

Abstract
The role of histone deacetylases (HDAC) and the potential of these enzymes as therapeutic targets for cancer, neurodegenerative diseases and a number of other disorders is an area of rapidly expanding investigation. There are 18 HDACs in humans. These enzymes are not redundant in function. Eleven of the HDACs are zinc dependent, classified on the basis of homology to yeast HDACs: Class I includes HDACs 1, 2, 3, and 8; Class IIA includes HDACs 4, 5, 7, and 9; Class IIB, HDACs 6 and 10; and Class IV, HDAC11. Class III HDACs, sirtuins 1–7, have an absolute requirement for NAD+, are not zinc dependent and generally not inhibited by compounds that inhibit zinc dependent deacetylases.
In addition to histones, HDACs have many nonhistone protein substrates which have a role in regulation of gene expression, cell proliferation, cell migration, cell death, and angiogenesis. HDAC inhibitors (HDACi) have been discovered of different chemical structure. HDACi cause accumulation of acetylated forms of proteins which can alter their structure and function. HDACi can induce different phenotypes in various transformed cells, including growth arrest, apoptosis, reactive oxygen species facilitated cell death and mitotic cell death. Normal cells are relatively resistant to HDACi induced cell death. Several HDACi are in various stages of development, including clinical trials as monotherapy and in combination with other anti-cancer drugs and radiation. The first HDACi approved by the FDA for cancer therapy is suberoylanilide hydroxamic acid (SAHA, vorinostat, Zolinza), approved for treatment of cutaneous T-cell lymphoma.
Keywords: Vorinostat, Histone deacetylase, apoptosis
Histone acetylation is a reversible process whereby histone and non-histone protein acetyl-transferases transfer the acetyl moiety from acetyl co-enzyme A to lysines and HDACs remove the acetyl groups reestablishing the positive charge in the proteins. The biological activities of the zinc dependent HDACs are not completely understood. Accumulating evidence indicates that these enzymes are not redundant in their activity. Class I HDACs are primarily localized in the nucleus (Table I). Class II HDACs are primarily cytoplasmic in location but may shuttle between the nucleus and cytoplasm (see reviews [Blackwell et al., 2008; Bolden et al., 2006; Dokmanovic et al., 2007a; Glozak and Seto, 2007; Jones and Baylin, 2007; Minucci and Pelicci, 2006; Shankar and Srivastava, 2008; Xu et al., 2007]. Based on selective gene knock-out and siRNA gene suppression studies, class I HDACs appear to play a primary role in cell survival and proliferation, while class II HDACs have tissue specific roles. HDAC 1knock-out mouse embryos have a general proliferation survival defect despite “compensatory” increases in levels of HDAC 2 and HDAC 3 activity. HDAC 1 and HDAC 3 associate with the transcription factor, hypoxia inducible factor-1α (HIF-1α) and modulate the expression of HIF-1α gene and, in turn, affect angiogenesis. HDAC 2 can modulate transcriptional activity through interaction with p53. HDAC 4 knock-out mice have defects in chondrocyte differentiation. HDAC 4, like other class IIa HDACs, shuttles between the nucleus and the cytoplasm. HDAC4 serves as a nuclear co-repressor involved in regulating bones and muscle development and plays an important role in promoting the survival of retinal neuron [Chen and Cepko, 2009]. HDAC 5 and HDAC 9 have a role in the development of cardiac muscle. HDAC 7 is a factor in the maintenance of T-cell integrity.
Table 1.
Zinc-Dependent Histone Deacetylases
| HDAC | Localization | Size (AA) | Chromosomal site | Tissue distribution* |
|---|---|---|---|---|
| Class I | ||||
| HDAC1 | Nucleus | 483 | 1p34.1 | ubiquitous |
| HDAC2 | Nucleus | 488 | 6p21 | ubiquitous |
| HDAC3 | Nucleus | 423 | 5q31 | ubiquitous |
| HDAC8 | Nucleus | 377 | Xq13 | ubiquitous |
| CLASS IIa | ||||
| HDAC4 | Nuc/Cyt | 1084 | 2q372 | H, SM, B |
| HDAC5 | Nuc/Cyt | 1122 | 17q21 | H, SM, B |
| HDAC7 | Nuc/Cyt | 855 | 12q13 | H, PL, PA, SM |
| HDAC9 | Nuc/Cyt | 1011 | 7p21-p15 | SM, B |
| Calss IIb | ||||
| HDAC6 | Mainly Cyt | 1215 | Xp11.22–33 | H, L, K, PA |
| HDAC10 | Mainly Cyt | 669 | 22q13.31–33 | L, S, K |
| Class V | ||||
| HDAC11 | Nuc/Cyt | 347 | 3p25.2 | B, H, SM, K |
SM=skeletal muscle; B=brain; PL=platelet; L=liver; K=kidney; S=spleen; H=heart; PA=pancreas.
HDAC 6 and 10 have two catalytic sides. HDAC 6 is a unique HDAC in having a ubiquitin binding site toward its C-terminal end which plays a critical role in aggresome formation in the pathway of proteolysis of misfolded proteins. HDAC 6is a specific deacetylase of several proteins including α-tubulin, cortactin, peroxiredoxins, chaperone protein, HSP90, but histones are not a substrate of this enzyme in vivo (Table II).
Table 2.
Substrates of histone deacetylases (partial list)*
| Pathway | Protein |
|---|---|
| HDACs | HDAC1 and ? other HDACs |
| Cell motility | α-tubulin, cortactin |
| Chaperones | HSAP90, HSP70 |
| Gene transcription factors and co-regulators | P53, p73, GATA-1, 2 and 3, MyoD, E2F1, 2 and 3, PLAG-1 and PLAGL-2, c-myc, BCL-2, Rb, PGC-α |
| Chromatin structure | HMG-A1, B1, B2, N1 and N2, SRY |
| Nuclear receptors (DNA) | Androgen receptor, glucocorticoid receptor, estrogen receptor α |
| Signaling mediators | SAT3, Smad7, β-Catenin |
| DNA repair | Ku70, WRN |
| Nuclear import | Importin-α7 |
| Inflammation mediator | HMGB1 |
| Viral protein | E1A, L-HDAg, S-HDAg |
See text for references.
All zinc dependent HDACs exist as components of large multiprotein complexes. Such complexes may contain more than one HDAC. HDACs do not bind to DNA directly but rather interact with DNA through multi protein complexes that include co-repressors and coactivators ([Blackwell et al., 2008; Bolden et al., 2006; Dokmanovic et al., 2007a; Xu et al., 2007]. The activity of HDACs is regulated on multiple levels including protein-protein interactions, post-translational modification by phosphorylation, acetylation, sumoylation and proteolysis, subcellular localization, and a variety of metabolic cofactors [Blackwell et al., 2008; Bolden et al., 2006; Dokmanovic et al., 2007a; Dokmanovic et al., 2007b; Sengupta and Seto, 2004; Xu et al., 2007]. Phosphorylation and subsequent association with the cytoplasmic protein 14-3-3, plays a role in the shuttling of the class II HDACs, HDAC 4, HDAC 5, HDAC 7 and HDAC 9 between cytoplasmic and nuclear localization. Tetradecapeptide repeat domains of HDAC 6 are involved in maintenance of the cytoplasmic localization of this enzyme. HDACi can selectively suppress the expression of HDAC 7 gene and, to a lesser extent, HDAC 4 gene [Dokmanovic et al., 2007b].
An increasing number of proteins are being identified as substrates of HDACs which alters the structure and, as a consequence, the activity of these target proteins (Table II) [Blackwell et al., 2008; Bolden et al., 2006; Dokmanovic et al., 2007a; Minucci and Pelicci, 2006; Xu et al., 2007]. Non-histone protein targets of HDACs include transcription factors, transcription regulators, signal transduction mediators, DNA repair enzymes, nuclear import regulators, chaperone proteins, structural proteins, inflammation mediators, and viral proteins (Table II). Acetylation can alter the stability of these proteins and affect protein-protein interactions. These HDAC substrates are directly or indirectly involved in numerous important cell pathways including control of gene expression, regulation of cell proliferation, differentiation, migration, and death. As a consequence, HDACi can have multiple mechanisms of inducing transformed cell growth arrest and cell death (Figure 1). This may be a key factor in the anti-cancer activity of HDACi against a broad spectrum of hematologic and solid neoplasms.
Figure 1.
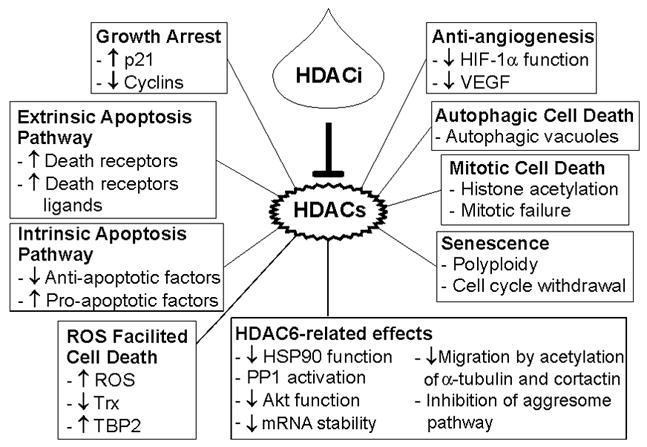
Zinc-dependent histone deacetylase inhibitors can induce transformed cell growth arrest and death by different pathways. HDAC6, histone deacetylase 6; HIF-1α, hypoxia induced factor-1α; HSP90, heat shock protein 90; PP1, protein phosphatase 1; ROS, reactive oxygen species; TBP2, thioredoxin binding protein 2; Trx, thioredoxin; VEGF, vascular endothelial growth factor.
Altered expression of HDACs has been reported in association with a number of human cancers [Blackwell et al., 2008; Dokmanovic et al., 2007a; Glozak and Seto, 2007; Jones and Baylin, 2007; Xu et al., 2007]. In ceratin transformed cells, HDAC 1 has been reported to interact directly with transcription repressors, and with pocket proteins, Rb, p107, p130, and with YY1, associated with deregulation of cell proliferation. HDAC 2 and HDAC 3 protein levels have been found increased in some colon cancers. HDAC 1 in gastric cancers and HDAC 5 and HDAC 10 in lung cancers with poor prognosis are reduced. HDAC 1 and HDAC 3 expression is elevated in some breast cancer tumors. Lower HDAC 1 expression has been associated with an invasive esophageal cancer phenotype. Increased expression of HDAC 6 has been reported to correlate with improved disease free and overall survival in patient with hormone sensitive breast cancer. HDAC 2 over expression has been found in precancerous lesions and colon cancers associated with abnormal adenomatosis polyposis colli tumor suppressor gene. It remains to be established whether these HDAC associations contribute to the malignant phenotype.
In addition to altered expression of HDACs, aberrant recruitment of these enzymes to specific loci occurs in certain malignancies. Aberrant recruitment of HDACs to transcription factors that involve oncogenetic DNA binding fusion proteins resulting from chromosomal translocation or over expression of repressive transcription factors, such as, the oncogenic PLML-RARα, or PLZF-RARα, and AML1-ETO fusion proteins are found in acute promyelocytic leukemia and acute myeloid leukemia respectively. The transcription factorBCL-6 is over expressed in large B cell lymphomas and recruits HDAC 2.
Structural mutations in HDACs appear to be rare in cancers. A truncation mutation of HDAC 2 has been discovered in human colon cancer and human endometrial cancer cell lines. A human prostate cell line has been found that does not express HDAC 6, with consequent constitutive accumulation of acetylated α-tubulin and other acetylated proteins that are targets of this deacetylase [Parmigiani et al., 2008].
Histone Deacetylase Inhibitors generally have in common three structural characteristics: a zinc binding moiety, an opposite capping group, and a straight chain alkyl, vinyl or aryl linker connecting the two (Tale III). Based on the co-crystal structures of hydroxamate inhibitors with HDAC4, HDAC 8 and an anaerobic bacterial HDAC like protein (HDLP), [Finnin et al., 1999; Somoza et al., 2004; Vannini et al., 2004] it is clear that these functional groups interact with three conserved regions of the active site. The first is the zinc ion that facilitates amide hydrolysis and is at the bottom of the narrow catalytic pocket. The second is a hydrophobic tunnel through which an acetyl-lysine substrate penetrates. The aliphatic chain with 6 carbons usually being an optimal link of the two functional groups. The third structure is at the channel opening where the rim interacts with the hydrophobic capping group. The rim has a greater degree of amino acid sequence diversity compared to the other two domains and, therefore, may have the most potential to be manipulated to develop selective HDACi.
Inhibitors of the zinc dependent HDACs have been discovered which are of several different structural classes, including hydroxamic acids, cyclic peptides, electrophilic ketones, short-chain fatty acid, and benzamides [Butler and Kozikowski, 2008; Dokmanovic et al., 2007a; Estiu et al., 2008; Jones et al., 2008a; Khan et al., 2008; Kozikowski et al., 2007; Marks and Breslow, 2007; Moradei et al., 2008; Schemies et al., 2009; Shankar and Srivastava, 2008; Xu et al., 2007] (Table III). The structural diversity among HDACi suggests that the mechanisms of action of these compounds may involve the interaction of the HDAC with other proteins independent of its deacetylase activity. Several HDAC inhibitors have been designed based on a rational approach using the structure of the catalytic site of HDACs. Assay of HDACi activity of the different compounds has been based on using purified recombinant HDACs and/or antiproliferative cell based assays with various transformed cells. Cell free assays with recombinant HDACs do not always reflect the activity of an inhibitor in cell based assays.
Table 3.
Histone Deacetylase Inhibitors in Clinical Trials (partial list)*
| Drug Name | Structure | Potency (in vitro) | Clinical Trials |
|---|---|---|---|
| Vorinosat (SAHA, Zolinza) | μMol/L | FDA approved - CTCL, PhII–III (p.o.) | |
| Panobinostat (LBH589) | 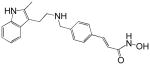 |
nMol/L | PhI–II (p.o.), PhII (i.v.) |
| Belinostat (PXD101) | 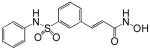 |
μMol/L | PhII (i.v.), PhI (p.o.) |
| ITF-2357 | nMol/L | PhI–II (p.o.) | |
| PCI-24781 (CRA-024781) | 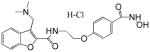 |
NA | PhI (p.o.) |
| Romidepsin (Depsipeptide, FK228) | 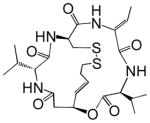 |
nMol/L | PhI–II (i.v.) |
| Valproic acid | 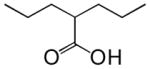 |
mMol/L | PhII–III (p.o.), PhII (p.o.) |
| Phenylbutyrate (VP-101, EL- 532) | 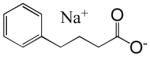 |
mMol/L | PhII (p.o.) |
| Pivanex (AN-9) | 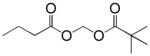 |
mMol/L | PhI–II (i.v.) |
| SB-939 | 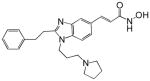 |
μMolL | PhI (p.o.) |
| Entinostat (MS-275, SNDX-275) | 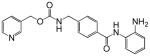 |
μMol/L | PhI–II (p.o.) |
| MGCD0103 | 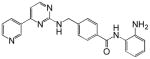 |
μMol/L | PhI–II (p.o.) |
| JHJ-26481585 | 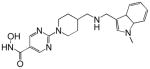 |
μMol/L | PhI–II (p.o.) |
See text for references.
A number of structurally diverse HDACi are in clinical trials as monotherapy and/or in combination with other anti-cancer agents (see below “Clinical Development”). Vorinostat (suberoylanilide hydroxamic acid, SAHA) is the first HDACi approved for cancer therapy-advanced cutaneous T-cell lymphoma [Marks and Breslow, 2007]. All HDACi reported to date have some isoform selectivity based on cell free assays with purified human recombinant HDACs (Table III) [Differences in the reported selectivity of HDACi may reflect differences in assay conditions ([Bolden et al., 2006; Butler and Kozikowski, 2008; Dokmanovic et al., 2007a; Estiu et al., 2008; Jones et al., 2008a; Khan et al., 2008; Kozikowski et al., 2007; Moradei et al., 2008; Rasheed et al., 2007; Schemies et al., 2009; Xu et al., 2007]. For example, vorinostat (SAHA) was found to inhibit class I HDACs, HDACs 1, 2, 3, and 8, class II HDACs, HDACs 6 and 10and HDAC11. Vorinostat does not inhibit HDAC 4, 5, 7, and9 at concentrations that are clinically relevant. N-carboxycinnamid acid bishydroxmic is a potent HDAC inhibitor and the structural bases for several derivatives including panobinostat (LBH589), and bleinostant (PXD-101) (Table III). These HDACi target a similar profile of HDACs as vorinostat. The cyclic peptides are a structurally complex group of HDAC inhibitors, such as, romidepsin (depsipeptide). Romidepsin inhibits primarily HDAC 1 and, more weakly, HDACs 2 and 3. In in vitro assays, the hydroxamic acid and cyclic peptide inhibitors are active at namamolar concentrations. Benzamide derivatives, including Entinostat (MS-275) and MGCD0103 are also active at nanomolar concentrations. Entinostat selectivity inhibits HDACs 1, 2, and 3. Butyrates and phenylbutyrate are drugs that have been on the market for non-oncologic uses for years and have recently been shown to have activity as HDAC inhibitors. These short chain fatty acids inhibit HDAC activity at millimolar concentrations. Valproic acid (VPA) an anticonvulsant has been shown to have HDACi activity also at millimolar concentrations. AN-9, pivaloyloxymethyl butyrate, is a novel prodrug of butyric acid. Tubacin is a small molecule which selectively inhibits HDAC 6 activity [Haggarty et al., 2003].
A number of HDAC isoform selective or specific inhibitors are currently in development [Butler and Kozikowski, 2008; Estiu et al., 2008; Haggarty et al., 2003; Jones et al., 2008a; Khan et al., 2008; Kozikowski et al., 2007; Moradei et al., 2008; Rasheed et al., 2007; Schemies et al., 2009; Somoza et al., 2004]. A question in the field of HDACi development which remains unanswered is whether selective inhibition of HDACs would be advantageous over the broader HDAC inhibitors as anti-cancer agents.
HDACi have multiple biologic effects consequent to alteration in patterns of acetylation of histones and many non-histone proteins which include proteins involved in regulation of gene expression, pathways of extrinsic and intrinsic apoptosis, cell cycle progression, redox pathways, mitotic division, DNA repair, cell migration, and angiogenesis (Figure 1) ([Blackwell et al., 2008; Bolden et al., 2006; Dokmanovic et al., 2007a; Glozak and Seto, 2007; Jones and Baylin, 2007; Minucci and Pelicci, 2006; Moradei et al., 2008; Rasheed et al., 2007; Shankar and Srivastava, 2008; Xu et al., 2007]. HDACi also have immunomodulatory activity that may contribute to mediating their anticancer effects. Further, in contrast to most cancer therapeutic agents, HDACi can induce death of transformed cells in both the proliferative and non-proliferative phases of the cell cycle [Burgess et al., 2004]. The mechanisms of action of HDACi are clearly complex and not completely elucidated. Nevertheless, the effect of HDACi on proteins that play a role in regulating many different cell pathways makes these agents attractive as potential anti-cancer therapeutics given the multiple defects that characterize most cancer cells [Jones et al., 2008b].
Various studies using cDNA arrays have found that between 2–20% of expressed genes are altered in transcription in the cells exposed to HDACi [Bolden et al., 2006; Dokmanovic et al., 2007a; Shankar and Srivastava, 2008]. HDACi increase the expression of about as many genes as are suppressed. The number and type of genes whose transcription is altered by HDACi is determined, in part, by the duration of exposure of the cells to the inhibitor, the HDACi to which the cells are exposed and the type of transformed cell studied [Ungerstedt et al., 2005]. Some changes in gene expression appear to be a direct effect of HDACi on the gene promoter and transcription factor complex while other changes in gene expression are downstream effects. While the patterns of alteration of gene expression are similar for different HDAC inhibitors, there are differences induced by different inhibitors [Peart et al., 2005]. The basis for the gene selective action of HDAC inhibitor in altering transcription is not well understood and may be determined, in part, by the composition of the transcription factor protein complexes [Gui et al., 2004].
HDACi induce cell cycle growth arrest in both normal and transformed cells [Ungerstedt et al., 2005; Xu et al., 2007]. Low concentrations of HDACi predominantly induce G1 arrest, while high concentrations induce both G1 and G2/M arrests. The increase of the cyclin dependent kinase (CDK) inhibitors and the decrease of cyclins induced by HDACi may work together to account for the reduced CDK activity and cause G1 and G2 arrest [Bolden et al., 2006; Xu et al., 2007]. CDK inhibitor p21 (WAF1/CIP1) is one of the most commonly induced genes by HDACi. HDACi induced p21expression is independent of p53 and correlates with the alterations of proteins associated with p21 promoter, including an increase in the acetylation of histones, a marked decrease in HDAC1 and Myc and recruitment of RNA polymerase II [Gui et al., 2004]. CDK inhibitor p27 (Kip1) is induced in some, but not all, tumor cells. HDACi can decrease the expression of cyclin D1 and cyclin A, and contribute to the G1 and G2 arrest, respectively.
HDACi can activate the extrinsic and/or extrinsic apoptotic pathways. HDACi up-regulate the expression of both death receptors and their ligands in vitro and in vivo in transformed cells, but not in normal cells. HDACi induced or increased expression of Fas and FasL, TRAIL and its receptor DR-5 and TNF-α. c-FLIP, an inhibitor of the death receptor pathway, was down-regulated by HDACi [Bolden et al., 2006; Rosato et al., 2006; Shankar and Srivastava, 2008; Xu et al., 2007].
The intrinsic or mitochondrial apoptotic pathway is a major mechanism of HDACi induced cell death. HDACi activate this pathway by causing the release of cytochrome c from mitochondrial intermembrane space and activation of caspase-9, while overexpression of Bcl-2 or Bcl-XL, which protect mitochondria, inhibits HDACi-induced apoptosis [Bolden et al., 2006; Xu et al., 2007]. Bid cleavage, which can initiate the intrinsic apoptotic pathway, occurs before mitochondrial disruption in CEM cells cultured with vorinostat. HDACi can up-regulate pro-apoptotic proteins of Bcl-2 family, such as Bim, Bmf, Bax, Bak and Bikand decrease antiapoptotic proteins of Bcl-2 family, such as Bcl-2, Bcl-XL, Bcl-w, Mcl-1, and XIAP and surviving ([Bolden et al., 2006; Robbins et al., 2005; Rosato et al., 2006; Xu et al., 2007]. The mechanism of these effects are not well understood. The basal level of these proteins vary dramatically in different tumor cells even of the same type of cancer, e.g. prostate, as do the changes in these proteins induced by HDACi [Xu et al., 2006].
HDACi can induce mitotic cell death of transformed cells by causing mitotic defects. HDACi increase histone acetylation that disrupts the structure and function of the centromere and the pericentric heterochromatin with loss of binding to heterochromatin binding proteins [Robbins et al., 2005]. Histone acetylation interferes with histone phosphorylation and disrupts the function of mitotic spindle checkpoint proteins, such as BubR1, hBUB1, CENP-F and CENP-E. HDACi cause degradation of two important mitotic serine/threonine kinases, aurora A and B, and survivin, which has dual role in apoptosis and mitosis [Robbins et al., 2005; Zhang et al., 2008]. As a result, the cells show a transient arrest at prometaphase, followed by aberrant mitosis such as missegregation and loss of chromosomes, resulting in cell death by either apoptosis or, mitotic cell death/catastrophe [Xu et al., 2007].
HDACi can induce autophagic cell death. Hela cells with Apaf-1 knockout or Bcl-XL over-expression were induced to autophagic cell death when cultured with vorinostat or butyrate [Oh et al., 2008; Xu et al., 2007; Yamamoto et al., 2008]. HDAC1 may play a role in autophagy because inhibition of HDAC1 with a specific inhibitor or siRNA can induce autophagy. HDACi can induce senescence in cultured fibroblast and in carcinoma cells associated with polyploidy cells and mitotic defects [Xu et al., 2006].
Accumulation of reactive oxygen species (ROS) occurs in transformed cells cultured with HDACi, such as vorinostat, TSA, butyrate or entinostat ([Ungerstedt et al., 2005; Xu et al., 2006]. Accumulation of ROS may be important in HDACi-induced cell death. ROS scavengers such as N-acetylcysteine can decrease HDACi-induced apoptosis. HDACi up-regulates the expression of TBP-2. TBP-2binds to and inhibits reduced Trx activity and contribute to ROS accumulation in transformed but not in normal cells [Ungerstedt et al., 2005]. Trx is also an inhibitor of apoptosis signal-regulating kinase 1 (ASK1). ASK1 promotes apoptosis by activation of SET1-JNK and MKK3/MKK6-p38 signaling cascades, and by enhancing the expression of pro-apoptotic protein Bim through a positive feedback on E2F1 activity [Tan et al., 2006]. Inhibition of Trx by TBP2 activates ASK1, which promotes apoptosis.
Specific inhibition of HDAC6 activity or its downregulation by siRNA increases accumulation of acetylated α-tubulin and cortactin and is associated with inhibition of cell migration[Zhang et al., 2007]. HSP90 acetylation disrupts its chaperone function and client proteins such as pro-survival and pro-proliferation proteins Akt, Bcr-Abl, c-Raf and ErbB2 can be poly-ubiquitinated and degradated via proteasome pathway associated with apoptosis. HDACi disrupt HDAC-protein phosphatase1 complexes and activates the phosphatase which, in turn, inactivates Akt by dephosphorylation. Inhibition of HDAC6 leads to disruption of the anti-apoptotic Akt pathway, by both Akt dephosphorylation and its degradation.
Androgen receptor is also a client protein of HSP90, and inhibition of HDAC6 can induce AR degradation [Chen et al., 2005]. HDACi can inhibit transcription of AR and block function of AR on its target genes [Welsbie et al., 2009]. These findings suggest that HDACi may be useful in combination treatment of both hormone-dependent and hormone-refractory prostate cancers.
Another effect of inhibition of HDAC6 is to disrupt the aggresome pathway. HDAC6 is a component of the aggresome, a cellular structure that constitutes a major site of degradation for misfolded protein aggregates, both non-ubiquitinated and ubiquinated misfolded proteins. HDAC6 can bind both mono and poly-ubiquitinated proteins. HDAC6 also binds p150, a component of dynein motor complex, and acts as a bridge between the dynein motors and the ubiquinated proteins leading to aggresome formation. Inhibition of HDAC6 function makes cells more sensitive to misfolded protein stress induced by protease inhibitor and, as a consequence, to cell death [Hideshima et al., 2005]. This is a mechanistic basis for a therapeutic strategy combining an HDACi and a proteome inhibitor to treat certain cancers.
HDACi can block tumor angiogenesis in vitro and in vivo by reducing expression of pro-angiogenesis factors including HIF-1α and VEGF ([Bolden et al., 2006; Minucci and Pelicci, 2006; Xu et al., 2007]. HDACi can disrupt HIF-1α function by several different mechanisms. HDACi can induce HIF-1α degradation by acetylation at Lys532. Class II HDACs, HDAC4 or HDAC6, physically associate with HIF-1α and their selective inhibition by siRNA induced HIF-1α degradation [Qian et al., 2006]. HIF-1α binds to HSP90 and HDACi can disrupt HSP90 chaperone function, exposing HIF-1α to proteasomal degradation. A decrease in HIF-1α activation can result from inhibiting HDAC7 activity, since under hypoxic conditions, HDAC7 translocates to the nucleus, interacts with HIF-1α and increases its transcriptional activity. HDACi can inhibit tumor angiogenesis by preventing endothelial cells from responding to the angiogenic stimulus generated by VEGF ([Bolden et al., 2006; Minucci and Pelicci, 2006; Xu et al., 2007]. TSA and vorinostat inhibit VEGF-induced expression of VEGF receptors and induce semaphorin III expression in endothelial cells. These observations support the use of HDACi in combination therapy with VEGF inhibitors for various cancers.
Mechanisms of Resistance to HDAC Inhibitors are not well elucidated. Resistance to HDACi may reflect drug efflux, epigenetic alterations, stress response mechanisms and anti-apoptotic, and pro-survival mechanisms [Fantin and Richon, 2007]. For example, elevated levels of anti-apoptosis factor, BCL-2 or the redox proteins, peroxiredoxins are reported in cancers resistant to chemotherapy and radiation therapy [Fantin and Richon, 2007]. Resistance to HDAC inhibitors can also be linked to factors, such as, poor pharmacokinetics and tumor cell micro-environment.
DNA hyper-methylation may cause resistance to HDACi, inducing compact nucleosomes with consequent block to access of acetylases which can result in silencing of tumor suppression genes. Increased activities of DNA damage repair mechanisms may be an adaptive mechanism for cancer resistance to HDACi.
In general, multi-drug resistance mediated by drug efflux mechanisms is not a major cause of cancer cell resistance to HDACi with a possible exception of romidepsin (depsipeptide). Romidepsin is the only HDACi identified to date which is a substrate of glycoprotein and multi drug resistant protein [Fantin and Richon, 2007].
Alterations in structure or expression of specific HDACs have been shown to be a possible mechanism of resistance to HDACi. For example, an inactivating mutation of HDAC2 has been identified in certain human colon and endometrial cancer cell lines which is associated with resistance to TSA.
Cellular polyamines are cationic molecules that have a role in regulating gene expression by affecting the chromatin microenvironment. Studies have shown that polyamines can modulate the response to structurally diverse HDACi and depletion in polyamines is associated with resistance to apoptosis induced by HDACi [Fantin and Richon, 2007].
Resistance to HDACi can be associated with an increased capacity of cancer cells to resist oxidative stress. HDACi cause increased production of ROS. This may be a significant factor in the pro-apoptotic affects of HDACi. Increased activity of thioredoxins and/or peroxiredoxins, redox proteins that play a role in protection of cells from ROS, may cause resistance to HDACi [Nonn et al., 2003; Parmigiani et al., 2008].
Identifying resistance markers, such as, high levels of BCL-2, peroxiredoxins, or polyaminesis an area of investigation that can lead to possible strategies to maximize the therapeutic efficacy of HDACi by combining them with agents that target factor(s) that contribute to resistance.
HDACi in combination therapy with targeted anticancer drugs, cytotoxic agents, anti-angiogenesis drugs or radiation is potentially very useful, based on results of preclinical studies and early clinical trials [Bolden et al., 2006; Dokmanovic et al., 2007a; Glozak and Seto, 2007; Jones and Baylin, 2007; Minucci and Pelicci, 2006; Nolan et al., 2008; Rasheed et al., 2007; Shankar and Srivastava, 2008; Xu et al., 2007]. In preclinical studies, HDACi have been shown to be synergistic with diverse chemical and biological therapeutic agents. For example, in acute promyelocytic leukemia the aberrant recruitment of HDACs to the fusion proteins, PLML-RARα or PLZF-RARα, is a rational for using HDACi in combination with all-trans retinoic acid as a therapeutic strategy and has been demonstrated effective both in in vitro and in in vivo preclinical models. There is a molecular rational for the combination of HDACi with DNA demethylating agents or the methyltransferase inhibitors since hypermethylation can lead to compact nucleosomes resistant to acetylation. In this combination therapy, the sequence of administration of the agents, 5-aza-cytidine or 5-aza-2-deoxyitidine, prior to the HDACi, appears to be important in achieving efficacy in treating various cancers.
HDACi have shown synergy when used in combination with chemotherapeutic agents, including, anthracyclines, carboplatinum, taxanes, topoisomerase inhibitors, the nucleoside analogs, gemcitabine and fludarabine antiangiogenic agents, azacytidine, tyrosine kinase inhibitor, imatinib, proteasome inhibitor, bortezomib, the apoptosis inducer, TRAIL, the heat shock protein (HSP90) antagonist 17-allylamino-17-demethoxy-geldanamycin, rituximab, trastuzumab, and the EGFR inhibitor, erlotinib. As we better understand the mechanisms of HDACi induced tumor cell death, particularly the downstream anti-tumor pathways affected by HDACi, development of more effective combination therapeutic stratgies will be developed. Indeed, it is likely that HDACi will be most effective therapeutically used in combination with other anti-cancer agents [Carew et al., 2008; Nolan et al., 2008].
Clinical Development of HDACi is an active area of investigation [Dokmanovic et al., 2007a; Glozak and Seto, 2007; Marks and Breslow, 2007; Xu et al., 2007]. Based on published reports, there are at least 15 structurally different HDACi in clinical trials as monotherapy and combination therapy for hematologic and solid neoplasms (Table III).
Vorinostat is the most advanced of the HDACi in clinical development. Vorinostat was the first HDACi approved by Federal Drug Administration for clinical use in treating patients with advance cutaneous T-cell lymphoma [Duvic and Vu, 2007; Marks and Breslow, 2007]. Panobinostat (LBH-589) is somewhat more potent than vorinostat. It is in phase I and II clinical trials for hematologic and solid tumors as monotherapy and combination therapy. Several other hydroxamic acid based HDACi including belinostat, ITF2357, and JHJ 26481585arein phase I and early phase II clinical trials (Table III). For each of these agents, preliminary evidence indicates anti-tumor activity including stable disease, partial response and, in a few incidences, completes responses of transient duration. At least two benzamides, entinostat (MS-275, SNDX-275), and MGCD103 are in clinical development. These agents, as with the hydroxamic based drugs, are in trials as single agents and in combination with other drugs.
Clinical trials with several of the HDACi include patients with a wide variety of hematologic and solid neoplasms including, chronic lymphatic leukemia, Hodgkin lymphoma, myeloproliferative disorder, B-cell lymphoma, acute myeloid leukemia, multiple myeloma, head and neck cancer, brain tumors, melanoma, lung cancer, hepatocellular cancer, breast cancer, ovarian cancer, renal cell carcinoma, and pancreatic adenocarcinoma (www.clinicaltrials.gov).
A useful marker of biological activity of HDACi is the accumulation of acetylated histones in normal peripheral mononuclear (PMN) cells. This marker does not correlate with clinical efficacy – rather it is an indication that a biologically active in vivo level of the HDACi is achieved. For example, a single oral dose of vorinostat, 400 mg (OD, P.O.) will induce PMN cells to accumulate acetylated histones in 2 to 4 hours which returns to pretherapy levels in 10–14 hrs.
The main adverse affects of HDACi include fatigue, nausea, dehydration, diarrhea, and thrombocytopenia. Generally, these side effects are reversible upon cessation of administration of the drug.
Definitive reports of results of phase II or phase III clinical trials with any of the HDACi other than vorinostat therapy for CTCL have not been published. The clinical development of HDACi remains very much a work in progress, but is clearly an area of intense investigation with different agents (Table III).
Perspectives and Conclusions
HDACi are a promising new group of anticancer agents. We anticipate that developing HDACi and increasing our understanding of the effect of targeted inhibitors of HDACs will lead to anti-cancer drugs that are safer and more effective than so called non-targeted agents. HDACi can induce cancer cell death across a broad spectrum of different types of hematologic and solid neoplasms. The mechanisms of HDACi induced tumor cell death require further elucidation. It is clear that these agents can cause cancer cell growth arrest and cell death by more than one pathway including: extrinsic and intrinsic apoptotic cell death, autophagy, and mitotic cell death, and inhibit cell migration, and tumor angiogenesis and alter immune response. HDACi have histone and many non-histone protein substrates. Normal cells are relatively resistant to HDACi induced cell death. The basis of normal cell resistance to HDACi is not understood. It may be speculated that cancer cells, with multiple defects, can not reverse the critical effects of HDACi as normal cells may do. A consequence of the relative resistance of normal cells to HDACi is the fact that HDACi, in general, are well-tolerated in clinical trials.
The HDACi developed to date are structurally diverse and generally inhibit several HDACs. While HDACi have demonstrated anti-tumor effect both pre-clinically and in clinical trials to date across a broad variety of different cancers, only a proportion of patients with a given diagnosis appear to respond to the HDACi. The accumulated evidence to date indicate that HDACi may be most useful in combination with other targeted or cytotoxic anti-cancer drugs. It is clearly important to have a better understanding of the mechanisms of action of HDACi and identify markers that may predict response. Our best chance to identify predictive biomarkers is to continue to gain better understanding of the targets of HDACs and, it follows, of HDACi.
The structural variety of active HDACi suggest that, in part at least, HDACs may have effects consequent to protein-protein interactions as well as, their catalytic deacetylase activities. The fact that many non-histone proteins are HDAC substrates suggest that these enzymes may more properly be referred to as “lysine deacetylases”. The development of isoforms HDACi will be very useful in elucidating the mechanism of action of each of the HDACs and understanding which isoforms are necessary for therapeutic efficacy and which contribute to off target toxicity. Among the questions that remain to be addressed is whether isoform specific HDACi may be clinically more useful than “pan” inhibitors.
Currently a number of clinical trials are going forward with HDACi as monotherapy but an increasing number are with HDACi in combination with different anticancer agents both chemotherapy, as well as, targeted agents and radiotherapy, against abroad spectrum of both hematologic and solid tumors.
Cancer cells typically contain multiple genetic deficits that disrupt key cell pathways including regulation of cell division, cell migration, and cell death. Recent studies have shown that tumors such as pancreatic cancer and glioblastomas have a large number of genetic defects revealing an extremely complex pattern of abnormalities which suggest that therapeutic strategies that target biological pathways and in particular, multiple biological pathways are likely to be more effective therapeutics than drugs targeted at a single gene or protein. This concept supports the need for continued development of HDACi, particularly, in combination therapeutic strategies.
Acknowledgments
We are grateful to Joann Perrone and Mabel Miranda for their excellent assistance in the preparation of this manuscript. Studies referred to in this review from our laboratory were supported in part by the National Institute of Health Grant, P30CA08748-41, the David Koch Foundation, the Jack and Susan Rudin Foundation, and Experimental Therapeutics Center at Memorial Sloan-Kettering Cancer Center (MSKCC).
MSKCC and Columbia University hold patents on SAHA and related compounds that were exclusively licensed to ATON Pharma, acquired by Merck, Inc in April 2004. PAM was a founder of ATON and has a financial interest in the further development of SAHA (vorinostat) by Merck.
References
- Blackwell L, Norris J, Suto CM, Janzen WP. The use of diversity profiling to characterize chemical modulators of the histone deacetylases. Life Sci. 2008;82:1050–8. doi: 10.1016/j.lfs.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Burgess A, Ruefli A, Beamish H, Warrener R, Saunders N, Johnstone R, Gabrielli B. Histone deacetylase inhibitors specifically kill nonproliferating tumour cells. Oncogene. 2004;23:6693–701. doi: 10.1038/sj.onc.1207893. [DOI] [PubMed] [Google Scholar]
- Butler KV, Kozikowski AP. Chemical origins of isoform selectivity in histone deacetylase inhibitors. Curr Pharm Des. 2008;14:505–28. doi: 10.2174/138161208783885353. [DOI] [PubMed] [Google Scholar]
- Carew JS, Giles FJ, Nawrocki ST. Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008;269:7–17. doi: 10.1016/j.canlet.2008.03.037. [DOI] [PubMed] [Google Scholar]
- Chen B, Cepko CL. HDAC4 regulates neuronal survival in normal and diseased retinas. Science. 2009;323:256–9. doi: 10.1126/science.1166226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Meng S, Wang H, Bali P, Bai W, Li B, Atadja P, Bhalla KN, Wu J. Chemical ablation of androgen receptor in prostate cancer cells by the histone deacetylase inhibitor LAQ824. Mol Cancer Ther. 2005;4:1311–9. doi: 10.1158/1535-7163.MCT-04-0287. [DOI] [PubMed] [Google Scholar]
- Dokmanovic M, Clarke C, Marks PA. Histone Deacetylase Inhibitors: Overview and Perspectives. Mol Cancer Res. 2007a;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- Dokmanovic M, Perez G, Xu W, Ngo L, Clarke C, Parmigiani RB, Marks PA. Histone deacetylase inhibitors selectively suppress expression of HDAC7. Mol Cancer Ther. 2007b;6:2525–34. doi: 10.1158/1535-7163.MCT-07-0251. [DOI] [PubMed] [Google Scholar]
- Duvic M, Vu J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin Investig Drugs. 2007;16:1111–20. doi: 10.1517/13543784.16.7.1111. [DOI] [PubMed] [Google Scholar]
- Estiu G, Greenberg E, Harrison CB, Kwiatkowski NP, Mazitschek R, Bradner JE, Wiest O. Structural origin of selectivity in class II-selective histone deacetylase inhibitors. J Med Chem. 2008;51:2898–906. doi: 10.1021/jm7015254. [DOI] [PubMed] [Google Scholar]
- Fantin VR, Richon VM. Mechanisms of resistance to histone deacetylase inhibitors and their therapeutic implications. Clinical cancer research. 2007;13:7237–42. doi: 10.1158/1078-0432.CCR-07-2114. [DOI] [PubMed] [Google Scholar]
- Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–93. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–32. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- Gui CY, Ngo L, Xu WS, Richon VM, Marks PA. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci U S A. 2004;101:1241–6. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100:4389–94. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, Anderson KC. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A. 2005;102:8567–72. doi: 10.1073/pnas.0503221102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Bottomley MJ, Carfi A, Cecchetti O, Ferrigno F, Lo Surdo P, Ontoria JM, Rowley M, Scarpelli R, Schultz-Fademrecht C, Steinkuhler C. 2-Trifluoroacetylthiophenes, a novel series of potent and selective class II histone deacetylase inhibitors. Bioorg Med Chem Lett. 2008a;18:3456–61. doi: 10.1016/j.bmcl.2008.02.026. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008b;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–9. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- Kozikowski AP, Chen Y, Gaysin A, Chen B, D’Annibale MA, Suto CM, Langley BC. Functional differences in epigenetic modulators-superiority of mercaptoacetamide-based histone deacetylase inhibitors relative to hydroxamates in cortical neuron neuroprotection studies. J Med Chem. 2007;50:3054–61. doi: 10.1021/jm070178x. [DOI] [PubMed] [Google Scholar]
- Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol. 2007;25:84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- Moradei O, Vaisburg A, Martell RE. Histone deacetylase inhibitors in cancer therapy: new compounds and clinical update of benzamide-type agents. Curr Top Med Chem. 2008;8:841–58. doi: 10.2174/156802608784911581. [DOI] [PubMed] [Google Scholar]
- Nolan L, Johnson PW, Ganesan A, Packham G, Crabb SJ. Will histone deacetylase inhibitors require combination with other agents to fulfil their therapeutic potential? Br J Cancer. 2008;99:689–94. doi: 10.1038/sj.bjc.6604557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonn L, Berggren M, Powis G. Increased expression of mitochondrial peroxiredoxin-3 (thioredoxin peroxidase-2) protects cancer cells against hypoxia and drug-induced hydrogen peroxide-dependent apoptosis. Mol Cancer Res. 2003;1:682–9. [PubMed] [Google Scholar]
- Oh M, Choi IK, Kwon HJ. Inhibition of histone deacetylase1 induces autophagy. Biochem Biophys Res Commun. 2008;369:1179–83. doi: 10.1016/j.bbrc.2008.03.019. [DOI] [PubMed] [Google Scholar]
- Parmigiani RB, Xu WS, Venta-Perez G, Erdjument-Bromage H, Yaneva M, Tempst P, Marks PA. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc Natl Acad Sci U S A. 2008;105:9633–8. doi: 10.1073/pnas.0803749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peart MJ, Smyth GK, van Laar RK, Bowtell DD, Richon VM, Marks PA, Holloway AJ, Johnstone RW. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:3697–702. doi: 10.1073/pnas.0500369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian DZ, Kachhap SK, Collis SJ, Verheul HMW, Carducci MA, Atadja P, Pili R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006;66:8814–21. doi: 10.1158/0008-5472.CAN-05-4598. [DOI] [PubMed] [Google Scholar]
- Rasheed WK, Johnstone RW, Prince HM. Histone deacetylase inhibitors in cancer therapy. Expert Opin Investig Drugs. 2007;16:659–78. doi: 10.1517/13543784.16.5.659. [DOI] [PubMed] [Google Scholar]
- Robbins AR, Jablonski SA, Yen TJ, Yoda K, Robey R, Bates SE, Sackett DL. Inhibitors of histone deacetylases alter kinetochore assembly by disrupting pericentromeric heterochromatin. Cell Cycle. 2005;4:717–26. doi: 10.4161/cc.4.5.1690. [DOI] [PubMed] [Google Scholar]
- Rosato RR, Maggio SC, Almenara JA, Payne SG, Atadja P, Spiegel S, Dent P, Grant S. The histone deacetylase inhibitor LAQ824 induces human leukemia cell death through a process involving XIAP down-regulation, oxidative injury, and the acid sphingomyelinase-dependent generation of ceramide. Mol Pharmacol. 2006;69:216–25. doi: 10.1124/mol.105.017145. [DOI] [PubMed] [Google Scholar]
- Schemies J, Sippl W, Jung M. Histone deacetylase inhibitors that target tubulin. Cancer Lett. 2009 doi: 10.1016/j.canlet.2009.01.040. [DOI] [PubMed] [Google Scholar]
- Sengupta N, Seto E. Regulation of histone deacetylase activities. J Cell Biochem. 2004;93:57–67. doi: 10.1002/jcb.20179. [DOI] [PubMed] [Google Scholar]
- Shankar S, Srivastava RK. Histone deacetylase inhibitors: mechanisms and clinical significance in cancer: HDAC inhibitor-induced apoptosis. Adv Exp Med Biol. 2008;615:261–98. doi: 10.1007/978-1-4020-6554-5_13. [DOI] [PubMed] [Google Scholar]
- Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure. 2004;12:1325–34. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Tan J, Zhuang L, Jiang X, Yang KK, Karuturi KM, Yu Q. Apoptosis signal-regulating kinase 1 is a direct target of E2F1 and contributes to histone deacetylase inhibitor-induced apoptosis through positive feedback regulation of E2F1 apoptotic activity. J Biol Chem. 2006;281:10508–15. doi: 10.1074/jbc.M512719200. [DOI] [PubMed] [Google Scholar]
- Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, Ngo L, Holmgren A, Jiang X, Marks PA. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:673–8. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkuhler C, Di Marco S. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci U S A. 2004;101:15064–9. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Xu J, Chen Y, Borsu L, Scher HI, Rosen N, Sawyers CL. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res. 2009;69:958–66. doi: 10.1158/0008-5472.CAN-08-2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Ngo L, Perez G, Dokmanovic M, Marks PA. Intrinsic apoptotic and thioredoxin pathways in human prostate cancer cell response to histone deacetylase inhibitor. Proc Natl Acad Sci U S A. 2006;103:15540–5. doi: 10.1073/pnas.0607518103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–52. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Tanaka K, Sakimura R, Okada T, Nakamura T, Li Y, Takasaki M, Nakabeppu Y, Iwamoto Y. Suberoylanilide hydroxamic acid (SAHA) induces apoptosis or autophagy-associated cell death in chondrosarcoma cell lines. Anticancer Res. 2008;28:1585–91. [PubMed] [Google Scholar]
- Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR, Yao TP, Lane WS, Seto E. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007;27:197–213. doi: 10.1016/j.molcel.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XH, Rao M, Loprieato JA, Hong JA, Zhao M, Chen GZ, Humphries AE, Nguyen DM, Trepel JB, Yu X, Schrump DS. Aurora A, Aurora B and survivin are novel targets of transcriptional regulation by histone deacetylase inhibitors in non-small cell lung cancer. Cancer Biol Ther. 2008;7:1388–97. doi: 10.4161/cbt.7.9.6415. [DOI] [PubMed] [Google Scholar]