

Abstract
Glycosyl hydrolase Family 4 (GH4) is exceptional among the 114 families in this enzyme superfamily. Members of GH4 exhibit unusual cofactor requirements for activity, and an essential cysteine residue is present at the active site. Of greatest significance is the fact that members of GH4 employ a unique catalytic mechanism for cleavage of the glycosidic bond. By phylogenetic analysis, and from available substrate specificities, we have assigned a majority of the enzymes of GH4 to five subgroups. Our classification revealed an unexpected relationship between substrate specificity and the presence, in each subgroup, of a motif of four amino acids that includes the active-site Cys residue: α-glucosidase, CHE(I/V); α-galactosidase, CHSV; α-glucuronidase, CHGx; 6-phospho-α-glucosidase, CDMP; and 6-phospho-β-glucosidase, CN(V/I)P. The question arises: Does the presence of a particular motif sufficiently predict the catalytic function of an unassigned GH4 protein? To test this hypothesis, we have purified and characterized the α-glucoside–specific GH4 enzyme (PalH) from the phytopathogen, Erwinia rhapontici. The CHEI motif in this protein has been changed by site-directed mutagenesis, and the effects upon substrate specificity have been determined. The change to CHSV caused the loss of all α-glucosidase activity, but the mutant protein exhibited none of the anticipated α-galactosidase activity. The Cys-containing motif may be suggestive of enzyme specificity, but phylogenetic placement is required for confidence in that specificity. The Acholeplasma laidlawii GH4 protein is phylogenetically a phospho-β-glucosidase but has a unique SSSP motif. Lacking the initial Cys in that motif it cannot hydrolyze glycosides by the normal GH4 mechanism because the Cys is required to position the metal ion for hydrolysis, nor can it use the more common single or double-displacement mechanism of Koshland. Several considerations suggest that the protein has acquired a new function as the consequence of positive selection. This study emphasizes the importance of automatic annotation systems that by integrating phylogenetic analysis, functional motifs, and bioinformatics data, may lead to innovative experiments that further our understanding of biological systems.
Keywords: glycosyl hydrolases, family GH4, phylogenetics, genomic annotation, enzyme specificity
Introduction
Family 4 of the glycosyl hydrolases (GH4) is unique among the 114 families that presently constitute the glycosyl hydrolase superfamily (see Carbohydrate-Active EnZymes database, http://www.cazy.org and Henrissat and Davies 1997; Coutinho and Henrissat 1999; Cantarel et al. 2009). First, and unlike all other families, GH4 enzymes have obligate requirements for NAD+, divalent metal ion and a reducing agent for catalytic activity (Burstein and Kepes 1971; Thompson et al. 1998, 1999; Raasch et al. 2000; Pikis et al. 2002). Second, family GH4 enzymes exhibit wide diversity in their specificity and selectivity for the α- or β-configurations of their substrates. Third, the members of the GH4 family include α-glucosidases (Raasch et al. 2000, 2002; Lodge et al. 2003); α-galactosidases (Burstein and Kepes 1971; Nagao et al. 1988); α-glucuronidases (Suresh et al. 2002, 2003); 6-phospho-α-glucosidases (Bouma et al. 1997; Thompson et al. 1998, 2004, 2008; Thompson, Robrish, Immel, et al. 2001) and 6-phospho-β-glucosidases (Thompson et al. 1999; Yip et al. 2004; Varrot et al. 2005). Finally, and in marked contrast with the two classic hydrolysis mechanisms proposed 55 years ago by Koshland (Koshland 1953; Rye and Withers 2000; Vocadlo and Davies 2008), cleavage of the glycosidic linkage by GH4 enzymes is catalyzed by a novel sequence of oxidation–elimination–addition and reduction reactions (Rajan et al. 2004; Yip et al. 2004, 2007; Yip and Withers 2006). In a recent medically important report, crystallographic analyses revealed an unsuspected (and tightly bound) molecule of NAD+ at the active site of α-N-acetylgalactosaminidase from Elizabethkingia meningosepticum (Liu et al. 2007). Kinetic studies provide convincing evidence that hydrolysis of substrates by α-N-acetylgalactosaminidase and related enzymes assigned to family GH109, proceeds via a mechanism similar to that employed by members of GH4 (Daniels and Withers 2007; Liu et al. 2007).
Family 4 GH4 is ancient and representatives are found in both the Archaea and the Bacteria. In January 2009, the CAZy web site listed 4 Archaeal and 379 Bacterial GH 4 enzymes representing 3 Archaeal and 99 Bacterial species. The number of different Family 4 enzymes in a genome ranged from one to eight (in Clostridium beijerincki), and many species include multiple enzymes that have the same putative hydrolytic activity.
As is typical in this age of rapid genomic sequencing only a small minority, 24, of those enzymes have been characterized experimentally, but structures of six of them, including all the substrate specificity groups except α-galactosidases, have been determined (Protein Data Bank: 1OBB, 1U8X, 3FEF, 1S6Y, 1UP4, and 1VJT). All include an active-site cysteine residue that is required to correctly position the divalent metal ion and that is part of a four amino acid motif whose sequence, upon inspection, appears to correlate with the putative function of the particular enzyme as reported in GenBank/GenProt annotations. Although many of the proteins are assigned specific functions such as α-galactosidase or 6-phospho-β-glucosidase, with or without experimental evidence, about one-third are assigned no specific function. Given the tiny fraction of Family 4 enzymes whose substrate specificities have been experimentally determined, it is appropriate to ask 1) whether specificity can confidently be assigned to those enzymes currently unassigned and importantly 2) whether the sequence of the cysteine-containing motif is, in itself, a predictor of enzyme specificity. We have approached these issues first by a phylogenetic analysis of 201 GH4 enzymes, followed by an experimental study in which the CHEI motif of an exemplar enzyme (α-glucosidase of Erwinia rhapontici) was manipulated by site-directed mutagenesis in order to assess the extent to which that sequence determines substrate specificity.
Consideration of the phylogeny led us to advance two hypotheses: First, that isoleucine and valine in this Cys motif may be interchanged without loss of enzymatic activity. Second, that changing the E. rhapontici CHEV motif to CHSV (the most common motif among the α-galactosidases) might convert the E. rhapontici enzyme from an α-glucosidase to an α-galactosidase. This report summarizes our tests of these hypotheses.
Materials and Methods
Phylogenetic Analysis
The sequences of 201 Family 4 enzymes from the 102 species represented on the CAZy GH4 web site were downloaded. With one exception, sequences were taken from only one strain of each species. That exception was Klebsiella pneumoniae in which strain MGH 78578 (ATCC 700721) contains seven Family 4 enzymes, but the enzyme from strain ATCC 23357 had been experimentally characterized. When sequences from multiple strains of the same species were available, the strain that included the largest number of Family 4 sequences was chosen. Protein sequences were aligned by ClustalW (Thompson et al. 1994) as implemented by MEGA 4 (Tamura et al. 2007). Pairwise gap opening penalties were 10.0 and gap extension penalties were 0.1, whereas multiple alignment gap opening penalties were 3.0 and gap extension penalties were 1.8. The Gonet protein weight matrix was used.
A phylogenetic tree was estimated by the Maximum Likelihood (ML) method as implemented by the PHYML–aLRT program (Guindon and Gascuel 2003; Anisimova and Gascuel 2006) using the JTT substitution model with InvGamma. The gamma shape parameter (1.395) and the proportion of invariant sites (0.004) were estimated from the data. The log likelihood of the data given that tree was −94442.1. Clade confidences were estimated using SH-like supports. The resulting unrooted tree was rooted by using the Archaea enzymes as an outgroup.
Materials
High purity sugars were obtained from Pfanstiehl Laboratories. Trehalulose was a generous gift from Südzucker (Germany). Maltulose, leucrose, and palatinose were obtained from TCI America, Fluka, and Wako Chemicals, respectively. Chromogenic compounds, Ultrogel-AcA 44, TrisAcryl M-diethylaminoethyl (M–DEAE), and other reagents were from Sigma–Aldrich. Phosphorylated derivates of chromogenic compounds and of O-α- and β-linked disaccharides were prepared in our laboratory as described previously (Thompson, Robrish, Pikis et al. 2001; Thompson et al. 2002). The mixture of glucose 6-phosphate dehydrogenase/hexokinase (G6PDH/HK) was obtained from Roche Molecular Biochemicals.
Analytical Methods
SDS-PAGE was performed in the Novex X-Cell mini system (Invitrogen), using NuPage (4–12%) bis-Tris gels, (2-(N-morpholino) ethane sulfonic acid) running buffer (pH 7.3) and Novex Mark 12 protein standards. Protein concentrations of cell extracts were measured with the bicinchoninic acid kit (Pierce), whereas purified PalH was determined spectrophotometrically using a theoretical molar absorption coefficient ϵ = 60,195 M (monomer)−1 cm−1, A(0.1%)280 nm = 1.20. The mass of PalH (monomer) was determined by electrospray in an HP1100 mass spectrometer, and the N-terminal amino acid sequence of the protein was obtained with an ABI 477A instrument (Applied Biosystems) connected in line to an ABI 120A phenylthiohydantoin analyzer.
Cloning of palH into Escherichia coli TOP 10 Cells
A plasmid (pPAL11) containing the palH gene from E. rhapontici DSM 4484 was kindly provided by Dr F. Boernke, Erlangen, Germany. Polymerase chain reaction (PCR) amplification of palH was carried out in a GeneAmp PCR system 9700 (PE Applied Biosystems) using high fidelity Pfu DNA polymerase (Stratagene). The electrophoretically purified amplicon (∼1.3 kb) was ligated into a pTrcHis2A expression vector (Invitrogen) to yield pTcrHis2ApalH. The recombinant plasmid was transformed into E. coli TOP 10 competent cells, and colonies were selected on Luria–Bertani (LB) agar plates containing 150 μg/ml ampicillin.
Site-Directed Mutagenesis of palH
The role(s) of amino acid residues 173E and 174I in the CHEI motif of PalH were assessed by site-directed mutagenesis of the gene using the QuickChange kit from Stratagene. Plasmids of pTrcHis2ApalH containing the desired mutations (E173S, I174V, and E173S/I174V) were transformed into competent cells of E. coli TOP 10. Verification of the relevant base change(s) was achieved by DNA sequence analysis. The MacVector suite of programs was used to assemble, edit, and analyze the data.
Expression and Purification of PalH
Cells of E. coli TOP 10 (pTrcHis2ApalH) were grown at 30 °C on a rotary shaker, in 2-l baffled flasks containing 800 ml of LB broth supplemented with 150 μg/ml ampicillin. At A600 nm ∼ 0.8, 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) was added to each flask, and after 1 h, the cultures were rapidly chilled in an ice–water bath. Cells were harvested by centrifugation, and the pellets were washed by resuspension and centrifugation from 25 mM Tris–HCl buffer (pH 7.5) containing 1 mM MnCl2, 0.1 mM NAD+, 1 mM dithiothreitol (DTT) and designated TMND buffer.
Preparation of cell extract. Washed cells (13 g wet weight) were resuspended in 32 ml of TMND buffer, and the organisms were disrupted by sonication at 0 °C. The cell extract was centrifuged (180,000 × g for 2 h at 5 °C), and the clarified high-speed supernatant was dialyzed against 4 l of TMND buffer (16 h at 4 °C).
Anion exchange chromatography. The dialyzed preparation was applied to a column (2.6 × 15 cm) of TrisAcryl M–DEAE that had been equilibrated in TMND buffer. Following removal of nonadsorbed material, PalH was eluted with 500 ml of a linear increasing concentration gradient of NaCl (0–0.4 M in TMND buffer). Fractions containing highest activity for pNPαGlc (pNP, para-nitro-phenyl) hydrolysis (see assays below) were pooled and concentrated to 12 ml by pressure filtration (Amicon PM-10 membrane, 50 psi). The concentrate was dialyzed against 4 l of 5 mM potassium phosphate buffer (pH 7.0) containing 0.1 mM NAD+ and 1 mM DTT (designated KPND buffer. Note: due to precipitation, MnCl2 was omitted from this buffer). The preparation was applied to a second column of TrisAcryl M–DEAE previously equilibrated with KPND buffer, and PalH was eluted with an increasing gradient of KPND buffer (5–300 mM). Fractions containing enzyme activity were pooled and concentrated.
Gel filtration chromatography. In the final stage of purification of PalH, trace contaminant polypeptides were removed by passage of the preparation through a gel filtration column (Ultrogel-AcA-44, 1.6 × 95 cm, flow rate 0.15 ml/min) equilibrated with TMND buffer.
PalH Activity Assays
The activity of PalH in cell extracts and column fractions was measured by a discontinuous colorimetric procedure using pNPαGlc as substrate. The reaction mixture (2 ml) contained 50 mM Tris–HCl (pH 7.5), 1 mM MnCl2, 1 mM NAD+, 5 mM DTT, and 0.5 mM pNPαGlc. After equilibration to 37 °C, the enzyme preparation was added and 0.25-ml aliquots were removed at 20-s intervals and immediately injected into 0.75 ml of 0.5 M Na2CO3 solution (pH 10.2) containing 0.1 M ethylenediaminetetraacetic acid (EDTA) to halt activity. The A400 nm was measured, and the rates of pNP formation were calculated from progress plots assuming a molar extinction coefficient ϵ = 18,300 for the yellow p-nitrophenolate anion. For determination of cofactor requirements, the basal 2-ml discontinuous assay contained 50 mM Tris–HCl buffer (pH 7.5) and additions were as follows: 1 mM NAD+, 1 mM MnCl2, 2 mM EDTA, and 5 mM DTT. PalH (10 μl sample; 47 μg protein) was added to the assay, and after 5 min of incubation at 37 °C, the reaction was initiated by the addition of 0.5 mM pNPαGlc. A continuous spectrophotometric assay was used for studies of substrate specificity and for kinetic analyses. This G6PDH/HK/NADP+ coupled assay monitored the release of glucose in a 1-ml reaction mixture that contained: 0.1 M Tris–HCl buffer (pH 7.5), 1 mM MnCl2, 1 mM MgCl2, 1 mM NAD+, 1 mM NADP+, 5 mM ATP, 5 mM DTT, 3 U G6PDH/HK, and desired substrate (0–20 mM for kinetic analyses). Reactions at 25 °C were initiated by the addition of 10 μl (46 μg) of purified PalH, and the increase in A340 nm was monitored in a Beckman DU 640 recording spectrophotometer. Initial rates of NADPH formation were determined using the kinetics program of the instrument. A molar extinction coefficient ϵ = 6,220 M−1 cm−1 was assumed for calculation of the rates of formation of NADPH (i.e., glucose generated) min−1 mg PalH−1. Kinetic parameters were determined from Eadie–Hofstee plots generated by the dogStar software program of D.G. Gilbert.
Results
Phylogenetic Analysis
Figure 1A shows the ML tree of 201 GH4 glucosidases that has been rooted by using the Archaea enzymes as an outgroup. To conserve space, four subgroup clades are shown as colored triangles. Figures 2–5 show the trees corresponding to each of the colored triangles. In each figure, sequence labels are followed by the active-site motif. Details, including genus and species name, accession number, Cys motif, function indicated in the GenBank annotation, and function that can confidently be estimated from the phylogeny are in supplementary table S1, Supplementary Material online. When enzymatic function and substrate specificity has been determined experimentally, the sequence name is shown in boldface italics. A scale bar shows the branch length scale in substitutions per site.
FIG. 1.—
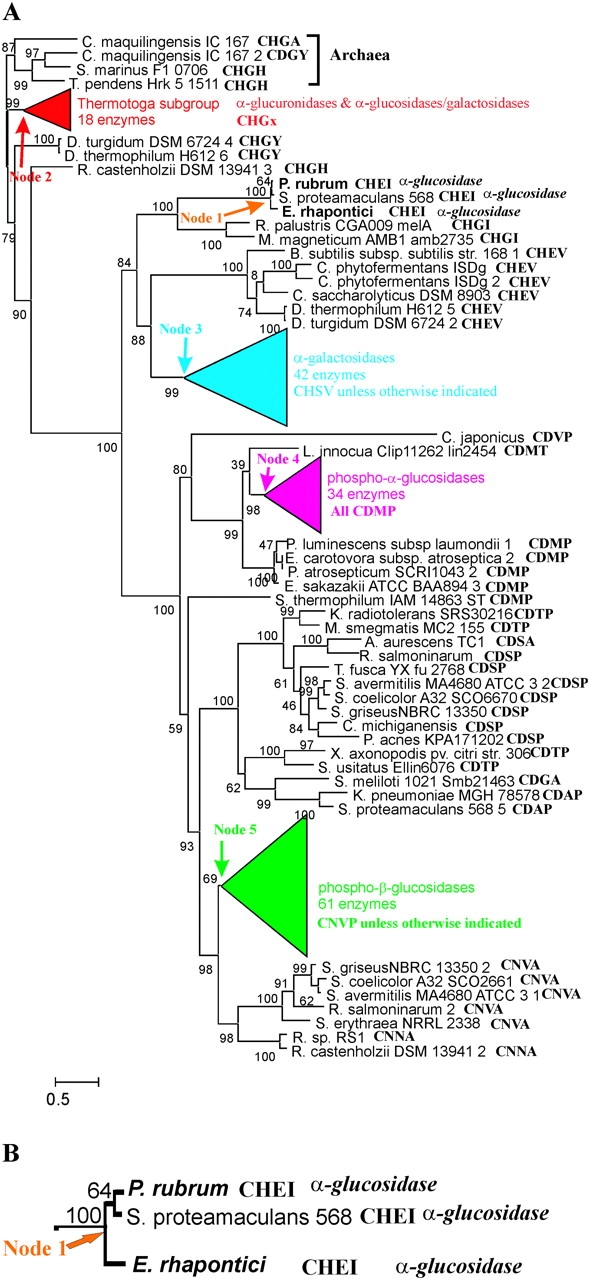
A) ML tree of 201 Family 4 glycosidases, (B) Enlarged view of α-glucosidase clade descended from Node 1. The active-site motif sequence is shown following each sequence label. Numbers at nodes are clade confidences. Branch lines are drawn proportional to branch lengths in substitutions per site as indicated by the scale bar. For clarity, four clades are drawn as colored triangles in which the horizontal width of the triangle is proportional to branch lengths within the clade and vertical height of the triangle is proportional to the number of sequences in the clade. Details of those clades are in figures 2–5. Enzymes whose functions have been experimentally determined are given in boldface italics.
FIG. 2.—

Thermotoga subgroup of Family 4 glycosidases. See legend to figure 1.
FIG. 3.—

α-Galactosidase clade. See legend to figure 1.
FIG. 4.—

6-Phospho-α-glucosidase clade. See legend to figure 1.
FIG. 5.—
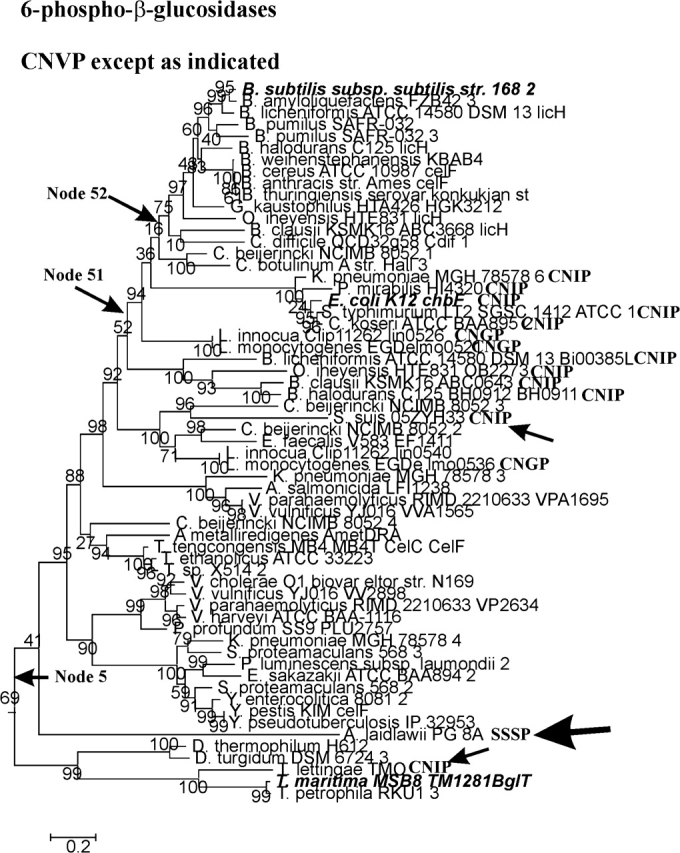
6-Phospho-β-glucosidase clade. See legend to figure 1.
Although substrate specificities have been determined experimentally for only 22 GH4 enzymes (Cantarel et al. 2009), it is often possible to deduce the catalytic activities of the remaining enzymes from the phylogeny. When both the upper and lower branches of a node lead to enzymes for which the same activities have been experimentally determined, it is safe to conclude that all of the enzymes descended from that node share the same substrate specificity. That conclusion is based on the parsimonious principle that the same function is more likely to have arisen once, at the node in question, and been passed on to all descendants than it is to have arisen independently multiple times, for example, the principle is that of Occam's Razor. That is, two of the three descendants of Node 1 (indicated by an orange arrow, detail in fig. 1B), the E. rhapontici and Protaminobacter rubrum enzymes have been shown to be α-glucosidases (this study and Mattes et al. 2005). It is therefore safe to conclude that the third enzyme, that of Serratia proteamaculans, is also an α-glucosidase. All three enzymes are therefore designated as α-glucosidases in figure 1. When it is not the case that an enzyme falls within such a clade, it is not possible to assign a function based on the phylogeny, and we consider the function to be unknown. On the basis of that conservative approach to functional assignment, the functions of all of the individual enzymes shown in figure 1 are unknown except for the three α-glucosidases mentioned above.
The Thermotoga Clade
Enzymes in the Thermotoga clade, shown in red in figure 1, include α-glucuronidases and α-glucosidase/galactosidases that share the active-site motif CHGx. The Thermotoga subgroup tree is shown in figure 2. The Thermotoga maritima TM0752 enzyme descends from the upper branch of Node 21, and the T. maritima TM0434 enzyme descends from the lower node. Both enzymes are experimentally determined α-glucuronidases; thus, it can be concluded that all of the descendants of Node 21 are α-glucuronidases. Similarly, the descendants of Node 23 are α-glucosidases/galactosidases. Those enzymes are usually described simply as α-glucosidases, but their activities toward α-galactosides are nearly as great as toward α-glucosides (Raasch et al. 2000). In contrast, the E. rhapontici enzyme in the Node 1 clade (fig. 1B) is highly specific for α-glucosides (this study). All of the descendants of Node 22 share the motif CHGH, so it is tempting to assume that all are α-glucosidase/galactosidases even though that conclusion cannot be phylogenetically justified. It is precisely that temptation that leads us to ask whether the active-site sequence determines substrate specificity.
The α-Galactosidase Clade
The α-galactosidases (fig. 1A, blue triangle) are descended from Node 3. The conclusion that all the enzymes shown in figure 3 are α-galactosidases is justified by the presence of the Sinorhizobium melloti 1021 agaL1 experimentally characterized enzyme in the top branch descending from Node 3 and the presence of four other experimentally determined α-galactosidases in the bottom branch descending from Node 3. The most parsimonious explanation for the configuration of the active-site motifs is that the sequence at Node 3 was CHGV, with a G>S substitution occurring along the branch leading to Node 31 to give the most common sequence, CHSV. It seems likely that with respect to α-galactosidase activity the G and S residues in the motif are equivalent. An H>Y substitution along the branch leading to Node 32, and a V>I substitution along the branch leading to Node 33, explain the remainder of the minority active-site motif sequences.
The Phospho-α-Glucosidase Clade
The phospho-α-glucosidases (fig. 1A, magenta triangle) are descended from Node 4. The conclusion that all the enzymes in figure 4 are phospho-α-glucosidases is justified by the presence of the experimentally characterized Clostridium acetobutylicum ATCC 824 glvG enzyme in the upper branch descending from Node 4 and the presence of five experimentally characterized enzymes in the lower branch descending from Node 4. The phospho-α-glucosidase clade is the most uniform of all the functional clades with respect to its active-site motif. Only one substitution, P>T, along the branch leading to Listeria innocua Clip11262 lin2454 occurs within the clade. Because the substituted enzyme has not been experimentally characterized, there is no assurance that it retains enzymatic activity.
The Phospho-β-Glucosidase Clade
The phospho-β-glucosidases (fig. 1A, green triangle) are descended from Node 5. The conclusion that all the enzymes in figure 5 are phospho-β-glucosidases is justified by the presence of the experimentally characterized T. maritima MSB8 TM1281BglT enzyme in the lower branch descending from Node 5 and the presence of the experimentally characterized enzymes in the upper branch descending from that node. It is clear that the active-site sequence at Node 5 is CNVP. A V>I substitution at Node 51 is reversed at Node 52. Those substitutions, together with the two independent V>I substitutions along the branches leading to Streptococcus suis 05ZYH33 and Thermotoga lettingae TMO, both indicated by arrows in figure 5, support the general notion that V and I are interchangeable within the active-site motif.
The Unusual GH4 Protein of A. laidlawii
The A. laidlawii enzyme in the 6-phospho-β-glucosidase clade (indicated by an arrow in fig. 5) deserves special mention. The active-site motif of this protein (SSSP) lacks the essential Cys residue that is required to correctly position the divalent metal ion that has been shown to be essential for Family 4 glycosidase activity (Rajan et al. 2004; Varrot et al. 2005). There is no question that the A. laidlawii protein is a legitimate member of the GH4 family, because it includes 1) a GAGS motif (residues 11–14) that is part of the nucleotide-binding domain that is conserved in all GH4 enzymes, 2) the NP motif in which N150 hydrogen bonds to the equatorial C3 and C4 OH groups of the nonreducing G6P moiety of the phosphorylated disaccharide, and 3) two arginyl residues (R89 and R271) that bond to the phosphate group of G6P (Rajan et al. 2004; Varrot et al. 2005). The four-residue sequence G(L/I)NH is conserved in all GH4 enzymes, and structural analyses of phospho-α-glucosidase (GlvA) and phospho-β-glucosidase (BglT) show that the His residue of this motif, as well as Cys in the Cys motif, are “both” coordinately linked to the catalytically essential Mn2+ ion. The loss of these metal-binding residues clearly makes glycoside hydrolysis by the GH4 mechanism impossible (Rajan et al. 2004; Yip et al. 2004; Varrot et al. 2005). Although the A. laidlawii protein is phylogenetically solidly within the 6-phospho-β-glucosidase clade, our recent cloning and expression studies have shown that this protein is not only devoid of phospho-β-glucosidase activity, but it also exhibits no detectable activity to pNP-β-glucopyranoside, α-glucopyranoside, α-galactopyranoside, or α-mannopyranoside (J. Thompson and A. Pikis, unpublished data).
Phylogenetic Estimation of Function and Substrate Specificity
Seventy seven of the 201 Family 4 enzymes we have studied are either unclassified in the GenBank annotations or are classified ambiguously as “glycosyl-hydrolase,” “sugar hydrolase,” “hypothetical protein,” or “α-galactosidase/6-phospho-β-glucosidase,” etc. Of those, 50 can be confidently assigned functions based on this phylogenetic analysis (see supplementary table S1, Supplementary Material online). There are 19 enzymes that are assigned specific functions in the annotations but whose functional assignments cannot be phylogenetically justified (supplementary table S1, Supplementary Material online). In addition, there are nine enzymes whose functions have been incorrectly specified in the annotations (italicized in column 4 of supplementary table S1, Supplementary Material online). These include an experimentally characterized 6-phospho-α-glucosidase of Lactobacillus casei ATCC 334, simA (Thompson et al. 2008) (described as a mannose-6-phosphate isomerase) and an experimentally characterized α-glucuronidase of T. maritima MSB8 TM0434 (designated an α-glucosidase).
Clearly, there is a strong association between the sequence of the active-site motif and enzyme function. Is the sequence of the Cys motif the major or sole determinant of substrate specificity? If so, function could be confidently assigned if one enzyme with that same motif had been characterized experimentally. Can we assume that the Symbiobacterium thermophilum IAM 14863 ST enzyme, which has the CDMP motif (fig. 1A), is a 6-phospho-α-glucosidase? Can we assume that all of the enzymes in the Thermotoga clade that have the CHGH motif are α-glucosidase/galactosidases? To answer such questions, we have purified and studied the E. rhapontici enzyme, a member of one of the smallest functional clades (fig. 1B).
Comparative Sequence Alignment of PalH
In E. rhapontici, palH is present in a cluster of nine genes whose products may facilitate the transport, and metabolism of palatinose (a 1→6, O-α-linked glucoside) in this plant pathogen (Bornke et al. 2001). Surprisingly, the amino acid sequence of the putative protein encoded by palH displayed greatest homology (22% identity) to a Family 4 α-galactosidase (MelA) from Bacillus subtilis. For some time, it appeared that palH was confined to E. rhapontici. However, subsequent DNA-sequencing projects have revealed a virtually identical “palatinase” gene in two other species, namely, S. proteamaculans (GenBank CP000826.1) and P. rubrum (Mattes et al. 2005). A comparative sequence alignment reveals >90% residue identity for these proteins (fig. 6), and in all cases, the catalytically essential Cys residue of these Family 4 members is found in a unique motif of four amino acids CHEI. The occurrence of this unusual active-site motif, allied with the questionable assignment of catalytic activity, encouraged us to purify and characterize PalH from E. rhapontici.
FIG. 6.—

Multiple sequence alignment (ClustalW2) and GenBank accession numbers of PalH proteins from: Erwinia rhapontici DSM 4484 (AAK28734, Bornke et al. 2001), Serratia proteamaculans 568 (ABV41624) and Protaminobacter rubrum (AAY69571, Mattes et al. 2005). Consensus symbols designate: *, identical residues; :, conserved substitutions and ., semiconserved substitutions. Abbreviations NBD and SIG. SEQ refer to residues that constitute the nucleotide-binding domain and the signature sequence of Family GH4 proteins, respectively. Red dots indicate catalytically important residues. Amino acids in red comprise the CHEI motif.
Expression and Purification of PalH
Cells of E. coli TOP 10, when transformed with pTrcHis2ApalH and induced by IPTG addition, yielded high-level expression of a polypeptide whose estimated molecular weight by SDS-PAGE was consistent with that expected of the full-length product encoded by palH. A three-stage chromatographic procedure, with pNPαGlc as substrate for detection of enzymatic activity, yielded 30 mg of highly purified PalH (Mr ∼ 50,000) from 13 g wet weight of cells (see, fig. S1, Supplementary Material online).
Physicochemical Characteristics
PalH eluted close to the void volume of a calibrated AcA-44 molecular sieve column (exclusion limit ∼ 200 K). Failure to detect polypeptides of intermediate molecular weight (50 or 100 K) suggests that in the solution state, PalH exists as a homotetramer comprised of 50-kDa subunits. The homogeneity of purified PalH was confirmed by the results of microsequence analysis that (except for the expected methionine residue in the first cycle) agreed precisely with the predicted translational sequence of palH: (M)ATKIVLVGAGSAQFGYGTLGDIFQS. The molecular mass of PalH (50,216 Da) determined by electrospray ionization/mass spectrometry was lower than the theoretical mass (50,340) by 124 Da. This result is reasonably consistent with the loss of a terminal (132 Da) methionyl residue.
Cofactor Requirements for PalH Activity
Previous studies of enzymes assigned to GH4 (Burstein and Kepes 1971; Thompson et al. 1998, 1999; Raasch et al. 2000, 2002; Pikis et al. 2002) have shown that a dinucleotide (NAD+), divalent metal ion (e.g., Mn2+) and a reducing agent DTT are prerequisites for activity. Data presented in table 1 reveal that PalH from E. rhapontici is also dependent upon these cofactors for catalytic activity. In the presence of 5 mM DTT and saturating concentrations of NAD+(1 mM) and Mn2+ (1 mM), PalH exhibited Michaelis–Menten saturation kinetics with respect to pNPαGlc hydrolysis, and the following parameters were determined: Km = 180 ± 20 μM and Vmax = 6.01 ± 0.70 μmol pNPαGlc hydrolyzed mg protein−1 min−1. The Km values for NAD+ and Mn2+ were estimated to be 104 ± 6.8 and 47.2 ± 11.4 μM, respectively. The stimulatory effects of other divalent ions were also investigated, but due to discoloration and precipitation of some ions (e.g., Co2+ and Ni2+) by DTT, the reducing agent was omitted from this series of assays. Under these conditions, the activation of PalH by selected metal ions, relative to that elicited by 1 mM Mn2+ (100%) were as follows: Co2+ (61%), Ca2+ (59%), Fe2+ (59%), Mg2+ (36%), Sr2+ (27%), and Ni2+ (18%). There was no detectable activation of PalH by other metals tested, including Cr2+, Cu2+, and Zn2+.
Table 1.
Cofactor Requirements of PalH for the Hydrolysis of Chromogenic pNPαGlca
| Additionsb | PalH Activityc |
| Control (PalH only) | NDAd |
| + NAD+ | 0.92 |
| + Mn2+ | NDA |
| + DTT | NDA |
| + EDTA | NDA |
| + NAD+ + Mn2+ | 1.27 |
| + NAD+ + DTT | 0.60 |
| + NAD+ + EDTA | NDA |
| + NAD+ + Mn2+ + DTT | 6.09 |
| + NAD+ + Mn2+ + DTT + EDTA | NDA |
PalH used in these experiments had previously been dialyzed for 18 h against 25 mM Tris–HCl (pH 7.5) buffer.
Additions to the basal 2-ml discontinuous assay were as described in Materials and Methods.
PalH activity expressed as μmol pNPαGlc hydrolyzed mg protein−1 min−1. Values are the mean of two separate assays.
NDA, No detectable PalH activity.
Substrate Specificity of PalH
In the presence of the requisite cofactors, pNPαGlc was readily hydrolyzed by PalH, as were maltose, sucrose, and several other natural α-glucosides, but there was no discernible hydrolysis of the phosphorylated derivative pNPαGlc-6P. In addition to the requirement of a free hydroxyl at C-6, PalH was highly specific toward the gluco-conformation and equatorial orientation of all –OH groups in the nonreducing glucose moiety of its substrates. Consequently, there was no detectable hydrolysis of the chromogenic C-2 and C-4 epimers, pNP-α-D-mannopyranoside and pNP-α-D-galactopyranoside, respectively. The obligate requirement of PalH for the O-α-glycosidic linkage was confirmed by the kinetic data for hydrolysis of a wide variety of naturally occurring α-linked disaccharides (table 2). This specificity is in marked contrast to the α-glucosidases of the Thermotoga subgroup that are almost as active toward α-galactosides as toward α-glucosides.
Table 2.
Kinetic Parameters for Substrate Hydrolysis by PalH from Erwinia rhapontici
| Substrate | Vmaxa | Km (mM) | kcat min−1 |
| Maltose: 4-O-α-D-glucopyranosyl-D-glucopyranose | 1.01 ± 0.02 | 8.58 ± 0.34 | 51 |
| Maltitol: 4-O-α-D-glucopyranosyl-D-sorbitol | 0.24 ± 0.01 | 7.31 ± 0.58 | 12 |
| Trehalose: 1-O-α-D-glucopyranosyl-α-glucopyranoside | 0.51 ± 0.01 | 6.59 ± 0.37 | 26 |
| Trehaluloseb: 1-O-α-D-glucopyranosyl-α-D-fructose | 0.47 ± 0.01 | 3.63 ± 0.31 | 24 |
| Sucrose: 1-O-α-D-glucopyranosyl-β-D-fructofuranoside | 0.31 ± 0.02 | 22.11 ± 3.37 | 16 |
| Turanose: 3-O-α-D-glucopyranosyl-D-fructose | 0.46 ± 0.03 | 10.04 ± 1.21 | 23 |
| Maltulose: 4-O-α-D-glucopyranosyl-D-fructose | 0.53 ± 0.01 | 1.75 ± 0.19 | 27 |
| Leucrose: 5-O-α-D-glucopyranosyl-D-fructose | 0.44 ± 0.03 | 5.31 ± 0.81 | 22 |
| Palatinose: 6-O-α-D-glucopyranosyl-D-fructofuranoside | 0.70 ± 0.01 | 2.52 ± 0.12 | 35 |
| Sophorose (2-O-β-D-glucopyranosyl-D-glucopyranose) NDHc | |||
| Laminaribiose (3-O-β-D-glucopyranosyl-D-glucopyranose) NDH | |||
| Cellobiose (4-O-β-D-glucopyranosyl-D-glucopyranose) NDH | |||
| Gentiobiose (6-O-β-D-glucopyranosyl-D-glucopyranose) NDH | |||
| Lactose (4-O-β-D-galactopyranosyl-D-glucopyranose) NDH | |||
| Melibiose (6-O-α-D-galactopyranosyl-D-glucopyranose) NDH | |||
Expressed as μmol substrate hydrolyzed mg protein−1 min−1. All assays were carried out spectrophotometrically at 25 °C.
Compounds in boldface are sucrose isomers.
NDH No detectable hydrolysis.
Site-Directed Mutagenesis of the CHEI Motif in PalH
All proteins in the GH4 family contain a conserved cysteine residue that is essential for catalytic activity. In PalH of E. rhapontici and in the homologous proteins from P. rubrum and S. proteamaculans, this crucial residue occurs in a unique motif of four amino acids: CHEI (see fig. 6). The effects of site-directed changes in residues of this motif, upon the activity and substrate specificity of PalH, were determined. Proteins present in similarly prepared extracts from cells containing the parental and mutated plasmids are shown in the SDS-PAGE gel (fig. 7). Laser densitometry of the SDS-PAGE gels revealed comparable amounts (±10%) of the highly expressed ∼50-kDa protein expected for full-length PalH. The activity of the enzyme was determined and the results are presented in figure 7, bottom. The residue change I174V had little effect upon the specific activity of PalH (2.24 vs. 2.29 μmol pNPαGlc hydrolyzed mg protein−1 min−1). However, those mutations involving change in 173E, namely, E173S and the double mutant E173S/I174V, resulted in the loss of all enzymatic activity. It is significant that mutant protein containing the motif CHSV (characteristic of GH4 α-galactosidases) failed to hydrolyze any of the O-α-linked galactosides tested, including the chromogenic pNPαGal and melibiose (data not shown).
FIG. 7.—

Effect of residue change in the CHEI motif upon the specific activity of PalH from Erwinia rhapontici. Plasmids of pTrcHis2ApalH containing the indicated site-directed changes in the CHEI motif were transformed, and the proteins were expressed in Escherichia coli TOP 10. Cell extracts (25 μg protein) were electrophoresed by SDS-PAGE to confirm, in each case, the high level expression of PalH. The preparations were assayed for enzymatic activity, and the results (μmol pNPαGlc hydrolyzed min−1 mg protein−1) are presented at the bottom of the figure.
Discussion
Phylogenetic analysis revealed an unusually strong relationship between the substrate specificity and the presence of a variable four-residue, Cys-containing motif, in the five subgroups of GH4 hydrolases. Biochemical analysis of the E. rhapontici enzyme showed that it is a highly specific α-glucosidase, in striking contrast with the enzymes of the Thermotoga clade that hydrolyze α-galactosides as effectively as they do α-glucosides. The E. rhapontici enzyme has the motif CHEI that, when changed to CHEV, has virtually no effect on specific activity. The most common active-site motif sequence for the α-galactosidases is CHSV. If substrate specificity is determined solely by the Cys-motif, then it would be expected that further changing CHEV of E. rhapontici to CHSV should convert the E. rhapontici enzyme from a strict α-glucosidase to a specific α-galactosidase. Contrary to expectation, changing the motif to CHSV resulted in a complete loss of catalytic activity. This finding makes it clear that substrate specificity and enzymatic activity, although clearly associated with a particular Cys motif, must also be determined by other factors including molecular structure and the architectural environment of the active site. In other words, it is not only the motif but also the context of the Cys motif that determines the substrate specificity of a particular subgroup of enzymes. In the context of the remainder of the E. rhapontici enzyme sequence, the CHSV motif results in inactivity, whereas in the context of the α-galactosidase sequences, it results in high activity.
With these caveats in mind, we cannot assume that the Symbiobacterium thermophilum IAM 14863 ST enzyme is a 6-phospho-α-glucosidase simply because it encodes a CDMP motif that it shares with all of the 6-phospho-α-glucosidases. Neither can we assume that the enzymes in the Thermotoga clade that carry the CHGH motif are all α-glucosidase/galactosidases. The “context” effect means that there is simply no substitute for direct, experimental evidence when assigning function and substrate specificity to an enzyme.
Although the Cys motif cannot be used by itself to estimate enzyme specificity, it can certainly serve as a red flag to direct our attention to likely unusual activities. The replacement of CNVP by SSSP in the A. laidlawii enzyme makes it very unlikely that the enzyme is a phospho-β-glucosidase; it certainly cannot utilize the normal GH4 mechanism to hydrolyze phospho-β-glucosides. Acholeplasma laidlawii lacks the phosphotransferase system enzymes II and cannot phosphorylate β-glucosides (Hoischen et al. 1993), making it even more unlikely that the enzyme is a phospho-β-glucosidase. It is possible that the enzyme is simply inactive and that it has accumulated substitutions in both of the metal-binding motifs, in what amounts to a pseudogene. Considering that several other motifs remain intact, it seems unlikely that “both” metal-binding motifs would have been mutated. It is also unlikely that the enzyme, whose length is typical for GH4 enzymes, would have escaped deletions, nonsense mutations, and frameshift mutations, along a branch that has an average of 2.2 substitutions per site unless it serves some function that is subject to purifying selection. Finally, A. laidlawii has an extremely small genome (∼1.5 Mb), and it is unlikely that a genome of this size would long retain a functionless gene. A more likely alternative is that the A. laidlawii enzyme has acquired a novel activity during a period of positive selection. The observation that A. laidlawii possesses a β-glucosidase that is inducible by growth on cellobiose (Hoischen et al. 1993) raises the intriguing possibility that the A. laidlawii GH4 enzyme has evolved β-glucosidase activity. We have initiated a study of the A. laidlawii enzyme in the hope of determining whether it possesses glycosyl hydrolase activity and (if so) determining the catalytic mechanism of that activity.
Our results are quite consistent with a similar study of the large GH13 family (Stam et al. 2006). GH13 is an enormous family of over 2,500 enzymes that include 26 different functions that are indicated by different EC numbers. The Stam study used a variety of methods to cluster those enzymes into 35 subfamilies that were entirely consistent with subtrees of an unrooted NJ tree of GH13. Most of the subfamilies were monospecific, that is, included only one experimentally determined EC number, whereas a few included enzymes with two closely related functions. They concluded that “assignment to a subfamily is a considerable step toward improved functional prediction. However, because not all subfamilies have a biochemical(ly) characterized member and because a significant number of sequences are not included in subfamilies, errors or imprecision are still possible during unsupervised automated genomic annotation.”
Practical considerations make it clear that sequencing will continue to outpace our ability to obtain direct experimental evidence for enzyme activities. It therefore behooves us to consider how best to use sequence information to maximize our confidence in estimating enzyme function and specificity. This study makes it clear that simple grouping on the basis of similarity of sequence motifs is an insufficient criterion. Automatic documentation algorithms sometimes make incorrect assignments, sometimes make unjustified assignments, and sometimes fail to make assignments that can be justified. Likewise active-site motif sequences (as we describe here) although perhaps indicative are nevertheless insufficient to assign enzyme function and specificity. Phylogenetic analysis is, so far, a reliable guide to function.
When phylogenetic analysis is used conservatively (as we have done) to assign specificity, there are no cases in which experimental evidence conflicts with phylogenetic assignment. Although we cannot hope to determine the functions of all the 201 GH4 enzymes considered here, consideration of the active-site motif together with phylogenetic analysis can serve as a guide to choosing enzymes for biochemical analysis. The speed and ease of modern DNA-sequencing technology can easily lead to a “stamp-collecting” approach in which genomes are sequenced for the sake of enlarging a collection, and the annotations serve as a dry catalog of that collection. Genome sequences and their annotations should serve as a guide to interesting problems and to the design of experiments, which will augment our understanding of biological processes. To achieve these aims, automated annotation “pipelines” will need to develop along the lines of expert systems that will include the phylogenetic analysis (not just clustering) of genes and that will integrate such analyses with functional motifs and other bioinformatic data.
Supplementary Material
Supplementary table S1 and supplementary figure S1 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
This work was supported by the Intramural Research Program of the NIDCR, National Institutes of Health, Bethesda, MD. We thank Frederik Boernke for kindly providing plasmid pPAL11containing the palH gene from E. rhapontici.
References
- Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol. 2006;55:539–552. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- Bornke F, Hajirezaei M, Sonnewald U. Cloning and characterization of the gene cluster for palatinose metabolism from the phytopathogenic bacterium Erwinia rhapontici. J Bacteriol. 2001;183:2425–2430. doi: 10.1128/JB.183.8.2425-2430.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouma CL, Reizer J, Reizer A, Robrish SA, Thompson J. 6-phospho-α-D-glucosidase from Fusobacterium mortiferum: cloning, expression, and assignment to family 4 of the glycosylhydrolases. J Bacteriol. 1997;179:4129–4137. doi: 10.1128/jb.179.13.4129-4137.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein C, Kepes A. The α-galactosidase from Escherichia coli K12. Biochim Biophys Acta. 1971;230:52–63. doi: 10.1016/0304-4165(71)90053-5. [DOI] [PubMed] [Google Scholar]
- Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The carbohydrate-active enZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho PM, Henrissat B. Carbohydrate-active enzymes: an integrated data base approach. In: Gilbert HJ, Davies G, Henrissat B, Svensson B, editors. Recent advances in carbohydrate bioengineering. Cambridge: Royal Society of Chemistry; 1999. pp. 3–12. [Google Scholar]
- Daniels G, Withers SG. Towards universal red blood cells. Nat Biotechnol. 2007;25:427–428. doi: 10.1038/nbt0407-427. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Henrissat B, Davies G. Structural and sequence-based classification of glycoside hydrolases. Curr Opin Struct Biol. 1997;7:637–644. doi: 10.1016/s0959-440x(97)80072-3. [DOI] [PubMed] [Google Scholar]
- Hoischen C, Dijkstra A, Rottem S, Reizer J, Saier Jr., MH Presence of protein constituents of the gram-positive bacterial phosphotransferase regulatory system in Acholeplasma laidlawii. J Bacteriol. 1993;175:6599–6604. doi: 10.1128/jb.175.20.6599-6604.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland DE. Stereochemistry and the mechanism of enzymatic reactions. Biol Rev Camb Phil Soc. 1953;28:416–436. [Google Scholar]
- Liu QP, Sulzenbacher G, Yuan H, et al. (16 co-authors) Bacterial glycosidases for the production of universal red blood cells. Nat Biotechnol. 2007;25:454–464. doi: 10.1038/nbt1298. [DOI] [PubMed] [Google Scholar]
- Lodge JA, Maier T, Liebl W, Hoffmann V, Strater N. Crystal structure of Thermotoga maritima α-glucosidase AglA defines a new clan of NAD+-dependent glycosidases. J Biol Chem. 2003;278:19151–19158. doi: 10.1074/jbc.M211626200. [DOI] [PubMed] [Google Scholar]
- Mattes R, Klein K, Schiweck H, Kunz M. Sequence 8 from Protaminobacter rubrum US Patent 6884611. 2005 [Google Scholar]
- Nagao Y, Nakada T, Imoto M, Shimamoto T, Sakai S, Tsuda M, Tsuchiya T. Purification and analysis of the structure of α-galactosidase from Escherichia coli. Biochem Biophys Res Commun. 1988;151:236–241. doi: 10.1016/0006-291x(88)90584-0. [DOI] [PubMed] [Google Scholar]
- Pikis A, Immel S, Robrish SA, Thompson J. Metabolism of sucrose and its five isomers by Fusobacterium mortiferum. Microbiology. 2002;148:843–852. doi: 10.1099/00221287-148-3-843. [DOI] [PubMed] [Google Scholar]
- Raasch C, Armbrecht M, Streit W, Hocker B, Strater N, Liebl W. Identification of residues important for NAD+ binding by the Thermotoga maritima α-glucosidase AglA, a member of glycoside hydrolase family 4. FEBS Lett. 2002;517:267–271. doi: 10.1016/s0014-5793(02)02641-8. [DOI] [PubMed] [Google Scholar]
- Raasch C, Streit W, Schanzer J, Bibel M, Gosslar U, Liebl W. Thermotoga maritima AglA, an extremely thermostable NAD+-, Mn2+-, and thiol-dependent α-glucosidase. Extremophiles. 2000;4:189–200. doi: 10.1007/pl00010711. [DOI] [PubMed] [Google Scholar]
- Rajan SS, Yang X, Collart F, Yip VL, Withers SG, Varrot A, Thompson J, Davies GJ, Anderson WF. Novel catalytic mechanism of glycoside hydrolysis based on the structure of an NAD+/Mn2+ -dependent phospho-α-glucosidase from Bacillus subtilis. Structure. 2004;12:1619–1629. doi: 10.1016/j.str.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Rye CS, Withers SG. Glycosidase mechanisms. Curr Opin Chem Biol. 2000;4:573–580. doi: 10.1016/s1367-5931(00)00135-6. [DOI] [PubMed] [Google Scholar]
- Stam MR, Etienne G, Danchin GJ, Rancurel C, Coutinho PM, Henrissat B. Dividing the large glycoside hydrolase family 13 into subfamilies: towards improved functional annotations of α-amylase-related proteins. Protein Eng Des Sel. 2006;19:555–562. doi: 10.1093/protein/gzl044. [DOI] [PubMed] [Google Scholar]
- Suresh C, Kitaoka M, Hayashi K. A thermostable non-xylanolytic α-glucuronidase of Thermotoga maritima MSB8. Biosci Biotechnol Biochem. 2003;67:2359–2364. doi: 10.1271/bbb.67.2359. [DOI] [PubMed] [Google Scholar]
- Suresh C, Rus'd AA, Kitaoka M, Hayashi K. Evidence that the putative α-glucosidase of Thermotoga maritima MSB8 is a pNP α-D-glucuronopyranoside hydrolyzing α-glucuronidase. FEBS Lett. 2002;517:159–162. doi: 10.1016/s0014-5793(02)02611-x. [DOI] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Thompson J, Hess S, Pikis A. Genes malh and pagl of Clostridium acetobutylicum ATCC 824 encode NAD+- and Mn2+-dependent phospho-α-glucosidase(s) J Biol Chem. 2004;279:1553–1561. doi: 10.1074/jbc.M310733200. [DOI] [PubMed] [Google Scholar]
- Thompson J, Jakubovics N, Abraham B, Hess S, Pikis A. The sim operon facilitates the transport and metabolism of sucrose isomers in Lactobacillus casei ATCC 334. J Bacteriol. 2008;190:3362–3373. doi: 10.1128/JB.02008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J, Lichtenthaler FW, Peters S, Pikis A. β-glucoside kinase (BglK) from Klebsiella pneumoniae. Purification, properties, and preparative synthesis of 6-phospho-β-D-glucosides. J Biol Chem. 2002;277:34310–34321. doi: 10.1074/jbc.M206397200. [DOI] [PubMed] [Google Scholar]
- Thompson J, Pikis A, Ruvinov SB, Henrissat B, Yamamoto H, Sekiguchi J. The gene glvA of Bacillus subtilis 168 encodes a metal-requiring NAD(H)-dependent 6-phospho-α-glucosidase. J Biol Chem. 1998;273:27347–27356. doi: 10.1074/jbc.273.42.27347. [DOI] [PubMed] [Google Scholar]
- Thompson J, Robrish SA, Immel S, Lichtenthaler FW, Hall BG, Pikis A. Metabolism of sucrose and its five linkage-isomeric α-D-glucosyl-D-fructoses by Klebsiella pneumoniae. Participation and properties of sucrose-6-phosphate hydrolase and phospho-α-glucosidase. J Biol Chem. 2001;276:37415–37425. doi: 10.1074/jbc.M106504200. [DOI] [PubMed] [Google Scholar]
- Thompson J, Robrish SA, Pikis A, Brust A, Lichtenthaler FW. Phosphorylation and metabolism of sucrose and its five linkage-isomeric α-D-glucosyl-D-fructoses by Klebsiella pneumoniae. Carbohydr Res. 2001;331:149–161. doi: 10.1016/s0008-6215(01)00028-3. [DOI] [PubMed] [Google Scholar]
- Thompson J, Ruvinov SB, Freedberg DI, Hall BG. Cellobiose-6-phosphate hydrolase (CelF) of Escherichia coli: characterization and assignment to the unusual family 4 of glycosylhydrolases. J Bacteriol. 1999;181:7339–7345. doi: 10.1128/jb.181.23.7339-7345.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varrot A, Yip VL, Li Y, Rajan SS, Yang X, Anderson WF, Thompson J, Withers SG, Davies GJ. NAD+ and metal-ion dependent hydrolysis by family 4 glycosidases: structural insight into specificity for phospho-β-D-glucosides. J Mol Biol. 2005;346:423–435. doi: 10.1016/j.jmb.2004.11.058. [DOI] [PubMed] [Google Scholar]
- Vocadlo DJ, Davies GJ. Mechanistic insights into glycosidase chemistry. Curr Opin Chem Biol. 2008;12:539–555. doi: 10.1016/j.cbpa.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Yip VL, Varrot A, Davies GJ, Rajan SS, Yang X, Thompson J, Anderson WF, Withers SG. An unusual mechanism of glycoside hydrolysis involving redox and elimination steps by a family 4 β-glycosidase from Thermotoga maritima. J Am Chem Soc. 2004;126:8354–8355. doi: 10.1021/ja047632w. [DOI] [PubMed] [Google Scholar]
- Yip VLY, Thompson J, Withers SG. Mechanism of GlvA from Bacillus subtilis: a detailed kinetic analysis of a 6-phospho-α-glucosidase from glycoside hydrolase family 4. Biochemistry. 2007;46:9840–9852. doi: 10.1021/bi700536p. [DOI] [PubMed] [Google Scholar]
- Yip VLY, Withers SG. Family 4 glycoside hydrolases are special: the first β-elimination mechanism amongst glycoside hydrolases. Biocatal Biotransform. 2006;24:167–176. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.