

Abstract
Genetic variation for pathogen infectivity is an important driver of disease incidence and prevalence in both natural and managed systems. Here, we use the interaction between the rust pathogen, Melampsora lini, and two host plants, Linum marginale and Linum usitatissimum, to examine how host–pathogen interactions influence the maintenance of polymorphism in genes underlying pathogen virulence. Extensive sequence variation at two effector loci (AvrP123, AvrP4) was found in M. lini isolates collected from across the native range of L. marginale in Australia, as well as in isolates collected from a second host, the cultivated species L. usitatissimum. A highly significant excess of nonsynonymous compared with synonymous polymorphism was found at both loci, suggesting that diversifying selection is important for the maintenance of the observed sequence diversity. Agrobacterium-mediated transient transformation assays were used to demonstrate that variants of both the AvrP123 and AvrP4 genes are differentially recognized by resistance genes in L. marginale. We further characterized patterns of nucleotide variation at AvrP123 and AvrP4 in 10 local populations of M. lini infecting the wild host L. marginale. Populations were significantly differentiated with respect to allelic representation at the Avr loci, suggesting the possibility of local selection maintaining distinct genetic structures between pathogen populations, whereas limited diversity may be explained via selective sweeps and demographic bottlenecks. Together, these results imply that interacting selective and nonselective factors, acting across a broad range of scales, are important for the generation and maintenance of adaptively significant variation in populations of M. lini.
Keywords: parasite, selection, virulence, effector, polymorphism, metapopulation
Introduction
Polymorphism for traits influencing the outcome of interactions between hosts and parasites is characteristic of nearly all species, and understanding how such polymorphisms are maintained within populations is of central importance for understanding the dynamics and distribution of disease. Associations between individual host and pathogen genotypes and disease outcomes have been demonstrated for a number of host–pathogen interactions (Buitkamp et al. 1996; Paterson et al. 1998; Lockett et al. 2001; Williams-Blangero et al. 2002; Ong and Innes 2006). Moreover, genetic variation for host resistance and various pathogen traits are likely to be critical factors influencing disease epidemiology (Thrall and Burdon 2000; Gonzalez et al. 2001) and the emergence and spread of new diseases (Prentice et al. 2001; Friesen et al. 2006; Piertney and Oliver 2006). However, we have little knowledge of how population-level ecological and evolutionary processes interact to influence the generation and maintenance of pathogenic variation at epidemiologically relevant loci.
One broadly used model to understand how polymorphism persists within different host and pathogen traits is the gene-for-gene (GFG) relationship (Laine and Tellier 2008). In the classic GFG model (Flor 1955), host resistance is governed by the recognition of pathogen virulence effectors (such recognized effectors are termed avirulence [Avr] genes) by corresponding host resistance (R) genes. A pathogen Avr allele is recognized by the host R allele, triggering responses in the host that prevent or largely reduce pathogen growth (Dangl and Jones 2001; Chisholm et al. 2006). Plants that lack the R allele are called susceptible, and pathogens with an unrecognized Avr allele are called virulent. This model suggests the potential for strong selection for polymorphisms at Avr loci that allow the pathogen to escape recognition by host R gene products. New recognition specificities may be generated through deletions or loss of function mutations, or sequence diversification. For bacterial pathogens, large population level repertoires of horizontally exchanged effectors can seemingly facilitate remarkable diversity in presence–absence polymorphisms at effector loci (Sarkar et al. 2006). In contrast, eukaryotic species are almost certainly more constrained with respect to the acquisition or loss of genes, and particularly where specific effectors play critical roles in the biology of the pathogen, protein diversification may be more common (e.g., van der Merwe et al. 2009).
Within such a mechanistic framework, different forms of natural selection may further result in distinct patterns of molecular divergence and diversity (Kniskern and Rausher 2001). For example, directional selection via continual selective sweeps and selective turnover of alleles may result in new Avr loci that are lacking in diversity (Bergelson et al. 2001; Ford 2002; Tiffin and Moeller 2006). Alternatively, the maintenance of genetic diversity within host–pathogen interactions may be promoted through different forms of heterogeneous selection. Negative frequency-dependent selection, where rare pathogen (or host) genotypes have higher fitness, is a common assumption in models of host–parasite interactions (Gillespie 1975; Clarke 1976; Anderson and May 1982) and might explain population level maintenance of resistance and virulence polymorphisms (Jayakar 1970; Hedrick 1974; Antonovics and Thrall 1994; Tellier and Brown 2007). Adding further complexity is the fact that many host–pathogen associations in natural ecosystems exist as interacting groups of small, geographically and genetically differentiated ephemeral populations (Burdon 1992). In such situations, spatially heterogeneous patterns of directional selection may occur in different populations, causing genetic divergence and local adaptation (Gandon et al. 1996; Gandon 1998; Hedrick 2006). Furthermore, spatially asynchronous frequency-dependent dynamics can maintain polymorphisms across wider metapopulation scales (Thrall and Burdon 2002), and many of these processes likely interact under natural conditions to create complex dynamics (Thompson 2005).
In this study, we examine the dynamics and evolution of Avr loci in the plant pathogen Melampsora lini. Melampsora lini is a highly specialized, obligate plant parasite able to infect a number of host species in the genus Linum (Lawrence et al. 2007). In particular, we focus on interactions between M. lini and two host species, the native Australian species Linum marginale and the widely cultivated flax species Linum usitatissimum. In L. usitatissimum, resistance to M. lini is controlled by genes segregating at five loci, whereas M. lini exhibits complex polymorphisms for virulence controlled by up to 20 avirulence genes (Lawrence et al. 2007). Avirulence genes have been identified from four loci in M. lini and are recognized by specific resistance genes in the cultivated flax host L. usitatissimum (Dodds et al. 2004; Catanzariti et al. 2006). Two of these Avr loci, AvrL567 and AvrM, contain more than one functional copy of these Avr genes, making it difficult to reliably determine allelic relationships. However, the AvrP4 locus contains a single gene, facilitating genetic sampling at the population level. The genetic structure of the fourth Avr locus, AvrP123, has not been characterized in detail. Although the exact function of these genes is not known, all encode novel, small secreted proteins that are generally thought to be transported into the host cytoplasm and play a role in establishing infection (Catanzariti et al. 2007). For the AvrL567 locus, changes in protein structure have been demonstrated to underlie changes in recognition specificity (Dodds et al. 2006).
Linum marginale is a widely distributed plant species endemic to Australia. Melampsora lini occurs throughout the entire range of L. marginale, and extensive phenotypic variation for host resistance and pathogen virulence both within and among populations has been demonstrated (Lawrence 1989; Lawrence and Burdon 1989; Thrall et al. 2002; Thrall and Burdon 2003). Melampsora lini has the potential to impose strong selection on L. marginale, causing 60–80% reductions in population size during severe epidemics (Jarosz and Burdon 1992). Although linked genetic studies in both host and pathogen have not been carried out for this interaction, Burdon (1994) demonstrated that L. marginale possesses single, major genes for resistance and that these genes are largely allelic or closely linked. Recently, isolates of M. lini infecting L. marginale in Australia have been shown to fall into two lineages (termed AA and AB; Barrett et al. 2007), which differ markedly in genetic structure, life-history traits, pathogenicity, and geographic distribution. Lineage AA isolates reproduce sexually, have low genetic diversity, and are homozygous at multiple microsatellite loci. In contrast, lineage AB isolates show a fixed pattern of heterozygosity (i.e., a different allele at each of the two nuclei) at corresponding microsatellite loci (Barrett et al. 2007), signifying they do not produce viable sexual offspring (Barrett, Thrall, et al. 2008). Lineage AB isolates consistently have one allele in common with lineage AA isolates and carry a second divergent allele not found in lineage AA. Despite extensive surveys, no intermediate genotypes have been identified to date.
Here, we use molecular and population genetic approaches to investigate the evolution of virulence polymorphisms in M. lini across a range of spatial scales. More specifically, we characterize in detail the genetic structure of the AvrP123 locus and perform transformation assays to examine whether allelic variants of the AvrP123 and AvrP4 genes are differentially recognized by resistance genes in L. marginale. For isolates collected across a broad geographic range, from both L. marginale and L. usitatissimum, we characterize patterns of nucleotide polymorphism at both the AvrP4 and AvrP123 loci and perform statistical tests to examine departures from models of neutral evolution. We further examine the extent and geographic distribution of Avr gene haplotype and nucleotide polymorphism in isolates collected from 10 local populations of the wild host L. marginale.
Materials and Methods
Melampsora lini
Melampsora lini is an obligate biotrophic species with multiple spore stages occurring on the same host species (autoecious). Mycelial cells growing in susceptible host plants each contain two haploid nuclei (dikaryotic) and produce dikaryotic urediospores that seed the clonal stage of the life cycle responsible for disease epidemics. Under ideal conditions, M. lini can propagate in the clonal state indefinitely. However, toward the end of the host-growing season dormant or resting teliospores are typically formed. During this stage, the two haploid nuclei can fuse and undergo meiosis, giving rise to several haploid spore stages at the start of the next growing season. The dikaryophase is reinitiated by the combination of two haploid pycniospores. F1 urediospores from a cross between different genotypes therefore carry discrete, haploid nuclei from each parent (Lawrence et al. 2007). Melampsora lini strains isolated from L. marginale and L. usitatissimum are closely related (van der Merwe et al. 2009), able to cross-infect some genotypes of both host species, and produce viable offspring when crossed (Lawrence 1989).
Collections
Isolates of M. lini were sampled at two distinct spatial scales (fig. 1). To examine broad geographic and species level patterns of polymorphism, we selected 29 M. lini isolates collected from L. marginale across the range of this host species in Australia and 4 from L. usitatissimum. Overall, 15 L. marginale-associated isolates belonged to lineage AA and 14 isolates belonged to lineage AB. Isolates from L. usitatissimum (cultivated flax) were obtained from the United States (2), South America (1), and New Zealand (1). All isolates displayed unique virulence phenotypes. For the sake of simplicity, L. usitatissimum isolates are referred to as “flax isolates” and the L. marginale isolates as “lineage AA’”and “lineage AB.” In what follows, haplotypes recovered from isolates collected from L. marginale are prefixed with “Lm,” and alleles recovered from isolates collected from L. usitatissimum are prefixed with “Lu.”
FIG. 1.—

Map of Australia showing locations of sampling sites and populations referred to in this study. Sampling locations for lineage AA and AB isolates (continental survey) are represented by open circles and closed triangles, respectively. The insert region shows locations of local populations sampled in two regions in New South Wales.
To further examine patterns of polymorphism at local and regional spatial scales, we sampled 238 isolates from 10 Australian populations associated with L. marginale (table 6). The pathogen populations occur in two geographically and environmentally distinct areas (termed the mountains and plain regions, respectively) that closely correspond to the distributions of lineage AA and lineage AB. The lineage identity of each of these isolates had been previously confirmed using amplified fragment length polymorphism (AFLP) markers (Barrett, Thrall, et al. 2008). For the five plains populations, we characterized 123 isolates belonging to lineage AA. For the mountain populations, we characterized 134 isolates belonging to lineage AB. For lineage AB isolates, given the very low variation in type “B” alleles at the continental scale, sequencing of B alleles was not performed for the local and regional samples (see Results).
Table 6.
Frequency Distribution of AvrP123 and AvrP4 Haplotypes within and among 10 Melampsora lini Populations Originating from Two Geographic Regions in Australia
| Locus | Allele | Plains Populations |
Mountains Populations |
||||||||
| Garra | Gundagai | LL | Marrar | Molong | B2 | B3 | Kiandra | N2 | SH2 | ||
| AvrP123 | Lm-1 | 0.93 | 0.88 | 0.97 | 0.59 | 0.88 | 0.21 | 0.90 | 0.00 | 0.79 | 0.18 |
| Lm-2 | 0.00 | 0.00 | 0.03 | 0.14 | 0.12 | 0.79 | 0.10 | 1.00 | 0.21 | 0.82 | |
| Lm-3 | 0.07 | 0.12 | 0.00 | 0.21 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| Lm-4 | 0.00 | 0.00 | 0.00 | 0.06 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| AvrP4 | Lm-1 | 0.94 | 0.70 | 0.13 | 0.86 | 0.09 | 0.19 | 0.93 | 0.00 | 0.88 | 0.00 |
| Lm-3 | 0.00 | 0.30 | 0.03 | 0.00 | 0.39 | 0.00 | 0.00 | 0.00 | 0.03 | 0.00 | |
| Lm-4 | 0.00 | 0.00 | 0.00 | 0.04 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| Lm-7 | 0.00 | 0.00 | 0.00 | 0.00 | 0.45 | 0.04 | 0.00 | 0.00 | 0.00 | 0.00 | |
| Lm-8 | 0.06 | 0.00 | 0.84 | 0.10 | 0.07 | 0.77 | 0.07 | 1.00 | 0.09 | 1.00 | |
Linum marginale Lines Used in Transient Transformation Assays
To assess whether Avr haplotype variants could act as Avr factors in the interaction with L. marginale, we used Agrobacterium infiltration to transiently express these genes in leaves of a standard set of 12 L. marginale differential host lines (AA, CC, HH, II, RR, UU, B, C, G, T, U, and V). Line “G” has no known resistance genes and was included as a negative control. These lines have been used extensively in previous work assessing pathogen population structure (Burdon and Jarosz 1991, 1992; Jarosz and Burdon 1991; Thrall et al. 2002).
Transient in Planta Expression Assays
We used Agrobacterium-mediated transient transformation to directly examine interactions between host plants and effector proteins. This technique permits the expression of individual pathogen genes directly inside the host cell. Transformation methods followed those implemented by Catanzariti et al. (2006). AvrP4 expression constructs contained the entire coding sequences including the signal peptide, whereas AvrP123 expression constructs omitted the 23 amino acid signal peptide. Agrobacterium tumefaciens cultures of strains containing the binary vector expression constructs were prepared at an OD600 of 1.0 in lysogeny broth containing 200 μM acetosyringone and infiltrated into either L. usitatissimum or L. marginale leaves by a syringe. Agrobacterium tumefaciens cultures containing an empty vector were used across all plants as a negative control. Host reaction phenotypes were scored after 14 days. Where transient expression of any of the AvrP123 or AvrP4 variants induced a hypersensitive (HR) type necrotic response in one of the differential host lines, this was interpreted as a specific recognition of the Avr allele by a corresponding R gene in the host. All host–allele combinations inducing a positive response were retested to confirm reproducibility.
Characterization of the AvrP123 Locus
A flax rust (M. lini) F2 family derived from a selfing rust strain CH5 (Lawrence et al. 1981) was used to characterize the AvrP123 locus. In this family, avirulence against the P, P1, P2, and P3 resistance genes segregates at a single locus, with AvrP encoded by one allele and AvrP1, AvrP2, and AvrP3 by the second allele (Lawrence et al. 1981). In a previous study (Catanzariti et al. 2006), a haustorially expressed secreted protein gene (HESP C51) was identified at this locus and shown to induce an HR in flax plants containing P1 or P2, confirming its avirulence function (Catanzariti et al. 2006). To identify allelic variants and additional homologs of this gene we used HESP C51 to screen a genomic DNA library derived from rust strain CH5 (Dodds et al. 2004) and also amplified this gene from CH5 F2 individuals homozygous for each of the two AvrP123 alleles. To test avirulence function, truncated versions of the AvrP123 proteins lacking the signal peptide were transiently expressed in flax lines containing the P, P1, P2, or P3 resistance genes backcrossed for 12, 10, 4, or 13 generations, respectively, into the variety Bison, which contains no recognized P locus resistance gene (Lawrence et al. 1981).
DNA Sequence Determination
Total genomic DNA was extracted from 100 mg of urediospores using a DNeasy plant mini kit (Qiagen). We used polymerase chain reaction (PCR) to amplify and subsequently sequence four nuclear regions, including two Avr loci, an 810-bp fragment of the β-tubulin locus, and the internal transcribed spacer (ITS) region. All sequence electropherograms were closely examined for double peaks (indicating heterozygosity). Amplicons containing two allelic variants were cloned using Promega pGEM-T vector system 1 cloning kits and followed standard protocols. Multiple PCR amplicons from cloned samples were sequenced until two identical copies of all cloned products were recovered. Sequence alignments were initially performed on the amino acid sequence, using conserved cysteine motifs as reference points and then backtranslated, using the software BioEdit (Hall 1999). Forward and reverse sequences were obtained for each locus in the species-level sample. Only forward primers were used to sequence the population-level samples; however, any novel variants identified were confirmed via reverse sequences and included in the species-level analyses.
AvrP4 encodes a 95 amino acid protein with a predicted 28 amino acid cleavable secretion signal peptide. PCR amplifications of this gene were performed on a Hybaid Express thermocycler under the following conditions: 95 °C for 3 min, 34 cycles at 94 °C for 30 s, 56 °C for 45 s, 72 °C for 90 s, followed by a 4 °C holding step. Two sets of primers were used for PCR amplification. For the broad scale survey, primers were designed to amplify 180 bp of 5′ flanking sequence, 285 bp of open reading frame, and 103 bp of 3′ flanking sequence (AvrP4_F1 5′-CATCAAAATCTAACCCGTAC-3′ and AvrP4_R1 5′-GTAGCATTGAGATCCATGG-3′). This survey consistently revealed two different allele types in lineage AB isolates and, with the exception of a single geographically distinct isolate, a lack of any polymorphism in the “B” type allele. Thus, to avoid cloning all lineage AB isolates, for the population level survey, we used an alternative reverse primer designed to amplify “A” type alleles only (AvrP4_R2 5′- TTGTTCAGGATAGATAGTGC-3′). Any novel variants uncovered were reamplified using the original primer set, sequenced, and cloned as required.
For AvrP123, primers were designed to amplify 119 bp of 5′ flanking sequence, 351 bp of open reading frame, and 128 bp of 3′ flanking sequence (AvrP123_F 5′-ATTGTGAACCTTTTGAAGGAC-3′ and AvrP123_R 5′CGCCATGGTATTGTTCAGAC-3′). As for AvrP4, we used alternative protocols for the species-level and population surveys. For the species-level surveys, PCR amplifications were performed as for AvrP4 except for a 54 °C annealing temperature. To avoid amplifying the “B” type allele (which had some mutations in the original priming sequence), we increased the annealing temperature to 58 °C for the population survey.
For β-tubulin and ITS genes, we investigated sequence polymorphism in the 29 species-level isolates only. Primers were designed to amplify an 810-bp fragment of the β-tubulin gene in M. lini (Ayliffe et al. 2001), incorporating 5′ flanking sequence and five introns and six exons (BT-793F 5′-AAAAACCCAAAACACTAAATCAAA-3′ and BT-793R 5′-GACCTTTGGCCCAGTTGTTA-3′). Only intronic regions were analyzed for polymorphism. The ITS region was amplified using the primer pair ITS1 and ITS4 (White et al. 1990). Only spacer regions 1 and 2 were analyzed for polymorphism.
Data Analyses
Gene Diversity and Phylogenetic Analyses
The DNAsp software package (Rozas and Rozas 1999) was used to calculate nucleotide diversity statistics. Maximum Likelihood (ML) phylogenetic trees were constructed for the Avr data in Paup* (Swofford 2001) with the AvrP4 and AvrP123 data partitioned according to the three codon positions. Substitution rates for each codon position were estimated from the data and included in the general time reversible (GTR) model with invariable sites and a gamma distribution (GTR + I + G). To obtain ML bootstrap support values (1,000 replicates) for nodes, we used the software RAxML v7.0.0 (Stamatakis et al. 2005) using the same parameters as above but without codon partitioning.
Tests for Selection
The average nonsynonymous/synonymous (dN/dS) rate ratio in pairwise comparisons among AvrP123 and AvrP4 allelic variants was estimated using the ML procedure of Yang and Nielsen (2000) as implemented in the yn00 module of the PAML software package (Yang 1997). We further estimated the nonsynonymous/synonymous rate ratio parameter ω through application of an ML method that takes into account phylogenetic structure when estimating likelihood scores, where ω was allowed to vary across codons. Neutral sites have ω = 1, whereas those under purifying selection have ω < 1 and those under positive selection ω > 1 (Nielsen and Yang 1998; Yang et al. 2000). This was done using codon substitution models in the program CODEML (Yang 1997). ML analyses were conducted using the AvrP123 and AvrP4 sequence alignments and ML trees as input data. For each gene, we compared models M7 (beta distribution) with M8 (beta distribution + positive selection). Likelihood ratio tests were performed through comparison of the test statistic 2Δl with the χ2 distribution (df = 2). Model M8 was used to estimate the dN/dS rate ratio parameter ω for amino acid sites and estimate the proportion of sites with ω > 1. The Bayes empirical Bayes procedure (Yang et al. 2005) was used to estimate the mean ω value for each codon site and the posterior probability that the site is under positive selection. Codons that were identified as having evolved under positive selection with high posterior probabilities (P > 0.95) were highlighted on an amino acid alignment of the two genes. Finally, we also calculated different ω ratios for different branches of the phylogeny and compared likelihood scores to a model where ω was held constant among all branches (ML trees: fig. 3). Likelihood estimates of the fit of these evolutionary models to the data were compared using likelihood ratio tests.
FIG. 3.—

Amino acid alignments for (A) AvrP123 and (B) AvrP4, with sites (clade A alleles only) under significant (P < 0.05) positive selection (M8) (shaded in dark gray). Alleles recovered from Linum marginale isolates are in black font and prefixed with Lm. Alleles recovered from L. usitatissimum isolates are in blue font and prefixed with Lu. Only those amino acids that differ from the top sequence are shown, with identical residues indicated by dots. Conserved Cysteine amino acid residues are highlighted in light gray. The names of the allelic variants used in transient transformation assays are underlined below.
In addition, the DNAsp software package (Rozas and Rozas 1999) was used to perform McDonald–Kreitman (MK) tests (McDonald and Kreitman 1991) on each Avr locus. Based on the high levels of divergence between the A and B genomes, we focused this test on polymorphism within the cluster of A alleles for each locus, using the divergent B alleles as a comparison. Statistical departure from neutrality was tested with a G-test on a 2 × 2 contingency table of synonymous and nonsynonymous mutations within and between species. The neutrality index (NI) (Rand and Kann 1996), which reflects the extent to which the levels of amino acid variation within species depart from the neutral model, is also reported. Under neutrality, an index of 1 is expected. Values greater than 1 indicate an excess of nonsynonymous substitutions within allelic classes, and values less than 1 indicate an excess of nonsynonymous substitutions among allelic classes, relative to the number of synonymous substitutions.
Recombination
Aligned sequence variants were tested for recombination using the software package RDP (Recombination Detection Program) version 2 (Martin et al. 2005). This software package implements a range of statistical algorithms for detecting recombination and uses a consensus approach to overcome concerns about the limitations of individual methods. The six methods applied here are RDP (Martin and Rybicki 2000), GENECONV (Padidam et al. 1999), Bootscan (Salminen et al. 1995), MaxChi (Smith 1992), Chimaera, and SiScan (Posada and Crandall 2001). The null hypothesis is that sequence variation is generated by nucleotide mutations. Recombination was deemed to occur at a locus if the null hypothesis was rejected by three or more tests at a significance level of P < 0.01.
Population Genetic Analyses
Within the 10 native Australian populations of M. lini, we generated data for the A type AvrP123 and AvrP4 alleles only. For both AvrP123 and AvrP4, we calculated allele frequencies and gene diversities (Nei 1987) for each population using the software GENALEX v6.0 (Peakall and Smouse 2005). To examine hierarchical partitioning of molecular variation among lineages and populations, we subjected AFLP and Avr gene data to analyses of molecular variance (AMOVA), using GENALEX. To explore relationships between genetic divergence and geographical distance among populations, matrices of pairwise genetic distance (ΦPT) and geographic distance were subjected to a Mantel test (Mantel 1967) using GENALEX with 10,000 permutations.
Results
Characterization of the AvrP123 Locus
Previously, we identified a haustorially expressed secreted protein gene (HESP C51) that detected restriction fragment length polymorphism (RFLP) cosegregating with the AvrP123 locus and showed that transient expression of a full-length version protein, including its signal peptide, induced an HR in flax plants containing the P1 or P2 resistance genes (Catanzariti et al. 2006). To further confirm its avirulence function, we generated an expression construct encoding a truncated version of the AvrP123 protein lacking the signal peptide. Transient expression of the nonsecreted AvrP123 protein in flax plants containing the corresponding R genes indicated that it could also induce HR in plants containing P3, as well as P1 and P2 (fig. 2). No response was induced in the cultivar Bison (which lacks a functional P locus resistance gene) or plants containing the P resistance allele. Thus, this gene is sufficient to explain the full avirulence phenotype associated with one allele of this Avr locus and we have designated the gene AvrP123 (Lu-1). HR induction by the intracellular version of this protein was stronger and more rapid than that induced by the secreted version (Catanzariti et al. 2006), confirming that the AvrP123 protein is recognized inside the plant cell. PCR amplification and sequencing from CH5 F2 rust lines confirmed that the cloned AvrP123 gene sequence was derived from the allele encoding avirulence on P1, P2, and P3 as expected. Individuals containing the allele for avirulence on P contained an alternative version of this gene, designated AvrP. The AvrP gene encodes a protein of 111 amino acids that is closely related to AvrP123, but contains 36 polymorphic amino acids and lacks the last 5 amino acids of AvrP123 due to an earlier stop codon (fig. 3A: Lu-4). Transient expression of a truncated version of this protein lacking the signal peptide induced an HR response that was specific to the P resistance gene but did not induce any response on plants containing the P1, P2, or P3 resistance genes (fig. 2). Hence, this gene is sufficient to explain the avirulence phenotype of this allele.
FIG. 2.—

Transient Expression of AvrP123 Genes in Flax Causes P locus Gene-Dependent Necrosis. Leaves of five different flax genotypes with different alleles of the P resistance gene locus were infiltrated with Agrobacterium tumefaciens cultures containing T-DNA plasmids encoding AvrP123 or AvrP lacking a signal peptide. Images were taken 10 days after infiltration. The variety Bison does not carry a functional P locus resistance gene, and the P, P1, P2, and P3 lines contain these resistance genes backcrossed into the variety Bison for 4–13 generations.
An AvrP123 probe had detected up to four RFLPs linked to the AvrP123/AvrP locus (Catanzariti et al. 2006), indicating that this locus likely contains an additional homologous gene. In fact, we isolated one additional related sequence from a CH5 genomic DNA library, but sequence analysis showed this gene contains a stop codon at amino acid position 43 and hence cannot encode a functional protein. PCR amplification from homozygous F2 individuals indicated that this pseudogene was present at both alleles of this locus, with just a single nucleotide substitution and a 4-bp indel in the 3′ untranslated region distinguishing the two allelic versions. Therefore, this gene does not contribute to the avirulence phenotype. Together with AvrP123 and AvrP, these genes are sufficient to account for all of the hybridizing fragments observed in DNA gel blots (Catanzariti et al. 2006). Thus, the AvrP123 locus consists of a single functional avirulence gene plus a second related pseudogene. The flanking sequences of these two genes differ, allowing specific PCR amplification of the single functional AvrP123 gene that we used in subsequent experiments to examine genetic diversity at this locus.
Phylogenetics and Molecular Diversity
Sequences from the ITS and β-tubulin loci were successfully sequenced for all 33 isolates collected from the two host species. Consistent with previous studies (Barrett et al. 2007), lineage AB isolates were fixed for heterozygosity at both loci (observed heterozygosity [Ho] = 1). One allele (clade A) in each isolate was identical to a single variant recovered from all lineage AA isolates (Ho = 0) collected from L. marginale. A second divergent class of alleles (clade B) was unique to lineage AB isolates. The AvrP4 and AvrP123 genes were successfully sequenced for all 33 isolates and displayed high levels of sequence variation compared with noncoding regions in ITS or β-tubulin (table 1). The relevant sequence data are deposited in GenBank (accession numbers EU642476–EU642503). Patterns of polymorphism were contrasting between alleles assigned to clades A and B (tables 2 and 3).
Table 1.
Gene Diversity Estimates for Nucleotide Sequence Data Collected from Avr Genes and Noncoding Regions within Housekeeping Genes in 33 Isolates of the Flax Rust Fungus Melampsora lini
| Locus |
||||
| AvrP123a | AvrP4a | ITS 1 & 2 | β-Tubulin Introns | |
| Length (bp) | 351 | 285 | 421 | 497 |
| No. of L.m. allelesb | 7 | 13 | 2 | 5 |
| No. of L.u. allelesc | 5 | 3 | 5 | 2 |
| Segregating sites | 118 | 91 | 11 | 9 |
| Gene diversity (π) | 0.124 | 0.079 | 0.004 | 0.006 |
| π clade A alleles | 0.034 | 0.016 | 0.002 | 0.004 |
Average statistics based on coding sequence only.
Isolates collected from Linum marginale (L.m.; n = 29).
Isolates collected from Linum usitatissimum (L.u.; n = 4).
Table 2.
Polymorphism and Gene Diversity Estimates for AvrP123 and AvrP4 Nucleotide Sequences across 33 Isolates of Melampsora lini
| Allelic Classes | Total Number of Alleles | No. of Protein Variants | No. of Segregating Sites | π Coding Sequence | πsyna | πnona | πnon/πsyna | |
| AvrP123 | L.m. Ab | 44 | 5 | 62 | 0.013 | 0.003 | 0.017 | 6.17 |
| L.u. Ac | 8 | 5d | 56 | 0.086 | 0.026 | 0.106 | 3.99 | |
| L.m. Be | 14 | 1 | 1 | 0.0004 | 0.0019 | 0 | — | |
| All | 66 | 9d | 118 | 0.124 | 0.099 | 0.129 | 1.31 | |
| AvrP4 | L.m. Ab | 44 | 11 | 17 | 0.014 | 0.009 | 0.015 | 1.70 |
| L.u. Ac | 8 | 3 | 7 | 0.010 | 0 | 0.012 | ∞ | |
| L.m. Be | 14 | 2 | 52 | 0.028 | 0.028 | 0.028 | 1.00 | |
| All | 66 | 16 | 91 | 0.078 | 0.085 | 0.076 | 0.89 |
syn., synonymous; non., nonsynonymous.
Clade A alleles sampled from isolates collected from Linum marginale (L.m.).
Clade A alleles sampled from isolates collected from Linum usitatissimum (L.u.).
One allele that presents a large internal deletion was not included in any population genetic or selection analyses.
Clade B alleles sampled from isolates collected from L. marginale.
Table 3.
dN/dS Rate Ratios and Parameter Estimates Generated by ML Model Comparisonsa for the AvrP123 and AvrP4 Genes
| Gene | Alleles | No. of Allelic Variants | Average dn/dsb | ps | ω | 2Δlc | Pc |
| AvrP123 | A alleles | 8 | 7.0 | 0.45 | 20.41 | 46.5 | <0.001 |
| Linum marginale A alleles | 5 | 7.95 | 0.47 | 29.96 | 36.0 | <0.001 | |
| All alleles | 9 | 2.19 | 0.35 | 11.12 | 59.42 | <0.001 | |
| AvrP4 | A alleles | 14 | 3.22 | 0.42 | 7.83 | 6.96 | <0.05 |
| L. marginale A alleles | 11 | 2.58 | 0.62 | 4.18 | 2.42 | NS | |
| All alleles | 16 | 1.00 | 0.18 | 6.67 | 16.54 | <0.001 |
For the model comparison, ω represents parameter estimates of dn/ds in the nonconstrained class and ps the proportion of sites estimated to belong to that class. A comparison between M7 (beta) versus M8 (beta & ω) is presented here. A comparison of models M1 (neutral) versus M2 (selection) produces highly similar results.
Average dn/ds ratio calculated over the entire coding sequence using the model M0 (assuming a single evolutionary rate among codons).
Likelihood Ratio Tests were performed with the nested models M7 and M8 by comparing the test statistic 2Δl with the χ2 distribution (df = 2). For model M8, ps = estimated proportion of sites with ω >1; ω = estimated dn/ds ratio for sites with ω > 1.
Lineage AB isolates were fixed for heterozygosity at all Avr and housekeeping loci. For AvrP123, a total of 12 nt and 10 amino acid alleles were recovered. The average pairwise nucleotide divergence was 18.7%, ranging between 0.3% and 39.2%. Most of the polymorphic sites were scattered across the mature protein, with some fixed polymorphisms between clade A and B haplotypes in the signal peptide (fig. 3A). One allele in a M. lini strain collected from L. usitatissimum was severely truncated (but occurred in a heterozygous state along with a full length variant). The truncated allele (Lu-5) has a large (186 bp) internal frame deletion (fig. 3A) and was excluded from phylogenetic analyses and tests for selection. Considering only amino acid variants, nine type A alleles and one divergent B allele were recovered (figs. 3A and 4A, table 2). The invariant B allele was present in all 14 lineage AB isolates. Five alleles identified in clade A (figs. 3A and 4A) were recovered from L. marginale isolates. With two exceptions, isolates collected from L. marginale carried one of three closely related alleles (Lm-1, Lm-2, and Lm-3) that differed at single amino acid sites. The remaining four alleles from clade A were recovered from samples collected from L. usitatissimum and constitute a diverse group (figs. 3A and 4A).
FIG. 4.—
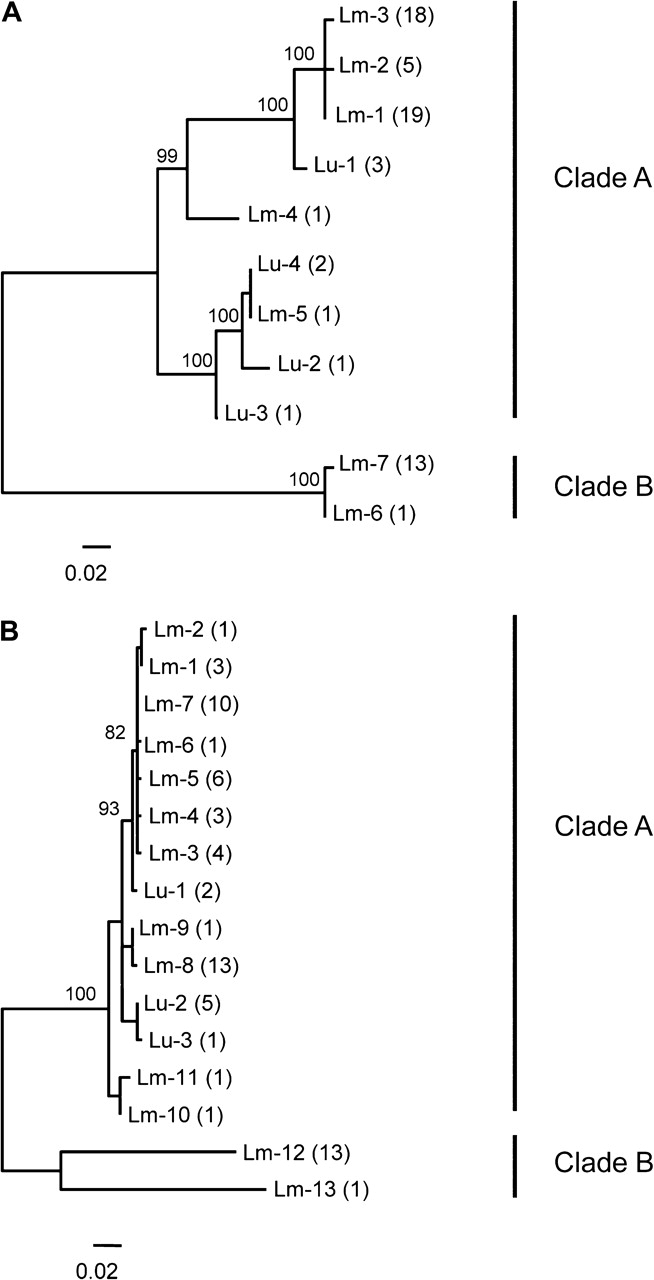
Unrooted ML trees constructed from the AvrP123 and AvrP4 sequence data. (A): AvrP123 and B: AvrP4. Bootstrap support values from 500 replicates higher than 70% are given in italics close to the nodes on top of the branches. Alleles from Melampsora lini isolates collected from Linum marginale are prefixed with Lm. Alleles from isolates collected from isolated collected from L. usitatissimum are prefixed with Lu. The number of times each allele was recovered is indicated in brackets after each allele code (with a diploid genotype inferred for each homozygote).
For the AvrP4 locus, we recovered a total of 16 different alleles. All variants at the DNA level also differed in terms of amino acids. The average pairwise nucleotide divergence was 7%, ranging between 0.4% and 24.8%. The largest proportion of polymorphic sites were clustered toward the 3′ end of the mature protein, with other polymorphisms scattered along its length, and some fixed polymorphisms between clade A and B haplotypes in the signal peptide (fig. 3B). For isolates collected from L. marginale, we recovered 11 relatively similar type A alleles and two highly divergent type B alleles (Lm-12 and Lm-13: from lineage AB individuals) (figs. 3B and 4B, table 2). Of the B alleles, one (Lm-13) was recovered from a single isolate collected in Tasmania. The second B allele (Lm-12) was present in all the remaining 13 hybrid isolates. Three clade A alleles were recovered from samples collected from L. usitatissimum (figs. 3B and 4B). Polymorphic sites were scattered across the entire protein, with relatively high levels of variation in the C terminal end of the protein (fig. 3B).
A phylogeny obtained from ML analyses of the AvrP123- and AvrP4-coding sequences is shown in figure 4. Alleles recovered from the 14 individuals belonging to lineage AB consistently grouped in two well-supported clades for both Avr loci, with one allele that clustered with alleles isolated from both lineage AA and L. usitatissimum isolates (the A clade), and a second divergent allele (the B clade). Only one allele was shared between L. usitatissimum and L. marginale isolates in clade A (AvrP123; Lm-5 and Lu-4) (fig. 4). Patterns of allelic diversity among the A and B clades were strongly contrasting, with much higher levels of nucleotide and amino acid polymorphism for clade A alleles (fig. 4, table 2). Both the AvrP123 and AvrP4 gene regions were tested for evidence of recombination using the six test algorithms included in the software RDP. No evidence of recombination was detected at either locus.
Considering levels of polymorphism at Avr loci compared with putatively neutrally evolving, noncoding regions, comparative analyses for the entire data set showed that haplotype and gene (π) diversities were more than an order of magnitude higher for AvrP123 and AvrP4 than for noncoding regions within ITS or β-tubulin, respectively (table 1). The magnitude of these differences in part reflects comparatively strong divergence between clade A and B alleles at Avr loci, although this pattern remains intact when clade B alleles are excluded from the analyses (table 1).
Selection
Tests comparing rates of synonymous (dS) and nonsynonymous (dN) substitution across the entire coding regions of each of the allelic variants, using an ML approach, were used to investigate the role of positive selection in driving nucleotide divergence across sequence variants. Parameter estimates for the two models for different subsets of the data are shown in table 3. In light of the unknown origin of clade B alleles, relatively high levels of allelic and nucleotide polymorphism within clade A, and strong divergence between clades A and B, the extent to which they are comparable in population genetic analyses is uncertain. Therefore, we focus below on analyses of variation in clade A, although results obtained from analyses of the entire data set are also reported.
Under models of variable ω values among sites, chi-square tests of twice the log-likelihood of differences among models show that model M8 (beta distribution + positive selection) fitted the clade A data set for both avirulence genes significantly better than the M7 (beta distribution) model (table 3). For AvrP123, 36 sites were under significant (P < 0.05) levels of positive selection, distributed throughout the length of the protein following the signal peptide region. For AvrP4, four sites were under significant levels of positive selection (P < 0.05), scattered across the length of the secreted protein (fig. 3).
We also conducted tests for selection using clade A variants recovered from isolates infecting L. marginale. Average dN/dS ratios calculated for this class of variants for the AvrP123 and AvrP4 genes were 7.95 and 2.58, respectively (table 3). For AvrP123, models of variable ω values among sites, with chi-square tests of twice the log-likelihood of differences among models show that model M8 (beta distribution + positive selection) fitted the entire data set significantly better than the M7 (beta distribution) model. In contrast, results for AvrP4 were not significant (P > 0.05) (table 3).
Average dN/dS ratios decreased markedly when all alleles (including clade B) were included in the analysis (AvrP123: 2.2; AvrP4: 1.0) (table 3). Regardless, models of variable ω values among sites, with chi-square tests of twice the log-likelihood of differences among models show that model M8 (beta distribution + positive selection) fitted the entire data set for both avirulence genes significantly better than the M7 (beta distribution) model (table 3). For both AvrP123 and AvrP4, likelihood estimates calculated for the branch models allowing ω to vary among branches did not fit the data significantly better than a model where ω was held constant among branches (P > 0.1).
We also used MK tests for positive selection. Based on the high level of divergence between the A and B clades, and the clear hybrid status of lineage AB, we conducted MK tests using sequences in clade A for the “intraspecific” comparison and both clade B alleles from AvrP123 for the “interspecific” comparison. Because of the high levels of nucleotide divergence (24%) between the common and the single, rare AvrP4 B alleles, only the common AvrP4 B allele was used in among-species comparisons. Patterns of synonymous and nonsynonymous variations and the NI in both Avr genes suggest a significant departure from neutrality for both loci and show a large excess of amino acid polymorphism within species (NI = 10.9 and 4.5 for AvrP123 and AvrP4, respectively) (table 4).
Table 4.
Summary of MK Tests
| Fixed Differences between Clades A & B | Polymorphic Sites within Clade A | G-Test Resultsa | NIb | ||
| AvrP123 | Synonymous | 19 | 2 | G = 15.57 | 10.92 |
| Nonsynonymous | 47 | 54 | P = <0.001 | ||
| AvrP4c | Synonymous | 17 | 2 | G = 4.55 | 4.50 |
| Nonsynonymous | 34 | 18 | P = 0.033 |
Probability determined by a G-test.
NI refers to the NI (Rand and Kann 1996).
The calculation of fixed differences between clades A and B for AvrP4 were performed using only the single clade B haplotype Lm-12 (see Materials and Methods).
Avr Diversity and Structure in Local Populations
We sequenced A type AvrP123 and AvrP4 amplicons from a total of 257 individual isolates, collected from 10 discrete localities across two geographic regions. Given the lack of variation in type B alleles at the continental scale, further sequencing of B alleles was not performed. The recovered alleles were a subset of those seen at the continental scale (figs. 3 and 4) with four AvrP123 and five AvrP4 haplotypes recovered from the 10 populations in the mountain and plain regions. Levels of nucleotide diversity (π) within local populations (population mean: AvrP123: 0.0029; AvrP4: 0.0041) was much reduced compared with the wider continental data set. The frequency and distribution of Avr haplotypes varied among both populations and lineages (tables 5 and 6). Population genetic analyses showed significant geographic structure in the AvrP123 and AvrP4 allelic data for clade A. For AvrP123, AMOVA (ΦPT = 0.29, P = 0.001) assigned 20% of the diversity to differences among plain and mountain regions, 9% between populations within a region, and the largest proportion of the variation among isolates within populations (71%). For AvrP4, AMOVA also revealed significant structure in the data (ΦPT = 0.49, P = 0.001). However, none of this variation could be attributed to differences among regions. Instead, 49% of the diversity was assigned to differences among populations and 51% of the variation among isolates within populations. Considering all 10 populations, for both AvrP123 and AvrP4, Mantel tests revealed no significant association between the degree of genetic population differentiation (ΦPT) and geographic distance (km) (AvrP123, P = 0.12; AvrP4, P = 0.18).
Table 5.
Gene Diversity Statistics for AvrP123 and AvrP4 in Local Populations
| Population | No. Isolates |
AvrP123 |
AvrP4 |
||||
| NA | Gene Div. | π Coding | NA | Gene Div. | π Coding | ||
| Garra | 30 | 2 | 0.23 | 0.00064 | 2 | 0.12 | 0.0030 |
| Gundagai | 26 | 2 | 0.30 | 0.00086 | 2 | 0.46 | 0.0032 |
| Larras Lee | 31 | 2 | 0.07 | 0.00000 | 3 | 0.32 | 0.0077 |
| Marrar | 21 | 4 | 0.58 | 0.02355 | 3 | 0.26 | 0.0053 |
| Molong | 21 | 2 | 0.26 | 0.00074 | 4 | 0.61 | 0.0052 |
| B2 | 25 | 2 | 0.38 | 0.00107 | 3 | 0.37 | 0.0086 |
| B3 | 26 | 2 | 0.19 | 0.00053 | 2 | 0.19 | 0.0045 |
| Kiandra | 33 | 1 | 0.00 | 0.00000 | 1 | 0.00 | 0.0000 |
| N2 | 28 | 2 | 0.33 | 0.00094 | 3 | 0.23 | 0.0057 |
| SH2 | 22 | 2 | 0.35 | 0.00099 | 1 | 0.00 | 0.0000 |
NA = number of alleles; Gene Div = Nei's Gene Diversity; and π = pairwise nucleotide diversity.
Transient Expression of AvrP123 and AvrP4 in L. marginale
Following sequencing of Australian M. lini isolates, we selected 6 of the 10 unique alleles recovered from the AvrP123 locus (Lm-1, Lm-3, Lm-4 [identical to Lu-5], Lm-5, Lm-6, and Lu-1) and 8 of the 16 unique alleles recovered from AvrP4 (Lm-1, Lm-5, Lm-7, Lm-8, Lm-10, Lm-12, Lu-1, and Lu-2) (fig. 2). Alleles (underlined in fig. 2) were selected so as to ensure that all major phylogenetic groups were represented, including alleles belonging to clade B (figs. 4 and 5). Each of these variants was transiently expressed in a set of 12 L. marginale differential lines using Agrobacterium-mediated transient expression. In this assay, resistance-gene mediated recognition of the expressed Avr protein is expected to lead to induction of an HR-like necrotic response.
FIG. 5.—

Transient expression of the Melampsora lini AvrP123 and AvrP4 genes in Linum marginale causes Avr gene-dependent necrosis. (A). Leaves of a single L. marginale genotype (differential line L) displaying a necrotic response following infiltration with Agrobacterium tumefaciens cultures containing the M. lini AvrP123 alleles Lm-1 and Lm-3. (B). Leaves of a single L. marginale genotype (differential line UU) displaying a necrotic response following infiltration with A. tumefaciens cultures containing the AvrP4 alleles Lm-1, Lm-5, Lm-7, Lm-8, and Lu-2.
Three AvrP123 variants (AvrP123-Lm-1, Lm-3, and Lm-5) recovered from L. marginale induced such a response in one L. marginale differential line (line L), indicating that this differential line has a resistance gene or allele capable of detecting these allelic variants (fig. 5). AvrP123-Lm-1 and AvrP123-Lm-3 differ by only a single amino acid, suggesting that they may be functionally identical with regards to recognition by the L differential line. The remaining AvrP123-differential combinations failed to elicit a necrotic response. Transient expression of four AvrP4 variants (AvrP4-Lm-1; AvrP4-Lm-5; AvrP4-Lm-7; and AvrP4-Lm-8) recovered from isolates infecting L. marginale, induced an HR-like necrotic response in one L. marginale differential line (line UU), indicating that this differential has the capacity to recognize multiple AvrP4 allelic variants (fig. 5). The remaining Avr-differential combinations failed to elicit a necrotic response, indicating the corresponding R gene is absent in these lines. The positive responses were reproducibly observed in multiple infiltration experiments, providing clear evidence that protein variation in AvrP123 and AvrP4 does indeed contribute to functional variation in the recognition of M. lini by L. marginale.
Discussion
The GFG system is an important model describing interactions between host resistance and parasite avirulence (antigenic) loci, yet the processes whereby polymorphisms are generated and maintained in GFG systems have remained puzzling to both theoretical and empirical scientists (Laine and Tellier 2008). Interactions between M. lini and Linum spp. provide an excellent forum to study the evolutionary forces that drive the evolution and maintenance of polymorphisms for specificity in GFG interactions. The native M. lini–L. marginale interaction provides the opportunity to gain an insight into how hosts and pathogens evolve under natural conditions and across multiple spatial scales. This is reinforced by several decades of research regarding the genetic basis of host specificity of M. lini in interactions with cultivated flax (L. usitatissimum). In particular, effector genes recognized by resistance genes in L. usitatissimum have recently been cloned from four loci in M. lini (Dodds et al. 2004; Catanzariti et al. 2006). All encode novel, small, secreted proteins that are recognized inside plant cells, making them obvious targets for host resistance genes. Two of these loci, AvrL567 and AvrM, contain more than one functional copy of these Avr genes, making it difficult to sample at the population level and reliably determine allelic relationships. In contrast, the AvrP4 locus contains a single gene. Here, we demonstrate that a fourth locus, AvrP123, partially characterized by Catanzariti et al. (2006), also contains a single functional gene, whose product is recognized inside plant cells and that allelic variants at this locus are differentially recognized by the P, P1, P2, or P3 resistance genes in L. usitatissimum. The characterization of these two single copy Avr genes facilitates broad genetic sampling and the opportunity to investigate evolution at the population level.
AvrP123 and AvrP4 Genes Are Conserved and Evolve under Strong Diversifying Selection
We demonstrate that in M. lini, Avr genes evolve by amino acid sequence diversification to avoid recognition by plant resistance proteins. AvrP123 and AvrP4 homologs were universally present in isolates collected across a wide range of spatial scales. Changes to the AvrP4 and AvrP123 proteins occurred almost exclusively via nonsynonymous mutations. We found no evidence for genetic recombination, nor did we find examples of entire gene deletion. None of the observed mutations obviously render the genes nonfunctional (e.g., frameshift mutations or early stop codons), with the possible exception of a single, large internal deletion of coding sequence in one AvrP123 allele, suggesting that there is likely selection to maintain a putative effector function of these proteins (Gabriel 1999).
Several lines of evidence suggest that selection processes associated with antagonistic host–pathogen interactions are important for driving the observed Avr gene diversification. First, variants of both AvrP123 and AvrP4 can differentially induce HR responses on both L. marginale and L. usitatissimum, demonstrating that resistance genes in both host species can confer specific recognition of Avr alleles. Secondly, patterns of nucleotide variation within the coding regions of the Avr genes demonstrate a significant and striking excess of nonsynonymous compared with synonymous polymorphism relative to expectations based on neutrality. Indeed, for AvrP123, mean dN/dS ratios across the entire gene (7.0), and ω rate ratio parameter values at the 36 codon sites identified as being under positive selection (20.4), were among the highest we could identify in the literature. Together, these results suggest that host–pathogen coevolution drives the emergence and maintenance of allelic diversity at loci that are involved in direct interactions with host R genes. The genetic diversity permits the pathogen to avoid host defences while conserving the innate function of the targeted protein. Similar genetic signatures have been detected at other pathogen effector loci where sequence variants differentially trigger host defence responses. For example, Ma et al. (2006) revealed positive selection acting on the diverse HopZ family of type III secreted effectors in the bacterial pathogen Pseudomonas syringae. For M. lini, high allelic diversity and a strong signature of positive selection are also characteristic of the AvrL567 locus (Dodds et al. 2006). Although R-gene imposed selection on allelic variants in all of these examples has likely contributed strongly to the accumulation of sequence diversity, selection for enhanced pathogenicity effector function (Alfano and Collmer 2004) may also be important.
Avr gene homologs in one host–pathogen interaction need not necessarily function as Avr elicitors in related hosts. Thus, the fact that the AvrP123 and AvrP4 genes identified from rust isolates of cultivated flax, L. usitatissimum, can elicit resistance responses in the native host, L. marginale, is of considerable interest. The congeneric status of L. usitatissimum and L. marginale suggests that the ability to recognize AvrP123 and AvrP4 proteins may be the result of shared, ancestral R gene homologs among the two hosts. If this is indeed the case, then the association between M. lini and Linum spp. is likely to be of long evolutionary standing, predating speciation in at least some species. For example, although L. marginale and L. usitatissimum are relatively closely related within the broader Linum genus they are geographically, morphologically and genetically distinct (McDill et al. 2009). Linum usitatissimum has its origin in Central Asia and the Mediterranean (Zeven and Zhokovsky 1975), whereas L. marginale is native to Australia. Furthermore, there are large differences in chromosomal number between L. marginale (2n = 84) and L. usitatissimum (2n = 30). Together, these data suggest a sustained period of genetic isolation between the two species. Such conservation of interactions between specific R and Avr gene homologs through speciation events has been demonstrated by Kruijt et al. (2005), who showed that the Cf-4 and Cf-9 R genes mediate recognition of the Avr4 and Avr9 genes of Cladosporium fulvum across multiple species in the genus Lycopersicon. An alternative explanation for the shared specificity of L. marginale and L. usitatissimum is that particular specificities may have evolved independently in the two species as a result of convergent evolution. Such convergent evolution underlies shared R gene specificities in Arabidopsis thaliana (RPM1) and Glycine max (Rpg1-b) to the Avr effector protein AvrRpm1 from P. syringae (Ashfield et al. 2004). Clearly, analogous investigation of R gene structure and diversity in populations of L. marginale is needed to better understand the evolution of plant disease resistance mechanisms.
The M. lini–L. marginale Interaction
Consistent with the discussion above, frequent polymorphism at AvrP123 and AvrP4 in M. lini isolates collected from across the range of L. marginale in Australia, differential induction of HR responses on L. marginale, and statistical evidence for positive selection (AvrP123) suggest that ongoing coevolutionary dynamics between M. lini and L. marginale drive pathogen diversification in Australia. However, the data further allow inference of other selective and nonselective processes that likely influence patterns of variation at AvrP123 and AvrP4. These provide additional insight into the complexity underlying the maintenance of adaptively significant variation in natural host–pathogen interactions.
Although expectations regarding patterns and levels of genetic variation in nonagricultural systems are not well developed, the distribution of sequence variation within both the continental and population data sets suggests that although diversifying selection is important for the maintenance of polymorphism, other processes may be acting to reduce the amount of genetic variation in the sample. For AvrP123, this can be seen in the rarity of the highly divergent allelic variants (Lm-4, Lm-5). Instead, the Australian sample is dominated by three clade A AvrP123 allelic variants (Lm-1, Lm-2, and Lm-3) that differ by only single nucleotides. For AvrP4, although allelic diversity is more evenly distributed throughout the tree, the overall nucleotide divergence among allelic variants is low compared with AvrP123, and evidence for diversifying selection is equivocal.
Different factors may limit the accumulation of variation within populations. As an adaptive process, the emergence of new resistance genes within populations can result in strong directional selection on local pathogen populations. Such a process may reflect a “coevolutionary arms race”—escalating patterns of adaptation and counteradaptation among host and pathogen. Such arms races can initiate a selective sweep favoring newly emerged genotypes, resulting in lower overall diversity at sites under selection (e.g., Wichmann et al. 2005). Of a more stochastic nature, genetic bottlenecks during epidemic cycles and the fragmented nature of host populations will also act to limit how much variation accumulates within populations (Barrett, Thrall, Burdon, and Linde 2008). Given that M. lini is a biotrophic foliar pathogen and occurs in small, fragmented and genetically diverse host populations, annual boom-and-bust epidemic dynamics promotes frequent population bottlenecks, with associated genetic drift and local extinctions (Burdon and Jarosz 1992). Heterogeneity in terms of host availability and environmental conditions may also limit opportunities for the establishment and persistence of migrants (Barrett, Thrall, Burdon, and Linde 2008).
Long-distance dispersal and gene flow has potential to introduce new genetic variants in the Australian M. lini population and may explain the existence of rare, divergent alleles. Isolates collected from L. marginale and L. usitatissimum produce viable offspring when crossed in the glasshouse (Lawrence, 1989) and given the aerially dispersed nature of M. lini, long-distance dispersal events and subsequent gene flow are plausible (Brown and Hovmoller 2002). Melampsora lini isolates from L. usitatissimum have been demonstrated to be virulent against some L. marginale host lines (Lawrence and Burdon 1989) (some L. marginale hosts are susceptible to all isolates they have been tested against), meaning that should long-distance spore dispersal take place, susceptible host substrate is potentially available. Furthermore, phylogenetic analysis of AvrP123 and AvrP4 shows that clade A variants collected from the two Linum hosts do not collectively form discrete evolutionary lineages and even have one allele in common. This suggests that the populations are recently diverged and possibly have been connected to some extent via gene flow.
Within local populations, variation is much reduced compared with the broader geographic sample. However, despite limited nucleotide divergence, allelic polymorphisms are common within local populations. Strong genetic divergence at the regional and population scales for both AvrP4 (ΦPT = 0.49), and to a lesser extent AvrP123 (ΦPT = 0.29), suggests that regional and local evolutionary outcomes contribute to maintenance of diversity at Avr loci in M. lini. Previous studies of the L. marginale–M. lini interaction suggest that selection for virulence has the potential to strongly influence local patterns of genetic variation in M. lini (Thrall and Burdon 2003) and comprehensive cross-inoculation trials have demonstrated strong local adaptation of the pathogen to host populations (Thrall et al. 2002). The current results, in combination with these previous studies, support the hypothesis that local adaptation and, more generally, geographically variable selection, maintains variation for virulence in M. lini. However, among-population genetic polymorphism in M. lini may be also strongly influenced by neutral genetic drift. “Boom-and-bust” epidemic dynamics, the potential for long-distance dispersal and frequent local extinction and recolonization result in highly stochastic demographic and genetic dynamics in populations of M. lini. Indeed, it is possible that selection for the same virulence phenotypes actually limits divergence among populations (Barrett, Thrall, et al. 2008).
Negative frequency-dependent selection within populations may also promote the maintenance of diversity at AvrP123 and AvrP4, particularly when virulence alleles influence different components of fitness (Jayakar 1970; Antonovics and Thrall 1994; Roy and Kirchner 2000; Thrall and Burdon 2002). With regard to the interaction between M. lini and L. marginale, there is no direct empirical evidence for these kinds of fluctuating genetic dynamics within local populations. However, the potential for frequency-dependent selection to maintain genetic polymorphisms within populations of M. lini is evidenced by 1) the existence of considerable diversity for virulence within populations (at both phenotypic and molecular levels) and 2) costs of pathogen virulence in the form of demonstrated trade-offs between spore production and virulence in M. lini isolates infecting the Australian host L. marginale (Thrall and Burdon 2003).
An important caveat for this discussion is that not all allelic variants identified are necessarily potential targets for host resistance. For example, the common allelic variants at the AvrP123 locus (Lm-1, Lm-2, and Lm-3) differ by only single amino acids and are all recognized by the same host differential line. Thus, there is no evidence that these variants have any functional consequence in terms of interactions with host R genes. However, single amino acid mutations can confer functional changes with regard to host recognition (Wang et al. 2007), and there may be other R variants that distinguish these alleles. Further experimentation examining the performance of different Avr allelic variants on different local hosts (i.e., local adaptation at the molecular level) will help to resolve the significance of these allelic differences and the extent to which geographic structure at Avr loci drives local adaptation.
Lineage AB
In Australia, a significant proportion of isolates sampled from wild L. marginale hosts are derived from a likely hybridization event (lineage AB) and are fixed in a heterozygous state for numerous molecular markers (Barrett et al. 2007; Barrett, Thrall, et al. 2008). Consistent with these previous results, we found fixed heterozygosity at both AvrP123 and AvrP4 in all lineage AB isolates. Lineage AB isolates always carried one allele similar to those found in lineage AA isolates (clade A alleles) and also carried a second highly divergent allele (clade B alleles) not found in lineage AA. The magnitude of the divergence between clades A and B is particularly striking given the relatively low levels of divergence between clade A and B alleles from the β-tubulin locus and the ITS region.
The extent to which it is appropriate to include sequences belonging to clade B in analyses examining population-level sequence diversification is unclear. To date, we have been unable to identify the putative BB parental lineage in Australia, despite sampling at wide geographic scales as well as more intensively in local populations. However, it seems likely that strong divergence between the clade A and B alleles occurred in discrete, ancestral populations prior to any hybridization event. Indeed, levels of nucleotide divergence between lineage A and B alleles are consistent with levels of divergence and diversity in AvrP4 sequence homologs for isolates of M. lini collected from three different host species (van der Merwe et al. 2009). In addition, (with one exception) clade B Avr alleles displayed no amino acid variation. These patterns of sequence variation and nucleotide polymorphism are consistent with a recent introduction of the B alleles into the population (through hybridization), and host–pathogen coevolutionary interactions with L. marginale occurring exclusively via clade A alleles at both the AvrP123 and AvrP4 loci. Although inconclusive, further support for this hypothesis comes from results obtained during the transformation assays, where no host recognition of clade B alleles could be demonstrated for either AvrP123 or AvrP4.
Concluding Remarks
This study was undertaken to investigate the evolutionary processes that maintain pathogenic variability using the model Linum–Melampsora interaction and provides a clear example of the potential for selective processes associated with antagonistic host–pathogen interactions to drive diversification in pathogen species. ML analyses of the ratio of nonsynonymous to synonymous polymorphism revealed very strong diversifying selection acting on alleles of the AvrP123 and AvrP4 effector genes, which are differentially recognized by host R genes. Sampling from natural populations across different spatial scales further reveals the potential for various other selective and nonselective factors to influence patterns of genetic variation in host–pathogen interactions. In particular, selective sweeps, long-distance dispersal, population bottlenecks and locally divergent patterns of selection are likely to be key determinants of pathogenic diversity in this system. A corresponding survey of R gene diversity and structure in populations of L. marginale would provide a deeper insight into reciprocal patterns of selection, coevolution, and the maintenance of diversity in resistance and virulence in natural populations.
Supplementary Material
Acknowledgments
We thank Caritta Eliasson for technical assistance with the glasshouse and laboratory studies and the New South Wales National Parks and Wildlife Service for access to field sites. Mark Kinnear and Joanna Kelly provided advice on PAML analyses. Jeff Ellis and Joel Kniskern provided useful comments on earlier drafts of this manuscript. This research was supported by funding from the National Institutes of Health (grant 5R01GM074265-01A2.).
References
- Alfano JR, Collmer A. Type III secretion system effector proteins: double agents in bacterial disease and plant defense. Annu Rev Phytopath. 2004;42:385–414. doi: 10.1146/annurev.phyto.42.040103.110731. [DOI] [PubMed] [Google Scholar]
- Anderson RM, May RM. Coevolution of hosts and parasites. Parasitology. 1982;85:411–426. doi: 10.1017/s0031182000055360. [DOI] [PubMed] [Google Scholar]
- Antonovics J, Thrall PH. Cost of resistance and the maintenance of genetic polymorphism in host-pathogen systems. Proc R Soc Lond B. 1994;257:105–110. [Google Scholar]
- Ashfield T, Ong LE, Nobuta K, Schneider CM, Innes RW. Convergent evolution of disease resistance gene specificity in two flowering plant families. Plant Cell. 2004;16:309–318. doi: 10.1105/tpc.016725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayliffe MA, Dodds PN, Lawrence GJ. Characterisation of a β-tubulin gene from Melampsora lini and comparison of fungal β-tubulin genes. Mycol Res. 2001;105:818–826. [Google Scholar]
- Barrett LG, Thrall PH, Burdon JJ. Evolutionary diversification through hybridization in a wild host–pathogen interaction. Evolution. 2007;61:1613–1621. doi: 10.1111/j.1558-5646.2007.00141.x. [DOI] [PubMed] [Google Scholar]
- Barrett LG, Thrall PH, Burdon JJ, Linde CC. Life history determines genetic structure and evolutionary potential of host–parasite interactions. Trends Ecol Evol. 2008;23:678–685. doi: 10.1016/j.tree.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett LG, Thrall PH, Burdon JJ, Nicotra AB, Linde CC. Population structure and diversity in sexual and asexual populations of the pathogenic fungus Melampsora lini. Mol Ecol. 2008;17:3401–3415. doi: 10.1111/j.1365-294X.2008.03843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergelson J, Kreitman M, Stahl EA, Tian DC. Evolutionary dynamics of plant R-genes. Science. 2001;292:2281–2285. doi: 10.1126/science.1061337. [DOI] [PubMed] [Google Scholar]
- Brown JKM, Hovmoller MS. Aerial dispersal of pathogens on the global and continental scales and its impact on plant disease. Science. 2002;297:537–541. doi: 10.1126/science.1072678. [DOI] [PubMed] [Google Scholar]
- Buitkamp J, Filmether P, Stear MJ, Epplen JT. Class I and class II major histocompatibility complex alleles are associated with faecal egg counts following natural, predominantly Ostertagia circumcincta infection. Parasitol Res. 1996;82:693–696. doi: 10.1007/s004360050187. [DOI] [PubMed] [Google Scholar]
- Burdon JJ. Host population subdivision and the genetic structure of natural pathogen populations. Adv Plant Path. 1992;8:81–94. [Google Scholar]
- Burdon JJ. The distribution and origin of genes for race-specific resistance to Melampsora lini in Linum marginale. Evolution. 1994;48:1564–1575. doi: 10.1111/j.1558-5646.1994.tb02196.x. [DOI] [PubMed] [Google Scholar]
- Burdon JJ, Jarosz AM. Host-pathogen interactions in natural populations of Linum marginale and Melampsora lini. I. Patterns of resistance and racial variation in a large host population. Evolution. 1991;45:205–217. doi: 10.1111/j.1558-5646.1991.tb05278.x. [DOI] [PubMed] [Google Scholar]
- Burdon JJ, Jarosz AM. Temporal variation in the racial structure of flax rust (Melampsora lini) populations growing on natural stands of wild flax (Linum marginale): local versus metapopulation dynamics. Plant Pathol. 1992;41:165–179. [Google Scholar]
- Catanzariti AM, Dodds PN, Ellis JG. Avirulence proteins from haustoria-forming pathogens. FEMS Microbiol Lett. 2007;269:181–188. doi: 10.1111/j.1574-6968.2007.00684.x. [DOI] [PubMed] [Google Scholar]
- Catanzariti A-M, Dodds PN, Lawrence GJ, Ayliffe MA, Ellis JG. Haustorially expressed secreted proteins from flax rust are highly enriched for avirulence elicitors. Plant Cell. 2006;18:243–256. doi: 10.1105/tpc.105.035980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm ST, Coaker G, Day B, Staskawicz BJ. Host–microbe interactions: shaping the evolution of the plant immune response. Cell. 2006;124:803–814. doi: 10.1016/j.cell.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Clarke B. The ecological genetics of host–parasite relationships. In: Taylor ER, Muller R, editors. Genetic aspects of host–parasite relationships. Oxford: Blackwell Scientific; 1976. [Google Scholar]
- Dangl JL, Jones JDG. Plant pathogens and integrated defence response to pathogens. Nature. 2001;411:826–833. doi: 10.1038/35081161. [DOI] [PubMed] [Google Scholar]
- Dodds PN, Lawrence GJ, Catanzariti AM, Ayliffe MA, Ellis JG. The Melampsora lini AvrL567 avirulence genes are expressed in haustoria and their products are recognized inside plant cells. Plant Cell. 2004;16:755–768. doi: 10.1105/tpc.020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds PN, Lawrence GJ, Catanzariti AM, Teh T, Wang CIA, Ayliffe MA, Kobe B, Ellis JG. Direct protein interaction underlies gene-for-gene specificity and coevolution of the flax resistance genes and flax rust avirulence genes. Proc Natl Acad Sci USA. 2006;103:8888–8893. doi: 10.1073/pnas.0602577103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flor HH. Host–parasite interaction in flax rust - its genetics and other implications. Phytopathology. 1955;45:680–685. [Google Scholar]
- Ford MJ. Applications of selective neutrality tests to molecular ecology. Mol Ecol. 2002;11:1245–1262. doi: 10.1046/j.1365-294X.2002.01536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen TL, Stukenbrock EH, Liu ZH, Meinhardt S, Ling H, Faris JD, Rasmussen JB, Solomon PS, McDonald BA, Oliver RP. Emergence of a new disease as a result of interspecific virulence gene transfer. Nat Genet. 2006;38:953–956. doi: 10.1038/ng1839. [DOI] [PubMed] [Google Scholar]
- Gabriel DW. Why do pathogens carry avirulence genes? Physiol Mol Plant Pathol. 1999;55:205–214. [Google Scholar]
- Gandon S. Local adaptation and host–parasite interactions. Trends Ecol Evol. 1998;13:214–216. doi: 10.1016/s0169-5347(98)01358-5. [DOI] [PubMed] [Google Scholar]
- Gandon S, Capowiez Y, Dubois Y, Michalakis Y, Olivieri I. Local adaptation and gene-for-gene coevolution in a metapopulation model. Proc R Soc Lond B. 1996;263:1003–1009. [Google Scholar]
- Gillespie JH. Natural selection for resistance to epidemics. Ecology. 1975;56:493–495. [Google Scholar]
- Gonzalez E, Dhanda R, Bamshad M, et al. (15 co-authors) Global survey of genetic variation in CCR5, RANTES, and MIP-1a: impact on the epidemiology of the HIV-1 pandemic. Proc Nat Acad Sci USA. 2001;98:5199–5204. doi: 10.1073/pnas.091056898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Hedrick PW. Genetic variation in a heterogeneous environment. I. Temporal heterogeneity and the absolute dominance model. Genetics. 1974;78:757–770. doi: 10.1093/genetics/78.2.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick PW. Genetic polymorphism in heterogeneous environments: the age of genomics. Ann Rev Ecol Evol Syst. 2006;37:67–93. [Google Scholar]
- Jarosz AM, Burdon JJ. Host–pathogen interactions in natural populations of Linum marginale and Melampsora lini. II. Local and regional variation in patterns of resistance and racial structure. Evolution. 1991;45:1618–1627. doi: 10.1111/j.1558-5646.1991.tb02667.x. [DOI] [PubMed] [Google Scholar]
- Jarosz AM, Burdon JJ. Host–pathogen interactions in natural populations of Linum marginale and Melampsora lini: III. influence of pathogen epidemics on host survivorship and flower production. Oecologia. 1992;89:53–61. doi: 10.1007/BF00319015. [DOI] [PubMed] [Google Scholar]
- Jayakar SD. A mathematical model for interaction of gene frequencies in a parasite and its host. Theor Pop Biol. 1970;1:140–164. doi: 10.1016/0040-5809(70)90032-8. [DOI] [PubMed] [Google Scholar]
- Kniskern J, Rausher MD. Two modes of host–enemy coevolution. Popul Ecol. 2001;43:3–14. [Google Scholar]
- Kruijt M, Kip DJ, Joosten M, Brandwagt BF, de Wit P. The Cf-4 and Cf-9 resistance genes against Cladosporium fulvum are conserved in wild tomato species. Mol Plant–Microbe Interact. 2005;18:1011–1021. doi: 10.1094/MPMI-18-1011. [DOI] [PubMed] [Google Scholar]
- Laine AL, Tellier A. Heterogeneous selection promotes maintenance of polymorphism in host–parasite interactions. Oikos. 2008;117:1281–1288. [Google Scholar]
- Lawrence GJ. Flax rust from Linum marginale - pathogenicity reactions on the Linum usitatissimum set of differential varieties. Can J Bot. 1989;67:3187–3191. [Google Scholar]
- Lawrence GJ, Burdon JJ. Flax rust from Linum marginale—variation in a natural host pathogen interaction. Can J Bot. 1989;67:3192–3198. [Google Scholar]
- Lawrence GJ, Dodds PN, Ellis JG. Rust of flax and linseed caused by Melampsora lini. Mol Plant Path. 2007;8:349–364. doi: 10.1111/j.1364-3703.2007.00405.x. [DOI] [PubMed] [Google Scholar]
- Lawrence GJ, Mayo GME, Sheperd KW. Interactions between genes controlling pathogenicity in the flax rust fungus. Genetics. 1981;71:12–19. [Google Scholar]
- Lockett SF, Robertson JR, Brettle RP, Yap PL, Middleton D, Brown AJL. Mismatched human leukocyte antigen alleles protect against heterosexual HIV transmission. JAIDS. 2001;27:277–280. doi: 10.1097/00126334-200107010-00010. [DOI] [PubMed] [Google Scholar]
- Ma WB, Dong FFT, Stavrinides J, Guttman DS. Type III effector diversification via both pathoadaptation and horizontal transfer in response to a coevolutionary arms race. Plos Genet. 2006;2:2131–2142. doi: 10.1371/journal.pgen.0020209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantel N. The detection of disease clustering and a generalised regression approach. Cancer Res. 1967;27:209–220. [PubMed] [Google Scholar]
- Martin D, Rybicki E. RDP: detection of recombination amongst aligned sequences. Bioinformatics. 2000;16:562–563. doi: 10.1093/bioinformatics/16.6.562. [DOI] [PubMed] [Google Scholar]
- Martin DP, Williamson C, Posada D. RDP2: recombination detection and analysis from sequence alignments. Bioinformatics. 2005;21:260–262. doi: 10.1093/bioinformatics/bth490. [DOI] [PubMed] [Google Scholar]
- McDill J, Repplinger M, Simpson BB, Kadereit JW. The phylogeny of Linum and Linaceae subfamily Linoideae, with implications for their systematics, biogeography, and evolution of heterostyly. Syst Bot. 2009;34:386–405. [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987. [Google Scholar]
- Nielsen R, Yang ZH. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong LE, Innes RW. AvrB mutants lose both virulence and avirulence activities on soybean and Arabidopsis. Mol Microbiol. 2006;60:951–962. doi: 10.1111/j.1365-2958.2006.05162.x. [DOI] [PubMed] [Google Scholar]
- Padidam M, Sawyer S, Fauquet CM. Possible emergence of new geminiviruses by frequent recombination. Virology. 1999;265:218–225. doi: 10.1006/viro.1999.0056. [DOI] [PubMed] [Google Scholar]
- Paterson S, Wilson K, Pemberton JM. Major histocompatibility complex variation associated with juvenile survival and parasite resistance in a large unmanaged ungulate population (Ovis aries L.) Proc Natl Acad Sci USA. 1998;95:3714–3719. doi: 10.1073/pnas.95.7.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peakall R, Smouse PE. GenAlEx v6: genetic analysis in Excel. Canberra (Australia): Australian National University; 2005. [Google Scholar]
- Piertney SB, Oliver MK. The evolutionary ecology of the major histocompatibility complex. Heredity. 2006;96:7–21. doi: 10.1038/sj.hdy.6800724. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc Natl Acad Sci USA. 2001;98:13757–13762. doi: 10.1073/pnas.241370698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice MB, James KD, Parkhill J, et al. (15 co-authors) Yersinia pestis pFra shows biovar-specific differences and recent common ancestry with a Salmonella enterica serovar typhi plasmid. J Bacteriol. 2001;183:2586–2594. doi: 10.1128/JB.183.8.2586-2594.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM, Kann LM. Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice and humans. Mol Biol Evol. 1996;13:735–748. doi: 10.1093/oxfordjournals.molbev.a025634. [DOI] [PubMed] [Google Scholar]
- Roy BA, Kirchner JW. Evolutionary dynamics of pathogen resistance and tolerance. Evolution. 2000;54:51–63. doi: 10.1111/j.0014-3820.2000.tb00007.x. [DOI] [PubMed] [Google Scholar]
- Rozas J, Rozas R. DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics. 1999;15:174–175. doi: 10.1093/bioinformatics/15.2.174. [DOI] [PubMed] [Google Scholar]
- Salminen M, Carr JK, Burke DS, McCutchan FE. Identification of recombination breakpoints in Hiv-1 by bootscanning. Aids Res Hum Retrovir. 1995;11:S119. doi: 10.1089/aid.1995.11.1423. [DOI] [PubMed] [Google Scholar]
- Sarkar SF, Gordon JS, Martin GB, Guttman DS. Comparative genomics of host-specific virulence in Pseudomonas syringae. Genetics. 2006;174:1041–1056. doi: 10.1534/genetics.106.060996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JM. Analyzing the mosaic structure of genes. J Mol Evol. 1992;34:126–129. doi: 10.1007/BF00182389. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005;21:456–463. doi: 10.1093/bioinformatics/bti191. [DOI] [PubMed] [Google Scholar]
- Swofford DL. PAUP*, Phylogenetic analysis using parsimony (*and other methods) Sunderland (MA): Sinauer Associates; 2001. [Google Scholar]
- Tellier A, Brown JKM. Stability of genetic polymorphism in host–parasite interactions. Proc R Soc Lond B. 2007;274:809–817. doi: 10.1098/rspb.2006.0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JN. The geographic mosaic of coevolution. Chicago: The University of Chicago Press; 2005. [Google Scholar]
- Thrall PH, Burdon JJ. Effect of resistance variation in a natural plant host–pathogen metapopulation on disease dynamics. Plant Pathol. 2000;49:767–773. [Google Scholar]
- Thrall PH, Burdon JJ. Evolution of gene-for-gene systems in metapopulations: the effect of spatial scale of host and pathogen dispersal. Plant Pathol. 2002;51:169–184. [Google Scholar]
- Thrall PH, Burdon JJ. Evolution of virulence in a plant host–pathogen metapopulation. Science. 2003;299:1735–1737. doi: 10.1126/science.1080070. [DOI] [PubMed] [Google Scholar]
- Thrall PH, Burdon JJ, Bever JD. Local adaptation in the Linum marginale–Melampsora lini host–pathogen interaction. Evolution. 2002;56:1340–1351. doi: 10.1111/j.0014-3820.2002.tb01448.x. [DOI] [PubMed] [Google Scholar]
- Tiffin P, Moeller DA. Molecular evolution of plant immune system genes. Trend Genet. 2006;22:662–670. doi: 10.1016/j.tig.2006.09.011. [DOI] [PubMed] [Google Scholar]
- van der Merwe MM, Kinnear MW, Barrett LG, Dodds PN, Ericson L, Thrall PH, Burdon JJ. Positive selection in AvrP4 avirulence gene homologues across the genus Melampsora. Proc R Soc Lond B. 2009; 276:2913–2922. doi: 10.1098/rspb.2009.0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CIA, Guncar G, Forwood JK, et al. (12 co-authors) Crystal structures of flax rust avirulence proteins AvrL567-A and -D reveal details of the structural basis for flax disease resistance specificity. Plant Cell. 2007;19:2898–2912. doi: 10.1105/tpc.107.053611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TJ, Lee S, Bruns TD, Taylor JW. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR protocols: a guide to methods and applications. San Diego (CA): Academic Press; 1990. [Google Scholar]
- Wichmann G, Ritchie D, Kousik CS, Bergelson J. Reduced genetic variation occurs among genes of the highly clonal plant pathogen Xanthomonas axonopodis pv. vesicatoria, including the effector gene avrBs2. Appl Environ Microbiol. 2005;71:2418–2432. doi: 10.1128/AEM.71.5.2418-2432.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams-Blangero S, McGarvey ST, Subedi J, Wiest PM, Upadhayay RP, Rai DR, Jha B, Olds GR, Wu GL, Blangero J. Genetic component to susceptibility to Trichuris trichiura: evidence from two Asian populations. Genet Epidemiol. 2002;22:254–264. doi: 10.1002/gepi.0187. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML: a computer package for phylogenetic analysis by maximum likelihood. Com App Biosc. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang ZH, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000;17:32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
- Yang ZH, Nielsen R, Goldman N, Pedersen AMK. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZH, Wong WSW, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- Zeven AC, Zhokovsky PM. Dictionary of cultivated plants and their centres of diversity. Wageningen (Netherlands): Centre for Agricultural Publishing and Documentation; 1975. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.