

Abstract
Background
TLR4 signaling mediates early inflammation after cold I/R. We hypothesized that the TLR4 co-receptor CD14, the intracellular adaptor proteins MyD88 and TRIF would be required for cold I/R induced inflammation. HMGB1 is a putative endogenous activator of TLR4. Therefore, we also assessed the contribution of HMGB1 in cold I/R induced inflammation.
Methods
Syngeneic heart transplants were performed in mice deficient in CD14, MyD88, TRIF, or wild-type mice. In other experiments, anti-HMGB1 neutralizing antibody or control IgG was administered at reperfusion. Donor hearts were subjected to 2 hours of cold ischemia and retrieved after 3 hours of reperfusion.
Results
After cold I/R, grafts revealed striking translocation of HMGB1 out of the nucleus in cardiac myocytes. Administration of an anti-HMGB1 neutralizing antibody resulted in reduced systemic IL-6 and TNFα and ICAM-1 mRNA levels (p≤0.05). Compared to controls, CD14KO mice exhibited significantly lower (p≤0.05) systemic IL-6 and JE/MCP-1 levels after cold I/R. Intra-graft TNFα and IL-1β mRNA levels were also significantly lower (p≤0.05) in CD14KO grafts. MyD88KO mice exhibited significantly lower (p≤0.05) systemic IL-6 levels compared to control mice after cold I/R. Intra-graft TNFα, IL-6, and ICAM1 mRNA levels were also significantly lower (p≤0.05) in MyD88KO grafts. Significantly lower levels (p≤0.05) of serum IL-6, MCP-1 as well as intragraft TNFα, IL-6, IL-1β, and ICAM1 were observed after cold I/R in TRIF deficient animals compared to controls.
Conclusions
CD14, MyD88, TRIF, and HMGB1 contribute to the inflammatory response that occurs after cold I/R. These results provide insight into the mechanisms of TLR4-mediated inflammation after cold I/R.
Keywords: Toll-like receptors, ischemia/reperfusion, transplant, inflammation, cold storage
Introduction
Toll-like receptors (TLRs) are a family of molecules that play a critical role in innate immunity. Members of this family of evolutionarily conserved transmembrane receptors serve as pattern recognition receptors (PRRs), which recognize conserved microbial motifs in molecules such as bacterial lipopolysaccharide (LPS), peptidoglycan, flagellin, unmethylated CpG DNA, double and single stranded RNA, and others(1,2). Activation of the corresponding TLR by any these molecules results in an inflammatory response, alerting the host to the presence of microbial invasion and initiating an immune response. Recent observations demonstrate that some TLR family members also alert the host to the presence of tissue damage and become activated by endogenous molecules released from damaged or ischemic tissues(2-4). Heparan sulfate, hyaluronic acid, fibrinogen, high mobility group box 1 (HMGB1), heat shock proteins, oxidized phospholipids and other endogenous molecules have been shown to initiate inflammatory pathways through TLR4 (5-13).
Accordingly, recent evidence has implicated TLR4 as a central mediator of inflammation and organ injury after I/R. Specifically, TLR4 has been shown to play a role in models of hepatic (14-18), pulmonary (19), brain(20,21), and renal (22) warm I/R injury. Furthermore, mice deficient in TLR4 signaling demonstrated reduced infarct sizes and diminished inflammatory responses in models of regional warm myocardial I/R mimicking myocardial infarction (23-25). Inhibition of TLR4 with a soluble inhibitor in the context of myocardial warm I/R resulted in similar findings(26). At least one study has also demonstrated that TLR4 signaling influences myocardial dysfunction after warm ischemic injury(27).
While these studies have clearly implicated TLR4 as an important mediator of inflammation and organ injury after warm I/R, it is well known that the molecular events that occur in the setting of cold I/R are markedly different. We have previously demonstrated that TLR4 signaling on both donor and recipient cell types plays a central role in mediating the robust early inflammatory response that occurs after cold I/R, as in the setting of solid organ transplantation(28). A remarkable dependency on TLR4 has also been reported in a model of hepatic injury after cold preservation and transplantation(29). Together, these studies strongly implicate TLR4 as a mediator of inflammation and organ injury after cold I/R.
The precise molecular mechanisms of TLR4 signaling have not been entirely elucidated and are currently under investigation. It has been well established, however, that CD14 cooperates with TLR4 at the cell surface to detect the presence of bacterial LPS(30). Further, it is known that after stimulation with LPS, TLR4 signaling is mediated by two distinct intracellular adaptor proteins: one known as myeloid differentiation factor 88 (MyD88) and another known as TIR domain-containing-adaptor inducing IFNβ (TRIF)(31,32). MyD88 and TRIF activate distinct, but partially overlapping intracellular signaling cascades that ultimately trigger an inflammatory response.
HMGB1 is an endogenous molecule originally identified as a DNA binding protein(33,34). It was later demonstrated that HMGB1 also serves as a late mediator of lethality in sepsis(35). Subsequent studies have demonstrated that neutralizing antibodies against HMGB1 ameliorate the inflammatory response and organ injury in several models of sterile injury including hemorrhagic shock(36), femur fracture(37), and hepatic warm I/R(18). In vitro studies have suggested that HMGB1 binds to the TLR4 signaling complex and activates TLR4(38,39). Other recent findings suggest that the interaction of HMGB1 with other molecules may be required for activity(40). Therefore, HMGB1 mobilized from the nucleus may serve as a key integrator of inflammatory signaling in both infection and injury.
Understanding the precise molecular events that trigger the early inflammatory response in the setting of cold I/R will be critical to ultimately developing therapeutics aimed at ameliorating this response, which may ultimately help prevent organ injury and possibly facilitate immune tolerance in solid organ transplantation. While our previous results implicate TLR4 as a central mediator of inflammation after cold I/R, it is unclear whether CD14 is also required to mediate this response in the setting of cold I/R. Furthermore, it is unknown whether TLR4 utilizes either the adaptor MyD88, the adaptor TRIF, or both of these adaptor molecules to mediate intracellular signaling in the setting of cold I/R. Lastly, the endogenous ligands that activate TLR4 in the setting of cold I/R have not been characterized.
In order to better understand the specific molecular pathways utilized by TLR4 in the setting of cold I/R, we hypothesize that the co-receptor CD14, the adaptor proteins MyD88 and TRIF, as well as the endogenous molecule HMGB1 contribute to the early inflammatory response after cold I/R in a murine cardiac transplant model. In this study, we utilized a heterotopic murine cardiac transplant model to test this hypothesis and accordingly found that each of these molecules plays a role in the inflammatory response that occurs in the setting cold I/R.
Materials and Methods
Animals
Male wild-type (C57/Bl6) mice (8-12 weeks old) were purchased from the Jackson Laboratory (Bar Harbor, ME). CD14-/- (CD14KO) were provided by Dr. Mason Freeman. MyD88-/- (MyD88KO) mice were provided by Dr. Ruslan Medzhitov. TRIF deficient (TrifLPS2/LPS2) mice were provided by Dr. Bruce Beutler. All mutant strains were backcrossed at least 10 times onto a C57/Bl6 background. Animals utilized in all experiments were maintained in laminar flow cages in a specific pathogen-free atmosphere at the University of Pittsburgh (Pittsburgh, PA). A standard diet and water were provided ad libitum. MyD88 deficient mice were provided water supplemented with trimethoprim (0.032 mg/ml) and sulfamethoxazole (0.16 mg/ml) until 8 weeks of age. Antibiotics were stopped for at least two weeks prior to use in experiments. Experimental protocols were approved by the Animal Care and Use Committee of the University of Pittsburgh and all experiments were performed in adherence to the National Institutes of Health Guidelines for the use of laboratory animals.
Heterotopic heart transplantation
Heterotopic heart transplantation was performed in wild-type and mutant strains as previously described (41). To summarize, hearts were procured with cold University of Wisconsin (UW) (Viaspan, Du Pont, Wilmington DE) solution and placed in cold UW solution for 2 hours at 4° C for preservation. Heart transplantation was performed in a heterotopic position by anastomosing the graft aorta and pulmonary artery to the recipient abdominal aorta and inferior vena cava (IVC) respectively. Each anastomosis was performed in an end-to-side fashion. At 3 hours post-reperfusion, grafts were subsequently harvested for analysis. Neutralizing anti-HMGB1 antibody was prepared as previously described (42). Intraperitoneal administration of 600 μg of either anti-HMGB1 neutralizing antibody (provided by Dr. Kevin J. Tracey) or control IgG (Sigma) was performed in experiments as indicated.
Serum analysis
At 3h post-reperfusion, blood samples were obtained from animals via IVC puncture. Serum was collected and stored at -80 °C until analysis was performed. Serum levels of cytokines, including IL-6 and JE/MCP-1, were measured using enzyme-linked immunosorbent assays (ELISAs) performed according to the manufacturer's instructions (R&D Systems; Minneapolis, Minnesota). In our previous study, the greatest differences among groups were observed when serum IL-6 and JE/MCP-1 were measured(28). For this reason, these cytokines were chosen for analysis in the current study.
SYBR Green real-time RT-PCR
Tissues were homogenized with a rotor-stator homogenizer and treated with Proteinase K (Qiagen, Valencia, CA) at 55° C for 20 min. RNA was prepared by utilizing a silica-gel based membrane method using the RNeasy Midi Kit (Qiagen) according to the manufacturer's instructions. An on-column DNase digestion using RNase-free DNase (Qiagen) was performed to rid the samples of genomic DNA. One μg of RNA was used to generate cDNA using oligo dT primers (Qiagen) and Omniscript (Qiagen) reverse transcriptase. PCR reaction mixtures were prepared using SYBR Green PCR master mix (PE Applied Biosystems, Foster City, CA). SYBR Green two-step real-time RT-PCR for TNFα, IL-6, IL-1β, ICAM-1, and β-actin was performed as described. All samples were run in duplicate. The level of gene expression for each sample was normalized to β-actin mRNA expression using the comparative Ct method(43).
Histopathology and Immunohistochemistry
Graft tissues were frozen in OCT (Optimal Cold Temperature; Sakura Finetek, Inc, Torrance, CA). The OCT-embedded tissues were then cut into 6 μm sections. Sections were then stained via immunohistochemistry.
Six μm sections on glass slides were rehydrated with two washes of phosphate buffered saline (PBS). After 3 washes with 0.5% bovine serum albumin (BSA) in PBS, non-specific binding was blocked with 2% BSA. After another wash with 0.5% BSA in PBS, rabbit anti-HMGB1 (Abcam, Cambridge, MA) antibody at a 1:100 dilution in 0.5% BSA was then applied and allowed to incubate for 60 min. at room temperature. The slides were washed with 0.5% BSA, and a secondary antibody (goat anti–rabbit Alexa Fluor 488, 1:500 in 0.5% BSA; Invitrogen) was applied. After washing, the slides were rinsed with PBS and coverslips were applied with mounting media (21 g polyvinylalcohol, 52 ml water, sodium azide, 106 ml 0.2-M Tris buffer). Slides were dried overnight and then visualized with a confocal microscope (Fluoview 1000; Olympus). Images were analyzed in a blinded fashion.
Statistical analysis
Results are expressed as mean +/- SEM. Student's t test was used for statistical analysis.
Results
HMGB1 is an endogenous molecule that contributes to the early inflammatory response after cold I/R
The endogenous molecule HMGB1 is predominantly confined to the nucleus of cells under normal conditions. In order to assess whether HMGB1 might act as a systemic inflammatory mediator in the setting of cold I/R, we first evaluated whether the sub-cellular localization of HMGB1 was altered after cold I/R using immunohistochemistry (Figure 1). In hearts from unmanipulated animals, HMGB1 was predominantly confined to the nucleus. However, after cold preservation and reperfusion, there was dramatic loss of HMGB1 from the nucleus of cardiomyocytes in the graft.
Figure 1. HMGB1 translocates out of the nucleus after cold I/R.
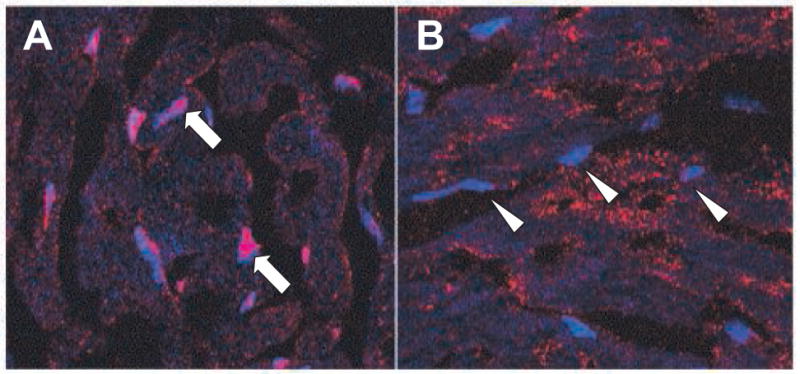
Unmanipulated hearts were procured from wild-type animals (A) Grafts that had undergone 2 hours of cold ischemia prior to transplantation were harvested after 3 hours of reperfusion (B). Frozen tissues were sectioned, and then stained using an anti-HMGB1 antibody (red), and counter-stained with a nuclear marker (DRAQ5, blue). Single arrows are used denote nuclei with intense staining for HMGB1. Arrowheads highlight nuclei with markedly less HMGB1 staining. Images representative of 3 independent experiments are shown. Magnification, 100×.
To further evaluate the role of HMGB1 as an inflammatory mediator in the setting of I/R, in a separate set of experiments, a polyclonal anti-HMGB1 neutralizing antibody was administered at the time of reperfusion. Administration of a neutralizing antibody against HMGB1 at the time of reperfusion resulted in significantly lower systemic levels of IL-6 after I/R compared to animals that received control IgG, although this difference was modest (Figure 2A). Significantly lower levels of intragraft TNFα and ICAM-1 mRNA were also observed in grafts from animals that received anti-HMGB1 antibody as compared to those that received control IgG (Figure2C, F). Trends toward lower levels of serum JE/MCP-1, as well as intragraft, IL-6, and IL-1β mRNA were also observed, but these differences did not reach statistical significance.
Figure 2. HMGB1 promotes systemic and intra-graft inflammation after cardiac cold IR.

Transplants were performed between wild-type mice. At the time of reperfusion, either a control IgG antibody or anti-HMGB1 antibody was administered (n= 4-6 per group). Serum and grafts were harvested after 3 hours of reperfusion. Serum levels of IL-6 (A) and JE/MCP-1 (B) were measured by ELISA. Intra-graft mRNA levels of TNFα (C), IL-6 (D), IL-1β (E), and ICAM-1 (F) were measured by quantitative RT-PCR. All values were normalized to transcript levels of β-actin. Results are displayed as mean ± SE, *p≤ 0.05 vs. control IgG transplant by student's t-test.
CD14 contributes to systemic and intragraft inflammation after cold I/R
Even in the absence of adaptive immunity, a systemic inflammatory response occurs early after cold I/R(44). Our previous data demonstrate that TLR4 is an essential component of the systemic and intra-graft inflammatory responses that occur in the setting of cold I/R(28). In order to determine whether the TLR4 co-receptor CD14 also plays a role in mediating this response, we performed syngeneic, heterotopic cardiac transplants in mice deficient in CD14 and their wild-type counterparts (C57/Bl6). As expected, in wild-type mice receiving cold-preserved cardiac grafts from wild-type mice, serum IL-6 levels were significantly elevated above known baseline levels. However, this response was significantly blunted in CD14 deficient mice that received hearts from CD14 deficient animals (Figure 3A). Similar results were obtained when the cytokine JE/MCP-1 was measured (Figure 3B).
Figure 3. CD14 contributes to systemic and intragraft inflammation after cardiac cold IR.

Serum samples were collected from both wild-type→wild-type and CD14 mutant→mutant animals (n =4-6 samples per group) 3 hours after reperfusion. Serum levels of (A) IL-6 and (B) JE/MCP-1 were measured by ELISA. Transplanted cardiac grafts were retrieved from both wild-type→wild-type and CD14 mutant→mutant animals (n =4-6 samples per group) 3 hours after reperfusion. Intragraft mRNA transcript levels of (C) TNFα, (D) IL-6 (E) IL-1β, and (F) ICAM-1 were measured by RT-PCR and normalized to β-actin transcript levels. Relative mRNA levels compared to the wild-type control group are displayed. Results are displayed as mean ± SE, *p≤ 0.05 vs. wild-type→wild-type transplant by student's t-test.
Quantitative RT-PCR was employed to measure the mRNA levels of intra-graft inflammatory cytokines that have been previously demonstrated to be upregulated after cold I/R. Intra-graft TNF mRNA levels were upregulated in transplanted grafts in the wild-type→wild-type group compared to control hearts. However, this response was blunted in transplanted hearts from the CD14 deficient group (Figure 3C). Similar results were observed when IL-1β mRNA levels were measured (Figure 3E). The leukocyte adhesion molecule ICAM-1 is also upregulated after I/R and directs leukocyte trafficking into tissues after reperfusion(45,46). ICAM-1 mRNA levels were dramatically up-regulated in wild-type grafts after transplant, while CD14 KO grafts exhibited significantly less ICAM-1 mRNA upregulation (Figure 3F). A trend towards lower levels of intragraft IL-6 mRNA in CD14 KO grafts compared to controls was observed, but this difference did not reach statistical significance.
MyD88 mediates systemic and intragraft inflammation after cold I/R
MyD88 is an intracellular adaptor protein that mediates signaling for most known TLRs, with the exception of TLR3. Upon activation, TLR4 signaling through the MyD88-dependent pathway leads to MAP kinase and NF-kB activation, resulting in pro-inflammatory cytokine production. MyD88 deficient animals exhibited substantially less inflammatory cytokine production and release, leukocyte infiltration in ischemic kidneys, and organ injury as measured by serum creatinine in a model of renal warm I/R(22). Further, blocking MyD88 signaling utilizing an adenoviral vector expressing a dominant negative mutant MyD88 resulted in reduced cardiomyocyte apoptosis, less NF-kB activation, and smaller infarct size compared to controls in a model of myocardial infarct model(47).
Based on these observations, we hypothesized that MyD88 would mediate the early inflammatory response observed after cold I/R. In order to test this hypothesis, we performed syngeneic, heterotopic cardiac transplants in MyD88 deficient mice and wild-type mice. We observed significantly lower levels of serum IL-6 in MyD88 deficient animals after cold I/R compared to wild-type controls (Figure 4A). There was also a clear trend toward lower levels of JE/MCP-1 after I/R in MyD88 deficient mice compared to wild-type mice (Figure 4B). TNFα, IL-6, and ICAM-1 mRNA levels were also lower in MyD88 deficient grafts compared to wild-type grafts (Figure 4C, D, F). In contrast, there was no appreciable difference in IL-1β mRNA levels in MyD88 deficient grafts compared to wild-type grafts (Figure 4E).
Figure 4. MyD88 mediates systemic and intra-graft inflammation after cardiac cold IR.

Serum samples were collected from both wild-type→wild-type and MyD88 mutant→mutant animals (n =4-6 samples per group) 3 hours after reperfusion. Serum levels of (A) IL-6 and (B) JE/MCP-1 were measured by ELISA. Transplanted cardiac grafts were retrieved from both wild-type→wild-type and MyD88 mutant→mutant animals (n =4-6 samples per group) 3 hours after reperfusion. Intragraft mRNA transcript levels of (C) TNFα, (D) IL-6 (E) IL-1β, and (F) ICAM-1 were measured by RT-PCR and normalized to β-actin transcript levels. Relative mRNA levels compared to the wild-type control group are displayed. Results are displayed as mean ± SE, *p≤ 0.05 vs. wild-type→wild-type transplant by student's t-test.
TRIF mediates systemic and intragraft inflammation after cold I/R
Among TLRs, TLR4 is unique as it can utilize the both the adaptor MyD88 and the adaptor TRIF. The role of TRIF-mediated signaling in cold I/R has not been previously investigated. To evaluate whether the adaptor TRIF also plays a role as a mediator of the early inflammatory response after cold I/R, we performed syngeneic, heterotopic cardiac transplants in mice deficient in TRIF and their wild-type counterparts. After cold I/R, serum levels of both IL-6 and JE/MCP-1 were significantly lower in TRIF deficient mice compared to wild-type mice (Figure 5A, B). There were also significantly lower levels of intragraft TNFα, IL-6, IL-1β and ICAM-1 mRNA in grafts from TRIF deficient animals compared to wild-type animals (Figure 5 C-F).
Figure 5. TRIF also mediates systemic and intra-graft inflammation after cardiac cold IR.

Serum samples were collected from both wild-type→wild-type and TRIF mutant→mutant animals (n =4-6 samples per group) 3 hours after reperfusion. Serum levels of (A) IL-6 and (B) JE/MCP-1 were measured by ELISA. Transplanted cardiac grafts were retrieved from both wild-type→wild-type and TRIF mutant→mutant animals (n =4-6 samples per group) 3 hours after reperfusion. Intragraft mRNA transcript levels of (C) TNFα, (D) IL-6 (E) IL-1β, and (F) ICAM-1 were measured by RT-PCR and normalized to β-actin transcript levels. Relative mRNA levels compared to the wild-type control group are displayed. Results are displayed as mean ± SE, *p≤ 0.05 vs. wild-type→wild-type transplant by student's t-test.
Discussion
The process of cold preservation of organs and subsequent reperfusion is of central importance in solid organ transplantation. The mechanisms of warm and cold ischemic injury, which have been reviewed in detail elsewhere, are complex and overlapping(48-51). In the ischemic phase of I/R injury, cellular anoxia results in a decrease in mitochondrial adenosine triphosphate production. The resultant energy deficit results in intracellular acidosis, cellular swelling, and increased intracellular calcium concentrations. Eventually, necrotic cell death ensues. While these processes occur to some extent during both warm and cold ischemia, cold preservation markedly decreases the rate at which these processes occur. Accordingly, experimental models have demonstrated that brief episodes of warm ischemia induce similar amounts of tissue damage as more prolonged periods of cold ischemia(52).
During reperfusion, an inflammatory response occurs and results in further tissue injury after both warm and cold ischemia. This phase is characterized by tissue infiltration by neutrophils and macrophages, cytokine release, and complement deposition. Endothelial activation and dysfunction also occurs, along with platelet activation and activation of the coagulation cascade. Another difference between warm and cold ischemia include increased susceptibility of endothelial cells to cell death during cold ischemia(53). Reperfusion also results in tissue damage through the production of reactive oxygen species. The magnitude of the inflammatory response and resultant tissue injury that occurs during reperfusion is in part dependent upon the degree of tissue injury during the anoxic phase and varies between warm and cold ischemia(54). Another unique feature of cold ischemia is cold-induced apoptosis, where apoptosis is triggered by the accumulation of chelatable iron that occurs during cold ischemia(55).
Our previous results demonstrated that the systemic and intra-graft inflammatory responses that occur in the setting of cold I/R are markedly reduced in the absence of TLR4 signaling. The data presented here yield insight into the mechanisms of TLR4-mediated signaling in the context of cold I/R. Specifically, these data implicate the TLR4 co-receptor CD14 and the TLR adaptor proteins MyD88 and TRIF as mediators of the inflammatory response after cold I/R. In addition, these data also indicate that HMGB1, one endogenous molecule thought to contribute to TLR4 signaling also contributes to cold I/R mediated inflammation.
Our observation that CD14 is involved in cold cardiac I/R induced inflammation is unique for a sterile injury model. CD14 is a GPI-linked surface molecule shown to be required for the recognition of LPS by the TLR4/MD2 complex. CD14 may also bind to oxidized lipids and therefore may be involved in inflammatory signaling in the setting of I/R. This dependency on CD14 may be unique to the heart, as we found no difference in I/R induced injury or cytokine levels when CD14-/- mice were subjected to warm I/R(18).
In a renal warm I/R model, there was significantly less pro-inflammatory cytokine production and cellular infiltration observed in kidneys from MyD88-deficient mice when compared to wild-type counterparts(22). In addition, by blocking the MyD88 dependent pathway with an adenoviral construct carrying a dominant-negative MyD88, Hua and colleagues found that inhibition of MyD88 resulted in less NF-kB activation, less cardiac myocyte apoptosis, and smaller infarct sizes in a myocardial warm I/R model(47). In contrast, previous observations by Zhai and colleagues suggest that TLR4 signaling after liver warm I/R is mediated through MyD88 independent mechanisms(17). Instead, these investigators implicated TRIF-dependent signaling by showing that mice deficient for IRF3 were protected from warm I/R induced injury. IRF3 is a transcription factor that is downstream of TRIF. Our results indicate that both MyD88 and TRIF play a role in the inflammatory response that occurs after cold I/R.
It should be noted that all known TLRs with the exception of TLR3 utilize MyD88. TLR3 exclusively utilizes the adaptor TRIF. TLR4 is unique among TLRs, as it utilizes both TRIF and MyD88-dependent intracellular signaling pathways. The central involvement of TLR4 as a key regulator of cold cardiac I/R induced inflammation is a one possible explanation for the involvement participation of both MyD88 and TRIF in our model. We have previously shown that both donor and recipient TLR4 contribute to cytokine production in cold cardiac I/R. Therefore it is possible that cells of different origins or types selectively utilize these TLR adaptors. Furthermore, we have not excluded roles for other TLRs in the inflammatory response to cold I/R. It is noteworthy that we have seen no role for TLR2 in the inflammatory cytokine production or organ injury seen in warm liver I/R(18).
Evidence is accumulating that suggests TLR activation plays an important role in promoting graft rejection. Administration of TLR agonists can abrogate the effects of costimulatory blockade and prevent tolerance induction(56-58). It is interesting to note that in the absence of MyD88, transplantation of skin grafts across minor histocompatibility antigens is possible(59). However, both TRIF and MyD88 must be absent to permit transplantation across major histocompatibility barriers. Recent in vitro evidence correlates with these findings by demonstrating that activation of MyD88- and TRIF-dependent pathways together accounts for maximal dendritic cell maturation in response to LPS(60).
Previous studies involving models of hepatic warm I/R have demonstrated a more prominent role for HMGB1 than what we have observed in our current study(18). This may indicate that other ligands, such as heparan sulfate, HSPs, fibrinogen, hyaluronan or oxidized phospholipids may play a more critical role as endogenous triggers of inflammation after cold I/R. Alternatively, organ specific differences may be accountable for this apparent difference.
These results provide insights into the mechanisms of TLR4-mediated inflammation after cold I/R. A better understanding of these mechanisms may ultimately lead to the development of therapeutics aimed at preventing graft injury and facilitating immunologic tolerance by ameliorating the inflammatory response that occurs in the setting of organ transplantation after cold I/R.
Acknowledgments
This work was supported by National Institutes of Health grants GM053789 (TRB) and GM050441 (TRB). D. J. Kaczorowski and R. Vallabhaneni are each recipients of American College of Surgeons Resident Research Scholarships.
Abbreviations
- BSA
bovine serum albumin
- HMGB-1
high mobility group box-1
- ICAM-1
intercellular adhesion molecule-1
- IL
interleukin
- I/R
ischemia-reperfusion
- IVC
inferior vena cava
- KO
knock-out
- LPS
lipopolysaccharide
- MCP-1
monocyte chemotractant protein-1
- MyD88
myeloid differentiation factor 88
- PBS
phosphate buffered saline
- PRR
pattern recognition receptor
- ROS
reactive oxygen species
- RT-PCR
reverse transcription-polymerase chain reaction
- TLRs
toll-like receptors
- TNF
tumor necrosis factor
- TRIF
TIR domain-containing-adaptor inducing IFNβ
- μm
micrometers
- UW
University of Wisconsin
References
- 1.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Miyake K. Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin Immunol. 2007;19:3–10. doi: 10.1016/j.smim.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Kaczorowski DJ, Mollen KP, Edmonds R, Billiar TR. Early events in the recognition of danger signals after tissue injury. J Leukoc Biol. 2008;83:546–552. doi: 10.1189/jlb.0607374. [DOI] [PubMed] [Google Scholar]
- 4.Mollen KP, Anand RJ, Tsung A, Prince JM, Levy RM, Billiar TR. Emerging paradigm: toll-like receptor 4-sentinel for the detection of tissue damage. Shock. 2006;26:430–437. doi: 10.1097/01.shk.0000228797.41044.08. [DOI] [PubMed] [Google Scholar]
- 5.Tang AH, Brunn GJ, Cascalho M, Platt JL. Pivotal advance: endogenous pathway to SIRS, sepsis, and related conditions. J Leukoc Biol. 2007;82:282–285. doi: 10.1189/jlb.1206752. [DOI] [PubMed] [Google Scholar]
- 6.Brunn GJ, Bungum MK, Johnson GB, Platt JL. Conditional signaling by Toll-like receptor 4. FASEB J. 2005;19:872–874. doi: 10.1096/fj.04-3211fje. [DOI] [PubMed] [Google Scholar]
- 7.Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–17084. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 9.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 10.Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001;167:2887–2894. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- 11.Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 12.Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–C924. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 13.Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van LG, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen XD, Ke B, Zhai Y, Gao F, Busuttil RW, Cheng G, et al. Toll-like receptor and heme oxygenase-1 signaling in hepatic ischemia/reperfusion injury. Am J Transplant. 2005;5:1793–1800. doi: 10.1111/j.1600-6143.2005.00932.x. [DOI] [PubMed] [Google Scholar]
- 15.Wu HS, Zhang JX, Wang L, Tian Y, Wang H, Rotstein O. Toll-like receptor 4 involvement in hepatic ischemia/reperfusion injury in mice. Hepatobiliary Pancreat Dis Int. 2004;3:250–253. [PubMed] [Google Scholar]
- 16.Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, et al. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol. 2005;175:7661–7668. doi: 10.4049/jimmunol.175.11.7661. [DOI] [PubMed] [Google Scholar]
- 17.Zhai Y, Shen XD, O'Connell R, Gao F, Lassman C, Busuttil RW, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004;173:7115–7119. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 18.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201:1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimamoto A, Pohlman TH, Shomura S, Tarukawa T, Takao M, Shimpo H. Toll-like receptor 4 mediates lung ischemia-reperfusion injury. Ann Thorac Surg. 2006;82:2017–2023. doi: 10.1016/j.athoracsur.2006.06.079. [DOI] [PubMed] [Google Scholar]
- 20.Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- 21.Cao CX, Yang QW, Lv FL, Cui J, Fu HB, Wang JZ. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem Biophys Res Commun. 2007;353:509–514. doi: 10.1016/j.bbrc.2006.12.057. [DOI] [PubMed] [Google Scholar]
- 22.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chong AJ, Shimamoto A, Hampton CR, Takayama H, Spring DJ, Rothnie CL, et al. Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J Thorac Cardiovasc Surg. 2004;128:170–179. doi: 10.1016/j.jtcvs.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 24.Oyama J, Blais C, Jr, Liu X, Pu M, Kobzik L, Kelly RA, et al. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–789. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 25.Hua F, Ha T, Ma J, Li Y, Kelley J, Gao X, et al. Protection against myocardial ischemia/reperfusion injury in TLR4-deficient mice is mediated through a phosphoinositide 3-kinase-dependent mechanism. J Immunol. 2007;178:7317–7324. doi: 10.4049/jimmunol.178.11.7317. [DOI] [PubMed] [Google Scholar]
- 26.Shimamoto A, Chong AJ, Yada M, Shomura S, Takayama H, Fleisig AJ, et al. Inhibition of Toll-like receptor 4 with eritoran attenuates myocardial ischemia-reperfusion injury. Circulation. 2006;114:I270–I274. doi: 10.1161/CIRCULATIONAHA.105.000901. [DOI] [PubMed] [Google Scholar]
- 27.Cha J, Wang Z, Ao L, Zou N, Dinarello CA, Banerjee A, et al. Cytokines link Toll-like receptor 4 signaling to cardiac dysfunction after global myocardial ischemia. Ann Thorac Surg. 2008;85:1678–1685. doi: 10.1016/j.athoracsur.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaczorowski DJ, Nakao A, Mollen KP, Vallabhaneni R, Sugimoto R, Kohmoto J, et al. Toll-like receptor 4 mediates the early inflammatory response after cold ischemia/reperfusion. Transplantation. 2007;84:1279–1287. doi: 10.1097/01.tp.0000287597.87571.17. [DOI] [PubMed] [Google Scholar]
- 29.Shen XD, Ke B, Zhai Y, Gao F, Tsuchihashi S, Lassman CR, et al. Absence of toll-like receptor 4 (TLR4) signaling in the donor organ reduces ischemia and reperfusion injury in a murine liver transplantation model. Liver Transpl. 2007;13:1435–1443. doi: 10.1002/lt.21251. [DOI] [PubMed] [Google Scholar]
- 30.Akashi S, Ogata H, Kirikae F, Kirikae T, Kawasaki K, Nishijima M, et al. Regulatory roles for CD14 and phosphatidylinositol in the signaling via toll-like receptor 4-MD-2. Biochem Biophys Res Commun. 2000;268:172–177. doi: 10.1006/bbrc.2000.2089. [DOI] [PubMed] [Google Scholar]
- 31.Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524–1525. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 32.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 33.Javaherian K, Liu JF, Wang JC. Nonhistone proteins HMG1 and HMG2 change the DNA helical structure. Science. 1978;199:1345–1346. doi: 10.1126/science.628842. [DOI] [PubMed] [Google Scholar]
- 34.Bustin M, Hopkins RB, Isenberg I. Immunological relatedness of high mobility group chromosomal proteins from calf thymus. J Biol Chem. 1978;253:1694–1699. [PubMed] [Google Scholar]
- 35.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 36.Yang R, Harada T, Mollen KP, Prince JM, Levy RM, Englert JA, et al. Anti-HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol Med. 2006;12:105–114. doi: 10.2119/2006-00010.Yang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levy RM, Mollen KP, Prince JM, Kaczorowski DJ, Vallabhaneni R, Liu S, et al. Systemic inflammation and remote organ injury following trauma require HMGB1. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1538–R1544. doi: 10.1152/ajpregu.00272.2007. [DOI] [PubMed] [Google Scholar]
- 38.Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 39.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 40.Sha Y, Zmijewski J, Xu Z, Abraham E. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol. 2008;180:2531–2537. doi: 10.4049/jimmunol.180.4.2531. [DOI] [PubMed] [Google Scholar]
- 41.Ono K, Lindsey ES. Improved technique of heart transplantation in rats. J Thorac Cardiovasc Surg. 1969;57:225–229. [PubMed] [Google Scholar]
- 42.Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal Biochem. 2000;285:194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- 44.He H, Stone JR, Perkins DL. Analysis of robust innate immune response after transplantation in the absence of adaptive immunity. Transplantation. 2002;73:853–861. doi: 10.1097/00007890-200203270-00005. [DOI] [PubMed] [Google Scholar]
- 45.Yamazaki T, Seko Y, Tamatani T, Miyasaka M, Yagita H, Okumura K, et al. Expression of intercellular adhesion molecule-1 in rat heart with ischemia/reperfusion and limitation of infarct size by treatment with antibodies against cell adhesion molecules. Am J Pathol. 1993;143:410–418. [PMC free article] [PubMed] [Google Scholar]
- 46.Jaakkola K, Jalkanen S, Kaunismaki K, Vanttinen E, Saukko P, Alanen K, et al. Vascular adhesion protein-1, intercellular adhesion molecule-1 and P-selectin mediate leukocyte binding to ischemic heart in humans. Journal of the American College of Cardiology. 2000;36:122–129. doi: 10.1016/s0735-1097(00)00706-3. [DOI] [PubMed] [Google Scholar]
- 47.Hua F, Ha T, Ma J, Gao X, Kelley J, Williams DL, et al. Blocking the MyD88-dependent pathway protects the myocardium from ischemia/reperfusion injury in rat hearts. Biochem Biophys Res Commun. 2005;338:1118–1125. doi: 10.1016/j.bbrc.2005.10.068. [DOI] [PubMed] [Google Scholar]
- 48.Jamieson RW, Friend PJ. Organ reperfusion and preservation. Front Biosci. 2008;13:221–235. doi: 10.2741/2672. [DOI] [PubMed] [Google Scholar]
- 49.Rauen U, de GH. New insights into the cellular and molecular mechanisms of cold storage injury. J Investig Med. 2004;52:299–309. doi: 10.1136/jim-52-05-29. [DOI] [PubMed] [Google Scholar]
- 50.Hoffman JW, Jr, Gilbert TB, Poston RS, Silldorff EP. Myocardial reperfusion injury: etiology, mechanisms, and therapies. J Extra Corpor Technol. 2004;36:391–411. [PubMed] [Google Scholar]
- 51.Buja LM. Myocardial ischemia and reperfusion injury. Cardiovasc Pathol. 2005;14:170–175. doi: 10.1016/j.carpath.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 52.Bigaud M, Gfeller P, Deveze S, Vogt G, Evenou JP, Bruns C, et al. Transplantation-induced ischemia/reperfusion injury in the rat heart. Transplant Proc. 1998;30:2311–2313. doi: 10.1016/s0041-1345(98)00635-6. [DOI] [PubMed] [Google Scholar]
- 53.Parolari A, Rubini P, Cannata A, Bonati L, Alamanni F, Tremoli E, et al. Endothelial damage during myocardial preservation and storage. Ann Thorac Surg. 2002;73:682–690. doi: 10.1016/s0003-4975(01)03029-6. [DOI] [PubMed] [Google Scholar]
- 54.Lutterova M, Szatmary Z, Kukan M, Kuba D, Vajdova K. Marked difference in tumor necrosis factor-alpha expression in warm ischemia- and cold ischemia-reperfusion of the rat liver. Cryobiology. 2000;41:301–314. doi: 10.1006/cryo.2000.2293. [DOI] [PubMed] [Google Scholar]
- 55.Rauen U, Petrat F, Li T, de GH. Hypothermia injury/cold-induced apoptosis--evidence of an increase in chelatable iron causing oxidative injury in spite of low O2-/H2O2 formation. FASEB J. 2000;14:1953–1964. doi: 10.1096/fj.00-0071com. [DOI] [PubMed] [Google Scholar]
- 56.Thornley TB, Brehm MA, Markees TG, Shultz LD, Mordes JP, Welsh RM, et al. TLR agonists abrogate costimulation blockade-induced prolongation of skin allografts. J Immunol. 2006;176:1561–1570. doi: 10.4049/jimmunol.176.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen L, Wang T, Zhou P, Ma L, Yin D, Shen J, et al. TLR engagement prevents transplantation tolerance. Am J Transplant. 2006;6:2282–2291. doi: 10.1111/j.1600-6143.2006.01489.x. [DOI] [PubMed] [Google Scholar]
- 58.Porrett PM, Yuan X, LaRosa DF, Walsh PT, Yang J, Gao W, et al. Mechanisms underlying blockade of allograft acceptance by TLR ligands. J Immunol. 2008;181:1692–1699. doi: 10.4049/jimmunol.181.3.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goldstein DR, Tesar BM, Akira S, Lakkis FG. Critical role of the Toll-like receptor signal adaptor protein MyD88 in acute allograft rejection. J Clin Invest. 2003;111:1571–1578. doi: 10.1172/JCI17573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen H, Tesar BM, Walker WE, Goldstein DR. Dual signaling of MyD88 and TRIF is critical for maximal TLR4-induced dendritic cell maturation. J Immunol. 2008;181:1849–1858. doi: 10.4049/jimmunol.181.3.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]