

Abstract
Lipotoxicity involves a series of pathological cellular responses after exposure to elevated levels of fatty acids. This process may be detrimental to normal cellular homeostasis and cell viability. The present study shows that nerve growth factor-differentiated PC12 cells (NGFDPC12) and rat cortical cells (RCC) exposed to high levels of palmitic acid (PA) exhibit significant lipotoxicity and death linked to an “augmented state of cellular oxidative stress” (ASCOS). The ASCOS response includes generation of reactive oxygen species (ROS), alterations in the mitochondrial transmembrane potential, and increase in the mRNA levels of key cell death/survival regulatory genes. The observed cell death was apoptotic based on nuclear morphology, caspase-3 activation, and cleavage of lamin B and PARP. Quantitative real-time PCR measurements showed that cells undergoing lipotoxicity exhibited an increase in the expression of the mRNAs encoding the cell death-associated proteins BNIP3 and FAS receptor. Cotreatment of NGFDPC12 and RCC cells undergoing lipotoxicity with docosahexaenoic acid (DHA) and bovine serum albumin (BSA) significantly reduced cell death within the first 2 hr following the initial exposure to PA. The data suggest that lipotoxicity in NGFDPC12 and cortical neurons triggers a strong cell death apoptotic response. Results with NGFDPC12 cells suggest a linkage between induction of ASCOS and the apoptotic process and exhibit a temporal window that is sensitive to DHA and BSA interventions.
Keywords: ASCOS, DHA, hypoxia/ischemia, lipotoxicity, neurotoxicity, traumatic brain injury
Lipotoxicity has been defined in terms of characteristic pathological changes in cells or organs exposed to elevated levels of fat in blood or tissues (Unger, 2003). This process is present in numerous pathological cellular processes and has been well described for the pancreas, kidney, and muscle (Lee et al., 1994; Unger and Orci, 2000). Lipotoxicity expresses common and distinct features particular for each cell type affected and can initiate events that trigger cell death in vitro or in vivo (Ulloth et al., 2003; Wu et al., 2003). This process has not been well studied in nerve cells, and there are limited studies examining its role in the pathology of trauma or neurodegeneration. This is of particular significance considering that elevated levels of free fatty acids (FFA) have been reported following traumatic brain injury (TBI) or hypoxic-ischemic insults in the central nervous system (CNS) and peripheral nervous system (PNS) and in the potential effects of chronic obesity on normal neuronal function (Dhillon et al., 1997; Rodriguez de Turco et al., 2002).
After hypoxic injury, FFA accumulate acutely, presumably as a result of membrane lipid degradation and activation of phospholipases (Bazan, 1970). Locally, there is a significant accumulation of FFA during ischemic injury to the heart and traumatic and hypoxic/ischemic injury to the CNS (Homayoun et al., 1997). Elevation of FFA species in these pathological conditions consists mainly of palmitic acid (PA; C16:0), stearic acid (SA; C18:0), oleic acid (OA; C18:1), and smaller amounts of arachidonic acid (AA; C20:4) and docosahexaenoic acid (DHA; C22:6; Rehncrona et al., 1982). For instance, after ischemia injury, the absolute concentration of these FFA increases in the following sequence: C16:0 > C18:0 > C18:1 > C20:4 > C22:6, all of them exhibiting FFA:BSA ratios in excess of 2:1 (Sun and Gilboe, 1994). The initial acute phase of FFA accumulation is followed by a second, longer lasting phase that occurs after the injury during postischemia reperfusion. This phase can last from days to weeks because of the sustained and time-dependent activation of phospholipase-mediated signaling pathways (Abe et al., 1987; Homayoun et al., 1997). However, determining the consequences of this local FFA accumulation after CNS injury and its potential connection with the generation of an “augmented state of cellular oxidative stress” (ASCOS) requires further research.
Understanding the effects of lipotoxicity on nerve cells requires uncovering cellular pathways activated by this process and determining their impact on cell viability. The present study investigates the link between PA-induced lipotoxicity and ASCOS. A previous study from our laboratory showed that exposing NGFDPC12 cells to high levels of SA or PA (SA:BSA or PA:BSA, 2:1 molar ratio), but not OA or AA, led to apoptosis and caspase-independent cell death (Ulloth et al., 2003). We report here that NGFDPC12 cells and rat cortical cells (RCC) exposed to PA exhibit lipotoxicity and death associated with a robust ASCOS response. Furthermore, our results show that potential therapeutic intervention to reverse this process has a distinct temporal window of opportunity.
Materials and Methods
Cell Culture
Undifferentiated PC12 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% horse serum, 5% fetal bovine serum (FBS), 2 mM L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin (Mediatech, Herndon, VA) at 37°C with 95% air/5% CO2 (Ulloth et al., 2003). Culture medium was replaced every 2–3 days. PC12 cells were differentiated by exposure to 50 ng/ml of 2.5S (grade II) nerve growth factor (NGF; Alomone Laboratories, Jerusalem, Israel) for 7–14 days in DMEM supplemented with 1% FBS, penicillin/streptomycin, and L-glutamine (low-serum medium). Twenty four to thirty-six hours prior to treatment with fatty acids, NGFDPC12 cells were replated at a density of 12,000 cells/cm2. E18 primary neuroPURE RCC were purchased from Genlantis (San Diego, CA) and were 99.9% glia free at the time of arrival but can exhibit higher levels of astrocytes after culture for several days. Cells were cultured according to company's instructions, using a serum-free medium consisting of Neurobasal with 1× B-27 supplement (Invitrogen, Carlsbad, CA) and 0.5 mM glutamine. Cells were cultured for 7–10 days before treatment with fatty acids.
Preparation of Fatty Acids
NGFDPC12 cells and RCC were exposed to PA (PA:BSA, 2:1 molar ratio) following procedures described previously by us (Ulloth et al., 2003) and by others (Lee et al., 1994; Yu-Poth et al., 2004, 2005). PA and/or DHA (Sigma, St. Louis, MO) was first dissolved/diluted in 100% ethanol and further diluted in warm low-serum medium for NGFDPC12 cells, or serum-free medium for RCC, with fatty acid-free BSA (EMD Biosciences, La Jolla, CA). The final concentration of ethanol was 0.1%. Prior to treatment, the PA/BSA-containing medium was supplemented with NGF (only for NGFDPC12 cells) and sterilized using a 0.22-μm filter. The concentration of unbound fatty acids was measured using the acrylodated intestinal fatty acid binding protein (ADIFAB) method (Molecular Probes, Eugene, OR), following the instructions of the manufacturer. In short, the concentration of unbound FFA was determined using the ratio of fluorescence intensities of bound and unbound ADIFAB indicator at 505 and 432 nm. With this method, the concentration of unbound free fatty acid in the media was about 11 nM at the PA:BSA 2:1 molar ratio (0.3 mM:0.150 mM).
Western Blot Analysis
Goat polyclonal antibodies to lamin B and PARP1 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Total protein extracts from NGFDPC12 cells (20 μg) were resolved on an SDS-PAGE and transferred to nitrocellulose membranes. After blocking with 7.5% milk in Tris-buffered saline with 0.05% Tween 20, pH 7.4 (TTBS), for 1 hr, the membranes were incubated with lamin B antibody (1:250) or PARP1 antibody (1:500) in 5% milk-TTBS at 4°C overnight. Subsequently, the membranes were washed three times with TTBS and incubated with HRP-goat anti rabbit IgG (1:3,000; GE Healthcare Bio-Science, Piscataway, NJ) for 1 hr, followed by three washed with TTBS. The signal was then detected by ECL-plus (GE Healthcare Bio-Science). To verify protein loading, the membranes were reblocked and analyzed for β-actin.
WST-1 Cell Viability Assay
NGFDPC12 cells were harvested and replated in 96-well tissue culture plates. The columns for the standard curve were plated at 500, 1,000, 2,000, 4,000, and 8,000 cells per well, and columns for experimental treatments were plated at 4,000 cells per well. Cells were cultured for 24 hr before treatment. At the end of treatments, culture medium was removed from the wells, followed by addition to each well of 100 μl of phenol red-free media containing 10 μl of WST-1 (Roche Applied Science, Indianapolis, IN). Optical density at 450 nm was determined after 3 hr.
Nuclear Morphology Analysis
Nuclear changes from nontreated and treated cells were assessed with Hoechst 33342 fluorescent dye. Hoechst was added to the culture medium at 1 ng/ml and incubated in the dark at 100% humidity for 10 min at 37°C. Nuclear morphology was visualized under fluorescent microscopy (×400 magnification). Apoptotic cells were identified by the presence of highly condensed chromatin or fragmented nuclei.
DNA Fragmentation Assay
DNA fragmentation associated with apoptosis was examined by the terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) method using a commercial assay kit (APO-BRDU Kit; BD Pharmingen, San Diego, CA). After treatment, cells were fixed with 1% paraformaldehyde in PBS (4°C, 30 min), washed in PBS, and permeabilized with ice-cold 70% ethanol. The BrdU-TUNEL assay was performed as described by the manufacturer. Briefly, fixed cells were washed twice using the kit wash buffer, and after centrifugation the supernatant was discarded. The DNA labeling solution (containing TdT enzyme and Br-dUTP) was added to the cell pellet, and the resuspended mixture was incubated for 1 hr at 37°C with occasional shaking. Cells were then labeled with FITC-conjugated mAb to BrdU, washed again, and resuspended in staining solution containing propidium iodide (PI) and RNase. Cells were incubated for 30 min at room temperature and immediately analyzed in a Becton-Dickinson FACSCalibur flow cytometer (BD Biosciences, San Francisco, CA). In total, 10,000 events were collected per test sample. The results were analyzed in CellQuest data analysis software. The percentage of cells with distinctive apoptotic DNA strand breaks and distinguished by a green fluorescent emission was calculated.
Analysis of Mitochondrial Membrane Potential
Disruption of the mitochondrial membrane potential (MMP) was assessed using the lipophilic cationic probe 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylben-zimidazol-carbocyanine iodide (JC-1 MitoScreen kit; BD Biosciences) as described previously (Singh et al., 2007). Briefly, unfixed cells were washed and resuspended in PBS supplemented with 10 lg/ml JC-1. Cells were then incubated for 15 min at room temperature in the dark, washed, and resuspended in PBS for immediate FACSCalibur flow cytometry analysis. The percentage of cells with disrupted MMP was calculated in the CellQuest software.
Measurement of Intracellular ROS
Detection of intracellular ROS was achieved using the 2′-7′-dichlorofluorescein (DCF) method. Briefly, NGFDPC12 cells were incubated with 10 μM of dichlorofluorescein diacetate (H2DCFDA, Invitrogen) for 30 min at 37°C. The intensity of DCF fluorescence, proportional to the amount of intracellular H2O2, was measured by excitation/emission at 488/530 nm using flow cytometry. As positive control, NGFDPC12 cells were treated with 300 μM H2O2 for 15 min.
Real-Time PCR Analysis
Total cellular RNA was extracted using Tri reagent (Molecular Research Center, Cincinnati, OH) and quantified by measuring the OD at 260 nm. RNA samples were stored at −80°C and used within 1 week of extraction. To measure gene expression changes at the transcriptional level, primers were designed for Bim, Bad, AIF, 14-3-3, BNIP-3, Fas-R, and GAPDH (internal reference), and one-step SYBR green real-time PCR (RT-PCR) was performed. PCR amplifications were done using the iCycler (Bio-Rad, Hercules, CA). Reactions were performed in a 50-μl mixture containing 1× SYBR green super mix PCR buffer [Bio-Rad; 100 mM KCl, 6 mM MgCl2, 40 mM Tris-HCl, pH 8.4, 0.4 mM of each dNTP (dATP, dCTP, dGTP, and dTTP), iTaq DNA polymerase 50 U/ml, SYBR green I, 20 mM fluorescein], containing 300 nM of each primer, 0.25 U/ml Multi Scribe Reverse Transcriptase (Promega, Madison, WI), and 0.4 U/ml RNase Inhibitor (Promega). The RT-PCR protocol started with 30 min at 42°C for the RT reaction. Prior to the PCR step, iTaq DNA polymerase was activated at 95°C for 10 min, followed by 30 sec denaturation at 95°C, 15 sec annealing at 57°C, and 1.5 min elongation at 72°C for 40 cycles. Fluorescence was detected at the end of every 72°C extension phase. To exclude contamination by nonspecific PCR products such as primer dimers, melting curve analysis was applied to all final PCR products after the cycling protocol. Also, PCRs without the RT reaction were performed for each sample to exclude DNA contamination.
The mRNA level of tested genes in the experimental group relative to that in the control group was determined by the comparative CT (threshold cycle) method, using arithmetic formulas: 2e − ΔΔCT. ΔCT is the average CT value for tested genes subtracted by the average CT value for GAPDH. ΔΔCT is the ΔCT for the experimental group, from which is subtracted the ΔCT for the control group.
Caspase-3 Activity Assays
NGFDPC12 cells were plated in black 96-well plates at a density of 4,000 cells per well. After treatment with FFA-BSA (2:1 ratio), caspase-3 activity was measured using the fluorogenic substrate Ac-DEVD-AMC (Alexis Biochemicals, Carlsbad, CA). Briefly, cells were lysed by the addition of 20 μl of 3× lysis buffer (150 mM HEPES, pH 7.4, 450 mM, NaCl, 150 mM KCl, 1.2 mM EGTA, 1.5% Nonidet P40, 0.3% CHAPS, 30% sucrose, 30 mM DTT, 3 mM PMSF), which included either substrate alone (150 mM) or substrate plus the caspases 3 inhibitor (2 mg/ml; DEVD-CHO; Alexis Biochemicals). After 2 hr of incubation for caspases 3, the released fluorochrome was measured with a plate-reading fluorimeter (Flx800; Bio-Tek Instrument Co.) using an excitation of 360 nm and an emission of 460 nm. The activity in wells treated with the inhibitor was subtracted from the activity in wells lacking the inhibitor. The resulting difference was expressed as a percentage of caspase activity in the untreated control cells.
Statistical Analysis
All the experiments were repeated independently at least three times. Values represent means ± SE. Statistical comparisons were made using one-way ANOVA, and significance was accepted at P < 0.05.
Results
PA-Induced Lipotoxicity Leads to Loss of Cell Viability and Apoptosis
We reported previously that PA and SA, but not OA or AA (FFA:BSA, 2:1 ratios), induced lipotoxicity and cell death in NGFDPC12 (Ulloth et al., 2003). The results shown in Figure 1 expand these observations. Lipotoxicity in NGFDPC12 triggers a significant loss in cell viability after 12 hr (70% viability) and 24 hr (10% viability) following exposure to PA compared with untreated controls (Fig. 1A). The viability values for NGFDPC12 cells after 24 hr of lipotoxicity were 90.8% ± 7.0%, 79.1% ± 1.5%, 54.9% ± 2.8%, and 16.0% ± 3.9% at PA/BSA ratios of 0.25:1, 0.5:1, 1:1, and 2:1, respectively (Fig. 1B). Untreated cells exhibited normal nuclear morphology (Fig. 1C, control), whereas NGFDPC12 cells exposed to PA showed evidence of typical apoptotic nuclear morphology, including chromatin condensation and fragmentation (Fig. 1C, PA). Next we evaluated the mRNA expression of genes associated with apoptosis and mitochondrial dysfunction in NGFDPC12 cells undergoing lipotoxicity. Quantitative real-time PCR experiments showed early FAS death receptor (FAS-R) up-regulation after exposure to PA, reaching a maximum of eightfold induction over control after 8 hr (Fig. 1D). Similarly, a threefold BNIP3 up-regulation was observed after 2 hr of treatment with PA, reaching eightfold induction at 6 hr. The mRNA levels of other apoptosis-related genes (Bim, Bad, AIF, and 14.3.30) did not show dramatic increases (Fig. 1D).
Fig. 1.
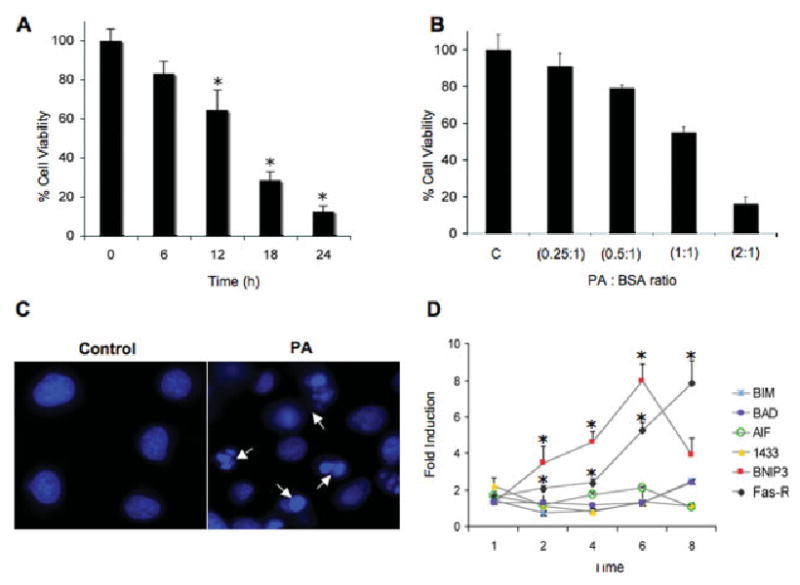
Palmitic acid-induced lipotoxicity in NGFDPC12 cells. A: NGFDPC12 cells were exposed to PA/BSA (2:1 ratio), and viability was determined using WST-1 assay at the indicated times. B: NGFDPC12 cells were exposed to different PA/BSA ratios (0.25:1, 0.5:1, 1:1, and 2:1 with BSA at a concentration of 0.150 mM) for 24 hr. Cell viability was measured by the WST-1 assay. C: NGFDPC12 cells were exposed to 0.150 mM BSA without PA (control) or PA/BSA (2:1; PA) for 12 hr. Cells were stained with Hoechst (10 ng/liter), and nuclei were visualized under fluorescent microscopy. Arrows indicate nuclei showing chromatin condensation and fragmentation. D: Regulation of apoptosis-asociated genes in NGFDPC12 cultures exposed to PA/BSA (2:1). Quantitative RT-PCR experiments show the time-dependent mRNA expression of Bim, Bad, AIF, BNIP-3, 14.3.3, and FAS-R. GAPDH mRNA expression was used as the internal control. Data represent mean 6 SEM of three independent experiments. ★P < 0.05 compared with control.
The APO-BRDU assay showed elevated DNA fragmentation in cells undergoing apoptosis (Fig. 2A,B). Quantification of DNA fragmentation indicated a threefold increase over control after 12 hr of exposure to PA and more than a fivefold increase after 24 hr (Fig. 2C). Consistently with the observed morphological apoptotic features, there was a significant increase in caspase-3 activity in cells exposed to PA (Fig. 2D). The next series of experiments examined the effect of decreasing the PA:BSA ratio to 0.5:1 by increasing the concentration of BSA to 0.6 mM, which results in a reduction of the concentration of unbound, free PA in the cultures. This approach would allow us to determine the minimum PA exposure time required to make lipotoxicity irreversible. Figure 2E shows that treating NGFDPC12 cells with 0.6 mM BSA at 6 hr (PA + BSA at 6 hr) after an initial PA (PA:BSA, 2:1) exposure resulted in lower protection (24.2% ± 2.8%) than treatment after 2 hr (PA + BSA at 2 hr; 88.3% ± 7.3%). These results suggest that PA-induced cell death is mostly BSA insensitive after 6 hr following PA exposure, but it is not fully developed and is reversible within the first 2 hr of treatment.
Fig. 2.
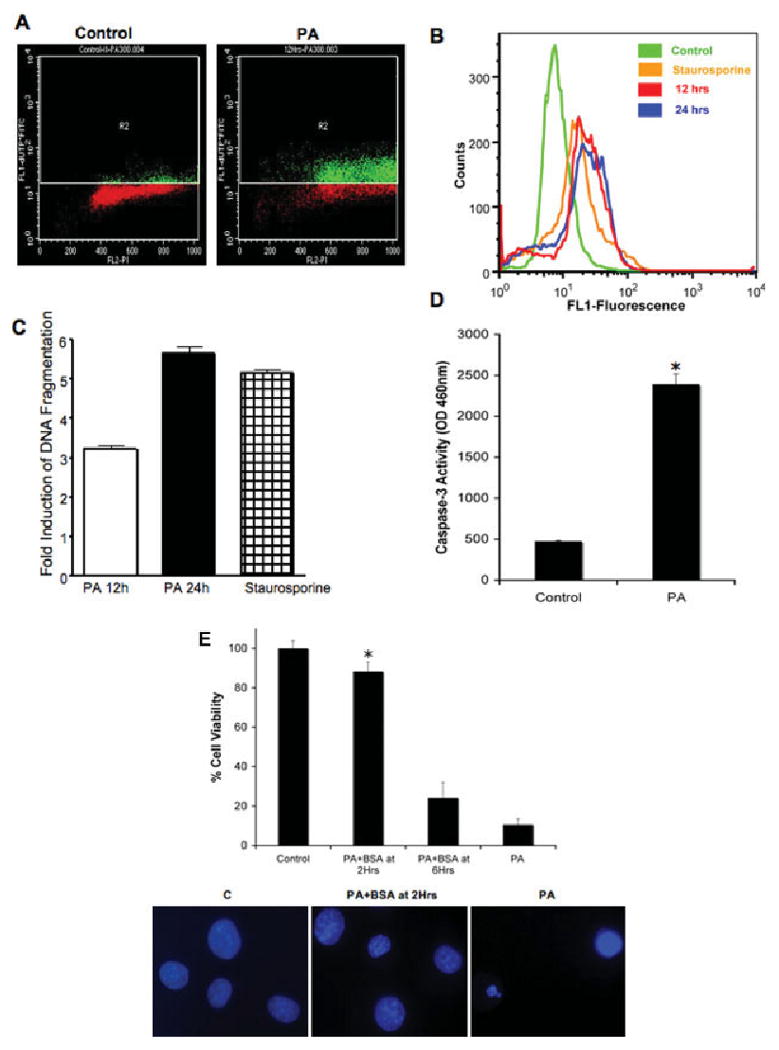
DNA fragmentation and caspase-3 activity of NGFDPC12 cells exposed to PA. A: Representative experimental results of BRDU-FITC plots (apoptotic cells) vs. PI (total DNA marker) for controls and cells exposed to PA/BSA (2:1) for 12 hr. The R2 quadrant shows cells exhibiting increased BrdU-FITC fluorescence (green). Nonapoptotic cells are indicated in red. B: Representative distribution diagram of positive apoptotic NGFDPC12 cells. Cells were treated with BSA for negative control (green), PA/BSA (2:1) for 12 hr (red) or 24 hr (blue), or staurosporine for positive control (orange). C: Quantitative analysis of positive apoptotic NGFDPC12 cells as shown in B. The data represent the mean ± SEM of three independent experiments done in triplicate. D: Caspase-3 activity in cell extracts was determined in control or NGFDPC12 cells treated with PA/BSA for 12 hr. E: Early posttreatment with BSA protects NGFDPC12 cells from PA-induced lipotoxicity. Additional BSA (0.6 mM final concentration) was added to NGFDPC-12 cells at 2 or 6 hr after initial PA/BSA (2:1 ratio) treatment. Percentage of viable cells was measured by the WST-1 assay at the end of a 24-hr incubation period. Nuclei in bottom panel were visualized with Hoechst staining. Data represent mean ± SEM of three independent experiments. ★P < 0.05 compared with control.
PA-Induced Lipotoxicity Leads to ASCOS
To study further the potential metabolic implications of lipotoxicity, we measured ASCOS in terms of ROS generation and alterations in mitochondrial membrane potential in NGFDPC12 cells exposed to PA (PA:BSA, 2:1). Flow cytometric analysis using DCF showed that PA-mediated lipotoxicity stimulated a sustained threefold increase in ROS generation from 3 to 12 hr after the initial PA exposure (Fig. 3A,B). The generation of ROS during PA-induced lypotoxicity was significantly inhibited by the antioxidant MCI-186 and BSA (Fig. 3C). However, DHA did not enhance the inhibitory effects of MCI-186. We next determined whether these cells exhibited an increase in mitochondrial membrane permeability, using the JC-1 assay. Figure 3D shows that control NGFDPC12 cells without PA displayed only a 5% decrease of FL2 (red) fluorescence (R2), indicative of JC-1 aggregate formation inside the intact mitochondria. By contrast, there was an 80% reduction of FL2 fluorescence in cells undergoing lipotoxicity after 24 hr of PA exposure, indicative of the presence of monomeric JC-1 in the cytoplasm resulting from disrupted mitochondrial membrane potential.
Fig. 3.
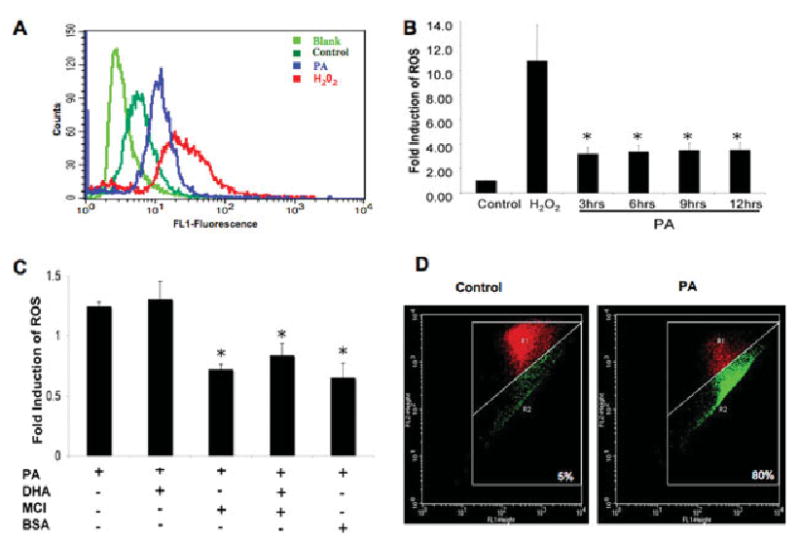
ROS generation and changes in mitochondrial membrane permeability in NGFDPC12 cells undergoing PA-induced lipotoxicity. A: ROS generation in NGFDPC12 cells was detected by the 2′,7′ -dichlorofluorescein diacetate (DCF) method at 12 hr after the initial exposure to PA using flow cytometry. NGFDPC12 cells were treated with no DCF (light green), DCF (dark green), PA + DCF (blue), or hydrogen peroxide + DCF (red). B: Quantification of ROS production in NGFDPC12 cells at 3, 6, 9, and 12 hr after PA/BSA treatment. C: ROS modulation after addition of DHA, MCI-186, or BSA to NCGDPC12 cells treated with PA for 6 hr. D: Mitochondrial depolarization in NGFDPC12 cells, control or PA/BSA treated, detected by JC-1 flow cytometric analysis. JC-1 was detected in the FL1 or FL2 channel depending on its aggregation status (see Mqaterials and Methods). The R2 quadrant defines cells with leaky mitochondria showing reduced FL2 readings (green). Cells with intact mitochondria are indicated in R1 (red). Data represent mean ± SEM sent of at least three independent experiments. ★P < 0.05 compared with control.
DHA Protects Against PA-Induced Lipotoxicity
These findings implicate the mitochondria in lipotoxicity injury. DHA has been shown to inhibit the expression of BAD preventing mitochondrial permeability (Mukherjee et al., 2004) and may function as a membrane stabilizer (Valentine and Valentine, 2004). It also has the ability to modulate fatty acid metabolism, probably by increasing the formation of neutral fats, an event that may decrease PA toxicity (Listenberger et al., 2003). Figure 4A shows that NGFDPC12 cells cotreated with PA and DHA (PA + DHA) from the start exhibited increased cell viability (91.75% ± 3.3% viability). Interestingly, NGFDPC12 cells preincubated in DHA for 12 hr before PA treatment (there was no DHA during PA treatment) showed a moderate decrease in viability (61.0% ± 4.2%) compared with control cells and cells undergoing lipotoxicity without previous DHA treatment (PA; 10.5% ± 1.0% cell viability). Further characterization of this phenomenon showed that cotreatment of cells with DHA and PA dramatically reduced the morphological features of apoptosis (Fig. 4B) as well as apoptotic nuclei (Fig. 4C). DHA cotreatment also inhibited caspase-3 activation in NGFDPC12 cells exposed to PA/BSA (2:1; Fig. 4D). In addition, DHA cotreatment inhibited the PA-induced apoptotic cleavage of PARP and lamin B (Fig. 4E).
Fig. 4.
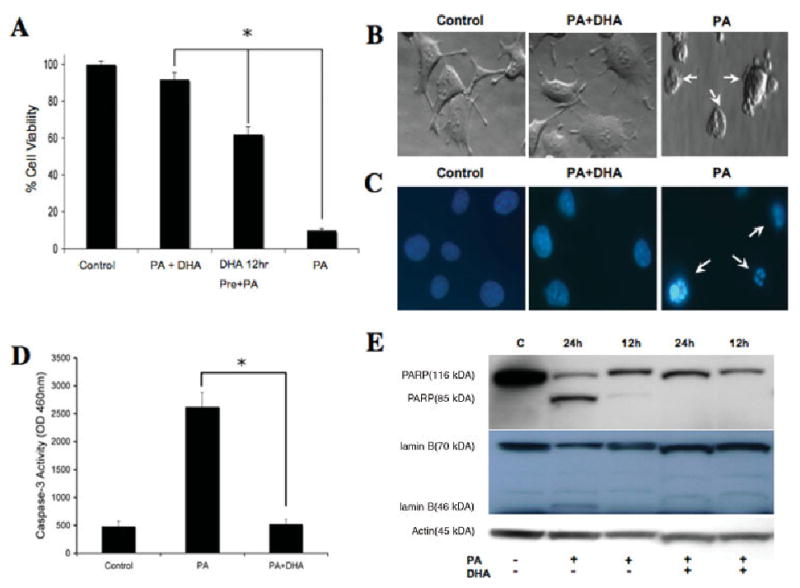
Inhibition of fatty acid-induced lipotoxicity by DHA. A: DHA inhibits induction of cell death by PA in NGFDPC12 cells. Cell viability was determined after treatment for 24 hr as follows: 0.150 mM BSA alone (control), PA/BSA 2:1 ratio (PA), PA/BSA 2:1 cotreated with DHA followed by an increase in BSA final concentration to 0.6 mM at 6 hr (PA + DHA + BSA at 6 hr), and DHA pretreatment for 12 hr followed by treatment with PA/BSA 2:1 (DHA 12 hr pre + PA). B: DHA treatment inhibits apoptotic features of NGFDPC12 cells treated with PA/BSA. Morphology of NGFDPC12 cells treated with BSA alone (control), PA/BSA 2:1 cotreated with DHA, or PA/BSA 2:1 for 24 hr. C: Nuclear morphology of NGFDPC12 cells treated with BSA alone (control), PA/BSA 2:1 cotreated with DHA or PA/BSA 2:1 for 24 hr. Arrows indicate nuclei exhibiting fragmentation. D: DHA cotreatment produced a significant inhibitory effect of caspase-3 activity in cellular extracts of NGFDPC12 cells exposed to PA/BSA (2:1). E: Cleavage of PARP and lamin B in NGFDPC-12 cells exposed to PA/BSA (2:1), as assessed by Western blotting. PA treatment induced the appearance of the signature apoptotic fragments of PARP (85 kDa) or lamin B (46 kDa). DHA inhibited PARP and lamin B cleavage. Data in A and D represent mean ± SEM of three independent experiments. ★P < 0.05.
We also investigated whether DHA stabilizes mitochondrial membrane potential, using the JC-1 flow cytometry assay. Figure 5A shows that nontreated control NGFDPC12 cells displayed only a 5% decrease in FL2 red fluorescence (R2) compared with 22% and 35% decreases in cells undergoing PA-induced lipotoxicity for 9 hr and 12 hr, respectively. This significant decrease in JC-1 florescence was completely abolished by DHA, as demonstrated by the observed 4% decrease in FL2 red fluorescence (Fig. 5A). Quantification results showed that cotreatment of cells with PA and DHA led to significant five- and sixfold reductions in mitochondrial membrane depolarization compared with the results obtained after 12 hr and 24 hr, respectively, of PA exposure in the absence of DHA (Fig. 5B). These results suggest that DHA is able to inhibit lipotoxicity-induced mitochondrial membrane depolarization completely.
Fig. 5.
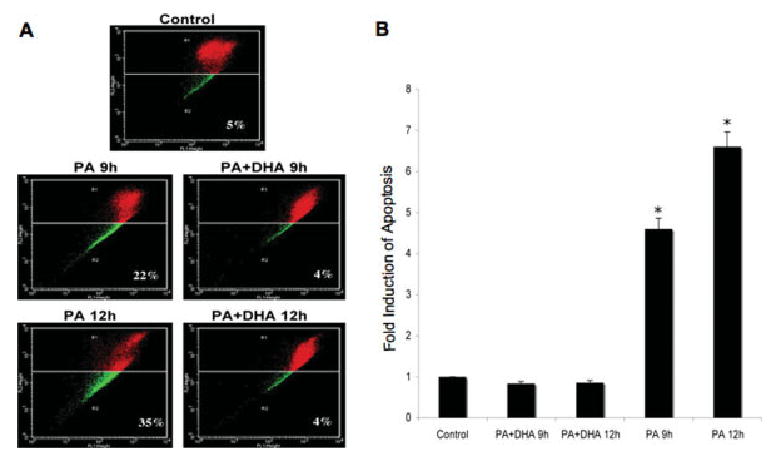
DHA reduces mitochondrial depolarization of NGFDPC12 cells undergoing PA-induced lipotoxicity. A: NGFDPC12 cells were treated with PA/BSA alone (PA) or in the presence DHA (PA + DHA) for 9 or 12 hr, and mitochondrial depolarization was determined by JC-1 flow cytometry assays. Representative flow cytometric plots are shown. B: Quantification of mitochondria depolarization (R2 in JC-1 flow cytometry) in NGFDPC12 cells undergoing PA-induced lipotoxicity. Data represent mean ± SEM of three independent experiments. ★P < 0.05.
PA Reduces the Viability of RCC
The last series of experiments evaluated whether PA-induced lipotoxicity has similar effects on RCC viability. We observed that RCC undergoing lipotoxicity exhibited cellular and nuclear features of apoptosis (Fig. 6A,B) similar to those observed in NGFDPC12 cells. RCC were also incubated in the presence of PA/BSA (2:1 ratio) with and without DHA for 48 hr, and cell viability was then evaluated by WST-1 assay. The survival rate of RCC undergoing lipotoxicity was 47.5% ± 6.7% in the absence of DHA and 75.2% ± 2.57% after cotreatment with PA and DHA (Fig. 6C).
Fig. 6.
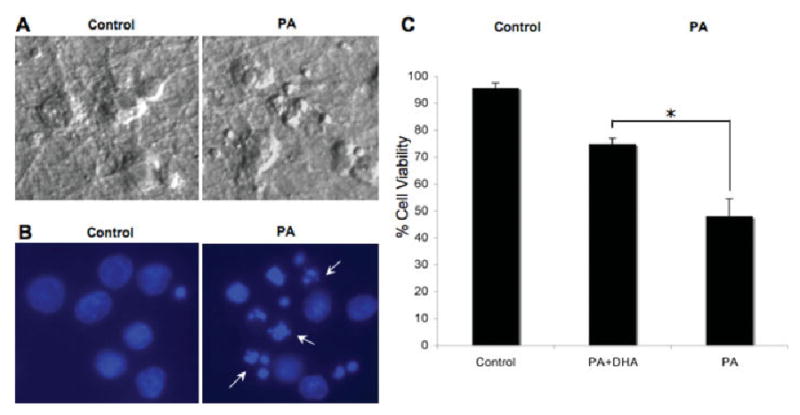
PA-induced lipotoxicity and DHA neuroprotective action on primary rat cortical cells (RCC). A: Cellular morphology of control and treated (PA/BSA, 2:1, 48 hr) RCC. B: Nuclear morphology of control and treated RCC. Arrows indicate fragmented nuclei. C: Viability of RCC treated for 48 hr with PA/BSA (2:1 ratio; PA) or in cotreatment with DHA (PA + DHA). Viability was determined using the WST-1 assay. Data represent mean ± SEM of three independent experiments. ★P < 0.05.
Discussion
The main findings of this study are that PA-mediated lipotoxicity: 1) induces apoptosis in NGFDPC12 and RCC cells; 2) increases the expression of the apoptosis associated genes BNIP3 and Fas-R; 3) elicits ASCOS, evident by increase in mitochondrial depolarization and ROS generation; and 4) is inhibited by DHA or BSA. The increase in the expression of key apoptosis-associated genes such as BNIP3 and Fas-R preceded the cell death observed following exposure to PA. Noticeable in our study was that BNIP3, associated primarily with caspase-independent cell death (Vande Velde et al., 2000), had a maximum eightfold induction following exposure to PA. BNIP3 mediates cell death in several different ways. For instance, the BNIP3 BH3 domain binds to Bcl-2, resulting in the sequestration of this protein and preventing its antiapoptotic action. BNIP3 has also been shown to integrate fully into the mitochondrial outer membrane after certain cell death insults, resulting in a rapid and profound mitochondrial depolarization (Vande Velde et al., 2000). Interestingly, ischemia-vulnerable CA1 neurons in the hippocampus exhibit BNIP3-positive granules in the nucleus 1–2 days after induction of cell death, which may be an early event preceding the neuronal damage observed at 3–7 days (Schmidt-Kastner et al., 2004). Up-regulation of BNIP-3 has also been associated with caspase-independent cell death (Vande Velde et al., 2000), which is consistent with our previous observations that FFA-induced neuronal lipotoxicity also occurs through a caspase-independent pathway (Ulloth et al., 2003).
Signaling through Fas-R typically leads to caspase-dependent cell death, although under certain circumstances it can also mediate caspase-independent cell death (Vercammen et al., 1998; Matsumura et al., 2000). The caspase-dependent pathway appears to dominate the caspase-independent pathway when caspases are functional (Matsumura et al., 2000). However, upon caspase inhibition, the caspase-independent pathway ensures that cell death proceeds. Based on our previous observations (Ulloth et al., 2003), we do not rule out that a similar phenomenon is occurring in NGFDPC12 cells during lipotoxicity. Thus, the mechanism of cell death from lipotoxicity seems to be multifaceted, involving the activation of alternative death pathways.
Lipotoxicity exhibits a robust induction of ASCOS in NGFDPC12 cells, as demonstrated by a threefold increase in ROS and a significant induction of mitochondrial depolarization. Elevated ROS production may lead to deleterious alterations in cell physiology (for review see Bazan et al., 2005). Among the most destructive effects of ROS is lipid peroxidation. ROS target fatty acid side chains, generating carbon-centered radicals, and subsequently lipoperoxyl radicals, capable of attacking adjacent fatty acids and the propagation of further membrane damage (Halliwell, 1991). A particular consequence is the accumulation of lipid hydroperoxides that alter membrane permeability and fluidity and oxidize membrane proteins, leading to alterations in ion transport, notably the intracellular flux of Ca2+. A remarkable influx of Ca2+ into mitochondria leads in turn to partial reduction of molecular oxygen and generation of the superoxide anion and its more toxic derivatives, H2O2 and OH (Dykens, 1994). Enzymatic sources of ROS under these conditions include the cyclooxygenases (COX), lipoxygenases, myeloperoxidase, and nitric oxide synthase (Bazan, 2006). The high unsaturated lipid content of neuronal membranes makes neurons excellent at propagating ROS (Bazan, 2006). Thus, our data suggest that the negative effects of ROS on the mitochondria might be responsible for the observed mitochondrial dysfunction and deporalization associated with lipotoxicity. This impairment of mitochondrial functions may possibly trigger apoptotic cell death (Mattson, 1998).
NGFDPC12 cells cultures exhibit 90% cell death following 24 hr of exposure to PA in comparison with 47% after 48 hr by RCC. These differences in cell viability can result from variability in the efficiency of incorporation of PA into the cell, the preferential mechanism of how different cells process fatty acids, and the effect of potential interactions between glial cells and neurons during lipotoxicity. For instance, primary neurons, oligodendrocyes, and astrocytes can exhibit differences in the incorporation of PA into ceramide (Blázquez et al., 2000; Kilkus et al., 2008). Differences in PA incorporation affect the level of the de novo synthesis of ceramide in the cell that may influence cell death and apoptosis (Hannun and Obeid, 1995; Hartfield et al., 1997). The RCC cultures used in our study were isolated and grown under culture conditions that select for neurons as opposed to glial cells (see Materials and Methods), but we cannot rule out the potential effect of these cells in the RCC cultures. Astrocytes can exhibit a higher rate of incorporation of PA than primary neurons (Blázques et al., 2000) and are the only cells in the brain capable of metabolizing fatty acids through β-oxidation (Edmond et al., 1987). The resulting ketone bodies produced by astrocytes can be used by RCC for energy and can increase their survival rate during lipotoxicity or other pathological conditions (Auestad et al., 1991).
BSA rescues cells from lipotoxicity, most likely because of its ability to sequester FFA, limiting their detrimental cellular effects. We observed that the neuroprotective effects of BSA decreased dramatically if the protein was added 6 hr after exposure to PA; however, significant protection was observed when the protein was added within 2 hr. Under certain pathological conditions, including traumatic injury, neurons can be exposed to albumin and other blood proteins resulting from breakdown of the blood–brain barrier (Cavanagh and Warren, 1985; Fukuda et al., 1995). In these situations, albumin can exert neuroprotective and neurotrophic effects associated with its function as a fatty acid binding protein capable of sequestering FFA (Tabernero et al., 2002; Guajardo et al., 2002). Albumin also induces mobilization of n-3 polyunsaturated fatty acids (PUFA), potentially contributing to replenishment of lost PUFA in the cell membrane after ischemia (Belayev et al., 2005). Human serum albumin has been shown to cross the blood–brain barrier, reaching intraneuronal sites and eliciting neuroprotection, when systemically injected during ischemia reperfusion in experimental animals (Remmers et al., 1999; Ginsberg, 2003). Albumin therapy has also been shown to protect parenchyma and vascular elements of the brain by diminishing brain edema, maintaining microvascular integrity, inhibiting endothelial cell apoptosis, and exerting antioxidant effects (Belayev et al., 2001, 2002). Interestingly, several studies have shown that there is a 4-hr therapeutic window during which the albumin treatment is highly neuroprotective in models of temporary focal cerebral ischemia and traumatic brain injury (Belayev et al., 1997, 1999, 2001). Delayed administration of high-dose human albumin failed to improve the outcome after transient focal cerebral ischemia in rats (Belayev et al., 2004). These studies are consistent with our results showing that the neuroprotective effect of BSA against lipotoxicity was time sensitive. It is reasonable to suggest that the neuroprotective effects of albumin in vivo may include interfering with lipotoxicity associated with traumatic/ischemic injury.
An important finding in our study was that treatment of NGFDPC12 and RCC cells with DHA inhibited PA-induced apoptosis. This neuroprotective activity of DHA is consistent with its potential therapeutic use during trauma and stroke (Marcheselli et al., 2003; Belayev et al., 2005). DHA is the major omega-3 fatty acid component of the neuronal membranes in the gray matter of the cerebral cortex and in retinal photoreceptors cells (Lauritzen et al., 2000). Insofar as DHA is highly unsaturated, it is expected to increase the fluidity of neuronal membranes, thereby playing an essential role in various neurochemical processes of the brain (Yehuda et al., 2002; Horrocks and Farooqui, 2004; Stillwell et al., 2005). Polyunsaturated FA deficiency has been reported in aging mouse neuronal membranes, probably leading to memory loss, learning disabilities, cognitive alterations, and impaired visual acuity that can be reversed by supplementing the diet with fish oil or DHA. Alterations in saturated/polyunsaturated fatty acid ratio have been established in the brain of patients with Alzheimer's disease, in whom the relative amounts of the saturated FA (PA and SA) are substantially increased and polyunsaturated fatty acids (DHA) decreased (Soderberg et al., 1991). This unbalanced in the FA content likely is detrimental to neurons and may influence cellular dysfunction and apoptosis.
Dietary DHA is incorporated into neuronal membrane phospholipids, but the extent of structural and functional changes associated with this incorporation is still unknown. DHA incorporation occurs mainly into the anionic phosphatidyl serine (PS) and phosphatidyl ethanolamine (PE; Aid et al., 2003). DHA addition to neuronal cells promotes PS biosynthesis (Garcia et al., 1998) and DHA enrichment, most likely because of the increased affinity of DHA for PS-synthesizing enzymes (Kim et al., 2004). The incorporation of DHA into phospholipids of cultured neurons could be a major component of the mechanism underlying DHA neuroprotective activity (Kim et al., 2000). The ability of neurons to concentrate DHA into PS and PE can lead to the build up of a reserve for neuroprotection. The antiapoptotic effects of DHA in PA-treated NGFDPC12 cells strongly suggest that DHA might be incorporated in the cell membrane phospholipids. This could lead to the development of preventive and therapeutic strategies based on loading phospholipids (PS/PE) with DHA. After a toxic event, phospholipases would then release the loaded DHA to exert its neuroprotective activity. This could have important implications for the prevention and treatment of stroke, TBI, diabetes, and other conditions in which increased levels of saturated FFA can induce irreparable neuronal damage.
It is also possible that DHA stabilizes the mitochondrial membrane of NGFDPC12 cells exposed to FFA. DHA is a precursor of neuroprotectin D1 (NPD1), a metabolite that has been proposed to have neuroprotective roles during stroke (Bazan, 2005). As with DHA, NPD1 rescues cells and augments the levels of the antiapoptotic proteins Bcl-xL and Bcl-2 while down-regulating the proapoptotic proteins Bax and Bad (Mukherjee et al., 2004). NPD1 seems to act upstream of caspase activation, modulating the expression of BCl-2 family members, and appears to be involved in the immediate and early response to injury (Mukherjee et al., 2004). Treatment with DHA may increase its incorporation into the mitochondrial membrane phospholipids, thereby augmenting the efficiency of electron transport by supporting lateral movement of proteins within the bilayer (Valentine and Valentine, 2004), thus influencing protein–protein interactions within the Bcl-2 family. This would be consistent with reports that DHA content in mitochondrial phospholipids correlates with membrane permeability (Hulbert, 2003). Further studies are necessary to investigate specific mechanisms by which DHA enrichment of the mitochondrial membrane results in neuroprotection.
Consistently with our findings, TBI and ischemic/hypoxic injuries exhibit significant ROS generation, which has been proposed to be a key component of the damaging effect (Jesberger and Richardson, 1991; Rokyta et al., 1996). As a polyunsaturated FA, DHA may be more susceptible to oxidation than saturated FA and could have a neuroprotective antioxidant role during FA-induced lipotoxicity. Arachidonic acid (AA) and its derivatives are potent mediators of the inflammation cascade leading to the production of inflammatory mediators that might be involved in ROS generation and lipotoxicity. After exposure to oxidative stress, oxidized omega-3 fatty acids such as DHA trigger the docosanoid pathway or DHA-derived messengers, including docosatrienes, neuroprotectins, and resolvins, as a response to oxidative stress (Marcheselli et al., 2003; Bazan, 2006). Thus, oxidative stress may lead to the activation of a DHA-dependent protective metabolic cascade of events with the ability to antagonize the proinflammatory cascade of AA and its derivatives.
Interestingly, our results indicated that treatment with DHA led to increased cellular protection against PA-induced lipotoxicity but did not inhibit PA-induced ROS. It was reported that treatment of human fibroblasts with DHA increased ROS but also induced a simultaneous strong antioxidant response, where the antioxidant enzymes GR, γ-GCL, and GST and their respective activities were up-regulated (Arab et al., 2006). It is possible that DHA or its oxygenated metabolites may exert neuroprotection also by a mechanism involving activation and transcriptional up-regulation of the antioxidant enzymes. Increasing the cellular resistance to oxidative stress by DHA supplementation could be invaluable for neuronal survival after injury.
In summary, the findings in the present study suggest a link between PA-induced lipotoxicity and ASCOS in NGFDPC12 cells. Current studies in our laboratories are evaluating whether lipotoxicity associated with ASCOS plays a significant role during TBI and stroke in vivo.
Acknowledgments
This work was supported in part by NIH awards P20MD001632 and R25GM060507 to MDL.
References
- Abe K, Kogure K, Yamamoto H, Imazawa M, Miyamoto K. Mechanism of arachidonic acid liberation during ischemia in gerbil cerebral cortex. J Neurochem. 1987;48:503–509. doi: 10.1111/j.1471-4159.1987.tb04121.x. [DOI] [PubMed] [Google Scholar]
- Aid S, Vancassel S, Poumes-Ballihaut C, Chalon S, Guesnet P, Lavialle M. Effect of a diet-induced n-3 PUFA depletion on cholinergic parameters in the rat hippocampus. J Lipid Res. 2003;44:1545–1551. doi: 10.1194/jlr.M300079-JLR200. [DOI] [PubMed] [Google Scholar]
- Arab K, Rossary A, Flourie F, Tourneur Y, Steghens JP. Docosahexaenoic acid enhances the antioxidant response of human fibroblasts by upregulating gamma-glutamyl-cysteinyl ligase and glutathione reductase. Br J Nutr. 2006;95:18–26. doi: 10.1079/bjn20051626. [DOI] [PubMed] [Google Scholar]
- Auestad N, Korsak RA, Morrow JW, Edmond J. Fatty acid oxidation and ketogenesis by astrocytes in primary culture. J Neurochem. 1991;56:1376–1386. doi: 10.1111/j.1471-4159.1991.tb11435.x. [DOI] [PubMed] [Google Scholar]
- Bazan NG., Jr Effects of ischemia and electroconvulsive shock on free fatty acid pool in the brain. Biochim Biophys Acta. 1970;218:1–10. doi: 10.1016/0005-2760(70)90086-x. [DOI] [PubMed] [Google Scholar]
- Bazan NG. Neuroprotectin D1 (NPD1): a DHA-derived mediator that protects brain and retina against cell injury-induced oxidative stress. Brain Pathol. 2005;15:159–166. doi: 10.1111/j.1750-3639.2005.tb00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan NG. The onset of brain injury and neurodegeneration triggers the synthesis of docosanoid neuroprotective signaling. Cell Mol Neurobiol. 2006;26:901–913. doi: 10.1007/s10571-006-9064-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan NG, Marcheselli VL, Cole-Edwards K. Brain response to injury and neurodegeneration: endogenous neuroprotective signaling. Ann N Y Acad Sci. 2005;1053:137–147. doi: 10.1196/annals.1344.011. [DOI] [PubMed] [Google Scholar]
- Belayev L, Busto R, Zhao W, Clemens JA, Ginsberg MD. Effect of delayed albumin hemodilution on infarction volume and brain edema after transient middle cerebral artery occlusion in rats. J Neurosurg. 1997;87:595–601. doi: 10.3171/jns.1997.87.4.0595. [DOI] [PubMed] [Google Scholar]
- Belayev L, Alonso OF, Huh PW, Zhao W, Busto R, Ginsberg MD. Posttreatment with high-dose albumin reduces histopathological damage and improves neurological deficit following fluid percussion brain injury in rats. J Neurotrauma. 1999;16:445–453. doi: 10.1089/neu.1999.16.445. [DOI] [PubMed] [Google Scholar]
- Belayev L, Liu Y, Zhao W, Busto R, Ginsberg MD. Human albumin therapy of acute ischemic stroke: marked neuroprotective efficacy at moderate doses and with a broad therapeutic window. Stroke. 2001;32:553–560. doi: 10.1161/01.str.32.2.553. [DOI] [PubMed] [Google Scholar]
- Belayev LS, Pinard E, Nallet H, Seylaz J, Liu Y, Riyamongkol P, Zhao W, Busto R, Ginsberg MD. Albumin therapy of transient focal cerebral ischemia: in vivo analysis of dynamic microvascular responses. Stroke. 2002;33:1077–1084. doi: 10.1161/hs0402.105555. [DOI] [PubMed] [Google Scholar]
- Belayev L, Khoutorova L, Belayev A, Zhang Y, Zhao W, Busto R, Ginsberg MD. Delayed post-ischemic albumin treatment neither improves nor worsens the outcome of transient focal cerebral ischemia in rats. Brain Res. 2004;998:243–246. doi: 10.1016/j.brainres.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Belayev L, Marcheselli VL, Khoutorova L, Rodriguez de Turco EB, Busto R, Ginsberg MD, Bazan NG. Docosahexaenoic acid complexed to albumin elicits high-grade ischemic neuroprotection. Stroke. 2005;36:118–123. doi: 10.1161/01.STR.0000149620.74770.2e. [DOI] [PubMed] [Google Scholar]
- Blázquez C, Galve-Roperh I, Guzmán M. De novo-synthesized ceramide signals apoptosis in astrocytes via extracellular signal-regulated kinase. FASEB J. 2000;14:2315–2222. doi: 10.1096/fj.00-0122com. [DOI] [PubMed] [Google Scholar]
- Cavanagh ME, Warren A. The distribution of native albumin and foreign albumin injected into lateral ventricles of prenatal and neonatal rat forebrains. Anat Embryol. 1985;172:345–351. doi: 10.1007/BF00318983. [DOI] [PubMed] [Google Scholar]
- Dhillon HS, Dose JM, Scheff SW, Prasad MR. Time course of changes in lactate and free fatty acids after experimental brain injury and relationship to morphologic damage. Exp Neurol. 1997;146:240–249. doi: 10.1006/exnr.1997.6524. [DOI] [PubMed] [Google Scholar]
- Dykens JA. Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated CA2+ and Na+: implications for neurodegeneration. J Neurochem. 1994;63:584–591. doi: 10.1046/j.1471-4159.1994.63020584.x. [DOI] [PubMed] [Google Scholar]
- Edmond J, Robbins RA, Bergstrom JD, Cole RA, de Vellis J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J Neurosci Res. 1987;18:551–561. doi: 10.1002/jnr.490180407. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Tanno H, Okimura Y, Nakamura M, Yamaura A. The blood–brain barrier disruption to circulating proteins in the early period after fluid percussion brain injury in rats. J Neurotrauma. 1995;12:315–324. doi: 10.1089/neu.1995.12.315. [DOI] [PubMed] [Google Scholar]
- Garcia MC, Ward G, Ma YC, Salem N, Jr, Kim HY. Effect of docosahexaenoic acid on the synthesis of phosphatidylserine in rat brain in microsomes and C6 glioma cells. J Neurochem. 1998;70:24–30. doi: 10.1046/j.1471-4159.1998.70010024.x. [DOI] [PubMed] [Google Scholar]
- Ginsberg MD. Adventures in the pathophysiology of brain ischemia: penumbra, gene expression, neuroprotection: the 2002 Thomas Willis Lecture. Stroke. 2003;34:214–223. doi: 10.1161/01.str.0000048846.09677.62. [DOI] [PubMed] [Google Scholar]
- Guajardo MH, Terrasa AM, Catala A. Retinal fatty acid binding protein reduce lipid peroxidation stimulated by long-chain fatty acid hydroperoxides on rod outer segments. Biochim Biophys Acta. 2002;1581:65–74. doi: 10.1016/s1388-1981(02)00121-x. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Reactive oxygen species in living systems: source, biochemistry, and role in human disease. Am J Med. 1991;91:14S–22S. doi: 10.1016/0002-9343(91)90279-7. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. Ceramide and the eukaryotic stress response. Biochem Soc Trans. 1997;25:1171–1175. doi: 10.1042/bst0251171. [DOI] [PubMed] [Google Scholar]
- Hartfield PJ, Mayne GC, Murray AW. Ceramide induces apoptosis in PC12 cells. FEBS Lett. 1997;401:148–152. doi: 10.1016/s0014-5793(96)01460-3. 20. [DOI] [PubMed] [Google Scholar]
- Homayoun P, Rodriguez de Turco EB, Parkins NE, Lane DC, Soblosky J, Carey ME, Bazan NG. Delayed phospholipid degradation in rat brain after traumatic brain injury. J Neurochem. 1997;69:199–205. doi: 10.1046/j.1471-4159.1997.69010199.x. [DOI] [PubMed] [Google Scholar]
- Horrocks LA, Farooqui AA. Docosahexaenoic acid in the diet: its importance in maintenance and restoration of neural membrane function. Prostaglandins Leukot Essent Fatty Acids. 2004;70:361–372. doi: 10.1016/j.plefa.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Hulbert AJ. Life, death and membrane bilayers. J Exp Biol. 2003;206:2303–2311. doi: 10.1242/jeb.00399. [DOI] [PubMed] [Google Scholar]
- Jesberger JA, Richardson JS. Oxygen free radicals and brain dysfunction. Int J Neurosci. 1991;57:1–17. doi: 10.3109/00207459109150342. [DOI] [PubMed] [Google Scholar]
- Kilkus JP, Goswami R, Dawson SA, Testai FD, Berdyshev EV, Han X, Dawson G. Differential regulation of sphingomyelin synthesis and catabolism in oligodendrocytes and neurons. J Neurochem. 2008;106:1745–57. doi: 10.1111/j.1471-4159.2008.05490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, Akbar M, Lau A, Edsall L. Inhibition of neuronal apoptosis by docosahexaenoic acid (22:6n-3). Role of phosphatidylserine in antiapoptotic effect. J Biol Chem. 2000;275:35215–35223. doi: 10.1074/jbc.M004446200. [DOI] [PubMed] [Google Scholar]
- Kim HY, Bigelow J, Kevala JH. Substrate preference in phosphatidylserine biosynthesis for docosahexaenoic acid containing species. Biochemistry. 2004;43:1030–1036. doi: 10.1021/bi035197x. [DOI] [PubMed] [Google Scholar]
- Lauritzen I, Blondeau N, Heurteaux C, Widmann C, Romey G, Lazdunski M. Polyunsaturated fatty acids are potent neuroprotectors. EMBO J. 2000;19:1784–1793. doi: 10.1093/emboj/19.8.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengqvist J, Mata De Urquiza A, Bergman AC, Willson TM, Sjovall J, Perlmann T, Griffiths WJ. Polyunsaturated fatty acids including docosahexaenoic and arachidonic acid bind to the retinoid X receptor alpha ligand-binding domain. Mol Cell Proteomics. 2004;3:692–703. doi: 10.1074/mcp.M400003-MCP200. [DOI] [PubMed] [Google Scholar]
- Lee YF, Hirose HF, Ohneda MF, Johnson JH, McGarry JD, Unger RH. Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte–beta-cell relationships. Proc Natl Acad Sci USA. 1994;91:10878–10882. doi: 10.1073/pnas.91.23.10878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr, Ory DS, Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA. 2003;100:3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronert K, Musto A, Hardy M, Gimenez JM, Chiang N, Serhan CN, Bazan NG. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and proinflammatory gene expression. J Biol Chem. 2003;278:43807–43817. doi: 10.1074/jbc.M305841200. [DOI] [PubMed] [Google Scholar]
- Matsumura H, Shimizu Y, Ohsawa Y, Kawahara A, Uchiyama Y, Nagata S. Necrotic death pathway in Fas receptor signaling. J Cell Biol. 2000;151:1247–1256. doi: 10.1083/jcb.151.6.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Modification of ion homeostasis by lipid peroxidation: roles in neuronal degeneration and adaptive plasticity. Trends Neurosci. 1998;21:53–57. doi: 10.1016/s0166-2236(97)01188-0. [DOI] [PubMed] [Google Scholar]
- Mukherjee PK, Marcheselli VL, Serhan CN, Bazan NG. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc Natl Acad Sci USA. 2004;101:8491–8496. doi: 10.1073/pnas.0402531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehncrona S, Westerberg E, Akesson B, Siesjo BK. Brain cortical fatty acids and phospholipids during and following complete and severe incomplete ischemia. J Neurochem. 1982;38:84–93. doi: 10.1111/j.1471-4159.1982.tb10857.x. [DOI] [PubMed] [Google Scholar]
- Remmers M, Schmidt-Kastner R, Belayev L, Lin B, Busto R, Ginsberg MD. Protein extravasation and cellular uptake after high-dose human-albumin treatment of transient focal cerebral ischemia in rats. Brain Res. 1999;827:237–242. doi: 10.1016/s0006-8993(99)01304-9. [DOI] [PubMed] [Google Scholar]
- Rodriguez de Turco EB, Belayev L, Liu Y, Busto R, Parkins N, Bazan NG, Ginsberg MD. Systemic fatty acid responses to transient focal cerebral ischemia: influence of neuroprotectant therapy with human albumin. J Neurochem. 2002;83:515–524. doi: 10.1046/j.1471-4159.2002.01121.x. [DOI] [PubMed] [Google Scholar]
- Rokyta R, Racek J, Holecek V. Free radicals in the central nervous system. Cesk Fysiol. 1996;45:4–12. [PubMed] [Google Scholar]
- Schmidt-Kastner R, Aguirre-Chen C, Kietzmann T, Saul I, Busto R, Ginsberg MD. Nuclear localization of the hypoxia-regulated proapoptotic protein BNIP3 after global brain ischemia in the rat hippocampus. Brain Res. 2004;1001:133–142. doi: 10.1016/j.brainres.2003.11.065. [DOI] [PubMed] [Google Scholar]
- Singh SK, Moretta D, Almaguel F, Wall NR, De Leon M, De Leon D. Differential effect of proIGF-II and IGF-II on resveratrol induced cell death by regulating survivin cellular localization and mitochondrial depolarization in breast cancer cells. Growth Factors. 2007;25:363–372. doi: 10.1080/08977190801886905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg M, Edlund C, Kristensson K, Dallner G. Fatty acid composition of brain phospholipids in aging and in Alzheimer's disease. Lipids. 1991;26:421–425. doi: 10.1007/BF02536067. [DOI] [PubMed] [Google Scholar]
- Stillwell W, Shaikh SR, Zerouga M, Siddiqui R, Wassall SR. Docosahexaenoic acid affects cell signaling by altering lipid rafts. Reprod Nutr Dev. 2005;45:559–579. doi: 10.1051/rnd:2005046. [DOI] [PubMed] [Google Scholar]
- Sun D, Gilboe DD. Ischemia-induced changes in cerebral mitochondrial free fatty acids, phospholipids, and respiration in the rat. J Neurochem. 1994;62:1921–1928. doi: 10.1046/j.1471-4159.1994.62051921.x. [DOI] [PubMed] [Google Scholar]
- Tabernero A, Velasco A, Granda B, Lavado EM, Medina JM. Transcytosis of albumin in astrocytes activates the sterol regulatory element-binding protein-1, which promotes the synthesis of the neurotrophic factor oleic acid. J Biol Chem. 2002;277:4240–4246. doi: 10.1074/jbc.M108760200. [DOI] [PubMed] [Google Scholar]
- Ulloth JE, Casiano CA, De Leon M. Palmitic and stearic fatty acids induce caspase-dependent and -independent cell death in nerve growth factor differentiated PC12 cells. J Neurochem. 2003;84:655–668. doi: 10.1046/j.1471-4159.2003.01571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger RH. Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends Endocrinol Metab. 2003;14:398–403. doi: 10.1016/j.tem.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Unger RH, Orci L. Lipotoxic diseases of nonadipose tissues in obesity. Int J Obes Rel Metab Disord. 2000;24(Suppl 4):S28–S32. doi: 10.1038/sj.ijo.0801498. [DOI] [PubMed] [Google Scholar]
- Valentine RC, Valentine DL. Omega-3 fatty acids in cellular membranes: a unified concept. Prog Lipid Res. 2004;43:383–402. doi: 10.1016/j.plipres.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–5468. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, Vandenabeele P. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med. 1998;188:919–930. doi: 10.1084/jem.188.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A, Molteni R, Ying Z, Gomez-Pinilla F. A saturated-fat diet aggravates the outcome of traumatic brain injury on hippocampal plasticity and cognitive function by reducing brain-derived neurotrophic factor. Neuroscience. 2003;119:365–375. doi: 10.1016/s0306-4522(03)00154-4. [DOI] [PubMed] [Google Scholar]
- Yehuda S, Rabinovitz S, Carasso RL, Mostofsky DI. The role of polyunsaturated fatty acids in restoring the aging neuronal membrane. Neurobiol Aging. 2002;23:843–853. doi: 10.1016/s0197-4580(02)00074-x. [DOI] [PubMed] [Google Scholar]
- Yu-Poth S, Yin D, Zhao G, Kris-Etherton PM, Etherton TD. Conjugated linoleic acid upregulates LDL receptor gene expression in HepG2 cells. J Nutr. 2004;134:68–71. doi: 10.1093/jn/134.1.68. [DOI] [PubMed] [Google Scholar]
- Yu-Poth S, Yin D, Kris-Etherton PM, Zhao G, Etherton TD. Long-chain polyunsaturated fatty acids upregulate LDL receptor protein expression in fibroblasts and HepG2 cells. J Nutr. 2005;135:2541–2545. doi: 10.1093/jn/135.11.2541. [DOI] [PubMed] [Google Scholar]