

Abstract
The extracellular matrix plays a key role in organ formation and tissue homeostasis. Recent studies have revealed that fibrillin assemblies (microfibrils) both confer tissue integrity and regulate signaling events that instruct cell performance and that perturbation of either function manifests in disease. These analyses have also indicated that fibrillin assemblies impart contextual specificity to TGFβ and BMP signaling. Moreover, correlative evidence suggests functional coupling between cell-directed assembly of microfibrils and targeting of TGFβ and BMP complexes to fibrillins. Hence, the emerging view is that fibrillin-rich microfibrils are molecular integrators of structural and instructive signals with TGFβs and BMPs as nodal points that convert extracellular inputs into discrete and context-dependent cellular responses.
Introduction
The interaction of bioactive ligands with the extracellular matrix (ECM) represents an important mechanism that establishes morphogen gradients in the developing embryo and that modulates instructive signals in the adult organism. In contrast to the well-established involvement of heparan sulfate proteoglycans (HSPG) in the extracellular regulation of several signaling pathways [1], a comparable role for the architectural elements of the ECM is only beginning to emerge. In Drosophila, genetic analyses have demonstrated that interaction between Decapentaplegic (Dpp) and collagen IV not only enhances Dpp signaling in the early embryo by promoting gradient formation, but also restricts the range of growth factor action in the ovary by sequestering Dpp complexes [2••]. Similar studies in mice have correlated mutations of fibrillin-1 with heightened TGFβ signaling in Marfan syndrome (MFS; OMIM #154700), and loss of fibrillin-2 with decreased BMP7 signaling in developing limbs [3, 4].
Fibrillin macro-aggregates perform two critical functions in organismal physiology; they provide a structural scaffold that imparts physical properties to connective tissue and they act as an instructive platform for soluble modulators of cell behavior. To generate morphologically and functionally diverse macro-aggregates, cells must tightly regulate the multiple protein interactions of fibrillins. Therefore, impaired microfibril biogenesis and/or cellular responses to abnormal matrices are probable contributors to dysregulated TGFβ and BMP signaling in fibrillin-deficient states. This review highlights recent findings that provide insights into how fibrillins control endogenous TGFβ and BMP signaling, and that suggest cell-mediated coupling of fibrillin microfibril nucleation at the plasma membrane with targeting of TGFβ and BMP complexes to fibrillins. These findings are discussed within the context of probable disease mechanisms resulting from fibrillin mutations that impair ECM integrity and growth factor sequestration.
Formation of microfibril scaffolds
Fibrillins are large cysteine-rich glycoproteins that form supramolecular arrays (microfibrils) that incorporate or associate with a variety of proteins, including TGFβ and BMP complexes, integrins, HSPGs, and insoluble elastin in elastic fibers [5•]. Assembly of fibrillins into microfibrils and microfibrils into elastic fibers are complex multistep processes that are yet to be fully delineated [5•]. Recent studies have focused on the mechanisms that control the initial polymerization of fibrillins into the characteristic beads-on-a-string microfibril structure in which individual molecules are organized in a head-to-tail arrangement and associate laterally (Fig. 1). Using recombinant polypeptides, Humbacher et al. [6••] demonstrated that the C-terminal half of fibrillin-1 drives formation of disulfide-bonded bead-like globular structures, which interact with N-terminal portion of fibrillin-1 to regulate linear assembly and inhibit microfibril formation when added to cell cultures. These investigators subsequently reported that the C-terminal globular multimers (but not fibrillin monomers) bind in vitro to a specific sequence of fibronectin, and that either silencing fibronectin biosynthesis or inhibiting fibronectin assembly disrupts microfibril formation (but not vice versa) [7••]. Additional analyses revealed that newly formed microfibrils localize to fibronectin fibrils, and that exogenously added fibrillin-1 can only assemble on a fibronectin network [7••]. Other investigators showed that antibody activation of β1 integrins, which mediate cell-driven assembly of fibronectin, could not restore microfibril formation in fibronectin-null cells [8]. These observations reiterate the central role of fibronectin in organizing early ECM assembly and in maintaining cell-matrix adhesion sites [9].
Figure 1.
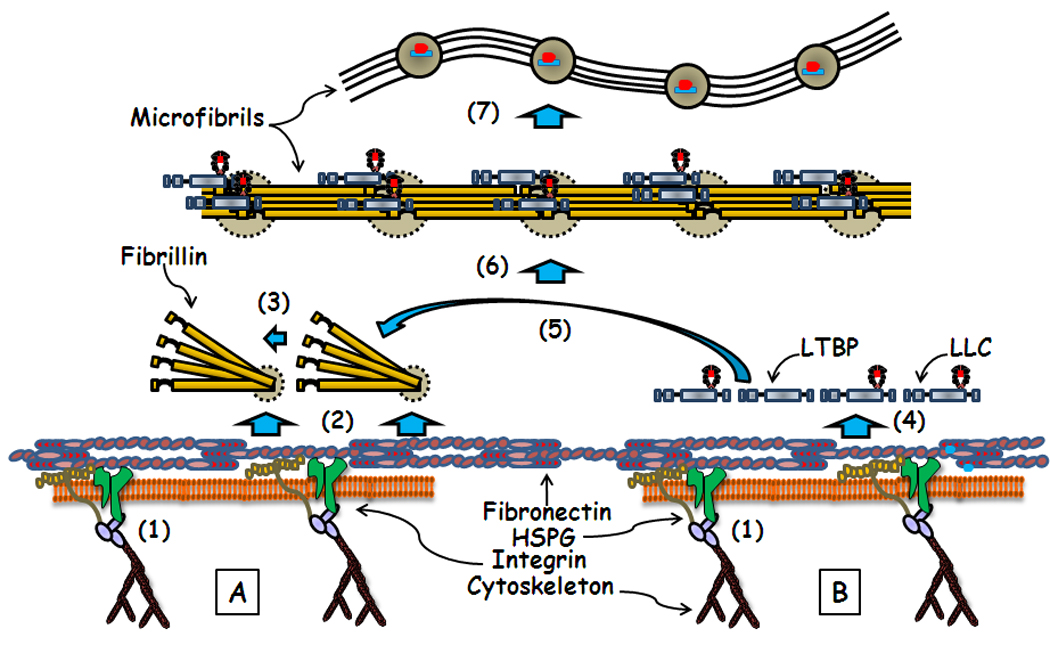
Schematic representation of the fibronectin-directed, HSPG-dependent processes of microfibril assembly (A) and LTBP and LLC incorporation into the ECM (B). Relevant steps discussed in the text include (1) promotion of fibronectin fibrillogenesis by the cytoskeleton/integrin complex; (2) fibronectin and HSPG supporting C-terminal association of fibrillin monomers into bead-like structures (grey circle with dotted outline), and (3) their subsequent linear assembly into microfibrils; (4) HSPG-dependent incorporation of LTBPs and LLCs onto fibronectin fibrils, and (5) their subsequent transfer to nascent microfibrils through ill-defined cellular activities. Also shown are (6) microfibrils decorated by LTBPs and LLC in the overlaps (beads in (8)) between the N- and C-terminal ends of fibrillins, and (8) a schematic rendition of the beads-on-a-string microfibril structure with LLCs bound to the beads [5].
Earlier work had shown that fibrillins contain several heparin-binding sites, and that HSPG inhibition reduces fibrillin polymerization [10–12]. Bax et al. [13] recently implicated a syndecan/glypican HS-binding site located immediately next to the Arg-Gly-Asp (RGD) sequence of fibrillin-1 as enhancing α5β1-mediated focal adhesion formation. Furthermore, Cain et al [14] correlated an N-terminal heparin-binding site with supporting the linear assembly of fibrillin molecules. Collectively, these data suggest that interactions with fibronectin fibrils and HSPGs control the initial steps of microfibril biogenesis on or close to the cell surface (Fig. 1). An attractive aspect of this model is that protein self-assembly is promoted by locally concentrating interacting molecules as they are secreted into the pericellular space. Similar involvement of fibronectin and HSGPs in targeting latent TGFβ complexes to nascent microfibrils (see below) raises the possibility that cells coordinate the formation of microfibril scaffolds with the sequestration of growth factors.
Targeting growth factors to microfibril scaffolds
TGFβs and BMPs specify a plethora of cellular activities, including ECM deposition and degradation, and are regulated at multiple levels, including by the storage in and release from the ECM [15••]. Control of matrix targeting of TGFβs initiates intracellularly with the covalent association between latent TGFβ-binding protein (LTBP) -1, -3 or 4 and small latent complexes (SLCs), which consist of bioactive TGFβ dimers non-covalently bound to their pro-domains (latency-associated protein or LAP). The resulting tripartite large latent complex (LLC) is secreted and binds to fibronectin and fibrillin assemblies through specific N- and C-terminal LTBP sequences, respectively (Fig. 1) [16]. Genetic evidence indicates that LTBPs also have a distinct structural role in the ECM [17,18].
Cell culture experiments suggest that initial interactions between LTBPs and ECM components (and thus sorting and targeting of LLCs to the ECM) occur at the plasma membrane with direct involvement of fibronectin fibrils and HSPGs [19, 20•, 21]. LTBPs subsequently localize to fibrillin-rich microfibrils through cell-directed reorganization of the provisional matrix (Fig. 1) [22]. Relocation of LTBPs and LLCs from fibronectin fibrils to nascent fibrillin polymers is associated with cells actively shunting ECM material from one location to another, in the absence of significant proteolytic activity [22]. Although less well understood, the mechanisms that modulate extracellular control of BMP bioavailability differ from those of TGFβ because of two features. First, BMPs are directly targeted to microfibrils by non-covalent association between their pro-domains and the N-terminal region of fibrillins [23•]. Second, BMP signaling can be activated through competitive displacement of the pro-domain by type II receptors [24•]. However, matrix-bound BMPs may still require release from fibrillins to signal. By analogy with Dpp regulation in Drosophila [2••], extracellular microfibrils may also sequester BMPs associated with soluble antagonists.
Release and activation of latent TGFβs from microfibril scaffolds
The prerequisite binding of TGFβs to microfibrils for proper function implies that growth factor action is modulated not only by the process of microfibril assembly, but also by the liberation and activation of ligands from the ECM [16]. Although earlier in vitro analyses suggested that proteases could release and activate TGFβs from the ECM, subsequent genetic studies failed to show diminished TGFβ signaling in mice lacking individual proteases probably because of proteolytic redundancy in the tissue environment [25]. The finding that mice without both BMP1 and Tolloid-like 1 activity (proteases that control both ECM formation and growth factors activity) display decreased TGFβ signaling supports this conclusion [26].
Unlike proteases, integrin-mediated activation of LLC is clearly a component of the extracellular control of TGFβ bioavailability [27]. Integrin αVβ6 activates latent TGFβs by binding the RGD sequence of LAP-β1 or -β3 and by associating with the cytoskeleton through the β6 subunit’s cytoplasmic tail [28]. LTBP-mediated binding of SLC to the matrix allows αVβ6 integrin to apply force and alter the latent complex structure. Furthermore, the finding that αVβ6 integrin activates only latent TGFβs associated with LTBP-1 suggests involvement of LTBP-specific interactions with the ECM [29]. The importance of αVβ6-mediated activation of latent TGFβs is underscored by recent studies showing that mice harboring mutations in the integrin-binding site or the LTBP-binding site of TGFβ1 phenocopy the abnormalities of Tgfβ1-null mice [30•, 31•]. Additionally, the demonstration that αVβ5 integrin and LTBP-1 cooperate in latent TGFβ activation by myofibroblasts indicates that, to promote activation, the ECM must resist the traction applied by the integrin/cytoskeletal complex [32••]. Myofibroblasts placed on compliant matrices fail to activate LLC, whereas those placed on more resistant matrices produce active TGFβs [32••]. Integrin αVβ8 activates latent TGFβs through a different mechanism that may require tethering of the cell-surface protease MT1-MMP to the LLC/integrin complex, but not integrin binding to the cytoskeleton [33]. Hence, integrins αVβ5 and αVβ6 must distort LLC conformation to either free or expose TGFβs to receptors, whereas αVβ8 integrin promotes protease activation of latent complexes.
Molecular consequences of microfibril scaffold disruption
If ECM incorporation is essential for proper control of TGFβ and BMP action, interference with matrix assembly should perturb growth factor signaling. Heterozygous mutations of fibrillin-2 result in congenital contractural arachnodactyly (CCA; OMIM # 121050), a connective tissue disorder akin to MFS, whereas heterozygous mutations of fibrillin-1 cause the pleiotropic manifestations of MFS, which principally involve the ocular, musculoskeletal and cardiovascular systems [5•]. Fbn1 or Fbn2 mutant mice exhibit distinct phenotypes associated with discrete perturbations of TGFβ and BMP signaling [5]. Syndactyly is the most evident phenotype of Fbn2-null mice and the unique trait of mice haploinsufficient for both Fbn2 and Bmp7 [3]. By contrast, Fbn1 mutant mice show normal digit formation but display abnormalities in the cardiovascular, respiratory, and muscular systems secondary to heightened TGFβsignaling [3, 4, 34–37•]. These phenotypic disparities in organs that accumulate large amounts of both fibrillin-1 and -2 imply that the mechanisms targeting TGFβ and BMP complexes to microfibrils are tissue-, stage-, and fibrillin-specific.
The realization that defective fibrillin-1 assembly promotes latent TGFβ activation has provided the rationale for using systemic TGFβ antagonism to mitigate MFS manifestations in mice [4, 34–36]. These proof-of-principle experiments have recently translated into a drug-based therapy (losartan, an angiotensin II type 1 receptor antagonist known to decrease TGFβ signaling) that reduces the rate of aortic growth in children with severe and rapidly progressive MFS [32••]. As a result, a model of MFS pathogenesis has emerged whereby FBN1 mutations preclude or decrease matrix sequestration of LLCs, rendering latent TGFβ more prone to or more accessible for activation with negative consequences to cell function and tissue remodeling [35, 4]. In this view, physiological levels of latent TGFβ activators are sufficient to drive disease progression in MFS irrespective of whether FBN1 mutations affect protein structure or expression (Fig. 2). In contrast to structural mutations in which phenotypic variability may reflect discrete perturbations of microfibril formation, clinical variability in MFS patients haploinsufficient or nearly haploinsufficient for FBN1 implicates additional factors in TGFβ-driven disease progression [33, 40]. One possibility is that the structurally abnormal ECM promotes cellular responses that contribute to MFS pathogenesis by heightening latent TGFβ activation through the action of integrins, proteases and/or other molecules (Fig. 2).
Figure 2.
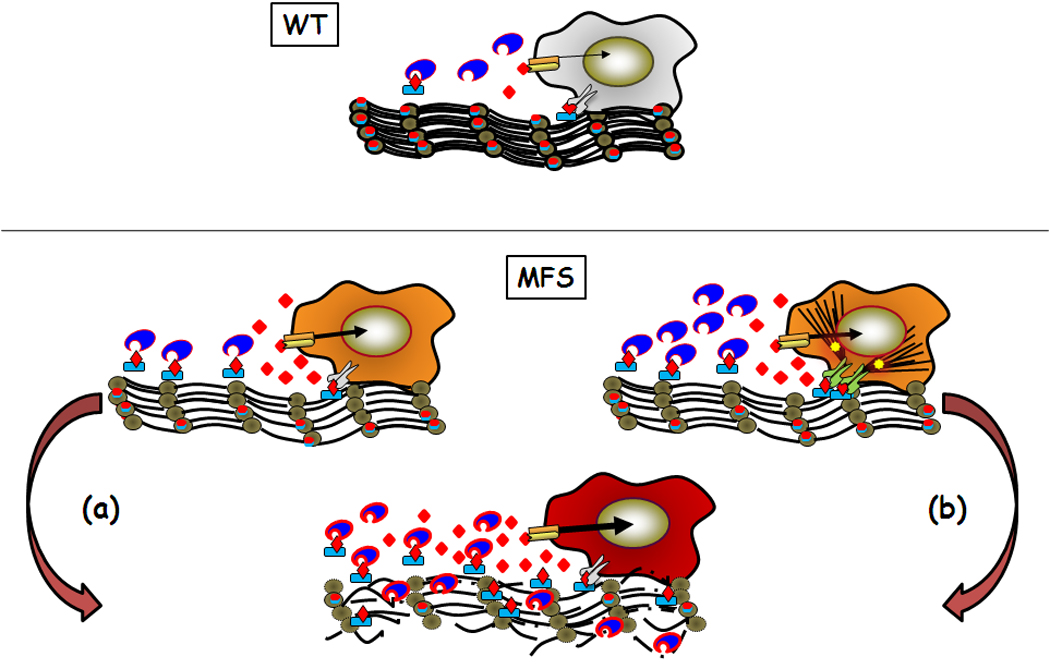
Model of normal regulation of TGFβ by microfibrils (WT) and perturbations associated with microfibril deficiency in Marfan syndrome (MFS). Two models of MFS pathogenesis leading to loss of tissue integrity (bottom) are shown. In the first model (a), physiological levels of activators (proteases (blue indented ovals) and integrins (grey symbols)) drive disease by increasing release of TGFβ (red diamonds) from matrix-free LLCs (blue rectangle with red diamond). In the second model (b), cells enhance TGFβ activity by producing more proteases, by increasing integrin-mediated LLC activation (also through heightened cytoskeletal tension (black lines)), and by stimulating stress-response signaling pathways (yellow stars). Although the two models focus on the onset of MFS pathogenesis, impaired LLC sequestration (model (a)) could be the immediate trigger of cellular responses to an abnormal matrix (model (b)).
Several lines of indirect evidence support the above hypothesis. These include the aforementioned observation that loss of LTBP association with microfibrils impairs elastogenesis [17, 18], the finding that interference with LLC matrix sequestration results in TGFβ over-activation [41], the discovery that reduced expression of microfibril-associated fibulin-4 increases TGFβ signaling and perturbs cardiovascular homeostasis [42], and the report that improper p38 MAPK signaling is an early determinant of constitutive Smad2/3 activity in Fbn1-null aortas [43•]. It is also noteworthy that vascular manifestations similar to those of MFS mutations are observed with mutations that affect smooth muscle cell contraction, such as β-myosin and α-actin, or the plasma membrane receptor LRP1, an integrator of PDGF and TGFβ signals in the aortic wall, [44–46•]. Additional factors connected with ECM remodeling that may further exacerbate TGFβ-driven aneurysm progression include a C-terminal fragment of fibrillin-1 that can displace LTBPs from microfibrils, and fibrillin-1 peptides that stimulate macrophage chemotaxis and MMP production [47, 48•, 49]. Consistent with the latter observation, systemic MMP inhibition improves aortic wall architecture and delays aneurysm rupture in Fbn1 mutant mice [50, 51].
Conclusions and perspectives
The studies we have described demonstrate that fibrillins play a vital role in organ formation and tissue homeostasis by imparting structural integrity to connective tissues and by modulating the activity of growth factors that regulate ECM formation and remodeling. Further investigations are required to determine the mechanistic relationships between microfibril biogenesis and TGFβ and BMP targeting to fibrillins, and between fibrillin interactions with secreted and cell-surface molecules and the formation of tissue- and stage-specific macro-aggregates. Likewise, additional effort is needed to delineate the pathophysiological sequence that links fibrillin-deficient matrices with perturbed growth factor bioavailability and cell performance. Biochemical, cellular and genetic approaches, together with new technologies to monitor the dynamics of ECM interactions in real time, should instruct progress of these studies.
Acknowledgements
Studies from the authors’ laboratories that are described in the review were supported by grants from the National Institutes of Health (AR-049698, AR-42044, CA034282, and AR49698) and the National Marfan Foundation. We thank Ms. Karen Johnson for organizing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and annotations
- 1.Häcker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 2005;6:530–541. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- ••2. Wang X, Harris RE, Bayston LJ, Ashe HL. Type IV collagens regulate BMP signaling in Drosophila. Nature. 2008;455:72–77. doi: 10.1038/nature07214. This elegant study implicates Drosophila collagen IV in the regulation of Dpp signaling, in addition to predicting a similar function for this basement membrane component in modulating BMP activity during vertebrate development.
- 3.Arteaga-Solis E, Gayraud B, Lee SY, Shum L, Sakai L, Ramirez F. Regulation of limb patterning by extracellular microfibrils. J. Cell Biol. 2001;154:275–281. doi: 10.1083/jcb.200105046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-β activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- •5. Ramirez F, Sakai LY, Rifkin DB, Dietz HC. Extracellular microfibrils in development and disease. Cell Mol Life Sci. 2007;64:2437–2446. doi: 10.1007/s00018-007-7166-z. A comprehensive and updated review of fibrillin structure, assembly and interactions, as well the roles of microfibrils in development and disease.
- ••6.Hubmacher D, El-Hallous EI, Nelea V, Kaartinen MT, Lee ER, Reinhardt DP. Biogenesis of extracellular microfibrils: Multimerization of the fibrillin-1 C terminus into bead-like structures enables self assembly. Proc. Natl. Acad. Sci. USA. 2008;105:6548–6553. doi: 10.1073/pnas.0706335105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••7. Sabatier L, Chen D, Fagotto-Kaufmann C, Hubmacher D, McKee MD, Annis DS, Mosher DF, Reinhardt DP. Fibrillin assembly requires fibronectin. Mol. Biol. Cell. 2009;20:846–858. doi: 10.1091/mbc.E08-08-0830. These two studies provide important new insight into the mechanism of microfibril nucleation. Together they show that specific domains in the C-terminal half of fibrillin-1 promote formation of a mutimeric structure that drives linear assembly of microfibrils, and that interaction with fibronectin fibrils is absolutely required for the formation of this fibrillin multimer.
- 8.Kinsey R, Willamson MR, Chaudhry S, Mellody KT, McGovern A, Takahashi S, Shuttleworth CA, Kielty CM. Fibrillin-1 microfibril deposition is dependent on fibronectin assembly. J. Cell Sci. 2008;121:2696–2704. doi: 10.1242/jcs.029819. [DOI] [PubMed] [Google Scholar]
- 9.Sottile J, Hocking DC. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol. Biol. Cell. 2002;13:3546–3559. doi: 10.1091/mbc.E02-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tiedemann K, Batge B, Muller PK, Reinhardt DP. Interactions of fibrillin-1 with heparin/heparin sulfate implications for microfibrillar assembly. J. Biol. Chem. 2001;276:36035–36042. doi: 10.1074/jbc.M104985200. [DOI] [PubMed] [Google Scholar]
- 11.Ritty TM, Broelmann TJ, Werneck CC, Mecham RP. Fibrillin-1 and -2 contain heparin-binding sites important for matrix deposition and that support cell attachment. Biochem. J. 2003;375:425–432. doi: 10.1042/BJ20030649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cain SA, Baldock C, Gallagher J, Morgan A, Bax DV, Weiss AS, Shuttleworth CA, Kielty CM. Fibrillin-1 interactions with heparin. J. Biol. Chem. 2005;280:30526–30537. doi: 10.1074/jbc.M501390200. [DOI] [PubMed] [Google Scholar]
- 13.Bax DV, Mahalingam Y, Cain S, Mellody K, Freeman L, Younger K, Shuttleworth CA, Humphries MJ, Couchman JR, Kielty CM. Cell adhesion to fibrillin-1: identification of an Arg-Gly-Asp-dependent synergy region and a heparin-binding site that regulates focal adheshion formation. J. Cell Sci. 2007;120:1383–1392. doi: 10.1242/jcs.003954. [DOI] [PubMed] [Google Scholar]
- 14.Cain SA, Baldwin AK, Mahalingam Y, Raynal B, Jowitt TA, Shuttleworth CA, Couchman JR, Kielty CM. Heparan sulfate regulates fibrillin-1 and C-terminal interactions. J. Biol. Chem. 2008;283:27017–27027. doi: 10.1074/jbc.M803373200. [DOI] [PubMed] [Google Scholar]
- ••15. Derynck R, Miyazono K. The TGFβ family. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2008. A must-have book written by an outstanding group of experts that covers all the pertinent aspects of signaling by TGFβ superfamily members in normal and disease conditions.
- 16.Rifkin DB. Latent transforming growth factor-β (TGF-β) binding proteins: orchestrators of TGF-β availability. J. Biol. Chem. 2005;280:7409–7412. doi: 10.1074/jbc.R400029200. [DOI] [PubMed] [Google Scholar]
- 17.Sterner-Kock A, Thorey IS, Koli K, Wempe F, Otte J, Bangsow T, Kuhlmeier K, Jin S, Keski-Oja J, von Melchner H. Disruption of the gene encoding the latent transforming growth factor-β binding protein 4 (LTBP-4) cause abnormal lung development, cardiomyopathy, and colorectal cancer. Genes Dev. 2002;16:2264–2273. doi: 10.1101/gad.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dabovic B, Chen Y, Choi J, Vassallo M, Dietz HC, Ramirez F, von Melchner H, Davis CC, Rifkin DB. Dual functions for LTBP in lung development: LTBP-4 independently modulates elastogenesis and TGF-β activity. J. Cell. Physiol. 2009;219:14–22. doi: 10.1002/jcp.21643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dallas SL, Sivakumar P, Jones CJP, Chen Q, Peters DM, Mosher DF, Humphries MJ, Kielty CM. Fibronectin regulates latent transforming growth factor-β (TGFβ) by controlling matrix assembly of latent TGFβ binding protein-1. J. Biol. Chem. 2005;280:18871–18880. doi: 10.1074/jbc.M410762200. [DOI] [PubMed] [Google Scholar]
- •20. Chen Q, Sivakumar P, Barley C, Peters DM, Gomes RR, Farach-Carson MC, Dallas SL. Potential role for heparan sulfate proteoglycans in regulation of transforming growth factor-β (TGF-β) by modulating assembly of latent TGF-β-binding protein-1. J. Biol. Chem. 2007;282:26418–26430. doi: 10.1074/jbc.M703341200. First in vitro evidence that HSPGs are required to incorporate LTBPs (and implicitly latent TGFβs) onto fibronectin fibrils.
- 21.Kantola AK, Keski-Oja J, Koli K. Fibronectin and heparin binding domains of latent TGF-β binding protein (LTBP)-4 mediate matrix targeting and cell adhesion. Exp Cell Res. 2008;314:2488–2500. doi: 10.1016/j.yexcr.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 22.Sivakumar P, Czirok A, Rongish BJ, Divakara VP, Wang YP, Dallas SL. New insights into extracellular matrix assembly and reorganization from dynamic imaging of extracellular matrix proteins in living osteoblasts. J. Cell Sci. 2006;119:1350–1360. doi: 10.1242/jcs.02830. [DOI] [PubMed] [Google Scholar]
- •23. Sengle G, Charbonneau NL, Ono RN, Sasaki T, Alvarez J, Keene DR, Bachinger HP, Sakai LY. Targeting of bone morphogenetic protein growth factor complexes to fibrillin. J. Biol. Chem. 2008;283:13874–13888. doi: 10.1074/jbc.M707820200. This paper describes the identification of sequences in fibrillin-1 and -2 that mediate non-covalent interactions between microfibrils and several BMP/GDF ligands.
- •24. Sengle G, Ono RN, Lyons KM, Bachinger HP, Sakai LY. A new model for growth factor activation: type II receptors compete with the prodomain for BMP-7. J. Mol. Biol. 2008;381:1025–1039. doi: 10.1016/j.jmb.2008.06.074. An interesting study that suggests a new model for pro-BMP7 signaling based on the ability of type II receptors to compete with the BMP prodomain for ligand binding.
- 25.Jenkins G. The role of proteases in transforming growth factor-β activation. Int. J. Biochem. Cell Biol. 2008;40:1068–1078. doi: 10.1016/j.biocel.2007.11.026. [DOI] [PubMed] [Google Scholar]
- 26.Ge G, Greenspan DS. BMP1 controls TGFβ1 activation via cleavage of latent TGFβ-binding protein. J. Cell Biol. 2006;175:111–120. doi: 10.1083/jcb.200606058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wipff P-J, Hinz B. Integrins and the activation of latent transforming growth factor β1-An intimate relationship. Eur. J. Cell Biol. 2008;87:601–615. doi: 10.1016/j.ejcb.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 28.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, et al. The integrin αvβ6 binds and activates latent TGF-β1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 29.Annes JP, Chen Y, Munger JS, Rifkin DB. Integrin αVβ6-mediated activation of latent TGF-β requires the latent TGF-β binding protein-1. J. Cell Biol. 2004;165:723–734. doi: 10.1083/jcb.200312172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •30. Yang Z, Mu Z, Dabovic B, Jurukovski V, Yu D, Sung J, Xiong X, Munger JS. Absence of integrin-mediated TGFβ1 activation in vivo recapitulates the phenotype of TGFβ1-null mice. J. Cell Biol. 2007;176:787–793. doi: 10.1083/jcb.200611044. This paper presents genetic evidence showing that LAP binding to integrins is responsible for most of TGFβ1 activity in the developing mouse.
- •31. Yoshinaga K, Obata H, Jurukovski V, Mazzieri R, Chen Y, Zilberberg L, Huso D, Melamed J, Prijatelj P, Todorovic V, et al. Perturbation of transforming growth factor (TGF)-β1 association with latent TGF-β binding protein yields inflammation and tumors. Proc. Natl. Acad. Sci. USA. 2008;105:18758–18763. doi: 10.1073/pnas.0805411105. The authors provide genetic confirmation that binding of TGFβ1 to LTBPs and integrins is required for optimal growth factor signaling during and after mouse development.
- ••32. Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 2007;17:1311–1323. doi: 10.1083/jcb.200704042. A seminal study that demonstrates how the balance between ECM pliability and cytoskeletal tension specify integrin-mediated activation of latent TGFβ. This new model of contraction-mediated TGFβ activation has important ramifications for our understanding of disease progression in fibrotic conditions.
- 33.Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. The integrin αvβ8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGFβ1. J. Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, Gabrielson KL, Hausladen JM, Mecham RP, Judge DP, et al. TGF-β-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J. Clin. Invest. 2004;114:1586–1592. doi: 10.1172/JCI22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carta L, Pereira L, Arteaga-Solis E, Lee-Arteaga SY, Lenart B, Starcher B, Merkel CA, Sukoyan M, Kerkis A, Hazeki N, et al. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J. Biol. Chem. 2006;281:8016–8023. doi: 10.1074/jbc.M511599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •37. Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, Gamradt M, ap Rhys CM, Holm TM, Loeys BL, et al. Angiotensin II Type 1 receptor blockade prevents TGFβ-induced failure of muscle regeneration in multiple myopathic states. Nature Med. 2007;13:204–210. doi: 10.1038/nm1536. Aside from confirming the importance of TGFβ signaling in MFS pathogenesis, this study indicates that losartan may also prove an effective strategy for diseases affecting muscle mass and regeneration.
- ••38. Brooke BS, Habashi JP, Judge D, Patel N, Loeys B, Dietz HC. Angiotensin II blockade and aortic-root dilation in Marfan's syndrome. N. Engl. J. Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. The first human study to report the efficacy of losartan therapy to mitigate progression of aortic dilation in MFS. The study also represents the first example in which life-threatening manifestations have been improved in a congenital disorder of the connective tissue. As such, this experience is bound to influence therapeutic efforts for other disorders in which gene- or cell-based strategies are difficult or impossible to implement.
- 39.Schrijver I, Liu W, Odom R, Brenn T, Oefner P, Furthmayr H, Francke U. Premature termination mutations in FBN1: Distinct effects on differential allelic expression and on protein and clinical phenotypes. Am. J. Hum. Genet. 2002;71:223–237. doi: 10.1086/341581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matyas G, Alonso S, Patrignani A, Marti M, Arnold E, Magyar I, Henggeler C, Carrel, Steimann B, Berger W. Large genomic fibrillin-1 (FBN1) gene deletions provide evidence for true haploinsufficiency in Marfan syndrome. Hum. Genet. 2007;122:23–32. doi: 10.1007/s00439-007-0371-x. [DOI] [PubMed] [Google Scholar]
- 41.Mazzieri R, Jurukovski V, Obata H, Sung J, Platt A, Annes E, Karaman-Jurukovska N, Gleizes PE, Rifkin DB. Expression of truncated latent TGF-β-binding protein modulates TGF-β signaling. J. Cell. Sci. 2005;118:2177–2187. doi: 10.1242/jcs.02352. [DOI] [PubMed] [Google Scholar]
- 42.Hanada K, Vermeij M, Garinis GA, de Waard MC, Kunen MGS, Myers L, Maas A, Duncker DJ, Meijers C, Dietz HC, Kanaar R, Essers J. Perturbations of vascular homeostasis and aortic valve abnormalities in fibulin-4 deficient mice. Circulation Res. 2007;100:738–746. doi: 10.1161/01.RES.0000260181.19449.95. [DOI] [PubMed] [Google Scholar]
- •43. Carta L, Smaldone S, Zilberberg L, Loch D, Dietz HC, Rifkin DB, Ramirez F. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J. Biol. Chem. 2009;284:5630–5636. doi: 10.1074/jbc.M806962200. The first report of the possible involvement of non-canonical Smad signaling in the progression of vascular disease in MFS.
- 44.Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc J-M, Brunotte F, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 45.Boucher P, Li W-P, Maz RL, Takayama Y, Auwerx J, Anderson RGW, Herz J. LRP1 functions as an atheroprotective integrator of TGFβ and PDFG signals in the vascular wall: implications for Marfan syndrome. PLoS One. 2007;16:e448. doi: 10.1371/journal.pone.0000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •46. Guo D-C, Pannu H, Tra-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, et al. Mutations in smooth muscle α-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. The identification of smooth muscle α-actin mutations in familial thoracic aortic aneurysm (TAA) that corroborates the importance of smooth muscle cell contraction in maintaining aortic wall integrity.
- 47.Booms P, Pregla R, Ney A, Barthel F, Reinhardt DP, Pletschacher A, Mundlow S, Robinson PN. RGD-containing fibrillin-1 fragments upregulate matrix metalloproteinase expression in cell culture: a potential factor in the pathogenesis of the Marfan syndrome. Hum. Genet. 2005;116:51–61. doi: 10.1007/s00439-004-1194-7. [DOI] [PubMed] [Google Scholar]
- •48. Chaudhry SS, Cain SA, Morgan A, Dallas SL, Shuttleworth CA, Kielty CM. Fibrillin-1 regulates the bioavailability of TGFβ1. J. Cell Biol. 2007;176:355–367. doi: 10.1083/jcb.200608167. Intriguing findings that strongly suggest the presence of a TGFβ-releasing fragment within fibrillin-1 that could enhance LLC activation during ECM degradation by inflammatory proteolytic enzymes.
- 49.Chung AWY, Yang HHC, Radomski MW, van Breemen C. Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in Marfan syndrome through the inhibition of matrix metalloproteinase-2 and -9. Circulation Res. 2008;102:e73–e85. doi: 10.1161/CIRCRESAHA.108.174367. [DOI] [PubMed] [Google Scholar]
- 50.Guo G, Booms P, Halushka M, Dietz HC, Ney A, Stricker S, Hecht J, Mundlos S, Robinson PN. Induction of macrophage chemotaxis by aortic extracts of the mgR Marfan mouse model and a GxxPG-containing fibrillin-1 fragment. Circulation. 2006;114:1855–1862. doi: 10.1161/CIRCULATIONAHA.105.601674. [DOI] [PubMed] [Google Scholar]
- 51.Xiong W, Knispel RA, Dietz HC, Ramirez F, Baxter BT. Doxycycline delays aneurysm rupture in a mouse model of Marfan syndrome. J. Vasc. Surg. 2008;47:166–172. doi: 10.1016/j.jvs.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]