ether ether ketone) tube from lipase dissolved in a 4:1 mixture of methyltrimethoxysilane and tetramethoxysilane. Alternatively, the monolith was prepared from the mixture of silanes without addition of the enzyme and then the pore surface of this generic monolith was coated with a sol obtained from butyltrimethoxysilane and tetramethoxysilane mixture containing lipase. The enzyme remains entrapped in the thin layer of the formed gel. Monolith treated with lipase entrapped in the butyltrimethoxysilane layer exhibited approximately ten times higher activity than lipase immobilized on monolith derived from the methyltrimethoxysilane-based silicate thus clearly demonstrating effect of the alkyl length on the lipase activity. Another explanation of this observation includes a better accessibility of the enzyme in the thin coating since a significant portion of lipase encapsulated during the single step preparation of the immobilized enzyme is likely buried within the matrix and not accessible. Transesterification of vinyl n-butyrate with glycidol in isooctane was used as the test reaction. Enantiomers in the formed racemic ester were resolved in a commercial 400 × 4.6 mm ID CHIRALPAK AD-H column attached to the outlet of the immobilized lipase reactor.
The most frequent silica precursors used for the preparation of monoliths are alkoxysilanes such as tetraethoxysilane and tetramethoxysilane. These precursors release methanol or ethanol upon hydrolysis. However, these alcohols can be detrimental to stability of the 3-D structure of enzymes. Therefore, new biocompatible silane precursors such as diglycerylsilane, and sodium silicate as well as new preparation methods have been developed recently. These precursors enable “alcohol-free” sol-gel process and often lead to a significant improvement in the activity of the entrapped enzyme. Use of glycerylsilanes also provides an additional benefit, i.e. the solid precursors can be dissolved directly in aqueous buffers at physiological pH thus avoiding the need to alter pH during the sol-gel process. Glycerol, the byproduct evolving throughout the diglycerylsilane hydrolysis maintains the entrapped enzyme in an active state during aging of the monolithic support.
Besanger et al. [34] reported monolithic 250 μm ID capillaries containing immobilized γ-glutamyl transpeptidase and their use in enzyme reactor chromatography. Monolithic silica support was prepared form a mixture of diglycerylsilane, poly(ethylene glycol) with a molecular weight of 10,000, which controls the porous structure of the monolith, and HEPES buffer containing enzyme. They studied in detail the effects of pH of the buffer and concentration of poly(ethylene glycol) in the reaction mixture on the morphology of the monolith and enzymatic activity towards a low molecular weight substrate L-glutamic acid p-nitroanilide. Mesoporous monolithic supports afforded reactors with the highest activity.
Even more biocompatible silica monoliths were prepared from silane precursors that have covalently attached sugar moieties such as those reported by Lin et al. [35]. They fabricated a miniaturized biosensor via a single step entrapment of horseradish peroxidase using the sol-gel method. The monolithic support was prepared from precursors diglycerylsilane and sodium silicate, respectively, and a sugar containing silane, N-(3-triethoxysilylpropyl)gluconamide. Compared to the “plain” silica monolith, the embedded sugar containing moieties significantly improved the activity of immobilized horseradish peroxidase. The sensors were then used for monitoring concentration of hydrogen peroxide with a detection limit of 6 μmol/L. The sensors were found very stable since they could be reused over multiple assay cycles for a period of several weeks with no apparent loss in sensitivity.
4 Organic polymer monolith
Monoliths prepared from organic polymers used as supports for enzyme immobilization and for the preparation of bioreactors are most often formed from either acrylamide derivatives or acrylate/methacrylate-based monomers. Generally, organic polymers offer wider variability in chemistries and better biocompatibility than silica. Also organic polymers are stable under extreme pH values at which the silica-based monoliths are likely to decompose. Interestingly, most of the polymer-based supports are prepared via polymerization of monomers in the presence of a mixture of dodecanol and cyclohexanol as porogens. It is worth mentioning that these porogens have been introduced in the mid 1970s for the preparation of macroporous methacrylate beads [36], and then re-applied to the preparation of monoliths in the late 1980s [37,38]. These porogens remain very popular ever since.
4.1 Acrylamide
Duan et al. [39] prepared a monolithic trypsin reactor in a 100 μm ID capillary by first copolymerizing acrylamide, N-acryloyloxysuccinimide and ethylene dimethacrylate in the presence of the “traditional” binary porogenic mixture comprising dodecanol and cyclohexanol, and azobisisobutyronitrile as an initiator. Acrylamide was used as the inert hydrophilic monomer to adjust hydrophilicity of the monolithic support. N-acryloyloxysuccinimide offers high reactivity towards amine group of an enzyme and enables the immobilization. The authors compared enzyme activity of they microreactor with that prepared in a single step using method developed previously by Palm and Novotny [40]. The latter involves the preparation of monolithic reactor from a buffered aqueous solution comprising trypsin, acrylamide, N,N-methylenebisacrylamide, poly(ethylene glycol) and acryloyloxysuccinimide polymerized at room temperature upon initiation with N,N,N′,N′-tetramethylethylenediamine and ammonium peroxodisulfate. Both Duan's and Palm's enzyme reactors were coupled with 75 μm ID capillary column with tapered end packed with 3 μm reversed phase silica enabling the separation of peptides and transfer in MS. A high voltage was applied at the inlet of the separation column to obtain the necessary electrical potential without any need to connect an additional spray emitter to the outlet end.
Although the Novotny's support prepared in an aqueous medium exhibited enhanced hydrophilicity compared to the matrix prepared in the presence of alcohols, reactor based on the latter was more active. The reason for that appears to be a higher content of the reactive monomer - acryloyloxysuccinimide - in the monolithic support due to the better solubility of all monomers in the organic porogenic solvents. Using this tryptic reactor, cytochrome c was digested at the flow rate of 1 μL/min affording the sequence coverage of 54.81% after a residence time of 7 s.
4.2 Glycidyl methacrylate
Although monoliths prepared from polyacrylamide-based monomers presented above were found useful for enzyme immobilization because of their good biocompatibility, these soft supports are difficult to use at high flow rates due to the ease of their deformation caused by the necessary pressure. In contrast, the mechanical strength of methacrylate-based monoliths is sufficient to withstand high pressures. The supports that are often used for the preparation of enzyme reactors are based on poly(glycidyl methacrylate-co-ethylene dimethacrylate) monolith. Epoxide groups in the monolithic structure enable a variety of routes to the immobilization of an enzyme. Fig. 4. presents a sampler of immobilization techniques that were used.
Fig. 4.

Enzyme immobilization pathways using the glycidyl methacrylate-based monolithic supports: (A) direct immobilization via epoxide functionality; (B) immobilization comprising aminolysis with a diamine, activation with glutaraldehyde, reaction with the enzyme, followed by hydrogenation of the aldimine bond with cyanoborohydride; (C) immobilization comprising acid-catalyzed hydrolysis to a diol, periodate oxidation to aldehyde, and reaction with the enzyme in presence of cyanoborohydride; (D) immobilization via acid-catalyzed hydrolysis to a diol, activation of hydroxyl groups with carbonyldiimidazole, followed by reaction with an enzyme.
4.2.1 Direct immobilization via epoxy functionalities
Commercial monolithic disks called Convective Interaction Media (CIM) produced by BIA Separations (Ljubljana, Slovenia) manufactured from a porous copolymer of glycidyl methacrylate and ethylene dimethacrylate are often used for immobilization of enzymes via their reaction with the epoxide functionalities as shown in Fig. 4A. This method has been developed for immobilization of enzymes on porous glycidyl methacrylate beads already in the late 1970s [41] and works just as well for monoliths. For example, a 3 mm thin CIM disk with 12 mm in diameter having a volume of 0.34 mL was used for immobilization of trypsin and deoxyribonuclease I (DNase) [42]. Disks with a pore size of 143, 634, and 2,900 nm, respectively, were used for the preparation of bioreactors and their biological activities were determined using benzoylarginine ethyl ester and DNA as substrates, respectively. The total amounts of incorporated enzymes differed almost five-fold with 0.29 mg of immobilized trypsin and 1.37 mg of DNase. Since both enzymes have a similar size, this difference cannot be attributed to a difference in accessible surface area but rather to a different number of amino groups available in the enzyme for attachment.
Although the highest amount of DNase could be immobilized on the monolith with the smallest pores, monolith with a pore size of 634 nm exhibited the highest activity due to much better access of DNA molecules into the larger pores. Specific activity increased as the pore size decreased, which is explained by a higher number of contacts between DNA and immobilized DNase. In contrast, penetration of low molecular weight substrate such as benzoylarginine ethyl ester in smaller pores of the trypsin reactor is easier. As a result, a higher activity of immobilized trypsin was found for monolithic supports with a smaller pore size. These results demonstrate the significance of correct choice of porous properties of the support selected for enzyme immobilization that is determined by the type of substrate and its accessibility to active sites of the immobilized enzyme.
Enzymes immobilized on CIM disks have been also found useful in biotechnology processes. Single step on-line production and purification of bioactive oligogalacturonides was achieved using CIM disk with pectin lyase from Aspergillus japonicus immobilized again via epoxy groups [14]. This conjugate was used for enzymatic degradation of polygalacturonic acid to oligogalacturonides in closed circuit flow-through mode. The amount of immobilized pectin lyase was estimated to be 1.2 mg per disk. During the on-line process, a polygalacturonic acid was converted to short oligogalacturonides with a degree of polymerization of up to five in less that 8 min (Fig. 5). The immobilized enzyme exhibited reasonable operation stability and could be reused at least 10 times without any significant change in activity. The good operational parameters indicate ability of this system to be scaled up to an industrial process.
Fig. 5.

On-line production and purification of oligogalacturonides using pectin lyase immobilized on monolithic epoxy-CIM disc combined with DEAE CIM disc, with both placed together in a single housing. Polygalacturonic acid solution (20 μL, 50 mmol/L in acetate buffer, pH 5) was injected and run through the cartridge at a flow rate of 4 mL/min. Elution from the DEAE disk was achieved using a gradient of 0–1 mol/L NaCl in acetate buffer and detection by UV at 235 nm. Numbers above the peaks refer to the number of galacturonide units in the oligomer as determined by comparison with individual standards. Reproduced with permission from [14]. Copyright 2008 Elsevier.
4.2.2 Aminolysis followed by activation with dialdehyde
Although the epoxides are often used in immobilizations, their reaction rate with amine groups of proteins is slow [43]. Therefore, a modification of the epoxide groups of the support is usually carried out to increase its reactivity. More than a decade ago, we demonstrated a multi-step in situ modification and immobilization process shown Fig. 4B that includes aminolysis of epoxide ring, activation with glutaraldehyde followed by enzyme immobilization, and reduction of the C=N bond [20]. Although we have meanwhile abandoned this tedious multistep process, it still remains a vital tool for other groups to immobilize enzymes and peptidic affinants.
Considering the high cost and difficulty facing over-expression, isolation and purification of enzymes from recombinant sources, the use of immobilized enzymes represents an extremely useful approach to preserve the activity of the small amount of enzyme typically available as well as to perform the kinetic and inhibition studies. For example, human recombinant acetylcholineesterase was immobilized on a EDA-CIM disk. EDA-CIM disk is a monolithic poly(glycidyl methacrylate-co-ethylene dimethacrylate) support obtained by reacting its epoxy groups with ethylenediamine leading to support containing amine functionalities. The EDA-CIM disk is first activated using glutaraldehyde, followed by reaction with acetylcholineesterase forming the Schiff base linkage that is then reduced by cyanoborohydride to a stable amine. This reactor was used for rapid evaluation of the thermodynamic and kinetic parameters and the mechanism of action of selected reversible inhibitors such as tacrine, donepezil, edrophonium, and ambenonium [13]. On-line studies performed by inserting the immobilized enzyme reactor in a HPLC system allowed evaluating the mechanism of action and determining the inhibition constant used then as key information to rational design of new drug candidates for treatment of Alzheimer's disease. Mere nanomoles of the immobilized enzyme have been used to perform more than 2,200 runs and the reactor has been in daily use for over seven months while maintaining most of its initial activity.
The same monolithic support and immobilization method were also applied to immobilization of human recombinant ß-secretase, which is a ß-site amyloid precursor protein cleaving enzyme (BACE-1). This protein is another enzyme associated with the pathogenesis of Alzheimer's disease [44]. The activity and kinetic parameters of the immobilized BACE-1 were investigated by combining the reactor with HPLC/MS system using JMV2236 peptide as substrate. Inhibition of the enzyme was demonstrated by simultaneous injection of donepezil and pepstatin A as reference inhibitors together with the peptidic substrate. Results of this inhibition study corresponded with those derived from the conventional method involving soluble enzyme. However, the immobilized counterpart exhibited both increased efficiency and stability, and enabled a significant decrease in time required for analysis. The long term stability demonstrated in these experiments qualifies the monolithic reactors for fast and reliable screening and detailed characterization of new drugs developed by pharmaceutical industry.
A similar monolithic support was prepared by copolymerization of acrylamide, glycidyl methacrylate, and ethylene dimethacrylate in the presence of dodecanol and cyclohexanol as porogens [45]. This monolithic support was again activated via the typical approach: first, it was treated with aqueous ammonia solution, then activated with glutaraldehyde to enable immobilization of trypsin and, finally, the aldimine link was reduced with a hydride. Digestion of a small protein cytochrome c using this column followed by an off-line CE or LC/MS/MS separation and detection of peptides afforded a sequence coverage of 57.7%, which is less impressive than that obtained by others [46-48]. The inclusion of hydrophilic acrylamide in the matrix was found to enhance activity of the tryptic reactor in experiments involving a low molecular weight substrate benzoylarginine ethylester. Although the activity determined for this substrate is not very relevant to that useful for digestion of large proteins, it indicated an improvement over activity of trypsin in solution. This study also claims an improvement in enzyme activity upon use of an aqueous-organic solvent. Digestion in a 20% aqueous ammonium carbonate-acetonitrile solution led to 62.5% peptide coverage and was better than 50% observed in the sole aqueous buffer with both values obtained after about 8 min residence time in the reactor.
In contrast, Zhang et al. [49] made the monolithic support more hydrophobic by copolymerizing glycidyl methacrylate, ethylene dimethacrylate, and butyl methacrylate again in the presence of the most popular porogenic mixture of cyclohexanol and dodecanol. Immobilization method based on glutaraldehyde activation was used again for the preparation of a dual function monolithic column containing immobilized trypsin. To further improve digestion efficiency of low abundant proteins, a C4 alkyl group was introduced into the monolith to combine on-column protein enrichment and digestion. This trypsin reactor was evaluated for bovine serum albumin digestion and analysis of digest in off-line mode by LC/ESI/MS and MALDI/MS, respectively. Compared with immobilized trypsin reactor prepared using support with no butyl groups, the trypsin immobilized on more hydrophobic monolith afforded according to the authors an improved digestion efficiency enabling identification of approximately 30% more peptide sequence. In addition, a significant increase in peak intensities was observed for peptides obtained by digestion in the reactor containing butyl methacrylate support. This reactor also exhibited good tolerance to organic solvents and maintained reproducible enzymatic activity for at least 30 days.
Most of the proteins, and in particular globular proteins, have to be denaturized before enzymatic digestion because of the inaccessibility of their internal peptide bonds to the active site of the proteolytic enzyme. Typically, high concentrations of denaturizing agent such as 8 mol/L urea or 6 mol/L guanidine hydrochloride are used. Feng et al. reported that immobilization of trypsin significantly improved its stability under denaturizing conditions and demonstrated an increase in peptide coverage in bovine serum albumin digest as the concentration of urea grew to 8 mol/L (Table 1) [50]. Unfortunately, no explanation of this observation is offered. These authors also used the dual mobile phase approach in which the digestion is carried out in an aqueous buffer at pH 8.1 and the separation in the reversed phase mode with MS detection using acetonitrile-water at pH less than 3. They found that activity of the tryptic reactor did not change even after several cycles of the mobile phases thus confirming our earlier findings [51].
While most of the reports on digestion of proteins using immobilized trypsin bioreactor demonstrate its performance with peptide mapping of small standard proteins, application of these monolithic microreactors for shotgun proteome analysis remains scarce. Zou's group prepared an immobilized tryptic reactor using again the aminolyzed poly(glycidyl methacrylate-co-ethylene dimethacrylate) monolith activated with glutaraldehyde and coupled this reactor with LC/MS/MS. They then digested the Saccharomyces cerevisiae yeast cell extract and identified a total of 1578 unique peptides corresponding to 541 proteins using mere 590 ng of original proteins and an incubation time of only 1 min [50].
4.2.3 Hydrolysis of epoxide groups followed by oxidation
The acid catalyzed hydrolysis of epoxide group of glycidyl methacrylate copolymer to a diol followed by its oxidation with periodate leading to polymer with aldehyde functionalities has been developed a long time ago [52] and later extended to enzyme immobilization [53]. This approach shown in Fig. 4C was recently used by Dovichi's group to functionalize poly(glycidyl methacrylate-co-ethylene dimethacrylate) monolith and immobilize pepsin [54]. The immobilized pepsin reactor was then integrated into a sophisticated, fully automated multi-dimensional system depicted in Fig. 6 designed for bottom-up protein analysis. Since the transfer of both charged proteins and peptides through the system occurs due to their electrophoretic mobility, no mechanical pumping is necessary. The concept appears simple. The protein mixture is first separated in CE mode. The individual proteins are then digested to peptides in immobilized pepsin reactor directly attached to the end of the separation capillary. This less specific proteolytic enzyme was used for digestion since it operates in acidic environment and allows application of a volatile acetic acid-ammonium acetate buffer. The peptides are then directed to a second CE capillary and once separated their mass is characterized by MS. The feasibility of this complex system for the desired application was successfully verified using a mixture of cytochrome c and myoglobin injected on just a few picomole level. The peak capacity of the first dimension, which is the separation of proteins, was 18.5, while it was 32 for the separation of peptides in the second dimension thus affording an overall peak capacity of 590. The respective sequence coverage of 48% and 22% for cytochrome c and myoglobin is acceptable taking into account the less than optimized conditions used in the study. An added benefit of this system is the sequential operation that can accelerate the process. While peptides generated in the previous digestion are separated in the second CE capillary, the next protein mixture can be digested in the microreactor since the voltage and thus the mobility in each unit is controlled independently. Although interesting, further tuning is necessary to obtain peak capacity comparable with those obtained in 2-D LC systems.
Fig. 6.

Experimental setup of the CE-immobilized pepsin reactor-CE-MS/MS analysis system. Reproduced with permission from [54]. Copyright 2007 American Chemical Society.
4.2.4 Hydrolysis of epoxides followed by activation with carbonyldiimidazole
Yet another approach to activation of epoxide groups typical of poly(glycidyl methacrylate-co-ethylene dimethacrylate) monolith has been developed by BIA Separations a few years ago and first used for the immobilization of trypsin and DNase [55]. This technique shown in Fig. 4D involves acid catalyzed hydrolysis followed by reaction with carbonyldiimidazole. Recently, ribonuclease was immobilized using this method in order to prepare reactor enabling removal of RNA from DNA. This purification step is critical to obtain DNA pure enough for gene therapy and vaccination [56]. Ribonuclease was immobilized either directly onto the epoxy-CIM disk monoliths or using the carbonyldiimidazole approach. The latter technique afforded a reactor with a six-fold higher activity determined with a low molecular weight substrate. Significant increase in activity was attributed to the difference in amount of accessible reactive groups at the pore surface within the support. This monolithic disk ribonuclease reactor was then tested in RNA degradation with detection of the residual RNA using agarose gel electrophoresis. The results shown in Fig. 7 revealed that immobilized ribonuclease reactor degraded RNA very efficiently. The complete removal of RNA from the mixture was also confirmed by reverse transcription coupled with polymerase chain reaction (PCR), which is an extremely sensitive method for detecting RNA. PCR product of RNA contaminant was detected only in untreated sample indicating that immobilized ribonuclease was able to efficiently remove all RNA. It should be emphasized that the residence time needed to achieve this result was only about 30 s at flow rates exceeding 1 column volume per min.
Fig. 7.

Removal of RNA from DNA sample with using ribonuclease immobilized on carbonyldiimidazole activated CIM disk. Mixture of total RNA and genomic DNA isolated from Aspergillus niger was injected in ribonuclease reactor (two injections each marked with +) and control reactor (marked −). Eluents from reactors were collected and the presence of RNA in genomic DNA samples was detected using 1.6% agarose gel electrophoresis and staining with ethidium bromide. Positions of genomic DNA (gDNA) and RNA (23S, 16S and 5S rRNA) are indicated at the right site of the electrophoretic image. Reproduced with permission from [56]. Copyright 2007 Elsevier.
4.3 Vinylazlactone copolymers
In the 1990s, 3M company used 1-vinyl-4,4-dimethylazlactone to prepare porous polymer beads formed via an inverse suspension process [57]. Heilmann et al. described evolvement of this chemistry in an excellent review [58]. These materials exhibit reactivity to amine and thiol groups and, among other applications, these reactive beds were also used for immobilization of various enzymes leading to conjugates featuring an enhanced activity [59,60]. Our early experiments carried out in 1999 led to poly(1-vinyl-4,4-dimethylazlactone-co-acrylamide-co-ethylene dimethacrylate) monoliths prepared by thermally initiated polymerization. These supports were found very suitable for immobilization of trypsin [61]. Later, we extended the choice of techniques and added functionalization of inert methacrylate monoliths via photografting as an option to the direct copolymerization [47,51,62]. The amine functionalities of enzymes react readily with the azlactone groups of the support in a ring opening reaction shown in Fig. 8 that leads to attachment of the protein through a dipeptide spacer, which positively affects the activity of the biocatalyst.
Fig. 8.

Scheme of the enzyme immobilization via azlactone functionality
Recently, we copolymerized 2-vinyl-4,4-dimethylazlactone and ethylene dimethacrylate in presence of 1,4-butanediol and 1-propanol as porogens to obtain reactive monolithic support. Using this monolith, we then prepared a capillary reactor with immobilized pepsin [63]. Although the reaction of azlactone functionalities is known to be sluggish at acidic pH, the immobilization of pepsin was carried out at pH 5 anyway to avoid the irreversible denaturing of enzyme that would occur at pH values exceeding 6 that would be favorable for the immobilization process. Additional benefit of this procedure is reduced activity of pepsin at this pH allowing immobilization in absence of an inhibitor that would otherwise be needed to prevent autodigestion. This immobilized pepsin reactor was connected to monolithic poly(lauryl methacrylate-co-ethylene dimethacrylate) column enabling preconcentration and separation of peptide fragments prior to the ESI/MS detection. Since pepsin is rapidly denatured by organic solvents, a 6-port switching valve was inserted between the reactor and the separation column to bypass the enzymatic reactor during the separation cycle and avoid contact of pepsin with acetonitrile containing mobile phase used for the reversed phase separation. Myoglobin, albumin and hemoglobin were digested in this system to demonstrate feasibility of the concept of two monoliths in-line. A typical chromatogram of peptides obtained by the digestion of myoglobin and separation of peptides exhibits 11 distinct peaks is shown in Fig. 9. This methodology combined with mass spectrometric detection of molar masses allowed identification of 15 peptides.
Fig. 9.
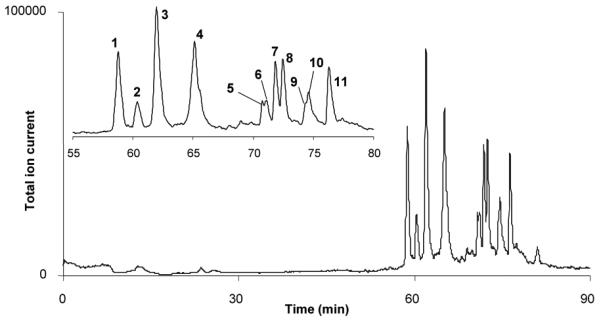
Separation of peptides from the in-line digestion of 2 pmol of myoglobin in 150×0.1 mm ID immobilized pepsin reactor using a 200×0.1 mm ID monolithic poly(lauryl methacrylate-co-ethylene dimethacrylate) column. Reproduced with permission from [63]. Copyright 2008 Elsevier.
In another project, we immobilized several enzymes in different, well-defined regions of monolith [64] using the previously developed photopatterning method [65]. Photoinitiated grafting of selected monomers through a photomask or photopatterning enables activation of the support located in a capillary only in defined areas. This concept was demonstrated with tandem immobilization of multiple enzymes. We used monolithic support prepared from a mixture consisting of butyl methacrylate, ethylene dimethacrylate, 1-decanol, and cyclohexanol in a 100 μm ID UV-transparent capillary [64]. To reduce non-specific adsorption of enzymes on the generic hydrophobic monolithic support, its pore surface was first modified by grafting with poly(ethylene glycol methacrylate), followed by surface activation via second step photografting with 2-vinyl-4,4-dimethylazlactone and immobilization of enzymes. The schematic diagram of the photopatterning and immobilization process is illustrated in Fig. 10.
Fig. 10.

Schematic diagram of the photopatterning process: (A) protein is immobilized to the surface of a polymer monolith in patterned region within a capillary; (B) PEG is grafted to the entire surface of the polymer monolith to prevent nonspecific protein adsorption. Then, vinyl azlactone is photopatterned onto the PEG surface through a mask and activates the surface is selected region for protein immobilization. Reproduced with permission from [64]. Copyright 2007 American Chemical Society.
Sequential multi-enzymatic reactions were demonstrated using the patterned assembly of two and three enzymes. Glucose oxidase and horseradish peroxidase were first patterned in separate regions of a single monolith. A solution of dextrose and Amplex Red was pumped through the reactor. Glucose oxidase oxidized dextrose while producing hydrogen peroxide later used by peroxidase for oxidation of Amplex Red and formation of fluorescent product – resorufin. The fluorescence of the eluent was quantified as a function of speed and flow direction. A significant resorufin formation occurred only when the substrate was introduced in the glucose oxidase to peroxidase direction. Then, another enzyme was added and a three-enzyme sequential reaction was performed using immobilized invertase, glucose oxidase, and peroxidase and a mixture of saccharose and Amplex Red as substrates. All six possible arrangements of the three enzymes were tested, but significant formation resorufin formation was only observed when the enzymes were in the correct sequential order [64].
5 Conclusions
This review covers works that have been produced in only about two years. Yet the number of papers published during this short period of time was quiet large. Some trends we see from analysis of the reports are clear: (i) Monolithic supports for immobilization of enzymes are gaining a significant attention in the academic circles. They enable fabrication of reactors with high local concentration of the enzyme and excellent mass transport characteristics thus considerably accelerating the enzyme catalyzed reactions; (ii) Majority of the studies concerns proteolytic enzymes for peptide mapping since their immobilization eliminates autodigestion during application and a rapid loss in activity; (iii) A substantial proportion of the supports are those that have been developed previously and were now used for new applications. It is very likely that monolithic support will enjoy high popularity even in the years to come since they offer a number of advantages not achievable with other means. Unfortunately, a broad use of these immobilized enzymes in routine applications in both academic and industrial labs is not yet in sight since neither the monolithic supports nor the enzymes immobilized on them are commercially available. Another distinct problem that needs to be addressed is a difficulty in comparing results from different laboratories. Some attempts to unify the evaluation of enzymes immobilized on monolithic supports have already been published [66]. However, it looks like that these recommendations are not yet widely accepted.
Acknowledgments
The work carried out at the Molecular Foundry was supported by the Director, Office of Science, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering, of the US Department of Energy under Contract No. DE-AC02-05CH11231. Support by a grant of the National Institute of General Medical Sciences, National Institutes of Health (GM-48364) is also gratefully acknowledged.
References
- 1.Nelson JM, Griffin EG. J. Am. Chem. Soc. 1916;38:1109–1115. [Google Scholar]
- 2.Messing RA. Immobilized Enzymes for Industrial Reactors. Academic Press; New York: 1975. [Google Scholar]
- 3.Chibata I, Tosa T. Applied Biochemistry and Bioengineering. 1976;1:329–357. [Google Scholar]
- 4.Chibata I. Advances in Molecular and Cell Biology. 1996;15A:151–160. [Google Scholar]
- 5.Arroyo M. Ars Pharmaceut. 1998;39:111–127. [Google Scholar]
- 6.Cao L, van Langen L, Sheldon RA. Curr. Opin. Biotechnol. 2003;14:387–394. doi: 10.1016/s0958-1669(03)00096-x. [DOI] [PubMed] [Google Scholar]
- 7.Turkiewicz M, Makowski K. Biotechnologia. 2004:113–128. [Google Scholar]
- 8.Novick SJ, Rozzell JD. Methods in Biotechnology. 2005;17:247–271. [Google Scholar]
- 9.Cao L. Curr.Opin. Chem. Biol. 2005;9:217–226. doi: 10.1016/j.cbpa.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 10.Brena BM, Batista-Viera F. Methods in Biotechnology. 2006;22:15–30. [Google Scholar]
- 11.Wang P. Curr. Opin. Biotechnol. 2006;17:574–579. doi: 10.1016/j.copbio.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 12.Mateo C, Palomo JM, Fernandez-Lorente G, Guisan JM, Fernandez-Lafuente R. Enzyme Microb. Technol. 2007;40:1451–1463. [Google Scholar]
- 13.Bartolini M, Cavrini V, Andrisano V. J. Chromatogr. A. 2007;1144:102–110. doi: 10.1016/j.chroma.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 14.Delattre C, Michaud P, Vijayalakshmi MA. J. Chromatogr. B. 2008;861:203–208. doi: 10.1016/j.jchromb.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 15.Kawakami K, Abe D, Urakawa T, Kawashima A, Oda Y, Takahashi R, Sakai S. J. Sep. Sci. 2007;30:3077–3084. doi: 10.1002/jssc.200700309. [DOI] [PubMed] [Google Scholar]
- 16.Mosbach K. Immobilized Enzymes. Academic Press; New York: 1976. [Google Scholar]
- 17.Mosbach K. Immobilized Enzymes. Academic Press; New York: 1987. [Google Scholar]
- 18.Svec F, Tennikova TB, Deyl Z, editors. Monolithic materials: preparation, properties, and applications. Elsevier; Amsterdam: 2003. [Google Scholar]
- 19.Abou-Rebyeh H, Koerber F, Schubert-Rehberg K, Reusch J, Josic D. J. Chromatogr. 1991;566:341–350. doi: 10.1016/0378-4347(91)80250-g. [DOI] [PubMed] [Google Scholar]
- 20.Petro M, Svec F, Fréchet JMJ. Biotech. Bioeng. 1996;49:355–363. doi: 10.1002/(SICI)1097-0290(19960220)49:4<355::AID-BIT1>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 21.Girelli AM, Mattei E. J. Chromatogr. B. 2005;819:3–16. doi: 10.1016/j.jchromb.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 22.Massolini G, Calleri E. J. Sep. Sci. 2005;28:7–21. doi: 10.1002/jssc.200401941. [DOI] [PubMed] [Google Scholar]
- 23.Josic D, Buchacher A. J. Biochem. Biophys. Meth. 2001;49:153–174. doi: 10.1016/s0165-022x(01)00195-6. [DOI] [PubMed] [Google Scholar]
- 24.Krenkova J, Foret F. Electrophoresis. 2004;25:3550–3563. doi: 10.1002/elps.200406096. [DOI] [PubMed] [Google Scholar]
- 25.Ma J, Zhang L, Liang Z, Zhang W, Zhang Y. J. Sep. Sci. 2007;30:3050–3059. doi: 10.1002/jssc.200700362. [DOI] [PubMed] [Google Scholar]
- 26.Ma J, Zhang L, Liang Z, Zhang W, Zhang Y. Anal. Chim. Acta. 2008 [Google Scholar]
- 27.Svec F. Electrophoresis. 2006;27:947–961. doi: 10.1002/elps.200500661. [DOI] [PubMed] [Google Scholar]
- 28.Wingard LB, editor. Enzyme Engineering. Interscience; New York: 1972. pp. 15–23. [Google Scholar]
- 29.Temporini C, Perani E, Mancini F, Bartolini M, Calleri E, Lubda D, Felix G, Andrisano V, Massolini G. J. Chromatogr. A. 2006;1120:121–131. doi: 10.1016/j.chroma.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 30.Temporini C, Calleri E, Campese D, Cabrera K, Felix G, Massolini G. J. Sep. Sci. 2007;30:3069–3076. doi: 10.1002/jssc.200700337. [DOI] [PubMed] [Google Scholar]
- 31.Temporini C, Perani E, Calleri E, Dolcini L, Lubda D, Caccialanza G, Massolini G. Anal. Chem. 2007;79:355–363. doi: 10.1021/ac0611519. [DOI] [PubMed] [Google Scholar]
- 32.Temporini C, Dolcini L, Abee A, Calleri E, Galliano M, Caccialanza G, Massolini G. J. Chromatogr. A. 2008;1183:65–75. doi: 10.1016/j.chroma.2007.12.091. [DOI] [PubMed] [Google Scholar]
- 33.Ma J, Liang Z, Qiao X, Deng Q, Tao D, Zhang L, Zhang Y. Anal. Chem. 2008;80:2949–2956. doi: 10.1021/ac702343a. [DOI] [PubMed] [Google Scholar]
- 34.Besanger TR, Hodgson RJ, Green JRA, Brennan JD. Anal. Chim. Acta. 2006;564:106–115. doi: 10.1016/j.aca.2005.12.066. [DOI] [PubMed] [Google Scholar]
- 35.Lin T-Y, Wu C-H, Brennan JD. Biosensors and Bioelectronics. 2007;22:1861–1867. doi: 10.1016/j.bios.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 36.Svec F, Hradil J, Coupek J, Kalal J. Angew. Macromol. Chem. 1975;48:135–143. [Google Scholar]
- 37.Tennikova TB, Svec F, Belenkii BG. J. Liquid Chromatogr. 1990;13:63–70. [Google Scholar]
- 38.Svec F, Fréchet JMJ. Anal. Chem. 1992;54:820–822. [Google Scholar]
- 39.Duan J, Sun L, Liang Z, Zhang J, Wang H, Zhang L, Zhang W, Zhang Y. J. Chromatogr. A. 2006;1106:165–174. doi: 10.1016/j.chroma.2005.11.102. [DOI] [PubMed] [Google Scholar]
- 40.Palm A, Novotny MV. Rapid Comm. Mass Spectr. 2004;18:1374–1382. doi: 10.1002/rcm.1500. [DOI] [PubMed] [Google Scholar]
- 41.Turkova J, Blaha K, Malanikova M, Vancurova J, Svec F, Kalal J. Biochim. Biophys. Acta. 1978;524:162–169. doi: 10.1016/0005-2744(78)90114-6. [DOI] [PubMed] [Google Scholar]
- 42.Bencina K, Bencina M, Podgornik A, Strancar A. J. Chromatogr. A. 2007;1160:176–183. doi: 10.1016/j.chroma.2007.05.034. [DOI] [PubMed] [Google Scholar]
- 43.Drobnik J, Saudek V, Svec F, Kalal J, Vojtisek V, Barta M. Biotech. Bioeng. 1979;21:1317. doi: 10.1002/bit.260210802. [DOI] [PubMed] [Google Scholar]
- 44.Mancini F, Naldi M, Cavrini V, Andrisano V. J. Chromatogr. A. 2007;1175:217–226. doi: 10.1016/j.chroma.2007.10.047. [DOI] [PubMed] [Google Scholar]
- 45.Duan J, Liang Z, Yang C, Zhang J, Zhang L, Zhang W, Zhang Y. Proteomics. 2006;6:412–419. doi: 10.1002/pmic.200500234. [DOI] [PubMed] [Google Scholar]
- 46.Qu H, Wang H, Huang Y, Wei Z, Lu H, Kong J, Yang P, Liu B. Anal. Chem. 2004;76:6426–6433. doi: 10.1021/ac049466g. [DOI] [PubMed] [Google Scholar]
- 47.Peterson DS, Rohr T, Svec F, Fréchet JMJ. J. Proteome Res. 2002;1:563–568. doi: 10.1021/pr0255452. [DOI] [PubMed] [Google Scholar]
- 48.Krenkova J, Bilkova Z, Foret F. J. Sep. Sci. 2005;28:1675–1684. doi: 10.1002/jssc.200500171. [DOI] [PubMed] [Google Scholar]
- 49.Zhang K, Wu S, Tang X, Kaiser NK, Bruce JE. J. Chromatogr. B. 2007;849:223–230. doi: 10.1016/j.jchromb.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 50.Feng S, Ye M, Jiang X, Jin W, Zou H. J. Proteome Res. 2006;5:422–428. doi: 10.1021/pr0502727. [DOI] [PubMed] [Google Scholar]
- 51.Peterson DS, Rohr T, Svec F, Fréchet JMJ. Anal. Chem. 2002;74:4081–4088. doi: 10.1021/ac020180q. [DOI] [PubMed] [Google Scholar]
- 52.Svec F, Hrudkova H, Kalal J. Angew. Macromol. Chem. 1978;70:101–108. [Google Scholar]
- 53.Svec F, Kalal J, Menyaylova I, Nakhapetyan LA. Biotech. Bioeng. 1978;20:1319. [Google Scholar]
- 54.Schoenherr RM, Ye M, Vannatta M, Dovichi NJ. Anal. Chem. 2007;79:2230–2238. doi: 10.1021/ac061638h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bencina K, Podgornik A, Strancar A, Bencina M. J. Sep. Sci. 2004;27:811–818. doi: 10.1002/jssc.200401800. [DOI] [PubMed] [Google Scholar]
- 56.Bencina M, Babic J, Podgornik A. J. Chromatogr. A. 2007;1144:135–142. doi: 10.1016/j.chroma.2006.12.083. [DOI] [PubMed] [Google Scholar]
- 57.Coleman PL, Walker MM, Milbrath DS, Stauffer DM, Rasmussen JK, Krepski R, Heilmann SM. J. Chromatogr. A. 1990;512:345–363. [Google Scholar]
- 58.Heilmann SM, Rasmussen JK, Krepski LR. J. Polym. Sci., Part A: Polym. Chem. 2001;39:3655–3677. [Google Scholar]
- 59.Drtina GJ, Haddad LC, Rasmussen JK, Gaddam BN, Williams MG, Moeller SJ, Fitzsimons RT, Fansler DD, Buhl TL, Yang YN, Weller VA, Lee JM, Beauchamp TJ, Heilmann SM. React. Funct. Polym. 2005;64:13–24. [Google Scholar]
- 60.Heilmann SM, Drtina GJ, Haddad C, Rasmussen JK, Gaddam BN, Liu JJ, Fitzsimons RT, Fansler DD, Vyvyan JR, Yang YN, Beauchamp TJ. J. Molec. Catal. B: Enzym. 2004;30:33–42. [Google Scholar]
- 61.Xie S, Svec F, Fréchet JMJ. Biotech. Bioeng. 1999;62:30–35. doi: 10.1002/(sici)1097-0290(19990105)62:1<30::aid-bit4>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 62.Peterson DS, Rohr T, Svec F, Fréchet JMJ. Anal. Chem. 2003;75:5328–5635. doi: 10.1021/ac034108j. [DOI] [PubMed] [Google Scholar]
- 63.Geiser L, Eeltink S, Svec F, Fréchet JMJ. J. Chromatogr. A. 2008;1188:88–96. doi: 10.1016/j.chroma.2008.02.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Logan TC, Clark DS, Stachowiak TB, Svec F, Fréchet JMJ. Anal. Chem. 2007;79:6592–6598. doi: 10.1021/ac070705k. [DOI] [PubMed] [Google Scholar]
- 65.Rohr T, Hilder EF, Donovan JJ, Svec F, Fréchet JMJ. Macromolecules. 2003;36:1677–1684. [Google Scholar]
- 66.Jiang HH, Zou HF, Wang HL, Ni JY, Zhang Q, Zhang YK. J. Chromatogr. 2000;903:77–84. doi: 10.1016/s0021-9673(00)00846-3. [DOI] [PubMed] [Google Scholar]