

SUMMARY
The constant attack on DNA by endogenous and exogenous agents gives rise to nucleobase modifications that cause mutations, which can lead to cancer. Visualizing the effects of these lesions on the structure of duplex DNA is key to understanding their biologic consequences. The most definitive method of obtaining such structures, X-ray crystallography, is troublesome to employ owing to the difficulty of obtaining diffraction-quality crystals of DNA. Here, we present a crystallization system that uses a protein, the DNA glycosylase AlkA, as a scaffold to mediate the crystallization of lesion-containing duplex DNA. We demonstrate the use of this system to facilitate the rapid structure determination of DNA containing the lesion 8-oxoguanine in several different sequence contexts, and also deoxyinosine and 1,N6-ethenoadenine, each stabilized as the corresponding 2′-flouro analog. The structures of 8-oxoguanine provide a correct atomic-level view of this important endogenous lesion in DNA.
INTRODUCTION
The ability of the genome to support normal cellular homeostasis and to transmit genetic information is directly dependent upon how well its covalent structure is preserved. Inside of cells, DNA is under constant attack by exogenous environmental toxins and cellular metabolites, insults that can produce various covalent nucleobase modifications such as methylation, hydrolytic deamination, oxidation, and alkylation (Friedberg et al., 1995; Lindahl, 2004). Left uncorrected, these lesions can cause mutations affecting nearly all aspects of genome function, including transcription, DNA replication, and recombination, and also nongenomic processes such as cell-cycle progression and apoptosis. The mutations that result from cellular mismanagement of such lesions are the cause of cancer (Engelward et al., 1998; Jelinsky and Samson, 1999).
To counter the threat posed by DNA lesions, all cells have evolved repair mechanisms charged with the responsibility for locating damaged sites and correcting them. Among the most common of these lesions are those in which one or, in some cases, two adjacent nucleobases become damaged; depending on the particular structures of the nucleobase lesions, they are corrected by one or more of four distinct pathways, namely, base-excision (BER), nucleotide-excision (NER), direct reversion (DRR), and mismatch excision (MMR). In all four pathways, structures of lesion-containing DNA bound to cognate repair proteins have provided a treasure trove of information regarding lesion-specific recognition and catalysis of repair. High-quality structures of the lesions alone in naked DNA have been harder to come by, however. The paucity of protons upon which to base intramolecular distance measurements in DNA limits the accuracy of NMR structures. The method most suited to obtaining atomic resolution, X-ray crystallography, has proven unreliable because of the difficulty of obtaining diffraction-quality crystals of DNA. Although structures of a number of important lesions have been solved despite these limitations, some of these “solved” structures provide only a partial structural answer, and many more yet remain to be elucidated at all. The major impediment to crystallization of DNA, indeed nucleic acids in general, is believed to be the high concentration of negative charge on its surface, which provides a repulsive barrier to side-on interactions in the crystal lattice. Even when crystals are obtained, the DNA is often found to adopt the unphysiologic A form conformation, owing to the high organic content and salt concentration of the crystallization media. In the case of RNA, Doudna and coworkers demonstrated that engineering a bound protein into the crystal lattice could substantially improve crystallization, and this method has since yielded numerous high-resolution structures of otherwise naked RNA (Ferre-D’Amare and Doudna, 2000). Georgiadis and coworkers have extended this host-guest concept to duplex DNA, using a fragment of the Moloney murine leukemia virus reverse transcriptase to host DNA oligonucleotides in the presence and absence of ligands (Cote and Georgiadis, 2001; Cote et al., 2000; Goodwin et al., 2005). Here, we report an alternative host-guest system that employs a base-excision DNA repair protein. In the studies described below, the DNA repair protein 3-methyladenine DNA glycosylase (AlkA) is shown to complex with the ends of a DNA duplex, thereby enabling facile formation of a three-dimensional crystal lattice that provides large, diffraction-quality crystals. We demonstrate the generality of this method as applied to single-base lesions by solving structures of DNA containing two important nucleobase lesions, 8-oxoguanine (oxoG) and 1,N6-ethenoadenine (εA). These structures provide the first, to our knowledge, definitive structures of oxoG in DNA, here in multiple sequence contexts.
RESULTS
Crystal Structure of the AlkA:Lesion DNA Complex
AlkA is an E. coli monofunctional DNA glycosylase that catalyzes the hydrolytic deglycosylation of a series of positively charged, but otherwise structurally diverse, lesions from DNA, including N7-methylguanine, N3-methyadenine, O2-methylthymine, and O2-methylcytidine (Bjelland et al., 1993, 1994). AlkA can also cleave neutral cyclic base adducts such as εA, deaminated bases such as hypoxanthine, and certain normal bases in mismatched base pairs (O’Brien and Ellenberger, 2004; Saparbaev et al., 1995; Saparbaev and Laval, 1994; Terato et al., 2002). Crystal structures of AlkA have been determined in unliganded form (Labahn et al., 1996) and bound to DNA containing a transition-state mimic of the glycosylic-bond cleavage, 1-azaribose (Hollis et al., 2000); however, as yet, there exists no structure of AlkA bound to lesion-containing DNA. Our original goal in this study was to visualize, at atomic resolution, AlkA bound to duplex DNA containing a bona fide lesion nucleobase.
In an attempt to crystallize a complex of AlkA bound to a lesion, we screened various hypoxanthine-containing oligonucleotides complexed with AlkA for the ability to form cocrystals. The hypoxanthine-bearing nucleoside in these oligonucleotides was synthetically 2′-β-fluorinated, a substitution known in other cases to forestall base excision while preserving lesion recognition (Barrett et al., 1999; Iwai et al., 1995; Scharer et al., 1997; Scharer and Verdine, 1995). Diffraction-quality cocrystals of one such complex were obtained, and the structure was solved by molecular replacement to 2.3 Å resolution (Table 1). The asymmetric unit in our crystals was found to contain four molecules of AlkA (Figure 1A), each being “dimerized” (AlkA is a monomer under physiologic conditions) to another in precisely the same way as in the structure of the AlkA:azaribose DNA complex (compare Figures 1A and 1B) (Hollis et al., 2000). However, the DNA-binding mode of AlkA observed in the present hypoxanthine structure is completely different from that seen previously in the azaribose structure. The azaribose structure is that of a lesion-recognition complex (LRC). In this structure, each AlkA protomer is bound at the middle of a single DNA duplex, the DNA is sharply bent, and the analog is extruded from the helical stack and inserted deep into the enzyme active site (Figure 1B). In our hypoxanthine structure, each AlkA protomer is bound to an end of a DNA duplex, and each DNA end contains a bound AlkA molecule (Figure 1C), such that the asymmetric unit consists of two AlkA dimers bridged by two DNA duplexes (Figure 1A). In contrast to the distorted, protein-invaded duplex DNA in the azaribose LRC (Hollis et al., 2000), the two duplexes in the hypoxanthine complex are nearly naked throughout their central portion, they lack any significant degree of bending, and they posses fully intact base pairs. The hypoxanthine-containing structure is therefore not an LRC, but instead can be described as a host/guest complex (HGC) in which two AlkA dimers host two bound duplex DNA guests. The DNA duplexes in the asymmetric unit of the hypoxanthine HGC are held by AlkA at nearly right angles with respect to each other (Figure 1A) and are separated by a water-filled chasm; the DNA molecules in this complex are completely devoid of intermolecular contacts to neighboring DNA molecules.
Table 1.
Data Collection and Refinement Statistics
| Speciesa | FdI:T | FdI:A | FεA:T | oxoG:A | oxoG1:C | oxoG6:C | oxoG8:C |
|---|---|---|---|---|---|---|---|
| Unique reflections | 67,627 | 61,455 | 36,783 | 60,184 | 60,019 | 52,651 | 64,696 |
| Completeness (%) | 99.5 (96.0) | 96.6 (96.6) | 97.2 (92.9) | 99.2 (93.6) | 98.1 (87.4) | 97.8 (84.5) | 95.1 (89.7) |
| Resolution (Å) | 50–2.3 (2.38–2.30) | 50–2.3 (2.37–2.26) | 50–2.8 (2.90–2.80) | 50–2.4 (2.49–2.40) | 50–2.4 (2.49–2.40) | 50–2.5 (2.59–2.50) | 50–2.3 (2.38–2.30) |
| Rsym (%)b | 5.0 (35.2) | 3.9 (14.9) | 6.5 (54.3) | 5.3 (36.1) | 4.2 (17.6) | 3.4 (22.6) | 6.1 (27.9) |
| <I>/σ <I> | 21.7 (2.2) | 26.8 (5.4) | 10.6 (2.0) | 17.6 (2.0) | 29.4 (3.6) | 25.0 (2.4) | 30.5 (3.1) |
| Rwork (%)c | 22.1 | 23.9 | 21.3 | 21.8 | 21.8 | 21.9 | 21.7 |
| Rfree (%)d | 25.5 | 28.0 | 27.4 | 27.1 | 26.0 | 27.3 | 25.9 |
| Rms deviation from ideal geometry | |||||||
| Rmsd bond (Å) | 0.007 | 0.007 | 0.008 | 0.007 | 0.007 | 0.007 | 0.007 |
| Rmsd angle (°) | 1.256 | 1.290 | 1.348 | 1.243 | 1.238 | 1.283 | 1.252 |
Values in parentheses are for the highest-resolution shell. Numbers for the oxoG species represent their position in the DNA duplex.
Rsym = Σ|I – <I>|/ΣI, where I is the integrated intensity of a given reflection.
Rwork = Σ|Fo – Fc|/ΣFo.
Rfree = Σ|Fo – Fc|/ΣFo., calculated with 10.0% of the data.
Figure 1. Structure of the AlkA:DNA Host/Guest Complex.

(A) Ribbon representation of the asymmetric unit in the AlkA:DNA host/guest complex (HGC) structure, with four AlkA monomers bookending two DNA duplexes. The two AlkA monomers in green are those that interact with the DNA duplex comprising chains G and H; this half of the structure is slightly more ordered than the other half and hence was used for the analyses throughout this work.
(B) Ribbon representation of the AlkA azaribose lesion-recognition complex (Hollis et al., 2000). One of the AlkA monomers is colored cyan. Note the difference in the DNA-binding mode between (A) and (B).
(C) Ribbon representation of the two AlkA monomers interacting with DNA chains G and H, (same as in [A]). Shown in red is the signature helix-hairpin-helix DNA-binding motif common to all members of the superfamily to which AlkA belongs. The loop colored in gold contains residues 249 and 251, which also participate in interactions with the DNA. The water molecule mediating protein:DNA interactions is colored in magenta, whereas the side chain of Leu125 is shown in blue.
(D) Schematic representation of the AlkA:DNA interactions. The prime sign (′) represents residues interacting from the second AlkA protomer.
Although distinct, the DNA-binding modes in the azaribose LRC and the hypoxanthine HGC are clearly related. Superposition of the protein component of the two complexes (rmsd of 0.37 Å for all Cα atoms) (Figure 2) reveals that the entire duplex segment on the 3′ side of the lesion in the LRC binds AlkA in the same way as a DNA end in the HGC. That is, the DNA end in the hypoxanthine HGC is behaving as a lesion, mimicking the 3′ duplex segment in the LRC, but altogether lacking a 5′ flank. Indeed, in both structures, the signature helix-hairpin-helix (HhH) motif is positioned so as to contact the DNA backbone immediately 3′ to the “lesion”; this motif is common to all members of the glycosylase superfamily to which AlkA belongs (Doherty et al., 1996; Guan et al., 1998; Nash et al., 1996). The intercalating probe residue, Leu125, which inserts itself into the DNA helical stack at the site of the lesion in the LRC, similarly abuts a helical segment in the HGC, with Leu125 in each protomer capping an end of the DNA duplex (Figures 1C and 1D). The 1 nt overhang present on one end of the DNA is disordered in the crystal and has therefore been omitted from our model (Figures 1C and 1D). The contacts made by AlkA to each end of the HGC are pseudosymmetric, with the side chain of Thr219 and the main chain amides of Gly214 and Gly216 forming hydrogen bonds to the phosphate backbone of the DNA. Additionally, density that we fit best to an ordered water molecule is apparent at the DNA interface; in the AlkA LRC, this was modeled as a sodium ion. The sole pseudosymmetric contact to the center of the DNA involves a single phosphate on each strand that appears to hydrogen bond simultaneously to the Thr249 side chain and the Ala251 main chain (Figures 1C and 1D); these interactions are not observed in the LRC, because the DNA duplex in that structure is not long enough to reach Thr249 and Ala251. Superposition with the HGC provides a zeroth-order model of elongating the DNA in the LRC, and, by this analysis (Figure 2, yellow arrow), the Thr249 and Ala251 phosphate contacts should be considered part of the contact repertoire used by AlkA to recognize lesion-containing DNA. The DNA duplexes in the hypoxanthine HGC DNA structure are normal B form DNA with a slight, delocalized bend of ~15°. This is in contrast to the azaribose LRC structure, in which the DNA has a pronounced 66° bend localized sharply at the site of the lesion (Hollis et al., 2000).
Figure 2. Cα Superposition of the AlkA Azaribose LRC Structure onto an AlkA/DNA Subunit from the AlkA HGC.

The rmsd of the LRC (the protein is colored cyan as in Figure 1B; DNA colored white) onto the HGC subunit (colored as in Figure 1C) is 0.37 Å. The crimson dot denotes the position of the phosphate that hydrogen bonds with the protein backbone at positions 249 and 251 (yellow loop); this phosphate is the only element of the otherwise naked central portion of the DNA that is contacted by the protein. The major sites of protein:DNA interaction are colored as in Figure 1C.
Although the hypoxanthine HGC does not yield any insight into recognition and removal of this mutagenic lesion by AlkA, the structure does provide valuable information on the structure of the lesion itself in a nearly naked B form duplex. As mentioned above, truly high-resolution structures of lesions in naked DNA have proven difficult to obtain; therefore, we turned our attention to the use of this host/guest system to illuminate the effects of lesions on DNA structure. The two guest duplexes in the HGC are nearly identical, with an average rmsd of 1.1 Å for all atoms. However, of the two guest duplexes in the hypoxanthine HGC, one (chains G+H) has better electron density and lower B factors than the other (E+F) (Figure 1A). Therefore, the analysis of the DNA structures to follow was performed on DNA strands G+H (Figure 1, duplex bound to green protomer).
Structure of 2′-Fluoro-2′-Deoxyinosine Base Paired with Adenine and Thymine
Hypoxanthine, the base in the nucleoside 2′-deoxyinosine (dI), is a product of the deamination of adenine and leads to misincorporation errors during transcription and replication given its ability to form stable base pairs with adenine, thymine, and cytosine (Singer and Kusmierek, 1982). The hypoxanthine HGC structure described above had the lesion (2′-fluoro,2′-deoxyinosine [FdI]) paired opposite A. To compare this with the structure of the lesion opposite T, we crystallized that complex in the same manner and solved its structure (Table 1), again obtaining an HGC. A structure of DNA containing hypoxanthine paired opposite A has been determined previously (Corfield et al., 1987), offering an opportunity to validate the AlkA scaffolded system. The structure of an oligonucleotide containing hypoxanthine paired opposite T has also been determined (Cruse et al., 1989); however, the conformation of the DNA in that structure is A form, precluding meaningful comparison with our structure.
In the FdI:A HGC, the FdI adopts a syn glycosidic torsion angle. N1-H of FdI hydrogen bonds with N7 of adenine, whereas O6 of FdI hydrogen bonds with N6-H of adenine (Figure 3A). In contrast, when FdI base pairs with T, both bases possess the anti glycosidic configuration; O6 of FdI hydrogen bonds with N3-H of dT, and N1 of FdI hydrogen bonds with O2 of dT (Figure 3B). The C1′-C1′ distances are 10.6 Å and 10.5 Å for the FdI:A and FdI:T base pair structures, respectively, typical values for B form DNA. There is no apparent distortion of the global geometry of the DNA duplex upon incorporation of the FdI DNA lesion. The average rise per residue is 3.3 Å, the sugar pucker conformation for most of the bases within the structure is C2′-endo, and the intrastrand phosphate distance averages are 6.7 Å for both structures, all values that are typical of normal B form DNA. The characteristics of the FdI-containing DNA structures are consistent in every respect with those seen in the previously reported structure of DNA containing 2′-deoxyinosine (Corfield et al., 1987).
Figure 3. Analysis of FdI:A and FdI:T Base Pairs.

(A and B) Structure of FdI base paired with (A) adenine and (B) thymine. The nucleotides are represented as sticks; atoms are colored as follows: carbon, green; nitrogen, blue; oxygen, red; phosphate, orange; and fluorine, cyan. The Fo − Fc electron density map of the base pair is represented as a mesh contoured at 3σ. Black, dotted lines represent hydrogen bonds.
(C) Effect of 2′-fluorine substitution on the deoxyribose sugar pucker of the FdI base. The color scheme is as in (A).
(D) Stick representation of the FdI base and the neighboring base, T9, illustrating the microenvironment of the 2′-fluorine atom. Dotted lines indicate distances from the 2′-fluorine to the three nearest atoms. All representations are from the FdI:A structure.
One point of difference with the previously reported structure is in the sugar pucker of the lesion, which was reported as C2′-endo for dI:dA but is here observed to be O4′-endo for FdI:A and FdI:T (Figure 3C). The O4′-endo sugar conformation is well accommodated within the overall B form DNA geometry. This same alternative sugar conformation was also seen in the crystal structure of DNA containing 2′-deoxy-2′-fluoro-arabinofuranosyl thymine (Berger et al., 1998). In that structure, potential clashes between the 2′-fluorine (F2′) and the hydrogen atom of the C6 carbon on the modified thymine as well as between the F2′ and the exocyclic methyl on the adjacent T nucleotide 3′ to the modified base greatly restrict the positioning of the fluorine atom and hence the accessible sugar puckers. In the structures of FdI presented here, and of FεA presented below, similar restrictions on the positioning of the fluorine substituent apply. For example, in the FdI:A structure, the distance between the F2′ and the C8 carbon on the purine ring is 3.2 Å (Figure 3D), which places the F2′ and C8-H in van der Waals contact; the F2′ is also in van der Waals contact with the C5-methyl on the 3′-T nucleoside (F-C distance, 3.0 Å, Figure 3D). Any change in the sugar pucker, for instance to generate the C2′-endo conformation, would result in a steric clash between F2′ and both of these contact partners. Therefore, it appears that the O4′-endo sugar conformation is that which minimizes steric clashes with F2′, yielding the most stable duplex overall.
Structure of 2′-Fluoro-2′-Deoxy-1,N6-Ethenoadenine Base Paired with Thymine
Metabolic epoxidation of the carcinogen vinyl chloride leads to the formation of chloroethylene oxide and chloroacetaldehyde (Guengerich et al., 1979), which can form mutagenic adducts with amine-containing nucleobases (Barrio et al., 1972; Kusmierek and Singer, 1982; Leonard, 1984). One such adduct that has received particular attention, εA, forms a stable Hoogsteen base pair with a misinserted dG (Leonard et al., 1994). The εA lesion also pairs with T (Singer et al., 1984; Singer and Spengler, 1986), despite the apparent absence of any suitable hydrogen-bond acceptor/donor functional groups on εA to stably interact with T. An NMR study on DNA containing an εA:T base pair was unable to arrive at an unambiguous structural solution, but proposed a model in which the T could be accommodated within the DNA duplex by displacement of the T nucleotide either 5′ or 3′ of the εA ring (Kouchakdjian et al., 1991).
Here, we have used the AlkA HGC system to determine the first, to our knowledge, unambiguous structure of DNA containing a εA:T base pair, solved by X-ray crystallography to 2.8 Å resolution (Table 1). To prevent repair of the lesion by AlkA, we 2′-flourinated the lesion nucleoside (FεA:T). The pairing of the FεA with T in this structure is remarkably similar to an A:T pair; both nucleosides have an anti glycosidic torsion angle (Figure 4A) and little local or global distortion of duplex geometry. To accommodate both the bulky tricyclic ring of the FεA and the pyrimidine ring of T within the DNA duplex, the T is displaced toward the base 3′ (T19) to it (Figure 4B). The displacement of the T nucleotide results in noncoplanarity in the FεA:T base pair (Figure 4C). The bulky etheno group is also accommodated in the εA:T pair by an increase in the mean distance separating the two nucleobases, and hence their attached sugars; the C1′-C1′ distance between FεA and T is 11.4 Å, slightly greater than the 10.85 Å seen in ideal B form DNA. An additional accommodation is provided by the T adopting the O1′-endo conformation, which though nonstandard is well accommodated in the overall B form duplex. The sugar pucker for the FεA ribose ring is O4′-endo, which results from the fluorine-dependent effects described above. The properties of the base pair (T19:A7) located 5′ to the FεA lesion also appear to be modestly perturbed by the displacement of T18. Though clearly engaged in a canonical Watson-Crick pairing interaction, the angle of propeller twisting for the T19:A7 base pair is −16.6°, substantially higher than the average propeller twist angle for the rest of the DNA duplex (−9.3°) (Figure 4C). The increase in propeller twist of this base pair might promote more favorable base-stacking interactions with the T18:FεA base pair, which could help to stabilize the FεA:T pair, devoid though it is of hydrogen-bonding interactions. In summary, modest conformational changes within the local environment of the bulky FεA lesion allow for its incorporation into DNA without major distortion of the global duplex geometry, even when positioned opposite a nucleobase with which it cannot pair through conventional hydrogen bonding.
Figure 4. Analysis of the FεA:T Base Pair.
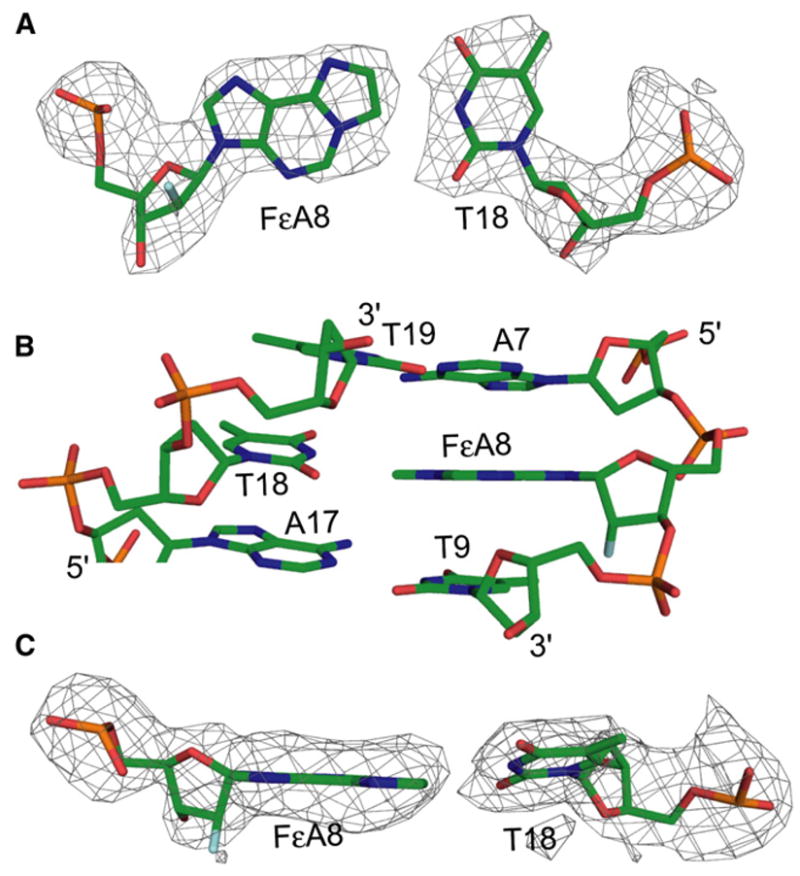
(A) Stick representation of the FεA:T base pair. The color scheme is the same as in Figure 3. The Fo − Fc electron density map of the base pair is represented as a mesh contoured at 3σ.
(B) FεA:T and the neighboring base pairs. The color scheme is as in Figure 3.
(C) The FεA:T base pair as viewed from the major groove, illustrating the propeller twist angle of T18. The color scheme and electron density map are as in (A).
Structures of 8-Deoxyguanine Base Paired with Cytosine and Adenine
Reactive oxygen species produced as the result of normal aerobic respiration covalently modify the nucleobases of DNA to produce genotoxic lesions. The most common product of oxidative damage to DNA, oxoG (Shigenaga et al., 1989), causes mutations by preferentially mispairing with dA during processive DNA replication, thereby giving rise to G → T transversion mutations (Shibutani et al., 1991). Consequently, organisms have evolved an array of repair enzymes to specifically recognize and remove oxoG from DNA. The process by which these enzymes search, recognize, and excise oxoG from DNA has been studied quite extensively by using a combination of biochemical and structural techniques (Banerjee et al., 2005, 2006; Banerjee and Verdine, 2006; Bruner et al., 2000; Coste et al., 2004; Fromme et al., 2003, 2004; Fromme and Verdine, 2002, 2003; Gilboa et al., 2002; Guan et al., 1998; Lingaraju et al., 2005; Massiah et al., 2003; Norman et al., 2003; Radom et al., 2007; Serre et al., 2002; Sugahara et al., 2000; Zharkov et al., 2000, 2002). Despite this rich body of data on recognition of oxoG while in the grasp of repair proteins, the structure of the lesion in naked DNA has not been determined unambiguously. A structure of naked B form DNA containing an oxoG:dC pair has been reported (Lipscomb et al., 1995) (PDB ID: 183D), and this has provided useful insights into base pairing by the lesion and its overall effect on helix geometry. However, errors in parameterization of the oxoG moiety have compromised the ability to interpret this structure in fine detail; specifically, the geometry around C8 is tetrahedral in the deposited structure, when in actuality, the geometry is trigonal planar, the geometry around C1′ is trigonal planar when it should be tetrahedral, and the purine ring of oxoG is buckled when it should be planar. These artifacts render the structure incapable of shedding light on the effect of the 8-oxo substituent on the pucker of its own sugar, a factor that appears to play a role in lesion recognition by the oxoG DNA glycosylase MutM (A. Banerjee and G.L.V., unpublished data). As the original reflection data for oxoG:C in naked DNA are no longer available, we decided to employ the AlkA HGC system to redetermine the structure, this time in multiple sequence contexts.
We determined three structures of oxoG:C located at base pairs 1, 6, and 8 in the DNA (Figure 1D; Table 1). The three crystal structures of oxoG opposite cytosine reveal that they interact in normal Watson-Crick fashion with both oxoG and C, and that they have prototypical anti glycosidic rotamers; the glycosidic torsion angles are −113.65°, −88.9°, and −103.47° for oxoG at the 1, 6, and 8 positions, respectively, as compared with an average between −84° and −144° for purines in ideal B form DNA. The hydrogen-bonding pattern observed for the oxoG:C base pairs in our structures is the same as for a normal G:C base pair (Figures 5A–5C). The hydrogen-bonding distances of the oxoG:C pair are similar in the three structures (Figures 5A–5C), and, indeed, the pairs superimpose with an rmsd of 0.191 Å. Interestingly, some slippage of the lesion base pair is observed when it resides at the 6 position, but not at the 1 or 8 positions (compare Figure 5B with Figures 5A and 5C). We note that one of the comparator G:C base pairs is also slipped somewhat (Figure 5A, right), but this pair resides at a different position in the duplex from the slipped oxoG:C; such slippage may therefore be within the normal range of variation for G:C and oxoG:C pairs. The present structures reveal unambiguously the sugar pucker of the oxoG nucleoside to be C2′-endo, the standard sugar pucker for nucleosides in B form DNA. The O8-C2′ distance for the three oxoG:C structures is ~3.0 Å (Figures 5A–5C), slightly greater than the van der Waals contact distance (~2.85 Å). This distance is long enough to avoid a steric clash between C2′ and O8, but it is short enough that a glycosidic bond rotation that shortens the distance could force a change in sugar pucker. The C1′-C1′ distances for the oxoG:C base pairs are 10.6 Å, as compared with 10.85 Å for ideal B form DNA, and no obvious distortion of the helix is observed.
Figure 5. B-Form DNA Containing oxoG:dA or oxoG:dC Base Pairs.

(A–C) The oxoG:C base pairs (left column) with oxoG in the (A) one position, (B) six position, and (C) eight position within the DNA duplex. In the right column, G:C structures in the same positions within the DNA as the respective lesion are shown.
(D) Structure of oxoG base paired with adenine. The color scheme, Fo − Fc electron density map and hydrogen bonding are represented as in Figure 3.
A correctly parameterized structure of DNA containing oxoG:A has been previously reported (McAuley-Hecht et al., 1994). To further validate the AlkA HGC system, we determined the structure of a duplex containing oxoG:A at base pair position 8 (Figure 5D). Our structure is fully consistent with the previously determined one, with oxoG adopting a syn glycosidic conformation so as to Hoogsteen pair with an anti A. The overall characteristics of the base pair, including its hydrogen-bonding distances (3.1 Å and 2.9 Å, Figure 5D) and its C1′-C1′ distance of 10.6 Å, closely match the reported literature values. No obvious perturbation of helical geometry is evident.
DISCUSSION
The present work began with an attempt to obtain crystals of an AlkA LRC. Instead, we obtained crystals of a complex in which the enzyme was not bound to the middle region of the duplex containing the lesion, but was bound at the ends. Such end-bound complexes have been observed for other DNA glycosylases (Barrett et al., 1998; Wibley et al., 2003). Interestingly, the DNA in these end-bound structures mimics the stretch of DNA in LRCs flanking the lesion on one side.
Here, we have used the fact that AlkA in our end-bound structure hosts an almost naked stretch of duplex DNA to solve several structures of important lesions. Several of these structures, notably those of the FdI:A and oxoG:A lesions, obtained by the AlkA HGC system are similar in all important respects to those obtained previously.
We also report the structures of a number of lesions for which previous structures were inconclusive, namely, FdI:T, FεA:T, and oxoG:C, in three different sequence contexts. The most important finding is that the sugar pucker in the oxoG:C base pair is the canonical C2′-endo, but that the C2′ and O8 are close enough to each other that even a modest rotation about the glycosidic bond could force a change in sugar pucker. Indeed, structures of MutM bound to non-lesion-containing DNA have revealed that the protein buckles the target base pair by precisely such a rotation about the glycosidic bond. These observations raise the formal possibility that MutM distinguishes an intrahelical G from oxoG by an 8-oxo-dependent change in sugar pucker induced by buckling of the target base pair.
We note the presence of a minor, delocalized 15° bend in all of the DNA duplexes that were structurally elucidated in this study. Given the uniformity in the nature of this bend in duplexes bearing different lesions, it is apparently not lesion specific, but instead is either an intrinsic property of the duplex sequence used here, or arises as the result of crystal packing forces. That being the case, we do not recommend the use of this system to characterize minor global deviations on DNA structure caused by the presence of lesions. On the other hand, lesions that dramatically distort the global DNA structure may not be capable of attaining the packing arrangement found in the HGC structures reported here. In such cases, it may be necessary to screen additional DNA constructs to obtain crystals that provide high-quality diffraction data.
EXPERIMENTAL PROCEDURES
AlkA Expression, Purification, and Crystallization
The gene encoding full-length E. coli AlkA (residues 1–282) was amplified by PCR and cloned into the expression vector pET30a (Novagen). The protein was expressed in E. coli Rosetta(DE3) cells at 16°C for 16 hr after induction with 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG). The cells were lysed by using sonication, and the protein was purified to near homogeneity by using a nickel NTA column. Proteolysis to remove the 6XHis tag was performed at 4°C for 48 hr with 1.5 U/ml thrombin (Novagen). To remove minor impurities, AlkA was further purified on a MonoS (Pharmacia) ion-exchange column, and selected fractions were pooled and dialyzed into a buffer containing 10 mM Tris-HCl (pH 7.4), 100 mM NaCl, and 4 mM DTT.
Oligonucleotides were synthesized on an Expedite Nucleic Acid Synthesis System and were purified by PAGE. The 2′-fluorinated phosphoramidites were synthesized by known procedures (Scharer and Verdine, 1995), whereas the oxoG phosphoramidite was purchased from Glen Research. The sequence of the lesion-containing DNA strand is 5′-GACATGAYTGCC-3′, where Y is the location of the different lesion bases. The sequence of the complementary strand is 5′-GGCAZTCATGTCA-3′, where Z is the base opposite the lesion. The strands were annealed in TE buffer (10 mM Tris-HCl [pH 7.4], 1 mM EDTA) plus 20 mM NaCl and were mixed with purified AlkA protein to yield a protein:DNA complex of 8 mg/ml protein.
Initial crystals were grown with AlkA:FdI-containing DNA at 25°C by using the hanging-drop vapor-diffusion method, with the drop consisting of a 1:1 ratio of the stock protein:DNA solution and a reservoir solution of 17% PEG 4000, 100 mM Na HEPES (pH 8.1), 100 mM NaCl, 50 mM MgCl2, and 8% ethylene glycol. Crystals of the other AlkA:lesion DNA complexes were grown by a combination of cross- and macro-seeding techniques into drops containing a 1:1 ratio of protein:DNA stock solution and a reservoir solution consisting of 15% PEG 4000, 100 mM Na HEPES (pH 7.9–8.1), 100 mM NaCl, 50 mM MgCl, and 8% ethylene glycol. Prior to data collection, the crystals were placed in a solution containing 18% PEG 4000, 100 mM Na HEPES (pH 8.0), 100 mM NaCl, 50 mM MgCl2, and 12% ethylene glycol for 3 hr, placed in the same solution with 20% ethylene glycol for 1 hr, and flash frozen in liquid nitrogen.
Structure Determination
Data for all of the AlkA:lesion DNA complexes were collected on the SBC-CAT beamline 19ID and the NE-CAT beamline 24-ID at the Advanced Photon Source (APS), Argonne National Laboratory. Data were processed with HKL2000 and were merged with SCALEPACK (Otwinowski and Minor, 1997). The crystals have four protein molecules and two DNA molecules in the asymmetric unit and belong to the monoclinic space group P21 with unit cell dimensions, on average, of a = 93.5 Å, b = 100.9 Å, c = 103.0 Å, and α = γ = 90° β = 93.8°. Molecular replacement, by using the unliganded AlkA monomer (Hollis et al., 2000) (PDB ID: 1DIZ) as a search model, was performed on crystals containing the AlkA:FdI DNA complex by using PHASER (Read, 2001) as part of the CCP4 program suite (CCP4, 1994). The best solution after rigid-body refinement with CNS (Brunger et al., 1998) had an R factor of 36.12%. 2Fo – Fc and Fo – Fc maps showed clear density for the DNA. The DNA was fitted to the density by using the program COOT (Emsley and Cowtan, 2004), and the model was refined in CNS interspersed with model building and fitting of water molecules. For crystals containing the other DNA lesion species, the above-described model without the lesion base pair was used as an initial model. After rigid-body refinement in CNS, Fo – Fc maps revealed clear density for the differing DNA lesion base pairs. These lesions were fitted to the electron density maps by using COOT and were refined in CNS. Crystallographic statistics for all of the lesion DNA crystal structures are presented in Table 1.
ACCESSION NUMBERS
Coordinates and structure factors have been deposited under PDB accession codes 3CVS, 3CVT, 3CW7, 3CWA, 3CWS, 3CWT, and 3CWU.
Acknowledgments
We thank the staff at the SBC-CAT and NE-CAT at the Advance Photon Source for assistance during data collection, and Hariharan Jayaram, Danaya Pakotiprapha, Marie Spong, Charisse Crenshaw, and members of the G.L.V. laboratory for useful discussions. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. This work was supported by a grant from the National Institutes of Health (CA100742 to G.L.V). B.R.B. is the recipient of a Ruth Kirschstein National Research Service Award fellowship (F32 GM077935-01). S.W. is the recipient of an undergraduate research fellowship from the Microbial Sciences Institute. Figures were computed by using PyMOL (DeLano Scientific LLC, 2008) as a renderer. All representations of the DNA and DNA lesion base pairs were made by using chains G and H of the PDB file.
References
- Banerjee A, Verdine GL. A nucleobase lesion remodels the interaction of its normal neighbor in a DNA glycosylase complex. Proc Natl Acad Sci USA. 2006;103:15020–15025. doi: 10.1073/pnas.0603644103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Yang W, Karplus M, Verdine GL. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature. 2005;434:612–618. doi: 10.1038/nature03458. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Santos WL, Verdine GL. Structure of a DNA glycosylase searching for lesions. Science. 2006;311:1153–1157. doi: 10.1126/science.1120288. [DOI] [PubMed] [Google Scholar]
- Barrett TE, Savva R, Barlow T, Brown T, Jiricny J, Pearl LH. Structure of a DNA base-excision product resembling a cisplatin inter-strand adduct. Nat Struct Biol. 1998;5:697–701. doi: 10.1038/1394. [DOI] [PubMed] [Google Scholar]
- Barrett TE, Scharer OD, Savva R, Brown T, Jiricny J, Verdine GL, Pearl LH. Crystal structure of a thwarted mismatch glycosylase DNA repair complex. EMBO J. 1999;18:6599–6609. doi: 10.1093/emboj/18.23.6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrio JR, Secrist JA, 3rd, Leonard NJ. Fluorescent adenosine and cytidine derivatives. Biochem Biophys Res Commun. 1972;46:597–604. doi: 10.1016/s0006-291x(72)80181-5. [DOI] [PubMed] [Google Scholar]
- Berger I, Tereshko V, Ikeda H, Marquez VE, Egli M. Crystal structures of B-DNA with incorporated 2′-deoxy-2′-fluoro-arabino-furanosyl thymines: implications of conformational preorganization for duplex stability. Nucleic Acids Res. 1998;26:2473–2480. doi: 10.1093/nar/26.10.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelland S, Bjoras M, Seeberg E. Excision of 3-methylguanine from alkylated DNA by 3-methyladenine DNA glycosylase I of Escherichia coli. Nucleic Acids Res. 1993;21:2045–2049. doi: 10.1093/nar/21.9.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelland S, Birkeland NK, Benneche T, Volden G, Seeberg E. DNA glycosylase activities for thymine residues oxidized in the methyl group are functions of the AlkA enzyme in Escherichia coli. J Biol Chem. 1994;269:30489–30495. [PubMed] [Google Scholar]
- Bruner SD, Norman DP, Verdine GL. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project Number 4) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Corfield PW, Hunter WN, Brown T, Robinson P, Kennard O. Inosine. adenine base pairs in a B-DNA duplex Nucleic Acids Res. 1987;15:7935–7949. doi: 10.1093/nar/15.19.7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste F, Ober M, Carell T, Boiteux S, Zelwer C, Castaing B. Structural basis for the recognition of the FapydG lesion (2,6-diamino-4-hydroxy-5-formamidopyrimidine) by formamidopyrimidine-DNA glycosylase. J Biol Chem. 2004;279:44074–44083. doi: 10.1074/jbc.M405928200. [DOI] [PubMed] [Google Scholar]
- Cote ML, Georgiadis MM. Structure of a pseudo-16-mer DNA with stacked guanines and two G-A mispairs complexed with the N-terminal fragment of Moloney murine leukemia virus reverse transcriptase. Acta Crystallogr D Biol Crystallogr. 2001;57:1238–1250. doi: 10.1107/s090744490100943x. [DOI] [PubMed] [Google Scholar]
- Cote ML, Yohannan SJ, Georgiadis MM. Use of an N-terminal fragment from moloney murine leukemia virus reverse transcriptase to facilitate crystallization and analysis of a pseudo-16-mer DNA molecule containing G-A mispairs. Acta Crystallogr D Biol Crystallogr. 2000;56:1120–1131. doi: 10.1107/s0907444900008246. [DOI] [PubMed] [Google Scholar]
- Cruse WB, Aymani J, Kennard O, Brown T, Jack AG, Leonard GA. Refined crystal structure of an octanucleotide duplex with I.T. mismatched base pairs. Nucleic Acids Res. 1989;17:55–72. doi: 10.1093/nar/17.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano Scientific LLC. 2008 PyMOL ( http://www.pymol.org)
- Doherty AJ, Serpell LC, Ponting CP. The helix-hairpin-helix DNA-binding motif: a structural basis for non-sequence-specific recognition of DNA. Nucleic Acids Res. 1996;24:2488–2497. doi: 10.1093/nar/24.13.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Engelward BP, Allan JM, Dreslin AJ, Kelly JD, Wu MM, Gold B, Samson LD. A chemical and genetic approach together define the biological consequences of 3-methyladenine lesions in the Mamm. Genome J Biol Chem. 1998;273:5412–5418. doi: 10.1074/jbc.273.9.5412. [DOI] [PubMed] [Google Scholar]
- Ferre-D’Amare AR, Doudna JA. Crystallization and structure determination of a hepatitis delta virus ribozyme: use of the RNA-binding protein U1A as a crystallization module. J Mol Biol. 2000;295:541–556. doi: 10.1006/jmbi.1999.3398. [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W. DNA Repair and Mutagenesis. Washington, D.C.: American Society for Microbiology; 1995. [Google Scholar]
- Fromme JC, Verdine GL. Structural insights into lesion recognition and repair by the bacterial 8-oxoguanine DNA glycosylase MutM. Nat Struct Biol. 2002;9:544–552. doi: 10.1038/nsb809. [DOI] [PubMed] [Google Scholar]
- Fromme JC, Verdine GL. DNA lesion recognition by the bacterial repair enzyme MutM. J Biol Chem. 2003;278:51543–51548. doi: 10.1074/jbc.M307768200. [DOI] [PubMed] [Google Scholar]
- Fromme JC, Bruner SD, Yang W, Karplus M, Verdine GL. Product-assisted catalysis in base-excision DNA repair. Nat Struct Biol. 2003;10:204–211. doi: 10.1038/nsb902. [DOI] [PubMed] [Google Scholar]
- Fromme JC, Banerjee A, Huang SJ, Verdine GL. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature. 2004;427:652–656. doi: 10.1038/nature02306. [DOI] [PubMed] [Google Scholar]
- Gilboa R, Zharkov DO, Golan G, Fernandes AS, Gerchman SE, Matz E, Kycia JH, Grollman AP, Shoham G. Structure of formamidopyrimidine-DNA glycosylase covalently complexed to DNA. J Biol Chem. 2002;277:19811–19816. doi: 10.1074/jbc.M202058200. [DOI] [PubMed] [Google Scholar]
- Goodwin KD, Long EC, Georgiadis MM. A host-guest approach for determining drug-DNA interactions: an example using netropsin. Nucleic Acids Res. 2005;33:4106–4116. doi: 10.1093/nar/gki717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH, Lloyd S, Tainer JA. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat Struct Biol. 1998;5:1058–1064. doi: 10.1038/4168. [DOI] [PubMed] [Google Scholar]
- Guengerich FP, Crawford WM, Jr, Watanabe PG. Activation of vinyl chloride to covalently bound metabolites: roles of 2-chloroethylene oxide and 2-chloroacetaldehyde. Biochemistry. 1979;18:5177–5182. doi: 10.1021/bi00590a023. [DOI] [PubMed] [Google Scholar]
- Hollis T, Ichikawa Y, Ellenberger T. DNA bending and a flip-out mechanism for base excision by the helix-hairpin-helix DNA glycosylase, Escherichia coli AlkA. EMBO J. 2000;19:758–766. doi: 10.1093/emboj/19.4.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai S, Maeda M, Shirai M, Shimada Y, Osafune T, Murata T, Ohtsuka E. Reaction mechanism of T4 endonuclease V determined by analysis using modified oligonucleotide duplexes. Biochemistry. 1995;34:4601–4609. doi: 10.1021/bi00014a013. [DOI] [PubMed] [Google Scholar]
- Jelinsky SA, Samson LD. Global response of Saccharomyces cerevisiae to an alkylating agent. Proc Natl Acad Sci USA. 1999;96:1486–1491. doi: 10.1073/pnas.96.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouchakdjian M, Eisenberg M, Yarema K, Basu A, Essigmann J, Patel DJ. NMR studies of the exocyclic 1,N6-ethenodeoxyadenosine adduct (ε dA) opposite thymidine in a DNA duplex. Nonplanar alignment of epsilon dA(anti) and dT(anti) at the lesion site. Biochemistry. 1991;30:1820–1828. doi: 10.1021/bi00221a014. [DOI] [PubMed] [Google Scholar]
- Kusmierek JT, Singer B. Chloroacetaldehyde-treated ribo- and deoxyribopolynucleotides. 1 Reaction products. Biochemistry. 1982;21:5717–5722. doi: 10.1021/bi00265a050. [DOI] [PubMed] [Google Scholar]
- Labahn J, Scharer OD, Long A, Ezaz-Nikpay K, Verdine GL, Ellenberger TE. Structural basis for the excision repair of alkylation-damaged DNA. Cell. 1996;86:321–329. doi: 10.1016/s0092-8674(00)80103-8. [DOI] [PubMed] [Google Scholar]
- Leonard GA, McAuley-Hecht KE, Gibson NJ, Brown T, Watson WP, Hunter WN. Guanine-1,N6-ethenoadenine base pairs in the crystal structure of d(CGCGAATT(ε dA)GCG) Biochemistry. 1994;33:4755–4761. doi: 10.1021/bi00182a002. [DOI] [PubMed] [Google Scholar]
- Leonard NJ. Etheno-substituted nucleotides and coenzymes: fluorescence and biological activity. CRC Crit Rev Biochem. 1984;15:125–199. doi: 10.3109/10409238409102299. [DOI] [PubMed] [Google Scholar]
- Lindahl T. Molecular biology: ensuring error-free DNA repair. Nature. 2004;427:598. doi: 10.1038/427598a. [DOI] [PubMed] [Google Scholar]
- Lingaraju GM, Sartori AA, Kostrewa D, Prota AE, Jiricny J, Winkler FK. A DNA glycosylase from Pyrobaculum aerophilum with an 8-oxoguanine binding mode and a noncanonical helix-hairpin-helix structure. Structure. 2005;13:87–98. doi: 10.1016/j.str.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Lipscomb LA, Peek ME, Morningstar ML, Verghis SM, Miller EM, Rich A, Essigmann JM, Williams LD. X-ray structure of a DNA decamer containing 7,8-dihydro-8-oxoguanine. Proc Natl Acad Sci USA. 1995;92:719–723. doi: 10.1073/pnas.92.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massiah MA, Saraswat V, Azurmendi HF, Mildvan AS. Solution structure and NH exchange studies of the MutT pyrophosphohydrolase complexed with Mg(2+) and 8-oxo-dGMP, a tightly bound product. Biochemistry. 2003;42:10140–10154. doi: 10.1021/bi030105p. [DOI] [PubMed] [Google Scholar]
- McAuley-Hecht KE, Leonard GA, Gibson NJ, Thomson JB, Watson WP, Hunter WN, Brown T. Crystal structure of a DNA duplex containing 8-hydroxydeoxyguanine-adenine base pairs. Biochemistry. 1994;33:10266–10270. doi: 10.1021/bi00200a006. [DOI] [PubMed] [Google Scholar]
- Nash HM, Bruner SD, Scharer OD, Kawate T, Addona TA, Spooner E, Lane WS, Verdine GL. Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr Biol. 1996;6:968–980. doi: 10.1016/s0960-9822(02)00641-3. [DOI] [PubMed] [Google Scholar]
- Norman DP, Chung SJ, Verdine GL. Structural and biochemical exploration of a critical amino acid in human 8-oxoguanine glycosylase. Biochemistry. 2003;42:1564–1572. doi: 10.1021/bi026823d. [DOI] [PubMed] [Google Scholar]
- O’Brien PJ, Ellenberger T. The Escherichia coli 3-methyladenine DNA glycosylase AlkA has a remarkably versatile active site. J Biol Chem. 2004;279:26876–26884. doi: 10.1074/jbc.M403860200. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromol Crystallgr A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Radom CT, Banerjee A, Verdine GL. Structural characterization of human 8-oxoguanine DNA glycosylase variants bearing active site mutations. J Biol Chem. 2007;282:9182–9194. doi: 10.1074/jbc.M608989200. [DOI] [PubMed] [Google Scholar]
- Read RJ. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D Biol Crystallogr. 2001;57:1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- Saparbaev M, Laval J. Excision of hypoxanthine from DNA containing dIMP residues by the Escherichia coli, yeast, rat, and human alkylpurine DNA glycosylases. Proc Natl Acad Sci USA. 1994;91:5873–5877. doi: 10.1073/pnas.91.13.5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saparbaev M, Kleibl K, Laval J. Escherichia coli, Saccharomyces cerevisiae, rat and human 3-methyladenine DNA glycosylases repair 1,N6-ethenoadenine when present in DNA. Nucleic Acids Res. 1995;23:3750–3755. doi: 10.1093/nar/23.18.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharer OD, Verdine GL. A designed inhibitor of base-excision DNA repair. J Am Chem Soc. 1995;117:10781–10782. [Google Scholar]
- Scharer OD, Kawate T, Gallinari P, Jiricny J, Verdine GL. Investigation of the mechanisms of DNA binding of the human G/T glycosylase using designed inhibitors. Proc Natl Acad Sci USA. 1997;94:4878–4883. doi: 10.1073/pnas.94.10.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre L, Pereira de Jesus K, Boiteux S, Zelwer C, Castaing B. Crystal structure of the Lactococcus lactis formamidopyrimidine-DNA glycosylase bound to an abasic site analogue-containing DNA. EMBO J. 2002;21:2854–2865. doi: 10.1093/emboj/cdf304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- Shigenaga MK, Gimeno CJ, Ames BN. Urinary 8-hydroxy-2′-deoxyguanosine as a biological marker of in vivo oxidative DNA damage. Proc Natl Acad Sci USA. 1989;86:9697–9701. doi: 10.1073/pnas.86.24.9697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer B, Kusmierek JT. Chemical mutagenesis. Annu Rev Biochem. 1982;51:655–693. doi: 10.1146/annurev.bi.51.070182.003255. [DOI] [PubMed] [Google Scholar]
- Singer B, Spengler SJ. Replication and transcription of polynucleotides containing ethenocytosine, ethenoadenine and their hydrated intermediates. IARC Sci Publ. 1986:359–371. [PubMed] [Google Scholar]
- Singer B, Abbott LG, Spengler SJ. Assessment of mutagenic efficiency of two carcinogen-modified nucleosides, 1,N6-ethenodeoxyadenosine and O4-methyldeoxythymidine, using polymerases of varying fidelity. Carcinogenesis. 1984;5:1165–1171. doi: 10.1093/carcin/5.9.1165. [DOI] [PubMed] [Google Scholar]
- Sugahara M, Mikawa T, Kumasaka T, Yamamoto M, Kato R, Fukuyama K, Inoue Y, Kuramitsu S. Crystal structure of a repair enzyme of oxidatively damaged DNA, MutM (Fpg), from an extreme thermophile, Thermus thermophilus HB8. EMBO J. 2000;19:3857–3869. doi: 10.1093/emboj/19.15.3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terato H, Masaoka A, Asagoshi K, Honsho A, Ohyama Y, Suzuki T, Yamada M, Makino K, Yamamoto K, Ide H. Novel repair activities of AlkA (3-methyladenine DNA glycosylase II) and endonuclease VIII for xanthine and oxanine, guanine lesions induced by nitric oxide and nitrous acid. Nucleic Acids Res. 2002;30:4975–4984. doi: 10.1093/nar/gkf630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wibley JE, Waters TR, Haushalter K, Verdine GL, Pearl LH. Structure and specificity of the vertebrate anti-mutator uracil-DNA glycosylase SMUG1. Mol Cell. 2003;11:1647–1659. doi: 10.1016/s1097-2765(03)00235-1. [DOI] [PubMed] [Google Scholar]
- Zharkov DO, Gilboa R, Yagil I, Kycia JH, Gerchman SE, Shoham G, Grollman AP. Role for lysine 142 in the excision of adenine from A:G mispairs by MutY DNA glycosylase of Escherichia coli. Biochemistry. 2000;39:14768–14778. doi: 10.1021/bi001538k. [DOI] [PubMed] [Google Scholar]
- Zharkov DO, Golan G, Gilboa R, Fernandes AS, Gerchman SE, Kycia JH, Rieger RA, Grollman AP, Shoham G. Structural analysis of an Escherichia coli endonuclease VIII covalent reaction intermediate. EMBO J. 2002;21:789–800. doi: 10.1093/emboj/21.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]