

SUMMARY
Notch proteins constitute the receptors of a highly conserved signaling pathway that influences cell fate decisions both during development and in adults. A proteolytic cascade induced by ligand stimulation results in release of the Notch intracellular domain from the cell membrane, allowing it to enter the nucleus and form a complex with a DNA-bound transcription factor called CSL and a coactivator of the Mastermind family. Assembly of this Notch nuclear complex is the key step in the transcriptional response to a Notch signal. In the studies reported here, we have mapped residues important for the stabilization of this multiprotein-DNA complex using site-directed mutagenesis, determined the affinity of the three-domain form of CSL for its various partners, and investigated sources of cooperativity in complex formation by monitoring the influence of various components of the complex on the interactions of CSL with its other partners. Our findings are consistent with a model for complex assembly in which the RAM domain of Notch increases the effective concentration of the ANK domain for its binding site on the Rel-homology region of CSL, enabling docking of the ANK domain and subsequent recruitment of the MAML co-activator.
Keywords: Signal transduction, Fluorescence resonance energy transfer, CSL, Mastermind, Protein-protein interaction
Introduction
Notch proteins constitute the receptors of a highly conserved signaling pathway that influences cell fate decisions both during development and in adults. Notch signals are used iteratively at different decision points and have functional outcomes that depend heavily on gene dose and context, regulating cell growth, differentiation, and death in a variety of tissue types 1; 2; 3. Both deficiencies and abnormal increases of Notch signaling are associated with human developmental anomalies and cancer, emphasizing the importance of precisely regulating Notch signal strength 4; 5; 6; 7; 8; 9; 10.
Notch receptors are single-pass transmembrane proteins that are normally activated by regulated intramembrane proteolysis (RIP) in response to transmembrane ligands expressed on adjacent cells (Figure 1A). Ligand binding induces proteolytic sensitivity to metalloprotease cleavage (S2) at a site just external to the plasma membrane 11; 12. This proteolytic step creates the substrate for subsequent cleavage by the multiprotein gamma-secretase complex (S3), which then releases the intracellular portion of Notch (ICN) from the membrane, allowing it to translocate to the nucleus where it induces the transcription of target genes.
Figure 1.
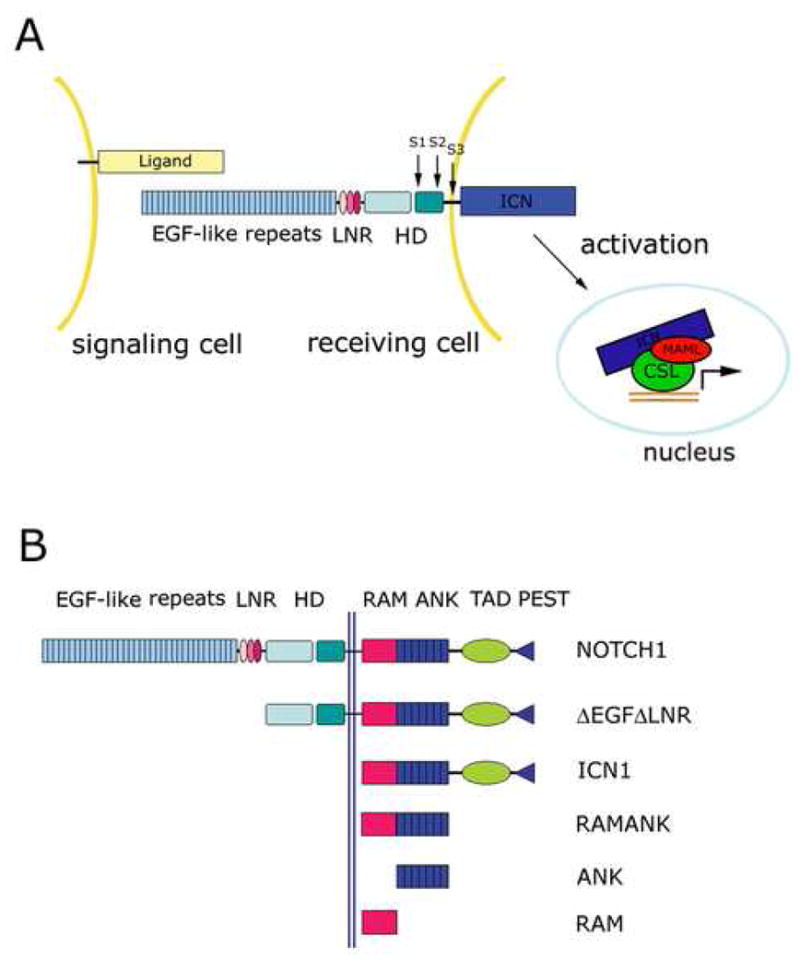
Overview of Notch signaling (A) and constructs used in this study (B). A. The mature Notch heterodimer is generated by cleavage at S1 by a furin-like protease. Activation of Notch by a ligand of the Delta/Serrate/Lag-2 family induces a proteolytic cascade (S2, S3) that releases the intracellular part of Notch (ICN) from the membrane, allowing it to enter the nucleus. ICN then forms a complex with a DNA-bound transcription factor called CSL and a co-activator of the Mastermind family to induce transcription of Notch target genes. B. Schematic illustrating Notch constructs used in this study. Abbreviations: LNR: LIN12-Notch repeat; HD: heterodimerization domain; ICN: intracellular Notch; RAM: RBP-Jκ-associated molecule; ANK: ankyrin-repeat domain.
The central effector of transcriptional activation in response to Notch signaling is the nuclear complex that controls gene expression. The core elements of this complex are ICN, a highly conserved transcription factor called CSL (for CBF-1/RBP-Jκ, Suppressor of Hairless and Lag-1; 13; 14; 15; 16), and a transcriptional co-activator of the mastermind-like (MAML) family 17; 18; 19. Once the core complex is assembled, the C-terminal portion of MAML not only contributes to transcriptional activation through association with p300, RNA polymerase II, and other unknown factors 20; 21; 22, but also limits the longevity of the assembled complex by promoting the phosphorylation of ICN by CDK8, which leads to rapid ICN turnover 23.
Previous genetic, biochemical and structural studies have implicated specific regions of CSL, ICN, and MAML in the physical and functional interactions of these proteins with one another. CSL is a highly conserved protein comprised of N-terminal and C-terminal Rel homology domains (RHR-n and RHR-c, respectively) and a central β-trefoil domain that binds DNA in a sequence-specific fashion 24; 25. CSL contacts its cognate DNA via its central, beta-trefoil domain and its RHR-n domain, but the RHR-c domain of CSL makes no contacts at all with the DNA (unlike other Rel-homology proteins such as NF-κB) and is instead positioned like an appendage in an unusual orientation next to the RHR-n domain 25; 26; 27. Binding of ICN to CSL depends in part on a high-affinity interaction between the β-trefoil domain of CSL and an N-terminal sequence of ICN known as the RAM (for RBP-Jκ-associated molecule) region 13; 28. In addition, the ankyrin domain (ANK) independently exhibits weak affinity for CSL through interactions with the Rel-homology region (RHR). Significantly, the ANK domain is capable of assembling complexes with CSL and MAML proteins on DNA because ANK and CSL cooperate to create a composite binding groove for a short 60–70 residue sequence at the N-terminal end of MAML 18; 19; 29. This N-terminal “handle” of the MAML coactivator adopts a kinked alpha-helical conformation in the X-ray structures of both worm and human MAML/ICN/CSL/DNA complexes 26; 27. Under conditions of enforced expression in cells, forms of ICN that contain ANK but not RAM also cause CSL-dependent Notch gain-of-function phenotypes 30; 31; 32, indicating that the essential domain for the effector function of Notch at target promoters is ANK.
Stable assembly of the MAML/ICN/CSL/DNA complex appears to depend on the cumulative contributions of a number of distinct protein-protein and protein-DNA interfaces. CSL contains distinct binding sites for DNA, RAM, and ANK, with the CSL-ANK interface then used for MAML binding. In the studies reported here, we have mapped residues important for the stabilization of this multiprotein-DNA complex using site-directed mutagenesis, determined the affinity of the three-domain form of CSL for its various partners, and investigated sources of cooperativity in complex formation by monitoring the influence of various components of the complex on the interactions of CSL with its other partners. Our findings are consistent with a model for complex assembly in which the RAM domain increases the effective concentration of the ANK domain for CSL, enabling docking of the ANK domain onto the RHR domain of CSL to allow subsequent recruitment of the MAML co-activator.
Results
To identify sites critical for stabilization of human Notch1 transcription complexes, we first performed an extensive mutational analysis of residues at different protein-protein interaction surfaces. These mutations were analyzed in a number of different assays using a range of different Notch1-derived proteins and fragments (Figure 1B). The crystal structure of the human MAML1/ANK/CSL/DNA complex 27 was used to guide the selection of substitution sites, which focused on conserved residues at the ANK-CSL, ANK-MAML1 and MAML1-CSL interfaces (Figure 2). In total, we examined 20 different mutants of the ANK domain of Notch1, including alanine, charge-reversal, and multisite substitutions, along with nine different mutated forms of the MAML1 polypeptide (Table 1).
Figure 2.
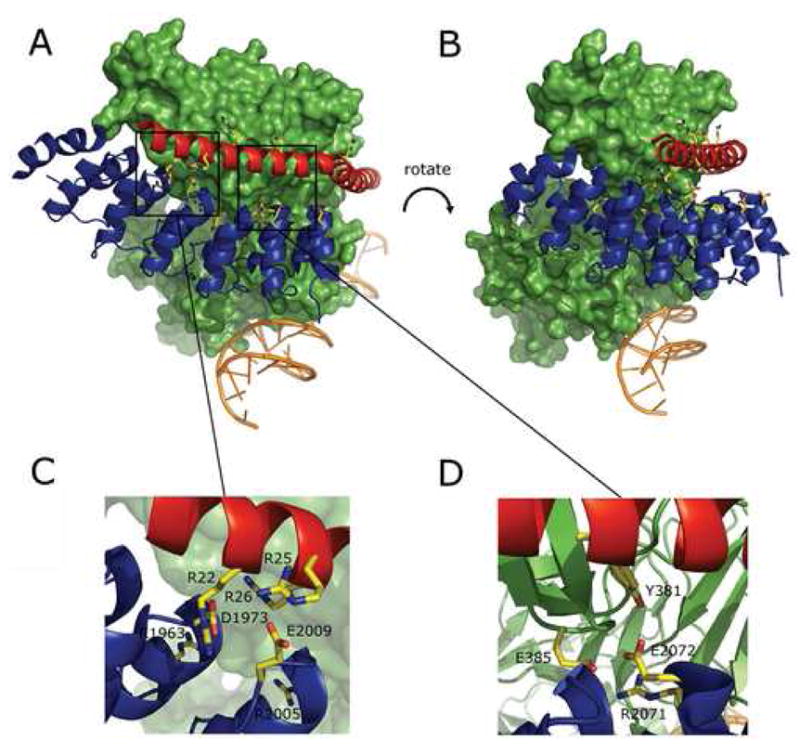
Sites of mutations mapped onto the X-ray structure of the human MAML-1/ANK/CSL/DNA complex. A, B. Overview of the structure (PDB ID code 2F8X; reference 27). Two views (rotated by approximately 20 degrees around the Y-axis and 5 degrees around the X-axis) are shown. CSL is rendered as a green molecular surface, while the DNA (orange), ANK domain (blue) and the MAML1 polypeptide (red) are illustrated as cartoons. Residues on ANK and MAML1 mutated in this study are shown in ball and stick form using CPK colors. C, D. Expanded regions showing sites where charge reversal mutations interfere with stable formation of complexes.
Table 1.
Summary of mutations and their effects on formation of MAML1/Notch/CSL complexes.
| MUTATED CONSTRUCT |
COMPLEX FORMATION |
MUTATED CONSTRUCT |
COMPLEX FORMATION |
MUTATED CONSTRUCT |
COMPLEX FORMATION |
|---|---|---|---|---|---|
| ANK | ANK | MAML | |||
| D1973A | + | N2040A | + | R22E | + |
| D1973R | − | N2040E | + | R25E | − |
| R2005A | + | N2040A E2072A | + | R26E | + |
| R2005E | + | R2071E | + | R22E R25E | − |
| E2009A | + | E2072A | + | R22E R25E R26E | − |
| E2009R | + | E2072R | − | R31E | + |
| D1973A E2009A | + | E2076A | + | E38R | + |
| W2035A | + | H2093R | + | R53E | + |
| W2035E | + | D2109A | + | T56E | + |
| W2035A N2040AE2072A |
+ | RAMANK | |||
| R1963A R2005E E2072R R2071E |
− |
The effect of each mutation on the assembly of MAML1/ANK/CSL complexes (without added DNA) was monitored by first mixing CSL, ANK (wild-type or mutant) and MAML1 (wild-type or mutant) in an approximately equimolar ratio, and then performing size-exclusion chromatography to resolve the complex. Although most individual point mutations did not prevent the formation of ternary complexes under standard conditions (Table 1), the D1973R and E2072R charge-reversal mutations of ANK and three charge-reversal and multisite mutations of MAML1 (R25E, R22E/R25E, and R22E/R25E/R26E) interfered with the formation of ternary complexes, as judged by the absence of the peak corresponding to the ternary complex on the size exclusion column (Figure 3A). In contrast to the D1973R and E2072R charge-reversal mutations, the D1973A and E2072A alanine substitutions in ANK do not prevent the formation of stable ternary complexes under standard conditions, suggesting that complex relies on numerous weak interactions rather than on an energetic “hot spot” 33; 34, and that disruption of the complex by charge reversal mutations is due to the introduction of unfavorable electrostatic interactions upon substitution.
Figure 3.
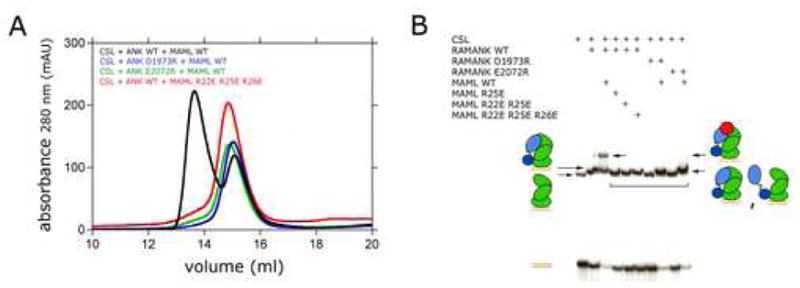
Analysis of the effects of ANK and MAML1 mutations on complex formation. A. Size exclusion chromatography. Various CSL (20 μM), ANK (50 μM), and MAML1 (50 μM) polypeptides were mixed as indicated, and formation of complexes was analyzed by passage through a Superdex 200 column, monitoring absorbance at 280 nm as a function of elution volume. Black trace: wild-type ANK, CSL, and MAML; blue trace: D1973R ANK with wild-type CSL and MAML; green trace: E2072R ANK with wild-type CSL and MAML; red trace: R22E/R25E/R26E MAML with wild-type ANK and CSL. B. Electrophoretic mobility shift assay. The indicated mixtures of normal or mutated CSL, Notch, and MAML proteins were tested for binding to a 32P-labeled oligonucleotide containing a consensus CSL binding site, as described in Methods. DNA/protein complexes were detected by autoradiography.
The mutations that abrogated ternary complex formation in gel-filtration assays without DNA were also evaluated for their effects on the formation of complexes in the presence of DNA in an electrophoretic mobility shift assay (EMSA). For these studies, the mutations in the ANK domain of Notch1 were introduced into proteins that contained both the RAM and ANK regions of Notch1 (hereafter designated RAMANK), and the mobility shifts resulting from accrual of the three protein components of the MAML1/RAMANK/CSL/DNA complex were monitored as previously described 29; 35. The EMSA data show that in the absence of MAML1, RAMANK proteins containing either the D1973R or E2072R mutations within the ANK domain still retain the ability to bind CSL via their RAM domains (Figure 3B), as anticipated based on previously reported studies 13; 28; 29; 36. On the other hand, recruitment of the MAML1 polypeptide into these complexes was either undetectable (D1973R) or strongly reduced (E2072R). The R25E, R25E/R26E and R22E/R25E/R26E forms of MAML1 abrogate the MAML1 dependent supershift in the EMSA assay, consistent with the observation that they prevent formation of stable ternary protein complexes in the gel filtration studies (Figure 3B).
To determine whether mutations that prevent formation of stable MAML1/ICN/CSL/DNA complexes in biochemical assays also interfere with Notch1-dependent transcriptional activation in cells, we tested the effects of the these mutations in a cell-based reporter assay 31. This assay monitors the expression of a luciferase reporter gene regulated by four iterated CSL-binding sites (4x CSL). We tested the D1973R and E2072R mutations of the ANK domain in two different contexts: (i) intracellular Notch1 (ICN1), which leads to strong ligand-independent induction of the luciferase reporter, and (ii) a form of the Notch1 receptor lacking the ligand-binding EGF repeats and the S2-protective LNR modules (ΔEGFΔLNR), which causes a more modest increase in ligand-independent reporter gene transcription (Figure 4A). Both the D1973R and E2072R mutations essentially eliminate the induction of reporter gene transcription in both the ICN1 and ΔEGFΔLNR context (Figure 4A). We also tested the effects of the R25E and R22E/R25E mutations in full-length MAML1 on the strength of the reporter gene signal when co-transfected with ΔEGFΔLNR. When co-transfected with wild-type ICN1, normal MAML1 enhances the transcriptional response at the 4x CSL promoter, whereas MAML1 with the R22E/R25E mutations does not (Figure 4B), exhibiting a dominant-negative effect instead. This finding is consistent with the failure of this mutated form of MAML1 to assemble efficiently into ICN/CSL/DNA complexes in the gel filtration and EMSA assays.
Figure 4.
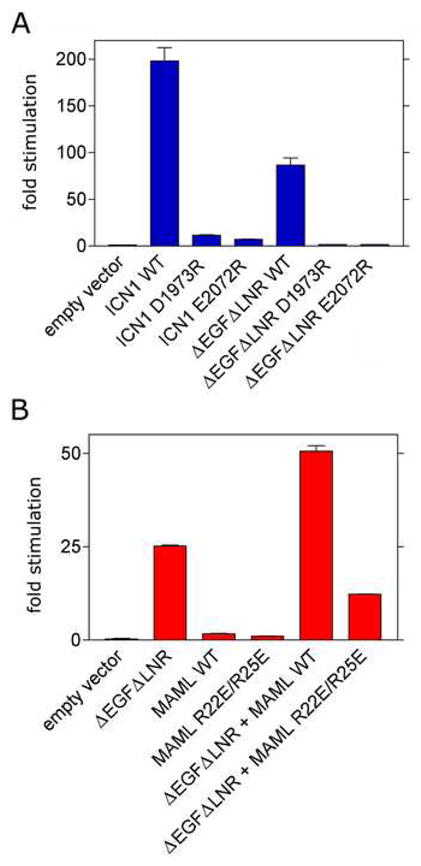
Effects of Notch1 (A) and MAML1 mutations (B) on transcription of a luciferase reporter gene. Luciferase assays were performed on U2OS cell lysates prepared from cells transfected in triplicate with empty pcDNA3 plasmid or plasmids encoding the indicated forms of Notch1 and/or MAML1, along with a luciferase reporter plasmid containing four iterated CSL-binding sites and an internal control plasmid expressing Renilla luciferase from a thymidine kinase promoter as described in Methods. Firefly luciferase activity, normalized for variation in Renilla luciferase activity, is expressed relative to the activity in extracts prepared from cells transfected with empty pcDNA3 vector, which is arbitrarily set to a value of 1.
Although the mutational studies provide a qualitative picture that identifies key residues in the different protein-protein interfaces of the complex, it does not define the affinities of different components for one another or give insight into the dynamics of the protein domains engaged in these different interfaces. To determine the affinity of different intracellular Notch1 fragments for CSL-DNA complexes and to explore whether or not the ANK domain is in contact with the Rel-homology region of CSL in the absence of MAML1, we developed a fluorescence resonance energy transfer (FRET) assay for the binding of ICN1 polypeptides to CSL-DNA complexes. In this assay, a donor fluorophore (Alexa Fluor 555) was covalently attached to various ICN1 polypeptides and an acceptor fluorophore (Alexa Fluor 594) was covalently added to the cognate DNA bound to CSL in a 1:1 complex. To examine RAM binding to CSL-DNA complexes, the native cysteine of RAM (C1872) was substituted with an alanine and a cysteine was introduced at the N-terminal end of the protein preceding the CSL binding motif. To introduce the donor fluorophore into ANK and RAMANK polypeptides, the naturally-occuring cysteine residues were replaced with alanine (C1891A for ANK, and both C1872A and C1891A for RAMANK), and a unique cysteine residue was introduced in place of F2085 (F2085C). This position is distant from the MAML1 and CSL contact interfaces of ANK, yet remains within the Förster distance of the acceptor site located about 37 Å away based on the structure of the MAML1/ANK/CSL/DNA complex (Figure 5A). It is important to note that the performance of the mutated, fluorescently labeled RAMANK protein in the formation of complexes is indistinguishable from the normal protein, as judged by EMSA (Figure 5B).
Figure 5.
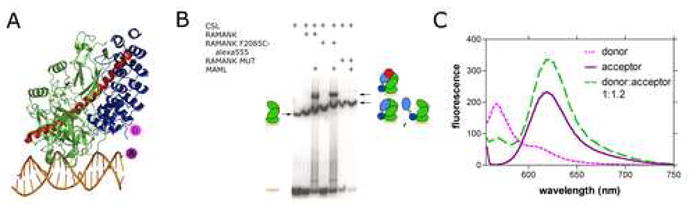
FRET assay design and implementation. A. Design of the FRET assay used to measure the affinity of various Notch polypeptides for CSL-DNA complexes. The fluorescent donor (Alexa Fluor 555) was installed onto one of three Notch polypeptides (ANK, RAMANK, or RAM), and the acceptor (Alexa Fluor 594) was installed onto one of the CSL-bound DNA strands. The diagram illustrates the assay for labeling of ANK or RAMANK at position 2085. B. EMSA showing CSL binding to DNA and RAMANK polypeptides used in the FRET assay. The RAMANK polypeptides used in the FRET assay were incubated with CSL and MAML proteins as indicated and tested for binding to a 32P-labeled oligonucleotide containing a consensus CSL binding site, as described in Methods. DNA/protein complexes were detected by autoradiography. RAMANK F2085C-Alexa Fluor 555 contains the mutation F2085C along with C1872A and C1891A. RAMANK-mut contains the mutations C1872A, C1891A, R1963A, R2005E, R2071E, E2072R and F2085C. C. Fluorescence emission spectra (excitation wavelength 555 nm) of donor alone (RAMANK F2085C~Alexa Fluor 555; dashed pink), acceptor alone (CSL-DNA~Alexa Fluor 594; solid purple), and of complexes exhibiting fluorescence resonance energy transfer from donor to acceptor (dashed green).
After demonstrating that complex formation could be monitored by FRET with this assay (Figures 5C and Supplementary Figure S1), we then used it to measure the binding affinity of RAM, RAMANK, and ANK for CSL (Figures 6 and S3). Titration of donor-labeled RAM with labeled CSL-DNA yielded a Kd of 1.1 ± 0.3 μM (Figure 6A), while the titration for RAMANK yielded a Kd of 0.51 ± 0.18 μM (Figure 6B). These affinity values are consistent with dissociation constants measured by isothermal titration calorimetry for binding of the RAM and RAMANK regions of ICN to the beta trefoil domain of CSL alone under different solution conditions 36. The data reported here also show that RAM makes the dominant thermodynamic contribution to the binding of the full three-domain form of CSL, a conclusion consistent with prior immunoprecipitation studies, electrophoretic mobility shift assays, and other studies suggesting that the affinity of ANK for CSL is weak when RAM is not present 13; 28; 29; 37.
Figure 6.
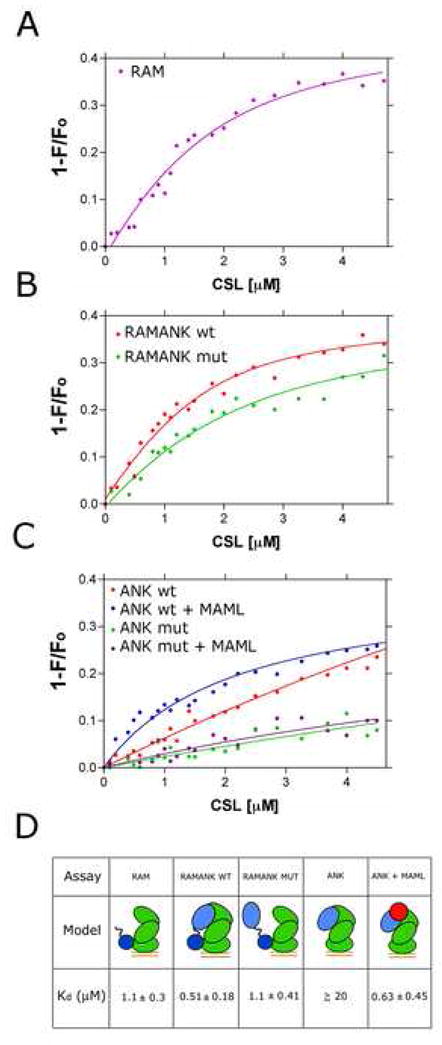
Affinity measurements for binding of various forms of intracellular Notch to CSL-DNA complexes. A. Isotherm for binding of RAM to CSL-DNA complexes. The data fit a single site model for binding with a value for Kd = 1.1 ± 0.3 μM. B. Isotherms for binding of RAMANK and RAMANK-mut to CSL-DNA complexes. The data for RAMANK give a value for Kd = 0.51 ± 0.18 μM, and the data for RAMANK-mut yield a value of Kd = 1.1 ± 0.4 μM. C. Isotherms for binding of ANK and ANK-mut to CSL-DNA complexes in the presence and absence of the MAML1 polypeptide. In the absence of MAML1, the affinity of ANK is too weak to determine a reliable value of Kd. When MAML1 is present, the apparent Kd of ANK for CSL-DNA is 0.63 ± 0.45 μM. When the control ANK-mut protein is used, binding to CSL-DNA complexes is undetectable regardless of whether or not MAML1 is present. D. Summary of binding data with cartoons illustrating interactions in the complexes tested. Notch polypeptides are blue, CSL is green, MAML is red, and bound DNA is represented by a pair of orange parallel lines.
The Kd value for ANK binding could not be reliably determined (≥ ~20 μM), because FRET measurements could not be carried out at the high concentrations needed to achieve saturation binding in the assay (Figure 6C). In contrast, saturating concentrations of MAML1 enhance the apparent affinity of ANK for CSL by at least 25-fold, with an apparent dissociation constant of 0.63 ± 0.45 μM (Figure 6C). Together, these findings show that inclusion of MAML1 in the complex dramatically stabilizes the interaction of ANK with CSL.
Because the binding of RAM is expected to increase the effective concentration of covalently attached ANK for its binding site on CSL, we next explored whether the ANK domain is positioned in its CSL-binding site when RAMANK binds to CSL-DNA complexes. To examine this question, we constructed control ANK and RAMANK proteins with multiple substitutions of residues in the ANK domain designed to prevent docking of the ANK domain into its binding site on CSL. Each protein contains four mutations, R1963A, R2005E, R2071E, and E2072R, along the surface of ANK contacting CSL in the human MAML1/ANK/CSL/DNA crystal structure, along with mutations to remove the native cysteines (C1872A in RAMANK, and C1891A in both proteins) and the F2085C substitution to introduce the label for the FRET binding assay These proteins are referred to as ANK-mut and RAMANK-mut. The four mutations were combined because of the extensive ANK-CSL interface area, and because even the E2072R protein exhibits some residual capacity to recruit MAML1 by EMSA (Figure 3B), suggesting that any given single-site mutation would be insufficient to entirely eliminate the ability of ANK to dock onto its CSL-binding site. RAMANK-mut completely prevented formation of a stable ternary complex as judged by EMSA (Figure 5B). Importantly, the circular dichroism spectrum of RAMANK-mut is nearly identical to that of wild-type RAMANK, indicating that the mutant proteins are properly folded under the conditions of the assay (Supplementary Figure S2).
The binding of the ANK-mut and RAMANK-mut proteins to CSL-DNA complexes was then evaluated by FRET. In contrast to wild-type ANK, which exhibits a weak intrinsic affinity for CSL that is enhanced by addition of MAML1, binding of the ANK-mut protein to CSL-DNA complexes is undetectable regardless of whether or not MAML1 is present (Figure 6C). Consistent with the observation that the ANK-mut protein fails to bind detectably to CSL, RAMANK-mut binds to CSL-DNA complexes with a dissociation constant of 1.1 ± 0.4 μM (Figure 6B), which is within the experimental error for the binding of RAM alone. In addition, the transfer efficiency from donor to acceptor fluorophore was reduced to 25 % for RAMANK-mut from a value of 41 % for wild-type RAMANK, indicating that the ANK domain is farther away from the acceptor fluorophore on DNA. Together, these FRET experiments (Figure 6D) support the conclusion that the ANK domain of intracellular Notch has sufficient intrinsic affinity for CSL to favor occupancy of its binding site upon delivery to CSL by RAM, even prior to MAML1 loading (Figure 7).
Figure 7.
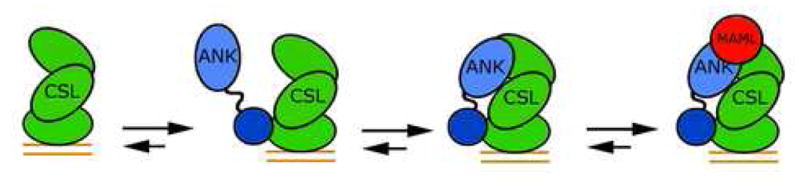
Model for complex assembly. High-affinity (~1 μM) binding of RAM to CSL-DNA complexes promotes docking of the ANK domain to the Rel-homology region of CSL. The MAML polypeptide then binds to the ANK-CSL interface to complete assembly of the core Notch transcription complex.
Discussion
The studies reported here focus on biochemical and mechanistic questions related to the assembly and stabilization of Notch transcriptional activation complexes. These complexes contain a number of different protein-protein interfaces, but we chose to place particular emphasis on the interactions of the ANK domain of ICN because of its central role in the transcriptional response to the induction of a Notch signal.
Initially, we embarked on a limited alanine scan of the residues of ANK at the CSL and MAML interfaces to identify side chains that make important contributions to stabilization of complexes. Four mutations were at the ANK-CSL interface (R2005A, W2035A, E2072A, and E2076A), and four were at the ANK-MAML1 interface (D1973A, E2009A, N2040A, and D2109A). However, each of these eight sites was tolerant to alanine substitution, forming ternary complexes with MAML1 and CSL stable to isolation by size exclusion chromatography. Other charged substitutions, such as R2071E and H2093R, as well as several multisite mutations, were also permissive for complex assembly. The two mutations tested that disrupted the formation of complexes were charge-reversal mutations: D1973R, which mutates a buried aspartate residue engaged in salt-bridges with the R22 and R26 side chains of the MAML1 peptide, and E2072R, which mutates a glutamate residue fully concealed from solvent in the complex engaged in a charged hydrogen bond with the side chain hydroxyl group of Y381 from CSL. The sensitivity of the MAML1-ANK-CSL complex to these charge reversal mutations, which are at sites insensitive to simple alanine substitution, indicates that the electrostatic complementarity of the different protein-protein interfaces plays an important role in assembly and stabilization of the MAML1-ANK-CSL complex. This conclusion is further supported by the deleterious effects of the charge-reversal mutations of the MAML1 peptide, which also prevented complex formation in the chromatographic assay.
Mutations in Notch1 that selectively affect either the ANK-CSL (E2072R) or ANK-MAML1 (D1973R) interface were used to test the effects of disrupting each of these two contact sites separately in cell-based reporter assays. Importantly, these studies used full-length CSL and MAML, and either intact ICN1 or a transmembrane receptor lacking the EGF and LNR repeats (ΔEGFΔLNR). Mutations disrupting the CSL binding site prevented transcription of a CSL reporter gene, as did mutations that interfered with MAML1 binding. The availability of Notch receptors with mutations highly selective for the CSL-binding interface of the ANK domain may also make it possible to clarify the importance of CSL-independent effects of Notch signaling in future studies.
The FRET assay we developed allowed us to quantify the affinity of various Notch polypeptides for CSL-DNA complexes. The Kd of the RAM polypeptide for the three-domain form of CSL is in close agreement with the reported affinity of different RAM peptides for the beta-trefoil domain of CSL reported previously by Kopan and Barrick’s groups 36, even though our measurements were performed at different pH and using an entirely different experimental approach. Several additional conclusions can be drawn from the FRET experiments carried out with the ANK and RAMANK proteins. Although the ANK domain exhibits a weak intrinsic affinity for CSL, when the RAM domain is bound to CSL and connected to ANK in cis by its native linker, the ANK domain appears to occupy its binding site on CSL preferentially even at concentrations below its intrinsic Kd. Moreover, the intrinsic affinity of the ANK domain for CSL is enhanced by at least 25-fold in the presence of MAML1, as predicted based on the ability of ANK to produce a electrophoretic mobility shift of CSL-DNA complexes in the presence, but not in the absence, of the MAML1 polypeptide 29.
Overall, the results reported here reinforce the current working model for assembly of Notch transcriptional activation complexes (Figure 7). The first event in assembly is likely to be the binding of RAM to its high-affinity site on the beta-trefoil domain of CSL, which facilitates docking of the ANK domain onto its binding site. Once the ANK-CSL interface is formed, MAML1 binds to the composite interface that is created to “flip on” the transcriptional switch. One issue that still remains unresolved is whether the different conformations of CSL seen in the crystal structures of Notch complexes formed with the human and worm proteins reflect an allosteric movement of CSL induced by RAM when it binds. If CSL exhibits intrinsic flexibility, this question may not be resolved only by comparing crystal structures of complexes containing RAM, RAMANK, and ANK polypeptides, because the conformation in the crystals may be dictated by lattice contacts or other forces rather than by intrinsic conformational preferences. We hope to clarify this issue in future studies by extension and refinement of the FRET assays developed here.
Materials and Methods
Mutagenesis and protein purification
Mutations and insertions were made either by Quikchange Site-directed Mutagenesis (Stratagene) or back-to-back PCR using Phusion Hot Start DNA Polymerase (New England Biolabs). The coding sequences of all mutated proteins were confirmed by DNA sequencing. CSL, RAMANK, ANK, MAML1 and RAM polypeptides were purified as previously described 27; 29
Gel Filtration
The formation of complexes was monitored by gel filtration on an AKTA chromatography workstation. CSL (20 μM), ANK (50 μM), and MAML1 (50 μM) polypeptides were mixed, and formation of complexes was analyzed by passage through a Superdex 200 column (Amersham Biosciences) in 20 mM Tris 8.5, 150 mM NaCl, 5 mM DTT. To assess complex formation, the absorbance at 280 nm was plotted as a function of elution volume; stable MAML1/ANK/CSL complexes elute from the column at approximately 14 mL, whereas uncomplexed CSL and ANK elute from the column in a peak centered around 15 mL (Figure 3A).
Electrophoretic Mobility Shift Assays
Oligonucleotides with 5′-overhangs were labeled with 32P-α-dCTP (Perkin-Elmer) by incubation with the Klenow fragment of Escherichia coli DNA polymerase I (New England Biolabs) for 15 min at room temperature. The labeled oligonucleotides were then separated from residual 32P-α-dCTP by passage through a Microspin G-50 Column (Amersham Biosciences). To test for complex assembly, 0.2 pmol of the DNA probe was incubated for 30 min at 30°C in binding buffer (20 μL final volume) containing 10% glycerol (w/v), 20 mM HEPES (pH 7.9), 60 mM KCl, 10 mM DTT, 5 mM MgCl2, dGdC (250 ng), and bovine serum albumin (0.2 mg/ml) in the presence or absence of CSL (300 ng), different RAMANK polypeptides (1000 ng) and various MAML1 polypeptides (100 ng). Samples were resolved by electrophoresis in 10% native gels at 4°C and 180 V. Following electrophoresis, gels were analyzed by autoradiography.
Reporter Gene Assays
Reporter gene assays were performed as previously described 31; 38. In reporter assays evaluating mutations of the ANK domain of Notch1, ICN1 and ΔEGFΔLNR constructs (with the normal Notch1 sequence or with mutations of the ANK domain) encoded in the plasmid pcDNA3 (10 ng) were transiently co-transfected (Lipofectamine Plus, Invitrogen) in triplicate into human U2OS cells, together with an internal Renilla luciferase control plasmid (Promega, Madison, Wis.) and a firefly luciferase reporter containing four tandem CSL binding sites 39. In studies examining normal and mutated forms of MAML1, the MAML1 protein of interest encoded in pcDNA3 (100 ng) was also co-transfected with ΔEGFΔLNR (10 ng), the Renilla luciferase control plasmid, and the luciferase reporter containing four tandem CSL binding sites. All dual luciferase assays were then performed and analyzed by using cell extracts prepared 48 hours after transfection, as described 31; 38. The data reported in Figure 4 are representative of at least three independent experiments (biological replicates). Although the absolute magnitude of the reporter signal for the active forms of Notch (wild-type ICN and ΔEGFΔLNR) may vary by several-fold from one experiment to the next, the relative activity of the mutated receptors compared to the wild-type and to one another is highly reproducible.
Fluorescent labeling
3′-oligonucleotides fluorescently labeled with the Alexa Fluor 594 were purchased from IDT (Coralville, IA) and used without further purification. Each mutated RAMANK, ANK and RAM protein was labeled by incubation with a 20-fold molar excess of Alexa Fluor 555 C2-maleimide (Molecular Probes, Invitrogen) for 2 hours at room temperature in a buffer containing 20 mM Tris (pH 7.4), 150 mM NaCl, and 5 mM TCEP. Each reaction was terminated by adding 5 mM β-mercaptoethanol, and the labeled protein was then separated from the free fluor by passage over a Sephadex G-25 (Amersham Biosciences) column preequilibrated with 20 mM Tris (pH 8.5), 150 mM NaCl, and 5 mM DTT, follwed by dialysis at 4°C against the same buffer until all excess dye was removed. Purified proteins were then concentrated to approximately 1 mg/mL, aliquoted, and stored at − 80 °C until use,
Measurements of Energy Transfer
Fluorescence measurements were carried out on a Varian Cary Eclipse at room temperature, monitoring transfer from the Alexa Fluor 555 donor on the ICN1 polypeptides to the Alexa Fluor 594 acceptor covalently attached to the DNA in CSL-DNA complexes. The decrease in emission of donor fluorescence was monitored between 556 and 750 nm (excitation wavelength at 555 nm), using excitation and emission slitwidths of 5 nm. Titrations were performed by addition of labeled CSL-DNA complexes to Notch1 polypeptides in 20 mM Tris-HCl, pH 8.5, 150 mM NaCl, 5 mM DTT in the presence or the absence of saturating concentration of MAML1. The initial protein concentrations of Notch1 and MAML1 polypeptides in the cuvette were 1–2 μM and 10 μM, respectively, and the titrating stock solution of the CSL-DNA-Alexa Fluor 594 complex was 10 μM. Samples were incubated for 2 minutes before each titration step. To ensure that the CSL protein remained bound to DNA throughout the titrations, a 10-fold excess of unlabeled DNA was included in all solutions. Errors reported refer to the goodness-of-fit error for the titration shown; each titration was performed at least twice, and the estimate of run-to-run error in each affinity measurement is approximately 30 % of the measured value. Transfer of resonance energy from the donor to the acceptor was also monitored as a function of increasing concentration of NaCl in control experiments. FRET efficiency (E) was calculated from the change of the fluorescence intensity of the donor molecule in the absence of the acceptor according to equation (1):
| (1) |
where FDA and FD are the fluorescence intensities of the donor molecule in the presence and the absence of the acceptor, respectively.
FRET Data Analysis
The decrease in the maximum emission (565 nm) of the donor was corrected for dilution, normalized and plotted against increasing concentration of the ligand (CSL-DNA complex). The value of the dissociation constant, Kd (for all titrations except binding to ANK alone), was determined by non linear least squares analysis according to equation (2), which applies to situations in which [R] ≪ Kd 40.
| (2) |
where [R] and [L] are the total concentration of receptor (i.e. Notch protein) and ligand (CSL-DNA). Ffree and Fbound are the value of fluorescence associated with free and bound receptor (i.e. Notch polypeptides), respectively. All data analysis was performed with the program GraphPad Prism (GraphPad Software, San Diego, CA).
Supplementary Material
| MUTATED NOTCH CONSTRUCT | TERNARY COMPLEX FORMATION | MUTATED MAML CONSTRUCT | TERNARY COMPLEX FORMATION |
|---|---|---|---|
| ANK | MAML | ||
| D1973A | + | R22E | + |
| D1973R | − | R25E | − |
| R2005A | + | R26E | + |
| R2005E | + | R22E R25E | − |
| E2009A | + | R22E R25E R26E | − |
| E2009R | + | R31E | + |
| N2040A | + | E38R | + |
| N2040E | + | R53E | + |
| R2071E | + | T56E | + |
| E2072A | + | ||
| E2072R | − | ||
| E2076A | + | ||
| H2093R | + | ||
| D2109A | + | ||
| D1973A E2009A | + | ||
| N2040A E2072A | + | ||
| W2035A N2040AE2072A | + | ||
| RAMANK | |||
| R1963A R2005E E2072R R2071E |
− |
Acknowledgments
We thank Luyan Song for constructing several of the mutated MAML1 polypeptides used for binding assays, and Gavin Histen for constructing the site-directed mutants of full-length MAML1 and help with reporter assays. We also thank Dr. Sheref S. Mansy for helpful suggestions on the FRET assay. CD is a recipient of a HFSP long-term postdoctoral fellowship. This work was supported by NIH grants R01 CA092433 (to SCB) and P01 CA119070 (to JCA and SCB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–89. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 2.Weng AP, Aster JC. Multiple niches for Notch in cancer: context is everything. Curr Opin Genet Dev. 2004;14:48–54. doi: 10.1016/j.gde.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–6. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 4.Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, Chandrasekharappa SC. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16:235–42. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- 5.Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, Costa T, Pierpont ME, Rand EB, Piccoli DA, Hood L, Spinner NB. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–51. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 6.Crosnier C, Driancourt C, Raynaud N, Hadchouel M, Meunier-Rotival M. Fifteen novel mutations in the JAGGED1 gene of patients with Alagille syndrome. Hum Mutat. 2001;17:72–3. doi: 10.1002/1098-1004(2001)17:1<72::AID-HUMU11>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 7.Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cecillion M, Marechal E, Maciazek J, Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier-Lasserve E. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–10. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 8.Garg V. Molecular genetics of aortic valve disease. Curr Opin Cardiol. 2006;21:180–4. doi: 10.1097/01.hco.0000221578.18254.70. [DOI] [PubMed] [Google Scholar]
- 9.Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–71. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 10.Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, Hui CC, Clevers H, Dotto GP, Radtke F. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33:416–21. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 11.Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, Israel A. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell. 2000;5:207–16. doi: 10.1016/s1097-2765(00)80417-7. [DOI] [PubMed] [Google Scholar]
- 12.Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, Ray WJ, Kopan R. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell. 2000;5:197–206. doi: 10.1016/s1097-2765(00)80416-5. [DOI] [PubMed] [Google Scholar]
- 13.Tamura K, Taniguchi Y, Minoguchi S, Sakai T, Tun T, Furukawa T, Honjo T. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H) Curr Biol. 1995;5:1416–23. doi: 10.1016/s0960-9822(95)00279-x. [DOI] [PubMed] [Google Scholar]
- 14.Fortini ME, Artavanis-Tsakonas S. The suppressor of hairless protein participates in notch receptor signaling. Cell. 1994;79:273–82. doi: 10.1016/0092-8674(94)90196-1. [DOI] [PubMed] [Google Scholar]
- 15.Christensen S, Kodoyianni V, Bosenberg M, Friedman L, Kimble J. lag-1, a gene required for lin-12 and glp-1 signaling in Caenorhabditis elegans, is homologous to human CBF1 and Drosophila Su(H) Development. 1996;122:1373–83. doi: 10.1242/dev.122.5.1373. [DOI] [PubMed] [Google Scholar]
- 16.Tani S, Kurooka H, Aoki T, Hashimoto N, Honjo T. The N- and C-terminal regions of RBP-J interact with the ankyrin repeats of Notch1 RAMIC to activate transcription. Nucleic Acids Res. 2001;29:1373–80. doi: 10.1093/nar/29.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu L, Aster JC, Blacklow SC, Lake R, Artavanis-Tsakonas S, Griffin JD. MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat Genet. 2000;26:484–9. doi: 10.1038/82644. [DOI] [PubMed] [Google Scholar]
- 18.Petcherski AG, Kimble J. Mastermind is a putative activator for Notch. Curr Biol. 2000;10:R471–3. doi: 10.1016/s0960-9822(00)00577-7. [DOI] [PubMed] [Google Scholar]
- 19.Petcherski AG, Kimble J. LAG-3 is a putative transcriptional activator in the C. elegans Notch pathway. Nature. 2000;405:364–8. doi: 10.1038/35012645. [DOI] [PubMed] [Google Scholar]
- 20.Wallberg AE, Pedersen K, Lendahl U, Roeder RG. p300 and PCAF Act Cooperatively To Mediate Transcriptional Activation from Chromatin Templates by Notch Intracellular Domains In Vitro. Mol Cell Biol. 2002;22:7812–9. doi: 10.1128/MCB.22.22.7812-7819.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fryer CJ, Lamar E, Turbachova I, Kintner C, Jones KA. Mastermind mediates chromatin-specific transcription and turnover of the Notch enhancer complex. Genes Dev. 2002;16:1397–411. doi: 10.1101/gad.991602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurooka H, Honjo T. Functional interaction between the mouse notch1 intracellular region and histone acetyltransferases PCAF and GCN5. J Biol Chem. 2000;275:17211–20. doi: 10.1074/jbc.M000909200. [DOI] [PubMed] [Google Scholar]
- 23.Fryer CJ, White JB, Jones KA. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol Cell. 2004;16:509–20. doi: 10.1016/j.molcel.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 24.Tun T, Hamaguchi Y, Matsunami N, Furukawa T, Honjo T, Kawaichi M. Recognition sequence of a highly conserved DNA binding protein RBP-J kappa. Nucleic Acids Res. 1994;22:965–71. doi: 10.1093/nar/22.6.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kovall RA, Hendrickson WA. Crystal structure of the nuclear effector of Notch signaling, CSL, bound to DNA. Embo J. 2004;23:3441–51. doi: 10.1038/sj.emboj.7600349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson JJ, Kovall RA. Crystal structure of the CSL-Notch-Mastermind ternary complex bound to DNA. Cell. 2006;124:985–96. doi: 10.1016/j.cell.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 27.Nam Y, Sliz P, Song L, Aster JC, Blacklow SC. Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell. 2006;124:973–83. doi: 10.1016/j.cell.2005.12.037. [DOI] [PubMed] [Google Scholar]
- 28.Kurooka H, Kuroda K, Honjo T. Roles of the ankyrin repeats and C-terminal region of the mouse notch1 intracellular region [published erratum appears in Nucleic Acids Res 1999 Mar 1;27(5):following 1407] Nucleic Acids Res. 1998;26:5448–55. doi: 10.1093/nar/26.23.5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nam Y, Weng AP, Aster JC, Blacklow SC. Structural requirements for assembly of the CSL. intracellular Notch1. Mastermind-like 1 transcriptional activation complex. J Biol Chem. 2003;278:21232–9. doi: 10.1074/jbc.M301567200. [DOI] [PubMed] [Google Scholar]
- 30.Roehl H, Bosenberg M, Blelloch R, Kimble J. Roles of the RAM and ANK domains in signaling by the C. elegans GLP-1 receptor. Embo J. 1996;15:7002–12. [PMC free article] [PubMed] [Google Scholar]
- 31.Aster JC, Xu L, Karnell FG, Patriub V, Pui JC, Pear WS. Essential roles for ankyrin repeat and transactivation domains in induction of T-cell leukemia by notch1. Mol Cell Biol. 2000;20:7505–15. doi: 10.1128/mcb.20.20.7505-7515.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeffries S, Robbins DJ, Capobianco AJ. Characterization of a High-Molecular-Weight Notch Complex in the Nucleus of Notch(ic)-Transformed RKE Cells and in a Human T-Cell Leukemia Cell Line. Mol Cell Biol. 2002;22:3927–41. doi: 10.1128/MCB.22.11.3927-3941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–6. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 34.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 35.Weng AP, Nam Y, Wolfe MS, Pear WS, Griffin JD, Blacklow SC, Aster JC. Growth suppression of pre-T acute lymphoblastic leukemia cells by inhibition of notch signaling. Mol Cell Biol. 2003;23:655–64. doi: 10.1128/MCB.23.2.655-664.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lubman OY, Ilagan MX, Kopan R, Barrick D. Quantitative dissection of the Notch:CSL interaction: insights into the Notch-mediated transcriptional switch. J Mol Biol. 2007;365:577–89. doi: 10.1016/j.jmb.2006.09.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aster JC, Robertson ES, Hasserjian RP, Turner JR, Kieff E, Sklar J. Oncogenic forms of NOTCH1 lacking either the primary binding site for RBP-Jkappa or nuclear localization sequences retain the ability to associate with RBP-Jkappa and activate transcription. J Biol Chem. 1997;272:11336–43. doi: 10.1074/jbc.272.17.11336. [DOI] [PubMed] [Google Scholar]
- 38.Sanchez-Irizarry C, Carpenter AC, Weng AP, Pear WS, Aster JC, Blacklow SC. Notch subunit heterodimerization and prevention of ligand-independent proteolytic activation depend, respectively, on a novel domain and the LNR repeats. Mol Cell Biol. 2004;24:9265–73. doi: 10.1128/MCB.24.21.9265-9273.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsieh JJ, Henkel T, Salmon P, Robey E, Peterson MG, Hayward SD. Truncated mammalian Notch1 activates CBF1/RBPJk-repressed genes by a mechanism resembling that of Epstein-Barr virus EBNA2. Mol Cell Biol. 1996;16:952–9. doi: 10.1128/mcb.16.3.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heyduk T, Lee JC. Application of fluorescence energy transfer and polarization to monitor Escherichia coli cAMP receptor protein and lac promoter interaction. Proc Natl Acad Sci U S A. 1990;87:1744–8. doi: 10.1073/pnas.87.5.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.