

Abstract
Parkinson’s disease (PD) is characterized by loss of dopaminergic neurons in the substantia nigra and formation of intracytoplasmic Lewy bodies (LBs). Loss-of-function mutations in parkin which encodes an E3 ubiqutin protein ligase contribute to a predominant cause of a familial form of PD termed autosomal recessive juvenile Parkinsonism (AR-JP). Drosophila parkin null mutants display muscle degeneration and mitochondrial dysfunction, providing an animal model to study Parkin-associated molecular pathways in PD. To define protein alterations involved in Parkin pathogenesis, we performed quantitative proteomic analyses of Drosophila parkin null mutants and age-matched controls utilizing both global internal standard technology (GIST) and extracted ion chromatogram peak area (XICPA) label-free approaches. A total of 375 proteins were quantified with a minimum of two peptide identifications from the combination of the XICPA and GIST measurements applied to two independent biological replicates. Sixteen proteins exhibited significant alteration. Seven of the dysregulated proteins are involved in energy metabolism, of which six were down-regulated. All five proteins involved in transporter activity exhibited higher levels, of which larval serum protein 1α, larval serum protein 1β, larval serum protein 1γ, and fat body protein 1 showed > 10-fold up-regulation and substantially higher level of fat body protein 1 was confirmed by Western blot analysis. These findings suggest that abnormalities in energy metabolism and protein transporter activity pathways may be associated with the pathogenesis of Parkin-associated ARJP.
Keywords: Parkin, Drosophila, Parkinson’s disease, Proteomics
Introduction
Parkinson’s disease (PD) is the second most common age-associated neurodegenerative disorder characterized by loss of dopaminergic neurons in the substantia nigra pars compacta and formation of intracytoplasmic Lewy bodies (LBs) and Lewy neurites. Although PD has been documented for about two centuries, the molecular mechanisms underlying dopamine neuron loss remain unclear and controversy remains about whether LBs are neuroprotective or neurotoxic in the pathogenesis of PD. Studies of PD-inducing factors including environmental exposures to neurotoxins1 (e.g., rotenone, paraquat, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) and gene mutations2 (e.g., parkin, PINK1, LARK2, UCH-L1, α-synuclein, and DJ-1) have suggested that oxidative stress,3 mitochondrial dysfunction,4 and ubiquitin-proteasome system (UPS) defects5 are three key players in the pathogenesis of PD. Although the prevalent PD cases are sporadic (i.e., PD with unknown causes), studies6–9 focused on the understanding of functions and roles of PD-linked genes in familial PD have provided invaluable insight into the etiology and pathology in PD.
Mutations in the gene parkin contribute to a predominant cause of a familial form of PD known as autosomal recessive juvenile Parkinsonism (AR-JP)10 and are also occasionally found in late-onset sporadic PD.11 Parkin is a 465 amino acid protein and functions as an E3 ubiquitin protein ligase involved in the UPS pathway for protein degradation.12 It has been suggested that loss-of-function mutations in parkin contribute to the pathogenesis in AR-JP,12 which has led to the assumption that accumulation of Parkin substrates causes the death of dopaminergic neurons. To date, at least nine Parkin substrates have been identified, including CDCrel-1,13 CDCrel-2,14 Cyclin E,15 Pael-R,16 synphilin-1,17 synaptotagmin XI,18 α/β-Tubulin,19 p38,20 and glycosylated α-synuclein.21 The diversity of Parkin substrates suggests a variety of roles that Parkin plays in the mechanisms of AR-JP.
To gain insight into Parkin-associated molecular pathways underlying AR-JP, the Drosophila melanogaster (commonly known as the fruit fly) parkin null model22 has been created. Like parkin-deficient mice models,23–25 Drosophila parkin null mutants do not show massive reduction of dopaminergic neurons; instead, these animals show shrinkage of the dorsomedial dopaminergic cell body, impaired flight and climbing ability, reduced longevity, and male sterility.22 The locomotor defects and male sterility were further explored and found to result from early mitochondrial dysfunction,22 supporting the correlation between mitochondrial defects and PD. A previous in vivo study26 showed that α-synuclein-induced mitochondrial damage was aggravated due to the lack of Parkin function. In support of this, Parkin was revealed to function as a multipurpose neuroprotective agent for dopaminergic neuron survival in the circumstance of various toxic insults.27 For instance, coexpression of Parkin suppresses toxicity induced by expression of mutant α-synuclein in Drosophila,28 suggesting a mechanistic link between α-synuclein and Parkin.
Our laboratory has performed proteome analyses29–32 on various forms of gain of function of α-synuclein Drosophila PD models to understand α-synuclein involved pathogenesis in PD. The previous work suggested that alterations in the actin cytoskeleton and mitochondrion may be associated with α-synuclein-mediated neurotoxicity that ultimately results in manifestation of the PD symptoms.29–32 To examine molecular events associated with the loss of function of Parkin and to gain a comprehensive view of protein alterations resulting from different PD-linked genes, we performed proteomic analyses of Drosophila parkin null mutants and age-matched controls utilizing multidimensional liquid chromatography (LC) and tandem mass spectrometry (MS/MS) techniques. Two well-established methods were employed for relative protein quantification, including a label-free approach based on extracted ion chromatogram peak area (XICPA)33 and an isotope labeling strategy based on global internal standard technology (GIST)31, 34.
Experimental Section
Drosophila genotypes and harvesting
The parkin null mutant genotype utilized was w; Df(3L)Pc-MK/park45. To obtain parkin null mutant flies, virgin females from a w; park45 /TM6B line (park45 was obtained from Leo J. Pallanck) were crossed to males from a w; Df(3L)Pc-MK/TM6B line or alternatively the cross was performed reciprocally. The w; Df(3L)Pc-MK/TM6B stock was obtained from the Bloomington Drosophila Stock Center (http://flystocks.bio.indiana.edu/). Mutant nomenclature and descriptions can be found at FlyBase (http://www.flybase.org/). Siblings carrying the following two genotypes: w; park45 /TM6B and w; Df(3L)Pc-MK/TM6B were used as controls. Control and parkin mutant flies were cultured on standard cornmeal medium, maintained under identical conditions (25 ± 1º C), and harvested at the same time. Only male flies were utilized to avoid variation associated with gender. A population of 200 adult fly heads was collected for each genotype at day 1 post-eclosion (within 24 h) for protein extraction, as described previously.31 A new batch of flies was raised for an independent replicate biological measurement.
Protein sample preparation
Fly samples were prepared as described previously29 with minor modifications. Briefly, fly head proteins were extracted using a mortar and an electric pestle in a 0.2 M phosphate buffer saline solution (pH 7.0) containing 8.0 M urea and 0.1 mM phenylmethylsulfonyl fluoride. After centrifugation (13,000 rpm) for 10 min, the supernatant was collected. A Bradford assay indicated that ~2.0 mg of protein was obtained from 200 adult fly heads. Human hemoglobin was spiked into equal amounts of control and parkin null fly samples at a 1:1 ratio. Protein mixtures were reduced, alkylated, and tryptically digested as described previously.29 Finally, tryptic peptides were desalted, dried, and stored at −80 ºC until future use.
GIST labeling of tryptic peptides
Equal amounts of dried control and parkin null fly tryptic peptides were processed in phosphate buffer (pH 7.5) to produce a 1 mg·mL−1 solution. The GIST labeling reaction was processed as described elsewhere.34 Briefly, a 100-fold molar excess of N-acetoxy-d0-succinimide (light) and N-acetoxy-d3-succinimide (heavy) was added to control or parkin null samples (in one experiment, the control sample was light-labeled and the parkin null sample was heavy-labeled, which was referred to as forward labeling; in a second independent biological replicate measurement, the control sample was heavy-labeled and the parkin null sample was light-labeled, which was referred to as reverse labeling). The reagents and protein mixtures were stirred for 5 h at room temperature. At the end of the reaction, the two differentially labeled samples were combined, treated with an excess amount of N-hydroxylamine as previously described,31 desalted, dried, and stored at −80 °C until further analysis.
Strong Cation Exchange (SCX) chromatography
Offline SCX chromatography was performed as described previously.31 Briefly, the isotopically labeled tryptic peptides were dissolved in a 5.0 mM potassium phosphate buffer solution in 75:25 water:acetonitrile at pH 3.0 (solvent A). The digest was then injected onto a javelin guard column (10 × 2.1 mm; PolyLC Inc., Southboro, MA) that preceded a polysulfoethyl aspartamide column (100 × 2.1 mm, 5 μm, 200 Å; PolyLC Inc., Southboro, MA). Mobile phases consisted of solvent A and B (solvent A with the addition of 350 mM potassium chloride). Binary gradients with respect to the percentage of solvent B were as follows: 0–5 min, 0%; 5–45 min, 0–40%; 45–90 min, 40–80%; 90–100 min, 80–100%; 100–110 min, 100%; 110–111 min, 100–0%; 111–121 min, 0%. The gradient was delivered at a flow rate of 0.2 mL·min−1 by a Waters 600 multisolvent delivery system (Waters, Milford, MA) and peptides were detected at 214 nm by a Waters 2487 dual λ absorbance detector (Waters, Milford, MA). One minute collections into 96 well plates over the 121 min gradient were combined into six fractions as follows: (1) 0–34 min, (2) 34–40 min, (3) 40–44 min, (4) 44–50 min, (5) 50–58 min, and (6) 58–121 min. Pooled fractions were desalted, dried, and stored at −80 ºC until further analysis.
Reversed-phase (RP)-LC-MS/MS analysis
RP-LC-MS/MS analysis was performed on an LTQ-FTICR mass spectrometer (ThermoElectron, San Jose, CA) coupled to an UltiMate 3000 LC system (Dionex Corporation, Sunnyvale, CA) which is interfaced through a Finnigan Nanospray II electrospray ionization source (ThermoElectron, San Jose, CA). The instrumental configuration has been described elsewhere.31 Briefly, peptides were separated on a 13.2 cm self-pack PicoFrit column (75 μm i.d.; New Objective, Woburn, MA) packed with Magic C18AQ (5 μm, 100 Å; Microm BioResources Inc., Auburn, CA). A sample volume of 6.4 μL was loaded onto a 300 μm i.d. × 5 mm precolumn cartridge (packed with C18 PreMap 100, 5 μm, 100 Å; LC Packings, a Dionex Company, Sunnyvale, CA) at a flow rate of 15 μL·min−1. Binary mobile phases consisting of 96.95:2.95:0.1 water:acetonitrile:formic acid (solvent A) and 99.9:0.1 acetonitrile:formic acid (solvent B) were delivered by an UltiMate micropump (Dionex Corporation, Sunnyvale, CA) at a flow rate of 250 nL·min−1. The same LC gradient was used as described previously.31 The instrument was operated to acquire a full FT-MS scan (m/z range of 300–2000) followed by MS/MS scans of the top three most intense ions in the linear ion trap using an automated data-dependent acquisition mode. The resolution was set to 100000 (at m/z 400) for the survey FT-MS scan. The data-dependent acquisition uses a 30 s exclusion duration time and a collision energy of 35%.
Data Analysis
Raw MS/MS spectra (.RAW) were processed using the Xcalibur software package BioWorks (version 3.3.1, Thermo Electron, San Jose, CA) to create a peak list (.DTA) with 50 ppm precursor ion tolerance. Individual.DTA files were grouped according to charge states of precursor ions using an algorithm written in-house. Charge-sorted.DTA files were submitted to MASCOT (Matrix Science, Version 2.1) and searched against the National Center for Biotechnology Information nonredundant (NCBInr) Drosophila database (28642 sequence entries) that is correlated to FBgn accession numbers in the FlyBase database for Drosophila protein identification. The same files were also searched against a home-built database containing protein sequences of human albumin, human hemoglobin, and horse heart myoglobin for assignment of the internal standard protein, human hemoglobin. The same parameters were utilized for the MASCOT search as previously described.31 Briefly, carbamidomethylation of cysteine residues was used as a fixed modification and acetylation (light or heavy) of lysine residues and the N-termini of peptides (in the case of the labeled samples) were defined as variable modifications. Additional parameters included a maximum of 2 missed trypsin cleavages, a precursor tolerance of 15 ppm and a MS/MS tolerance of 0.8 Da. Spectra that led to scores at or above the MASCOT assigned homology score (the homology score defines a spectral match at a 95% confidence level) are assigned to specific peptide sequences; peptide sequences matching to multiple FBgn accession numbers were discarded (i.e., only peptides having sequences that are unique to a single protein are considered). To estimate the false positive rate of peptide identifications in the present study, one of the samples (the parkin null sample was used for the label-free approach, and the most peptide-rich SCX fraction 3 was used for the GIST method) was searched against the reverse NCBInr Drosophila database (28642) using the same parameters described above. The false positive rate calculated according to the formula by Gygi and co-workers35 was 3.3% for the label-free approach and 1.3% for the GIST method.
Abundance ratios of light- and heavy-labeled peptides were computed using peak intensities from extracted ion chromatograms with an in-house written algorithm, as previously described by our lab.36 For peptides identified in multiple SCX fractions or charge states, peak intensities were summed prior to calculating relative abundance ratios. For peptides identified in the label-free approach, relative abundance ratios were obtained by comparing the XICPAs of the same peptides identified in parkin null mutants and controls using the ProteinQuant Suite.37 Relative quantification of proteins was obtained by averaging the ratios of multiple derived peptides from the protein, and then was normalized to the observed ratio of hemoglobin for each corresponding measurement. Only proteins with a minimum of two peptide sequence identifications from the two independent experiments were considered in the present study. Abundance ratios for proteins reported as differentially expressed were confirmed by manual inspection of the peaks observed in the XICPA label-free approach and the GIST method using the criteria described elsewhere.31
Western blot analysis
For Western blot analysis, a new batch of male control and parkin null flies was raised and harvested to obtain a total of 200 adult fly heads each. Proteins were extracted in 400 μL ice-cold 50 mM Tris buffer (pH 7.5) containing 10 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 50 mM dithiothreitol, and 4% CHAPS, as previously described.31 Protein concentrations were determined with a Bradford assay. Equal amounts of proteins were separated by SDS-PAGE with 8% polyacrylamide gels and electroblotted onto polyvinylidene difluoride membranes. Membranes were blocked overnight at 4°C in Odyssey blocking buffer (LI-COR, Lincoln, NE), then simultaneously incubated with anti-beta-tubulin monoclonal antibody (loading control, 1:5000 dilution; Developmental Studies Hybridoma Bank, Iowa City, IA) and anti-fat body protein 1 polyclonal antibody (kindly provided by Qisheng Song) for 2 h at room temperature with gentle shaking. Next, infrared dye 700-labeled goat anti-mouse IgG (LI-COR, Lincoln, NE) and infrared dye 800 conjugated affinity purified goat anti-rabbit IgG (LI-COR, Lincoln, NE) secondary antibodies were added and allowed to incubate for 2 h at room temperature. Immunoblotting bands were detected and quantified utilizing a LI-COR Odyssey Infrared imaging system (simultaneous two-color targeted analysis) and software.
Results
Overview of the experimental strategy
The present study utilized LC-MS/MS coupled to GIST and XICPA approaches for protein identification and quantification (Figure 1). Human hemoglobin was spiked into equal amounts of protein extracts from parkin null mutants and control flies at a 1:1 ratio. Approximately 14.3% (1/7) of the tryptic peptides from control and parkin null mutants were directly subjected to one-dimensional (1D) RPLC-MS/MS analysis for protein identification and quantification using the XICPA label-free approach; this is referred to as a 1D-XICPA approach. The remaining tryptic peptides from the control and parkin null mutant samples were differentially isotopically labeled using the GIST approach and then were combined at a 1:1 ratio. Approximately 14.3% of the combined GIST-labeled peptides were directly subjected to RPLC-MS/MS analysis; this is referred to as a 1D-GIST approach. The remaining labeled sample was pre-fractionated using an offline SCX chromatography before it was subjected to an online RPLC-MS/MS analysis; this is referred to as a 2D-GIST approach. Two biological replicates were performed; one was labeled in the forward direction and the other in the reverse fashion.31
Figure 1.

Experimental design for quantitative proteomic analysis. Standard protein hemoglobin was spiked into equal amounts of control and pakin null mutants at a 1:1 ratio. Three approaches including a 1D-XICPA, a 1D-GIST, and a 2D-GIST were employed for the analyses of each biological dataset. For measurements with isotope labeling, two independent experiments were carried out by isotopically labeling biological replicate samples in forward and reverse directions.
Example data for relative peptide quantification using the XICPA and GIST approaches
To increase protein coverage, both XICPA and GIST approaches were utilized in this study. Figure 2 shows examples of extracted ion chromatogram and mass spectrum for relative peptide quantification from the two methods. Human hemoglobin was spiked into equal amounts of control and parkin null mutant samples at a 1:1 ratio; the peptide ion [FFESFGDLSTPDAVMGNPK+2H]2+ belonging to hemoglobin that was doped into the control sample eluted at 64.06 min (Figure 2a, black trace) and the same peptide eluted at 64.04 min in the parkin null mutant sample (Figure 2a, red trace). The observed ratio of this peptide calculated from the XICPA approach is 1.00, faithfully representing the theoretical value (we note that observed peptide ratios differ and are not always 1.00, as shown in Table 1). Another peptide ion [IDEPLMFEEWIK+2H]2+ belonging to Drosophila larval serum protein 1γ (LSP-1γ) eluted at 76.36 min in the control sample and at 76.35 min in the parkin null mutant sample (Figure 2b). However, the XICPA obtained in parkin null mutants (red trace) is approximately five times of that in the control animals (black trace), suggesting up-regulation of LSP-1γ in parkin null flies (We note that the peak at 30 min corresponds to a different ion with the same m/z, 775.39 as the Drsophila LSP-1γ peptide [IDEPLMFEEWIK+2H]2+. Although the two ions have the same m/z, they were assigned to different peptides from their fragmentation spectra.) Consistently, the relative intensities of the light- and heavy-labeled peptide ion [KVADALTNAVAHVDDMPNALSALSDLHAHK+3H]3+ derived from hemoglobin do not show substantial differences between control and parkin null mutants (Figure 2c), whereas the relative intensities of the peptide ion [FLFEIVHR+2H]2+ derived from Drosophila LSP-1γ clearly indicates its up-regulation (4.7-fold, Figure 2d) in parkin null mutants.
Figure 2.

Examples of extracted ion chromatogram and mass spectrum for relative peptide quantification using the XICPA label-free method and the GIST isotope labeling approach. a) extracted ion chromatograms of a doubly charged peptide ion [FFESFGDLSTPDAVMGNPK+2H]2+ derived from hemoglobin spiked into the parkin null mutant sample (trace in red) and into the control fly sample (trace in black). b) extracted ion chromatograms of a doubly charged peptide ion [IDEPLMFEEWIK+2H]2 derived from Drosophila LSP-1γ from the parkin null mutant sample (trace in red) and the control fly sample (trace in black). c) mass spectra for light- and heavy- labeled peptide ion [KVADALTNAVAHVDDMPNALSALSDLHAHK+3H]3+ pairs derived from hemoglobin. d) mass spectra for light- and heavy- labeled peptide [FLFEIVHR+2H]2+ ion pairs from Drosophila LSP-1γ. The acetylation site is indicated in parenthesis with Ac.
Table 1.
Quantification of hemoglobin spiked into parkin null mutants and control flies.
| Biological replicate | Expected ratio (parkin null mutants/control) | Observed ratio (parkin null mutants/control) | ||
|---|---|---|---|---|
| 1D-XICPAa | 1D-GISTa | 2D-GISTa | ||
| I | 1.00 | 0.80 ± 0.15 (8) | 0.98 ± 0.09 (12) | 0.97 ± 0.12 (13) |
| II | 1.00 | 1.01 ± 0.18 (6) | 1.13 ± 0.12 (11) | 1.04 ± 0.12 (14) |
The numbers in the parentheses indicate the total number of peptides used for protein quantification. The observed ratio ± SD represents the average ratio and standard deviation of multiple peptides detected for a protein.
Quantification of internal standard proteins
Hemoglobin was spiked into control and parkin null mutants to assess variations associated with sample handling and instrumental analysis. In the measurement of the first biological replicate, a total of 8, 12, and 13 peptides were quantified from the 1D-XICPA, 1D-GIST, and 2D-GIST approach, respectively. The observed average ratio ± standard deviation (SD) was 0.80 ± 0.15, 0.98 ± 0.09, and 0.97 ± 0.12, respectively (Table 1). Similar results were obtained from the analyses of the second biological replicate; the observed ratio was 1.01 ± 0.18, 1.13 ± 0.12, and 1.04 ± 0.12 from 1D-XICPA, 1D-GIST, and 2D-GIST measurements, respectively (Table 1).
Quantitative proteome analysis of Drosophila parkin null mutants
Three approaches including 1D-XICPA, 1D-GIST, and 2D-GIST were utilized to analyze two biological replicates to increase confidence in the identification and quantification of proteins. Only proteins identified with a minimum of two unique peptides from the two biological replicates were considered for quantification; this criterion drastically increased the overlap between the two biological replicate measurements. A total of 131 proteins were quantified from the 1D-XICPA approach, among which 125 (95%) proteins were common between the two replicates (Figure 3a); 74 proteins were quantified from the 1D-GIST measurements, of which 67 (91%) proteins overlapped (Figure 3b); 329 proteins were quantified from the 2D-GIST analyses and 291 (88%) proteins were detected in both replicates (Figure 3c). The correlation coefficients of the commonly quantified proteins between the two replicates were 0.81, 0.90, and 0.89 in the 1D-XICPA, 1D-GIST, and 2D-GIST measurements, respectively, suggesting good protein quantification reproducibility between the two replicate experiments.
Figure 3.

Venn diagrams comparing the number of proteins quantified from two independent biological replicates from the a) 1D-XICPA, b) 1D-GIST, and c) 2D-GIST approach, respectively.
For the analysis of the first biological replicate, a total of 352 proteins were quantified from the combination of the three approaches; 46 proteins were commonly detected from all the three approaches; additionally, 6 proteins were in common between the 1D-GIST and 1D-XICPA measurements, 14 proteins were in common between the 1D-GIST and 2D-GIST approaches, and 34 proteins were in common between the 1D-XICPA and 2D-GIST experiments; there was 1, 40, and 211 proteins uniquely quantified utilizing the 1D-GIST, 1D-XICPA, and 2D-GIST approach, respectively (Figure 4a). Similar results were obtained in the analysis of the second biological replicate. A total of 361 proteins were quantified (three proteins quantified with peptides showing singleton light-labeled peaks in the 2D-GIST were not included in Figure 4b, therefore only 358 proteins are shown); 214 proteins were uniquely contributed from the 2D-GIST approach and 51 proteins were in common to all the three approaches (Figure 4b). For the commonly identified proteins, the three approaches yielded fairly consistent results, with a relative standard deviation (RSD) in the range of 2–33% for the first biological replicate and 5–27% for the second replicate.
Figure 4.

Quantification profiles of proteins with a minimum of two peptide identifications from the combination of 1D-GIST, 1D-XICPA, and 2D-GIST measurements on a logarithm scale. a) protein quantification profile obtained from the analysis of the first biological replicate; b) protein quantification profile obtained from the analysis of the second biological replicate. Proteins were classified into seven groups: Group a, proteins quantified from all three approaches; group b, proteins that uniquely overlap between the 1D-GIST and 1D-XICPA approaches; group c, proteins that uniquely overlap between the 1D-GIST and 2D-GIST approaches; group d, proteins that uniquely overlap between the 1D-XICPA and 2D-GIST measurements; group e, proteins quantified only in the 1D-GIST experiment; group f, proteins quantified only in the 1D-XICPA experiment; and group g, proteins quantified only in the 2D-GIST experiment. Along the x axis are proteins identified in each group (see above) that were sorted in an ascent order of relative protein abundance (parkin mutant/control).
In the present study, a total of 375 proteins were quantified (Supplementary table SI) from the combination of three independent measurements of two biological replicates. According to the quantification results for hemoglobin, a protein is determined to be significantly differentially expressed if it exhibits a well-established 1.5-fold change31, 33 in both biological replicates and if the change is observed in the same direction from a minimum of two peptide identifications. Four, eight, and fourteen proteins were significantly altered in 1D-GIST, 1D-XICPA, and 2D-GIST measurements, respectively, contributing to a total number of 18 proteins. Ubiquitin activating enzyme 1 (Uba-1) and the protein encoded by CG3731 showed down-regulation from the 1D-XICPA approach, however, the 2D-GIST measurements from a set of different peptides indicated unobvious change of the two proteins and therefore they were not considered altered. The 16 differentially expressed proteins are shown in Table 2. Half of the proteins (i.e., eight) were up-regulated and the other half were down-regulated in parkin null mutants relative to age-matched controls. Six proteins exhibited significantly differential expression from two of the approaches, including fat body protein 1 (FBP-1), LSP-1γ, LSP-2, glycogen phosphorylase, photoreceptor dehydrogenase, and oligomycin sensitivity-conferring protein. Five proteins showed up- or down-regulation from a minimum of five unique peptides, including LSP-2, FBP-1, glycogen phosphorylase, regucalcin, and photoreceptor dehydrogenase. Four proteins exhibited strong up-regulation (>10-fold), including LSP-1α, LSP-1β, LSP-1γ, and FBP-1, as shown from the relative intensities of the example peptides for each of the proteins from both forward and reverse labeling experiments (Supplementary Figure S1). Though closely related to LSP-1α, LSP-1β, and LSP-1γ, LSP-2 only exhibited mild up-regulation (~2-fold), as exemplified from the relative intensities of the peptide ions identified as [VHLEAGVNHIK+2H]2+ (Supplementary Figures S2a and S2b), [HDYYFDVHNFK+2H]2+ (Supplementary Figures S2c and S2d), and [TIVSHYWHLMETYPEYHKK+3H]3+ (Supplementary Figures S2e and S2f), from both forward and reverse labeling experiments. A total of 12 peptides (23% protein sequence coverage) were identified and quantified for LSP-2, strongly supporting its mild up-regulation in parkin null mutants.
Table 2.
Differentially expressed proteins observed in parkin null mutants relative to controls.
| Gene name or IDa | Biological replicate Ib | Biological Replicate IIb | Total peptides | Experimental approach |
|---|---|---|---|---|
| Fat body protein 1 (Fbp-1) | 8.51 ± 5.27 (4) | 7.78 ± 4.92 (4) | 6 | 1D-GIST |
| Fat body protein 1 (Fbp-1) | >10 (5) | >10 (9, 6 singleton L) | 10 | 2D-GIST |
| Fat body protein 1 (Fbp1) | 4.40 ± 1.23 (2) | 8.11(1) | 2 | 1D-XICPA |
| Larval serum protein 1 γ (Lsp1γ) | 7.17 (1) | 15.18 ± 13.23 (2) | 2 | 1D-GIST |
| Larval serum protein 1 γ (Lsp1γ) | 7.22 ± 4.31 (3) | >10 (3, 2 singleton L) | 4 | 2D-GIST |
| Larval serum protein 1 γ (Lsp1γ) | 4.97 (1) | 5.52 (1) | 1* | 1D-XICPA |
| Larval serum protein 2 (Lsp2) | 1.95 ± 0.75 (5) | 2.19 ± 1.75 (5) | 5 | 1D-GIST |
| Larval serum protein 2 (Lsp2) | 1.68 ± 0.37 (8) | 1.64 ± 0.33 (11) | 11 | 2D-GIST |
| Glycogen phosphorylase (GlyP) | 0.62 ± 0.06 (5) | 0.64 ± 0.12 (6) | 7 | 2D-GIST |
| Glycogen phosphorylase (GlyP) | 0.65 ± 0.05 (4) | 0.63 ± 0.10 (5) | 7 | 1D-XICPA |
| Photoreceptor dehydrogenase (Pdh) | 0.44 ± 0.09 (2) | 0.67 (1) | 2 | 1D-GIST |
| Photoreceptor dehydrogenase (Pdh) | 0.65 ± 0.11 (5) | 0.67 ± 0.20 (5) | 5 | 1D-XICPA |
| Oligomycin sensitivity-conferring protein (Oscp) | 0.41 (1) | 0.51 ± 0.22 (2) | 2 | 2D-GIST |
| Oligomycin sensitivity-conferring protein (Oscp) | 0.34 (1) | 0.53 ± 0.07 (2) | 2 | 1D-XICPA |
| Larval serum protein 1 α (Lsp1α) | 9.10 ± 7.26 (2) | >10 (3, 1 singleton L) | 4 | 2D-GIST |
| Larval serum protein 1 β (Lsp1β) | 9.19 (1) | 14.29 (1) | 2 | 2D-GIST |
| Cytochrome c proximal (Cyt-c-p) | 1.73 ± 1.08 (2) | 2.18 (1) | 2 | 2D-GIST |
| Trehalose-6-phosphate synthase 1 (Tps1) | 0.60 (1) | 0.60 ± 0.10 (3) | 3 | 2D-GIST |
| Rpn1 | 0.56 (1) | 0.55 ± 0.23 (2) | 3 | 2D-GIST |
| Regucalcin | 1.62 ± 0.38 (5) | 1.72 ± 0.48 (3) | 6 | 1D-XICPA |
| CG11198 | 0.47 ± 0.03 (2) | 0.50 ± 0.14 (2) | 2 | 2D-GIST |
| CG15674 | 2.43 ± 1.02 (2) | NA | 2 | 2D-GIST |
| UGP | 0.53 ± 0.03 (2) | 0.46 (1) | 2 | 2D-GIST |
| CG33138 | 0.57 ± 0.05 (3) | 0.56 ± 0.19 (4) | 4 | 2D-GIST |
Gene name or IDs were obtained from the FlyBase database (http://www.flybase.org/).
The numbers in parentheses indicate the total number of peptides used for protein quantification. The observed ratio ± SD represents the average ratio and standard deviation of multiple peptides detected for a protein. Detailed information on peptide identification is in supplemental Table SI.
A total of six unique peptides were identified for this protein, the other five were only identified in parkin null mutants and therefore were not included in the relative protein quantification.
Validation of protein abundance change by Western blot analysis
To validate protein expression alterations observed in parkin null mutants, we applied Western blot analysis to one of the differentially expressed proteins, i.e., FBP-1. Specifically, our proteomic measurements revealed that FBP-1 was strongly up-regulated in parkin null mutants relative to age-matched controls averaged from a number of peptides with obtained ratios spanning a wide range (Table 2). We note that the large ratio differences within the detected peptides may be associated with several reasons, such as the well-known post-translational modifications and post-translational cleavages associated with FBP-1.38 Consistently, Western blot analysis (with anti-FBP-1 polyclonal antibodies) detected several bands (Figure 5) that correspond to FBP-1 fragments resulting from immediate post-translational cleavage by 20-hydroxyecdysone (20E).38 Western blot analysis indicated that the fragments at 50 kDa and 19 kDa are the two most abundant species and all the fragments were up-regulated, with the 19 kDa fragment (446–603) showing a 12.7-fold up-regulation in parkin null mutants compared to that in controls, supporting the proteomic measurement.
Figure 5.
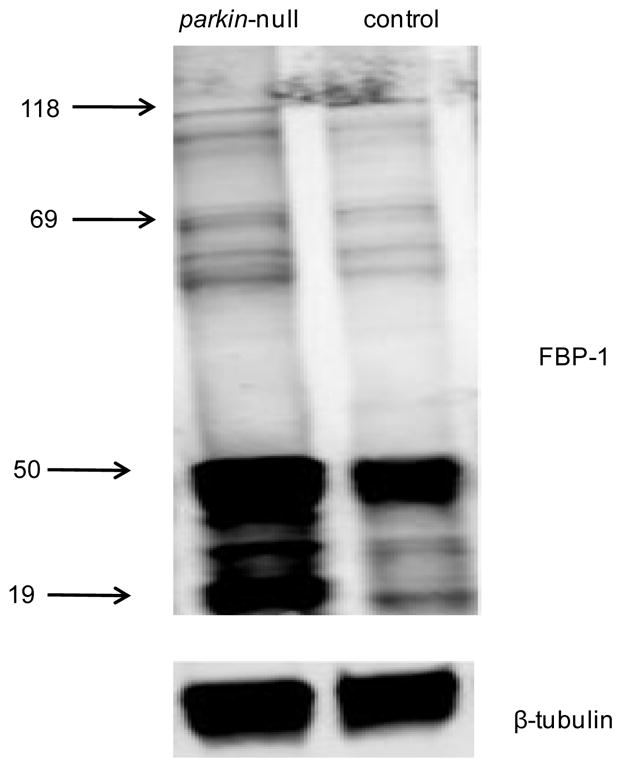
Western blot analysis of FBP-1 using anti-FBP-1 polyclonal antibodies (top) with beta-tubulin (bottom) as the loading control. Many bands corresponding to the FBP-1 protein (118 kDa) and several FBP-1 fragments were detected. The fragments at 50 kDa and 19 kDa are the two most abundant species.
Discussion
Overview of proteomic measurements
The present study describes a quantitative proteomic analysis of Drosophila parkin null mutants and age-matched controls utilizing both label-free and isotope labeling approaches on two independent biological replicates. Results from the internal standard protein hemoglobin indicated that both the XICPA label-free method and the GIST isotope labeling approach provided reasonable accuracy (≤20% error in this study) for relative protein quantification. The three measurements exhibited inherent consistency (negative errors in the first biological replicate and positive errors in the second replicate). Incorporation of an additional SCX separation significantly increased proteome coverage as well as quantification confidence compared to a 1D-LC separation. Expectably, the number of proteins quantified from the GIST coupled to the 1D LC-MS/MS analysis was lower (~56%) than that obtained from the 1D LC-MS/MS label-free method because the GIST approach acetylates the primary amines (N-termini and lysine residues), which effectively removes protonation sites, and thus makes the lysine-containing peptides harder to detect. On the other hand, the isotopic labeling approach generally yields more accurate and more reproducible quantification measurements. Overall, the two methods provide complementary information (i.e., the number of proteins quantified from the combination of the two methods is greater than that from either of the methods alone).
A total of 1203 proteins were identified in this study with the consideration of those assigned with one single peptide. To lower the false discovery rate (refer to the experimental section) associated with protein identification and to increase the reliability of protein quantification, only proteins observed with a minimum of two unique peptides were considered for quantitative analysis. A total of 375 proteins were quantified from assignments of at least two peptide sequences from two independent biological measurements. Pallanck and co-coworkers39 reported that at the transcriptional level ~1000 (16.7%) out of the ~6000 genes on the cDNA microarrays showed significantly altered expression at 1-day-old parkin null mutants using the whole adult flies, whereas 26 (0.22%) mRNAs out of the ~12000 genes were significantly differentially expressed at 48 h parkin null mutant pupae. In contrast, the present study on 1-day-old parkin null mutant heads revealed that 16 (4.3%) out of the total 375 quantified proteins were significantly regulated (We note here that the proteins identified in this study are primarily the most abundant ones because the whole cell lysates were analyzed without prior depletion of high abundant proteins. Also only water soluble proteins were extracted for the analysis. Thus the majority of low abundant proteins and membrane-associated proteins were not readily identified.). Up-regulation of regucalcin and LSP-1γ and down-regulation of UGP and CG33138 were also observed at the mRNA level from the whole fly, whereas, photoreceptor dehydrogenase was observed oppositely regulated at the mRNA level.39 Flybase database (http://www.flybase.org/) search indicates that nine out of the 16 proteins have human homologs/orthologs, including glycogen phosphorylase, photoreceptor dehydrogenase, Oligomycin sensitivity-conferring protein, cytochrome C proximal, Rpn1, regucalcin, UGP, CG11198, and CG 33138. Currently, nine human Parkin substrates have been identified and only two (i.e., cyclin E and α/β-tubulin) of them have known Drosophila homologues. In this work Cyclin E was not identified and α/β-tubulin was not included due to the redundancy of the observed peptides. Four proteins (LSP-1α, LSP-1β, LSP-1γ, and FBP-1) exhibited substantially higher expression level in parkin null mutants and strong up-regulation of FBP-1 was confirmed by Western blot analysis. Gene ontology analysis40 indicates that the altered proteins are primarily associated with energy metabolism, transporter activity, and protein degradation (Figure 6). A brief discussion of the relevance of these dysregulated proteins to the loss of function of Parkin in PD is presented below.
Figure 6.

Bar chart representation in the annotation of biological process for the 16 dysregulated proteins in parkin null mutants relative to age-matched controls. Bars in black and white represent up-regulated and down-regulated proteins, respectively.
Energy metabolism associated proteins
The majority of the proteins (i.e., cytochrome C proximal, glycogen phosphorylase, olycomycin sensitivity conferring protein, trehalose-6-phosphate synthase 1, UGP, and two unnamed proteins encoded CG33138, and CG11198) are involved in the functional category of energy metabolism. Specifically, it includes the glycolytic pathway and the mitochondrial electron transport chain. Trehalose-6-phosphate synthase 1 is the sole protein in Drosophila that catalyzes the biosynthesis of trehalose, a major sugar in the hemolymph that provides energy during flight and reduces protein aggregation during heat shock.41 Glycogen phosphorlyase is the enzyme involved in the first step of the glycogen degradation.42 The protein encoded by UGP, UTP:glucose-1-phosphate uridylytransferase is involved in polysaccharide metabolism and it shows down-regulation in old (> 25-day) flies relative to young flies at the transcriptional level.43 The protein encoded by an unknown gene, CG33138 is also involved in carbohydrate metabolism as inferred from electronic annotation.44 Reduced levels of these carbohydrate metabolism-associated proteins suggest substantial metabolic abnormalities, which appear to be supported by the observed flight muscle degeneration22 as well as the reduced mass and cell size in Drosophila parkin null mutants relative to age-matched controls.45 Similar to what was observed in Drosophila parkin null mutants, parkin-deficient mice also exhibited reduced body weight gain in comparison to age-matched controls.46 Proteomic analyses of parkin knockout mice also revealed a high portion of dysregulated proteins involved in energy metabolism.47, 48
Two dysregulated genes are involved in the mitochondrial respiratory chain, including oligomycin sensitivity-conferring protein and cytochrome C proximal. Oligomycin sensitivity-conferring protein is one of the protein components present at the stalk region connecting F0 and F1 segments of the mitochondrial ATP synthase.49 It has been demonstrated that oligomycin sensitivity-conferring protein plays a pivotal role in transmitting the energy of the electrochemical gradient to the catalytic F1 segment for the synthesis of ATP.49 A reduced expression level of oligomycin sensitivity-conferring protein may affect the proper function of the stalk attached to the F0 rotor and thereby the production of ATP. The cytochrome C proximal and cytochrome C distal are the only two cytochrome C genes found in Drosophila, while the cytochrome C proximal is predominantly expressed over the cytochrome C distal.50 Early studies50, 51 suggested that cytochrome C proximal functions in respiration whereas cytochrome C distal is required for caspase activation necessary for spermatid individualization. However, one recent study52 demonstrates that the distinct roles of the two cytochrome C proteins are interchangeable. Specifically, the loss of function of cytochrome C distal leads to male fly sterility and expression of cytochrome C proximal can rescue all the sterile phenotypes, demonstrating the function of cytochrome C proximal in caspase activation. Because the male sterile phenotype in parkin null mutants arises from a late defect at the individualization stage22 and spermatid individualization requires caspase activity, the perturbed expression level of cytochrome C proximal may be involved in the molecular mechanism responsible for male sterility in parkin null mutants. Notably, cytochrome c proximal is the only protein involved in energy metabolism that is up-regulated in parkin null mutants. It is possible that the increased expression level of cytochrome c proximal may be utilized to counterbalance the energy deficiency caused by the defects in ATP synthase and the glycolytic pathway.
Transporter activity associated proteins
Five proteins are associated with transporter activity, including LSP-1α, LSP-1β, LSP-1γ, LSP-2, and FBP-1. Gene functional classification utilizing the Database for Annotation, Visualization and Integrated Discovery (DAVID) tool53 indicates that the five genes form one cluster and are genetically positively associated (Figure 9). Though previous studies reported exclusive expression of LSP-1 during the larval and pupal stage,54 our proteomic measurements provide evidence of its adult expression at 1-day-old flies. Particularly, LSP-1α, LSP-1β, and LSP-1γ were all drastically up-regulated at similar levels (Table 2 and Figure 5); these results are corroborated by the finding that these three proteins equimolarly constitute the heterohexamer hemolymph protein LSP-1.55 In contrast to LSP-1, another major hemolymph protein, LSP-2 (a homohexamer), exhibited much less perturbation. DAVID analysis indicated that LSP-2 differs from LSP-1 only in one gene-term association, i.e., hemocyanin related larval serum protein arylphorin related (Figure 7). It is interesting to mention that both LSP-1 and LSP-2 are synthesized during the larval development and their amounts significantly decline in late larval life; however, the abundance of LSP-1 declines at a faster rate than LSP-2,56 suggesting that LSP-1 and LSP-2 regulation may undergo different pathways at the late larval stage. This appears to support our findings of the drastic regulation differences between LSP-1 and LSP-2. Similarly, previous studies38 also demonstrated that FBP-1 is uniquely expressed at the end of the third larval instar; however, our results indicated that FBP-1 was expressed at fairly high abundance at 1-day-old adult flies and its expression level was strongly elevated in parkin null mutants. Notably, Lepesant and coworkers38 have demonstrated that FBP-1 serves as the receptor of both LSP-1 and LSP-2 and the uptake of the storage proteins is 20E-dependent. After translation, FBP-1 undergoes immediate cleavage in three subsequent steps,38 which supports our observation of several bands in the Western blot analysis (Figure 7). The finding of the strong (with the exception of LSP-2) up-regulation of these hormone-responsive genes involved in transporter activity in parkin null mutants raises two possibilities: i) 20E is present at a higher concentration level in parkin null mutants compared to that in controls, and ii) degradation of these proteins was impaired due to the loss of function of Parkin. Nevetherless, furhter studies are warranted to test these hypotheses. One of these future directions may be to study whether these transorter activity-associated proteins are Parkin substrates.
Figure 7.

One gene cluster composing of five genes revealed by gene functional analysis using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) tool 53. The box in grey indicates that the corresponding gene-term association is positively reported and the box in black indicates that the corresponding gene-term association is not reported yet.
Comparison of parkin null mutants with α-synuclein transgenic flies
A comparison of the three α-synuclein Drosophila PD models29–31 with the parkin null AR-JP model shows five commonly disturbed proteins including FBP-1, LSP-2, LSP-1γ, regucalcin and trehalose-6-phosphate hydrolase. However, all five proteins show opposite regulation directions between the gain of function of α-synuclein Drosophila models and the loss of function of Parkin null AR-JP model. For example, FBP-1 and LSPs were down-regulated in the α-synuclein PD models30–32 whereas they were up-regulated in the parkin null AR-JP model (Table 2, Figure 5, and Figures S1&S2). The findings of the commonly dysregulated proteins with opposite regulation directions between α-synuclein- and Parkin-linked Drosophila PD models are intriguing. It raises the possibility that these two genes may be involved in certain common molecular pathways leading to the alteration of the same proteins and possibly, consequently similar phenotypes including locomotor dysfunction. On the other hand, because the gain of function of α-synuclein is associated with LB pathology while the loss of function of Parkin is not, the other hypothesis is that the disease symptoms progress through different pathways that lead to opposite regulation patterns of the same perturbed proteins as well as the uniquely regulated proteins. In view of previous studies57–60 on the possible pathological interactions between α-synuclein and Parkin, for instance, co-expression of Parkin rescues the toxicity induced by overexpression of α-synuclein in rats61 and by expression of mutant α-synuclein in Drosophila,60 it appears to support the former hypothesis that there is a mechanistic link between the two genes that may shed light on common molecular mechanisms underlying PD. One strategy to test this speculation is to study whether Parkin and α-synuclein are involved in certain common protein interaction networks. The report that Parkin interacts with and ubiquitinates synphilin-1, an α-synuclein-interacting protein, provides strong evidence that Parkin and α-synuclein are associated with a common pathologic pathway leading to PD.17
Supplementary Material
Acknowledgments
The authors gratefully acknowledge financial support from the National Institute of Heath (NIH #RO1-AG-024547-3); additional partial support is provided from the Analytical Node of the Indiana University Metabolomics and Cytomics Initiative (funded by the Lilly Endowment). We also acknowledge the following invaluable contributions: Melissa Phelps (Indiana University, Bloomington) for generating parkin null flies; Dr. Qisheng Song (University of Missouri, Columbia) for providing the fat body protein 1 antibody; Dr. Kenneth P. Nephew and Xinghua Long (Department of Medical Sciences, Indiana University, Bloomington, IN) for providing assistance with Western blot analysis. The anti beta-tubulin antibody developed by Michael Klymkowsky was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA.
References
- 1.Di Monte DA. Lancet Neurol. 2003;2:531–538. doi: 10.1016/s1474-4422(03)00501-5. [DOI] [PubMed] [Google Scholar]
- 2.Thomas B, Beal MF. Hum Mol Genet. 2007;16:R183–R194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- 3.Sohmiya M, Tanaka M, Tak NW, Yanagisawa M, Tanino Y, Suzuki Y, Okamoto K, Yamamoto Y. J Neurolog Sci. 2004;223:161–166. doi: 10.1016/j.jns.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 4.Keeney PM, Xie J, Capaldi RA, Bennett JP. J Neurosci. 2006;26:5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McNaught KSP, Olanow CW, Halliwell B, Isacson O, Jenner P. Nat Rev Neurosci. 2001;2:589–594. doi: 10.1038/35086067. [DOI] [PubMed] [Google Scholar]
- 6.Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JMG, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 7.Autere JM, Hiltunen MJ, Mannermaa AJ, Jakala PA, Hartikainen PH, Majamaa K, Alafuzoff I, Soininen HS. Eur J Neurol. 2002;9:479–483. doi: 10.1046/j.1468-1331.2002.00458.x. [DOI] [PubMed] [Google Scholar]
- 8.Burke RE. Neurologist. 2004;10:75–81. doi: 10.1097/01.nrl.0000117822.90759.83. [DOI] [PubMed] [Google Scholar]
- 9.Solano SM, Miller DW, Augood SJ, Young AB, Penney JB. Ann Neurol. 2000;47:201–210. [PubMed] [Google Scholar]
- 10.Abbas N, Lucking CB, Ricard S, Durr A, Bonifati V, De Michele G, Bouley S, Vaughan JR, Gasser T, Marconi R, Broussolle E, Brefel-Courbon C, Harhangi BS, Oostra AB, Fabrizio E, Bohme GA, Pradier L, Wood NW, Filla A, Meco G, Denefle P, Agid Y, Brice A. Hum Mol Genet. 1999;8:567–574. doi: 10.1093/hmg/8.4.567. [DOI] [PubMed] [Google Scholar]
- 11.Pilcher H. Lancet Neurol. 2005;4:798–798. doi: 10.1016/s1474-4422(05)70237-4. [DOI] [PubMed] [Google Scholar]
- 12.von Coelln R, Dawson VL, Dawson TM. Cell Tiss Res. 2004;318:175–184. doi: 10.1007/s00441-004-0924-4. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Gao J, Chung KKK, Huang H, Dawson VL, Dawson TM. Proc Natl Acad Sci USA. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi P, Snyder H, Petrucelli L, Theisler C, Chong M, Zhang Y, Lim K, Chung KKK, Kehoe K, D’Adamio L, Lee JM, Cochran E, Bowser R, Dawson TM, Wolozin B. Mol Brain Res. 2003;117:179–189. doi: 10.1016/s0169-328x(03)00318-8. [DOI] [PubMed] [Google Scholar]
- 15.Staropoli JF, McDermott C, Martinat C, Schulman B, Demireva E, Abeliovich A. Neuron. 2003;37:735–749. doi: 10.1016/s0896-6273(03)00084-9. [DOI] [PubMed] [Google Scholar]
- 16.Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- 17.Chung KKK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM. Nat Med. 2001;7:1144–1150. doi: 10.1038/nm1001-1144. [DOI] [PubMed] [Google Scholar]
- 18.Huynh DP, Scoles DR, Nguyen D, Pulst SM. Hum Mol Genet. 2003;12:2587–2597. doi: 10.1093/hmg/ddg269. [DOI] [PubMed] [Google Scholar]
- 19.Ren Y, Zhao JH, Feng J. J Neurosci. 2003;23:3316–3324. doi: 10.1523/JNEUROSCI.23-08-03316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corti O, Hampe C, Koutnikova H, Darios F, Jacquier S, Prigent A, Robinson JC, Pradier L, Ruberg M, Mirande M, Hirsch E, Rooney T, Fournier A, Brice A. Hum Mol Genet. 2003;12:1427–1437. doi: 10.1093/hmg/ddg159. [DOI] [PubMed] [Google Scholar]
- 21.Shimura H, Schlossmacher MC, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ. Science. 2001;293:263–269. doi: 10.1126/science.1060627. [DOI] [PubMed] [Google Scholar]
- 22.Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. P Natl Acad Sci. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu NP, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- 24.Itier JM, Ibanez P, Mena MA, Abbas N, Cohen-Salmon C, Bohme GA, Laville M, Pratt J, Corti O, Pradier L, Ret G, Joubert C, Periquet M, Araujo F, Negroni J, Casarejos MJ, Canals S, Solano R, Serrano A, Gallego E, Sanchez M, Denefle P, Benavides J, Tremp G, Rooney TA, Brice A, de Yebenes JG. Hum Mol Genet. 2003;12:2277–2291. doi: 10.1093/hmg/ddg239. [DOI] [PubMed] [Google Scholar]
- 25.von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM. Proc Natl Acad Sci USA. 2004;101:10744–10749. doi: 10.1073/pnas.0401297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stichel CC, Zhu XR, Bader V, Linnartz B, Schmidt S, Lubbert H. Hum Mol Genet. 2007;16:2377–2393. doi: 10.1093/hmg/ddm083. [DOI] [PubMed] [Google Scholar]
- 27.Feany MB, Pallanck LJ. Neuron. 2003;38:13–16. doi: 10.1016/s0896-6273(03)00201-0. [DOI] [PubMed] [Google Scholar]
- 28.Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR. Neuron. 2002;36:1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- 29.Xun ZY, Sowell RA, Kaufman TC, Clemmer DE. J Proteome Res. 2007;6:348–357. doi: 10.1021/pr060488o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xun ZY, Sowell RA, Kaufman TC, Clemmer DE. J Proteome Res. 2007;6:3729–3738. doi: 10.1021/pr0700504. [DOI] [PubMed] [Google Scholar]
- 31.Xun ZY, Sowell RA, Kaufman TC, Clemmer DE. Mol Cell Proteomics. 2008;7:1191–1203. doi: 10.1074/mcp.M700467-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xun ZY, Kaufman TC, Clemmer DE. J Proteome Res. 2008;7:3911–3921. doi: 10.1021/pr800207h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bondarenko PV, Chelius D, Shaler TA. Anal Chem. 2002;74:4741–4749. doi: 10.1021/ac0256991. [DOI] [PubMed] [Google Scholar]
- 34.Chakraborty A, Regnier FE. J Chromatogr A. 2002;949:173–184. doi: 10.1016/s0021-9673(02)00047-x. [DOI] [PubMed] [Google Scholar]
- 35.Peng JM, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. J Proteome Res. 2003;2:43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 36.Liu XY, Miller BR, Rebec GW, Clemmer DE. J Proteome Res. 2007;6:3134–4142. doi: 10.1021/pr070092s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mann B, Madera M, Sheng Q, Tang H, Mechref Y, Novotny MV. Rapid Commun Mass Spectrom. 2008;22:3823–3834. doi: 10.1002/rcm.3781. [DOI] [PubMed] [Google Scholar]
- 38.Burmester T, Antoniewski C, Lepesant JA. Eur J Biochem. 1999;262:49–55. doi: 10.1046/j.1432-1327.1999.00315.x. [DOI] [PubMed] [Google Scholar]
- 39.Greene JC, Whitworth AJ, Andrews LA, Parker TJ, Pallanck LJ. Hum Mol Genet. 2005;14:799–811. doi: 10.1093/hmg/ddi074. [DOI] [PubMed] [Google Scholar]
- 40.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Q, Behar KL, Xu T, Fan C, Haddad GG. J Biol Chem. 2003;278:49113–49118. doi: 10.1074/jbc.M308652200. [DOI] [PubMed] [Google Scholar]
- 42.Tick G, Cserpan I, Dombradi V, Mechler BM, Torok I, Kiss I. Biochem Bioph Res Co. 1999;257:34–43. doi: 10.1006/bbrc.1999.0396. [DOI] [PubMed] [Google Scholar]
- 43.Zou S, Meadows S, Sharp L, Jan LY, Jan YN. Proc Natl Acad Sci USA. 2000;97:13726–13731. doi: 10.1073/pnas.260496697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson RJ, Goodman JL, Strelets VB. Nucleic Acids Res. 2008;36:D588–D593. doi: 10.1093/nar/gkm930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G. Development. 2004;131:2183–2194. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- 46.Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 47.Periquet M, Corti O, Jacquier S, Brice A. J Neurochem. 2005;95:1259–1276. doi: 10.1111/j.1471-4159.2005.03442.x. [DOI] [PubMed] [Google Scholar]
- 48.Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 49.Joshi S, Javed AA, Gibbs LC. J Biol Chem. 1992;267:12860–12867. [PubMed] [Google Scholar]
- 50.Arama E, Agapite J, Steller H. Dev Cell. 2003;4:687–697. doi: 10.1016/s1534-5807(03)00120-5. [DOI] [PubMed] [Google Scholar]
- 51.Limbach KJ, Wu R. Nucleic Acids Res. 1985;13:631–644. doi: 10.1093/nar/13.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arama E, Bader M, Srivastava M, Bergmann A, Steller H. Embo J. 2006;25:232–243. doi: 10.1038/sj.emboj.7600920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 54.Roberts DB, Wolfe J, Akam ME. J Insect Physiol. 1977;23:871–878. doi: 10.1016/0022-1910(77)90013-0. [DOI] [PubMed] [Google Scholar]
- 55.Wolfe J, Akam ME, Roberts DB. Eur J Biochem. 1977;79:47–53. doi: 10.1111/j.1432-1033.1977.tb11782.x. [DOI] [PubMed] [Google Scholar]
- 56.Powell D, Sato JD, Brock HW, Roberts DB. Dev Biol. 1984;102:206–215. doi: 10.1016/0012-1606(84)90185-4. [DOI] [PubMed] [Google Scholar]
- 57.Kawahara K, Hashimoto M, Bar-On P, Ho GJ, Crews L, Mizuno H, Rockenstein E, Imam SZ, Masliah E. J Biol Chem. 2008;283:6979–6987. doi: 10.1074/jbc.M710418200. [DOI] [PubMed] [Google Scholar]
- 58.Moran LB, Croisier E, Duke DC, Kalaitzakis ME, Roncaroli F, Deprez M, Dexter DT, Pearce RKB, Graeber MB. Acta Neuropathol. 2007;113:253–263. doi: 10.1007/s00401-006-0181-6. [DOI] [PubMed] [Google Scholar]
- 59.Yasuda T, Miyachi S, Kitagawa R, Wada K, Nihira T, Ren YR, Hirai Y, Ageyama N, Terao K, Shimada T, Takada M, Mizuno Y, Mochizuki H. Neurosci. 2007;144:743–753. doi: 10.1016/j.neuroscience.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 60.Haywood AFM, Staveley BE. Genome. 2006;49:505–510. doi: 10.1139/g06-011. [DOI] [PubMed] [Google Scholar]
- 61.Yamada M, Mizuno Y, Mochizuki H. Hum Gene Ther. 2005;16:262–270. doi: 10.1089/hum.2005.16.262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.