

Abstract
Background
Polycystic ovary syndrome (PCOS) is a heterogenic, complex common genetic disease. Multiple pathways are involved in its pathogenesis, including the androgen signaling pathway and insulin signaling pathway. Small glutamine-rich tetratricopeptide repeat (TPR)-containing protein alpha (SGTA) is a putative member of the androgen receptor-chaperone-cochaperone complex, and may play a role in androgen signaling as a co-chaperone. Polymorphisms in the SGTA gene have not been evaluated for a role in PCOS.
Methods
Women with and without PCOS (287 cases, 187 controls) were genotyped for three single nucleotide polymorphisms (SNPs) in SGTA. SNPs and haplotypes were determined and tested for association with PCOS and component traits of PCOS.
Results
For SNP rs1640262, homozygotes for the minor allele were protected against PCOS (P=0.009). Haplotype 1 (G-A-T) was associated with increased risk of PCOS (P=0.015). In women with PCOS, haplotype 2 (A-G-C) was associated with increased insulin resistance (P=0.013), consequently resulting in increased insulin secretion (P=0.014).
Conclusions
This study presents genetic evidence suggesting a potential role of SGTA in the pathogenesis of PCOS. SGTA may provide a connection between multiple pathways in PCOS.
Keywords: Polycystic ovary syndrome, small glutamine-rich tetratricopeptide repeat-containing, alpha, single nucleotide polymorphism, haplotype, association
Introduction
Polycystic ovary syndrome (PCOS) is a heterogenic, complex common genetic disease, affecting approximately 6-8% of reproductive age women (Azziz et al. 2004). PCOS is characterized by hyperandrogenism, menstrual dysfunction, and polycystic ovarian morphology (Goodarzi and Azziz 2006). Patients with PCOS may also present with infertility, obesity, insulin resistance, and often have a high burden of cardiovascular risk factors.
The molecular basis for PCOS is poorly understood. Candidate genes for PCOS have been chosen from logical pathways, such as the insulin signaling pathway or androgen biosynthetic pathway (Escobar-Morreale et al. 2005, Franks and McCarthy 2004, Nam Menke and Strauss 2007, Urbanek 2007). Hypotheses regarding these pathophysiologic pathways have led to the implication of several genes in the development of PCOS. However, since PCOS is a complex common disease, which is likely due to both genetic and environmental factors, more evidence is needed to explain the molecular mechanisms of PCOS.
Although the etiology of PCOS remains unknown, most patients with PCOS present with hyperandrogenism. The phenotype of PCOS may have hirsutism and ovulatory dysfunction without significantly increased androgens (Azziz et al. 2006). Up to 25% of PCOS patients have normal levels of circulating androgens, which suggests sensitivity to androgens is increased in this type of PCOS (Chang et al. 2005). Even in those with hyperandrogenemia, the degree of androgen elevation is typically mild. Therefore, components of androgen signaling may play a key role in the pathophysiology of PCOS, in some or all subjects. Unbound androgen receptor (AR) is inactive in the cytoplasm as a large dynamic heterocomplex which is composed of heat shock proteins (such as Hsp70 and Hsp90) and their co-chaperones (Pratt and Toft 1997). Three main families of co-chaperones include Bag-1 homology (Bcl-2 binding athanogene or Bag domains), DnaJ homology (or J domains), and tetratricopeptide repeat (TPR) containing proteins (Young et al. 2003). The complex of chaperones and their co-chaperones binds to the hormone binding domain of AR, and assists AR to function properly at each step of its functional cycle (Pratt and Toft 1997). Abnormal function or expression of co-chaperones is proposed to affect AR activity. For example, in tumor cells of prostate cancer, the abnormal Hsp70 and Bag-1L expression in the secretory epithelium enhances transcriptional activity of the AR (Shatkina et al. 2003). This suggests that genes encoding components of the co-chaperone complex can be candidates for study of the androgen signaling pathway, and may further help us understand the role of androgen signaling in conditions such as PCOS.
The current study focused on a new candidate gene for PCOS—small glutamine-rich tetratricopeptide repeat (TPR)-containing protein alpha (SGTA), as one of the co-chaperones binding to the AR. It is located on chromosome 19p13, 5.3 MB telomeric to the D19S884 microsatellite implicated by several studies in PCOS susceptibility (Stewart et al. 2006, Tucci et al. 2001, Urbanek et al. 1999, Urbanek et al. 2005). SGTA restrains androgen signaling by holding the androgen receptor in the cytoplasm (Buchanan et al. 2007). However, whether SGTA is involved in the pathophysiology of PCOS is not known. Since this gene is expressed ubiquitously in various tissues, this protein may serve a housekeeping function. Alternatively, SGTA may affect multiple processes in addition to androgen signaling, making it a good candidate gene for PCOS, a syndrome characterized by dysfunction in multiple systems.
We studied the association between SGTA and PCOS, and investigated its role in androgen- and insulin-related traits in affected women. We analyzed single nucleotide polymorphisms (SNPs) as well as haplotypes in SGTA, to capture common variation across the entirety of the gene. We found that 1) variation in SGTA was associated with PCOS risk, and 2) one of the haplotypes was associated with increased insulin resistance, resulting in increased beta cell function.
Patients and Methods
Subjects
A total of 287 consecutive White patients with PCOS, aged 13-47 years, and 187 healthy White control women, aged 14-60, were recruited from the Birmingham, Alabama area. All subjects were unrelated. PCOS subjects were recruited consecutively from the reproductive endocrine practice of one of the investigators (RA) at the University of Alabama at Birmingham (UAB). Participation in research studies was offered to patients meeting inclusion criteria (premenopausal, non-pregnant, on no hormonal therapy, including oral contraceptives, for at least three months, and meeting diagnostic criteria for PCOS). In order to ensure the inclusion of women with the classic disorder, the presence of PCOS was defined by the 1990 NIH consensus criteria (Zawadzki and Dunaif 1992), including: (i) clinical hyperandrogenism and/or hyperandrogenemia, (ii) oligo-ovulation, and (iii) the exclusion of related disorders, including androgen-producing tumors, nonclassic 21-hydroxylase-deficient adrenal hyperplasia (NCAH), hyperprolactinemia, active thyroid disease, or Cushing's syndrome. The specific parameters for defining hirsutism, hyperandrogenemia, ovulatory dysfunction, and exclusion of related disorders were previously reported (Azziz et al. 2004).
Controls were healthy women, with regular menstrual cycles or a history of regular menstrual cycles before menopause, and without family history of hirsutism. These women had no evidence of hirsutism, acne, or alopecia, or endocrine dysfunction and had not taken hormonal therapy (including oral contraceptives) for at least three months prior to testing. Controls were recruited by word of mouth and advertisements in the Birmingham, Alabama area, through a call for “healthy women” without detailing further the nature of the studies.
All subjects gave written informed consent, and the study was performed according to the guidelines of the Institutional Review Boards of UAB and Cedars-Sinai Medical Center.
Phenotyping
Subjects underwent a brief physical examination, hirsutism scoring using a modification of the Ferriman-Gallwey method (mFG) (Hatch et al. 1981), and underwent blood sampling. Hormonal measures, including total and free testosterone (T), dehydroepiandrosterone sulfate (DHEAS), 17α-hydroxyprogesterone (17-HP), and sex hormone binding globulin (SHBG), were obtained between days 3 and 8 (follicular phase) following a spontaneous menstrual cycle or progesterone-induced withdrawal bleed, as described (Azziz et al. 2004). Total T was measured after serum extraction by an in-house RIA method, SHBG activity was measured by competitive binding analysis, using Sephadex G-25 (Sigma-Aldrich Corp., St. Louis, MO) and [3H]T as the ligand, and the free T was calculated as previously described (Boots et al. 1998, Pearlman et al. 1967). The SHBG method gives values of approximately 100-300 nmol/L in normal adult women. DHEAS and 17-HP were measured by direct RIA using commercially available kits (from Diagnostic Products Corp., Los Angeles, CA). The intra- and interassay variations for the hormonal assays have been previously reported (Knochenhauer et al. 1998). The same laboratory assays were employed for all subjects. For these androgen-related traits measured in the women with PCOS, completeness of data was over 98%. The total and free testosterone values of three cases were statistical outliers; therefore, these values were deleted from analysis.
Fasting glucose and insulin were also obtained in a subset of the cohort (∼70%). The computer-based homeostasis model assessment (HOMA, www.dtu.ox.ac.uk/homa) utilizes fasting glucose and insulin to calculate indices of insulin resistance (HOMA-IR) and insulin secretion (HOMA-%B) (Levy et al. 1998, Wallace et al. 2004). An ideal, normal-weight person less than 35 yr of age has a HOMA-IR=1 and HOMA-%B=100% (Matthews et al. 1985). For the insulin-related traits only, subjects with diabetes (n=6) were excluded because the hyperglycemia of diabetes may induce secondary changes in insulin-related traits that reduce their utility for genetic analyses.
Genotyping and haplotype determination
We selected three single nucleotide polymorphisms (SNPs), rs2238614, rs741103, and rs1640262, which span the 28.6 kb genomic length of SGTA. These were selected because they are predicted to tag the haplotypes (across the entire gene, plus 2 kb upstream) occurring at >1% frequency in the Caucasian population of the HapMap database (The International HapMap Consortium 2003). The three SNPs were genotyped using the 5′-exonuclease assay (TaqMan MGB, Applied Biosystems, Foster City, CA) described previously (Goodarzi et al. 2003, Livak 1999); duplicate genotyping of 96 samples for one SNP yielded 100% concordance. The PCR primers and TaqMan MGB probes are presented in Table I. The genotyping success rate was 94.4%.
Table I.
Primers and probe sequences used in the 5′-exonuclease assay.
| Variant | PCR primers | TagMan MGB Probes |
|---|---|---|
| rs2238614 | GGAACTCTTTAGTGGCAGACACCTA GAAAAGTCACAGAAACTGGTCCTATG |
CGCCTCCGtGGCT ACGCCTCCGcGGC |
| rs741103 | TGCACAACGCTGAGTGAACA CCGGGCAGCGTTTCTG |
ACAGGCAGGcACGT CACAGGCAGGtACGT |
| rs1640262 | AACCGCCCCCAGCTGTA TTCCAGCCTTTCTTCCCACTT |
ATCACTGAcGGCCAC AATCACTGAtGGCCACT |
Primers for PCR are listed 5′ to 3′ and were synthesized by Invitrogen (Carlsbad, CA). Probes are listed 5′ to 3′ and were synthesized by Applied Biosystems (Foster City, CA). The probes are labeled at the 5′ end with 6FAM or VIC (laser-activated fluorescent dyes), and at the 3′ end with a quencher/minor groove binder. In the probe sequences, variant nucleotides are indicated in lower case.
Haploview 3 (Barrett et al. 2005) was used to determine haplotypes as well as haplotype blocks. Haploview constructs haplotypes using an accelerated expectation maximization algorithm similar to the partition/ligation method (Qin et al. 2002). Haploview was used to calculate linkage disequilibrium (LD, the D′ statistic and r2) between each pairwise combination of SNPs. Haploview is able to determine haplotype blocks using different block partition algorithms or user-defined blocks (Barrett et al. 2005). As the various algorithms gave slightly different haplotype blocks, we chose to consider a block spanning the entirety of the gene. Haplotypes were assigned to individual subjects only when the assignment could be made with a greater than 95% certainty.
Statistical Analysis
For all analyses, quantitative trait values were log- or square root-transformed as appropriate to reduce non-normality. Unpaired T-tests and chi-square tests were used to compare clinical characteristics between women with and without PCOS. Quantitative data are presented as median (interquartile range).
Association of SNPs or haplotypes with presence/absence of PCOS was evaluated using logistic regression, adjusting for body mass index (BMI) and age. Association with quantitative phenotypic variables utilized analysis of covariance (ANCOVA), again adjusting for age and BMI.
To confirm any significant associations, we estimated empirical P values by permutation analysis. For each significant association, the samples were permuted by shuffling genotypic data 1000 times, and subsequent association analyses were carried out to obtain the distribution of the test statistic under the null hypothesis of no association. The empiric P values were obtained as the proportion of the 1000 replicates that had a P value less than or equal to the nominal ones obtained from the actual (unshuffled) data. These empiric P values are reported in the Results.
As a further measure to handle multiple testing, significance was taken at P<0.017, considering that we analyzed one linkage disequilibrium group of SNPs against three families of traits (PCOS diagnosis, androgens, metabolic traits), yielding a correction factor of three (i.e. three independent comparisons). Analyses were carried out using Statview 5.0 (SAS Institute, Cary, NC).
Results
Clinical characteristics of the study cohort are shown in Table II. We genotyped three SNPs spanning the SGTA gene (Table III and Figure 1). Linkage disequilibrium (D′) among the SNPs in our subjects ranged from 0.70 to 0.99, with an average D′ of 0.81. The r2 ranged from 0.36 to 0.91 (average 0.55). The overall high degree of linkage disequilibrium confirmed the possibility of constructing haplotypes across the entire gene. Table IV displays the SGTA haplotypes and their frequencies. The haplotypes observed in our White subjects matched those predicted for Caucasians in HapMap, differing only moderately in frequency. The three haplotypes of frequency >5% were tested for association with PCOS and its component traits.
Table II.
Clinical Characteristics.
| Control (n=187) | PCOS (n=287) | |
|---|---|---|
| Age (yr) | 33.0 (17.0) | 27.5 (11.5)* |
| BMI (kg/m2) | 24.1 (6.4) | 34.7 (13.5)* |
| WHR | 0.78 (0.08) | 0.83 (0.10)* |
| mFG score | 0 (0) | 7.0 (5.0)* |
| Hirsute (%) | 0 | 73.9* |
| Total testosterone (nmol/l) | 1.42 (0.92) | 2.77 (1.07)* |
| Free testosterone (pmol/l) | 12.1 (9.0) | 29.1 (16.3)* |
| DHEAS (μmol/l) | 2.58 (2.03) | 5.66 (4.61)* |
| SHBG (nmol/l) | 220.0 (120.0) | 150.0 (70.0)* |
| Insulin (pmol/l) | 49.5 (45.9) | 129.15 (129.2)* |
| Glucose (mmol/l) | 4.77 (0.56) | 4.77 (0.72) |
| HOMA-IR | 0.92 (0.83) | 2.29 (1.93)* |
| HOMA-%B | 103.9 (59.5) | 175.3 (99.3)* |
Data are median (interquartile range).
P < 0.001 compared to control group, by unpaired T-tests or chi-square tests as appropriate; quantitative data were transformed to approximate normality.
WHR, waist to hip ratio; mFG: modified Ferriman–Gallwey; DHEAS, dehydroepiandrosterone sulfate; SHBG, sex hormone-binding globulin; HOMA-IR, homeostasis model assessment of insulin resistance; HOMA-%B, homeostasis model assessment of insulin secretion
Table III.
Frequency and location information on SGTA variants.
| Variant Designation | Alleles (major/minor) | Location | Overall MAF | PCOS MAF | Control MAF |
|---|---|---|---|---|---|
| rs2238614 | G/A | exon 14 (3′UTR) | 0.195 | 0.168 | 0.237 |
| rs741103 | A/G | intron 1 | 0.150 | 0.135 | 0.173 |
| rs1640262 | T/C | intron 1 | 0.208 | 0.183 | 0.249 |
MAF = minor allele frequency.
Figure 1.
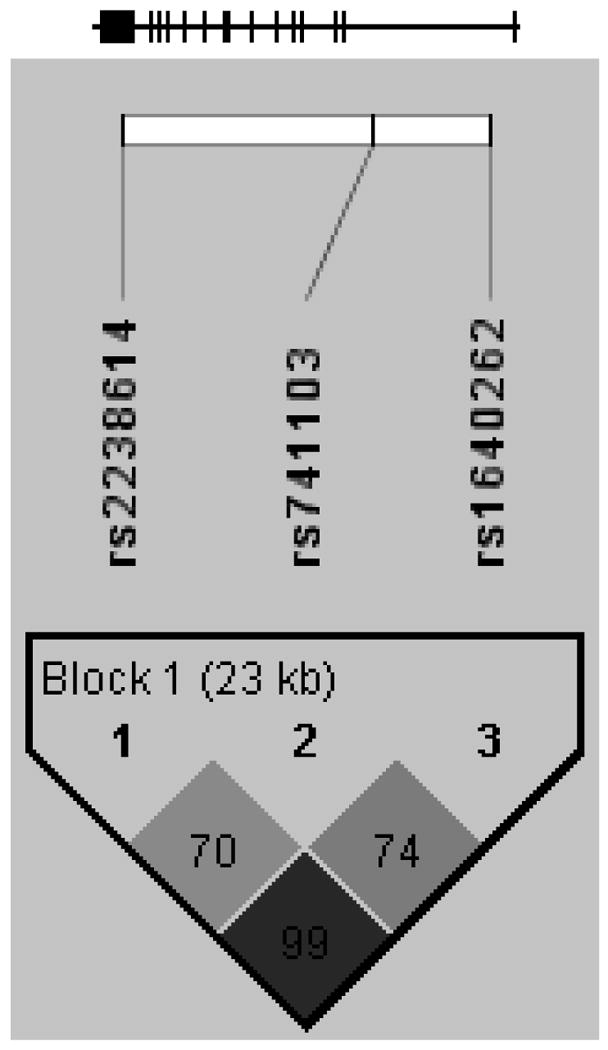
Gene structure and linkage disequilibrium plot for SGTA. The gene structure of SGTA is shown at top; the gene has 14 exons (represented by vertical bars) and is located on the reverse strand of chromosome 19 (19p13.3). The locations of the genotyped SNPs relative to the exons are indicated. The linkage disequilibrium plot at the bottom displays D′ values (%) for each pair of SNPs in the box at the intersection of the diagonals from each SNP. The SNPs were considered together in one haplotype block as indicated.
Table IV.
SGTA haplotypes and haplotype frequencies.
| Haplotype | Overall Frequency | PCOS Frequency | PCOS Count* | Control Frequency | Control Count |
|---|---|---|---|---|---|
| G-A-T | 0.761 | 0.790 | 437 | 0.715 | 246 |
| A-G-C | 0.115 | 0.102 | 57 | 0.135 | 46 |
| A-A-C | 0.079 | 0.066 | 36 | 0.101 | 35 |
| G-G-T | 0.031 | 0.028 | 16 | 0.035 | 12 |
Order of SNPs in SGTA haplotypes is rs2238614, rs741103, and rs1640262.
Count represents number of chromosomes assigned a particular haplotype by the expectation maximization algorithm.
Homozygotes for the minor allele of SNP rs1640262 were protected against PCOS, with an age- and BMI- adjusted OR of 0.18 (95% CI 0.039-0.82, P=0.009). Haplotype 1 (the most common haplotype), G-A-T, was associated with increased risk of PCOS, with an age- and BMI-adjusted OR of 4.12 (95% CI 1.20-14.10, P=0.015). SNP rs1640262 and haplotype 1 were not associated with quantitative component traits (see Supplementary Tables I-IV for trait medians by genotype).
In women with PCOS, haplotype 2 (A-G-C, second most common haplotype) was associated with increased insulin resistance (HOMA-IR: haplotype 2 carriers: 2.32 (1.48), non-carriers: 2.19 (2.00); P=0.013), and also associated with increased insulin secretion (HOMA-%B, haplotype 2 carriers: 185.80 (95.25), non-carriers: 171.15 (109.75); P=0.014). No associations were observed with the other quantitative traits in women with PCOS. No quantitative trait associations with SGTA markers were observed within the control women (see Supplementary Tables V and VI for trait medians by haplotype 2 carrier status).
To further determine whether the increase in beta cell function was a compensatory mechanism triggered by the increase in insulin resistance or an independent genetic effect of haplotype 2, we reanalyzed association of haplotype 2 with HOMA-%B, adjusting for insulin resistance (by including HOMA-IR as a covariate). In this model, the haplotype 2 was no longer associated with HOMA-%B (P=0.56), suggesting the SGTA gene may affect primarily insulin resistance, and that the apparent association with beta cell function represents compensatory insulin hypersecretion. In other words, increased beta cell function was a consequence of increased insulin resistance.
Discussion
This is the first study evaluating association of variants in the SGTA gene with PCOS. Homozygotes for the minor allele of SNP rs1640262 were protected against PCOS. Haplotype 1 was associated with increased risk of PCOS. Haplotype 2 was associated with increased insulin resistance, and consequently increased beta cell function. SGTA was initially selected based on its probable role in androgen signaling, which has recently been verified experimentally (Buchanan et al. 2007).
Hyperandrogenemia due to excessive ovarian and adrenal androgen production is widely regarded as the predominant feature of PCOS (Rosenfield 1999). Even PCOS patients with biochemical normoandrogenemia may have increased androgen levels at both the systemic and the tissue level, albeit not consistently detectable in the laboratory evaluation. To evaluate the responsiveness or sensitivity of the androgen receptor at the cellular level, it is a prerequisite to estimate the concentrations of androgens (its specific ligand) acting upon the receptor. However, this estimate is precluded by our inability to measure serum andogen levels accurately, as well as by the fact that circulating androgen levels do not equate to local tissue levels. In any case, the degree of hyperandrogenemia in PCOS is typically not severe. With this in mind, we believe androgen hypersensitivity may play an important contributing role in the development of PCOS. Indeed, there is already some genetic evidence to support this hypothesis: specifically, associations of 5-alpha reductase and androgen receptor gene variants with PCOS (Goodarzi et al. 2006, Hickey et al. 2002).
The SGTA protein has 313 amino acids and contains three TPR motifs in tandem. The TPR domain, which mediates protein-protein interactions, is a degenerate 34–amino acid motif, containing eight loosely conserved consensus residues: W-L-G-Y-A-F-A-P (Fang et al. 1969, Lamb et al. 1995). It has been found in many proteins from bacteria to humans, and is involved in many processes such as cell cycle control, transcriptional repression, and protein kinase inhibition (Goebl and Yanagida 1991, Lamb et al. 1995). Interacting with the chaperones Hsp70 and Hsp90, TPR co-chaperones (e.g. Hip (Hsp70 interacting protein), Hop (Hsp organizer protein), and FKBP52 (member of the FK-506 binding protein family)) participate in AR function, by facilitating steroid receptor maturation, maintaining the inactive cytoplasmic receptor in a state of high binding affinity to hormones, and assisting in nuclear translocation and degradation of receptors (Fang et al. 1969, Pratt and Toft 1997, Prescott and Coetzee 2006, Smith 2004).
SGTA, which interacts with Hsp70 and Hsp90 (Liou and Wang 2005, Liu et al. 1999), has recently been shown to be a key participant in AR function. SGTA binds to the hinge region of the AR (Buchanan et al. 2007). Knockdown of SGTA resulted in increased AR activity and promiscuous activation of the AR by non-classical ligands (e.g. progesterone), while SGTA overexpression inhibited AR activity in response to dihydrotestosterone (Buchanan et al. 2007). SGTA was shown to restrain AR function by promoting AR localization in the cytoplasm (Buchanan et al. 2007). While these experiments were performed in cultured cell lines, we assume they provide compelling evidence that SGTA modulates androgen sensitivity in human female physiology.
SGTA may affect PCOS risk via a role in apoptosis, as well as androgen signaling. Overexpression and knockdown experiments suggested that SGTA promotes apoptosis by enhancing DNA fragmentation and nuclear breakdown (Wang et al. 2005, Yin et al. 2006). Another group found that SGTA knockdown led to apoptosis via misaligned chromosomes and mitotic arrest (Winnefeld et al. 2006). Recent work comparing gene expression profiles in ovarian tissue from women with and without PCOS has implicated apoptosis as a possible pathway contributing to PCOS (Hughes et al. 2006, Jansen et al. 2004, Wood et al. 2007). Genes for Hsp70 and Hsp90, which participate in apoptosis and are known binding partners of SGTA, also exhibited reduced expression in PCOS ovaries (Arya et al. 2007, Jansen et al. 2004). Clearly further work is needed to define the molecular role of SGTA in androgen signaling and apoptosis in PCOS. Given that PCOS is a syndrome with multiple features, SGTA is an ideal candidate given that it may influence multiple processes.
Our results suggest yet another pathway which may be affected by SGTA, that of insulin signaling. In our data, variation in the SGTA gene was related to insulin resistance, and consequently increased insulin secretion in PCOS. This is a novel finding, and suggests SGTA may be a connection between hormone action and metabolic signaling pathways in the pathogenesis of PCOS.
Our results provide preliminary evidence that SGTA genetic variants may be associated with PCOS risk. Because the three studied SNPs do not change the SGTA amino acid sequence, their functional significance remains to be determined in future study. They may affect splicing of the SGTA transcript or expression of SGTA and/or other genes, or may be in linkage disequilibrium with SGTA coding variants yet to be discovered. Because one SNP in these haplotypes is located in the 3′ untranslated region, an effect on transcript stability or translational efficiency is possible (Mazumder et al. 2003). Considering the limits of linkage/association approaches, the association of SGTA with PCOS should be subject to replication efforts in larger sample sizes and independent cohorts (Escobar-Morreale et al. 2005).
In conclusion, the current genetic study of SGTA provides preliminary data implicating SGTA as a genetic determinant of PCOS. SGTA is a potentially exciting candidate for the multi-faceted syndrome of PCOS, because it may play roles in multiple pathways that contribute to the whole picture of PCOS, including androgen action, the insulin axis and apoptosis.
Supplementary Material
Appendix Table I. Clinical Characteristics by rs1640262 genotype in controls.
Appendix Table II. Clinical Characteristics by rs1640262 genotype in PCOS.
Appendix Table III. Clinical Characteristics by haplotype 1 carrier state in controls.
Appendix Table IV. Clinical Characteristics by haplotype 1 carrier state in PCOS.
Appendix Table V. Clinical Characteristics by haplotype 2 carrier state in controls.
Appendix Table VI. Clinical Characteristics by haplotype 2 carrier state in PCOS.
Acknowledgments
National Institutes of Health grants (R01-HD29364 and K24-HD01346 to R.A.); General Clinical Research Center Grant from the National Center for Research Resources (M01-RR00425); an endowment from the Helping Hand of Los Angeles, Inc.
Footnotes
Publisher's Disclaimer: This is a pre-copy-editing, author-produced PDF of an article accepted for publication in Human Reproduction following peer review. The definitive published-accepted version [Goodarzi MO, Xu N, Cui J, Guo X, Chen Y-DI, Azziz R. Small glutamine-rich tetratricopeptide repeat-containing protein alpha (SGTA), a candidate gene for polycystic ovary syndrome. Human Reproduction 2008;23:1214-1219] is available online at: http://humrep.oxfordjournals.org/cgi/reprint/23/5/1214
References
- Arya R, Mallik M, Lakhotia SC. Heat shock genes - integrating cell survival and death. J Biosci. 2007;32:595–610. doi: 10.1007/s12038-007-0059-3. [DOI] [PubMed] [Google Scholar]
- Azziz R, Carmina E, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Futterweit W, Janssen OE, Legro RS, Norman RJ, Taylor AE, et al. Positions statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society guideline. J Clin Endocrinol Metab. 2006;91:4237–4245. doi: 10.1210/jc.2006-0178. [DOI] [PubMed] [Google Scholar]
- Azziz R, Woods KS, Reyna R, Key TJ, Knochenhauer ES, Yildiz BO. The prevalence and features of the polycystic ovary syndrome in an unselected population. J Clin Endocrinol Metab. 2004;89:2745–2749. doi: 10.1210/jc.2003-032046. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Boots LR, Potter S, Potter D, Azziz R. Measurement of total serum testosterone levels using commercially available kits: high degree of between-kit variability. Fertil Steril. 1998;69:286–292. doi: 10.1016/s0015-0282(97)00464-0. [DOI] [PubMed] [Google Scholar]
- Buchanan G, Ricciardelli C, Harris JM, Prescott J, Yu ZC, Jia L, Butler LM, Marshall VR, Scher HI, Gerald WL, et al. Control of androgen receptor signaling in prostate cancer by the cochaperone small glutamine rich tetratricopeptide repeat containing protein alpha. Cancer Res. 2007;67:10087–10096. doi: 10.1158/0008-5472.CAN-07-1646. [DOI] [PubMed] [Google Scholar]
- Chang WY, Knochenhauer ES, Bartolucci AA, Azziz R. Phenotypic spectrum of polycystic ovary syndrome: clinical and biochemical characterization of the three major clinical subgroups. Fertil Steril. 2005;83:1717–1723. doi: 10.1016/j.fertnstert.2005.01.096. [DOI] [PubMed] [Google Scholar]
- Escobar-Morreale HF, Luque-Ramirez M, San Millan JL. The molecular-genetic basis of functional hyperandrogenism and the polycystic ovary syndrome. Endocr Rev. 2005;26:251–282. doi: 10.1210/er.2004-0004. [DOI] [PubMed] [Google Scholar]
- Fang S, Anderson KM, Liao S. Receptor proteins for androgens. On the role of specific proteins in selective retention of 17-beta-hydroxy-5-alpha-androstan-3-one by rat ventral prostate in vivo and in vitro. J Biol Chem. 1969;244:6584–6595. [PubMed] [Google Scholar]
- Franks S, McCarthy M. Genetics of ovarian disorders: polycystic ovary syndrome. Rev Endocr Metab Disord. 2004;5:69–76. doi: 10.1023/B:REMD.0000016125.05878.96. [DOI] [PubMed] [Google Scholar]
- Goebl M, Yanagida M. The TPR snap helix: a novel protein repeat motif from mitosis to transcription. Trends Biochem Sci. 1991;16:173–177. doi: 10.1016/0968-0004(91)90070-c. [DOI] [PubMed] [Google Scholar]
- Goodarzi MO, Azziz R. Diagnosis, epidemiology, and genetics of the polycystic ovary syndrome. Best Pract Res Clin Endocrinol Metab. 2006;20:193–205. doi: 10.1016/j.beem.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Goodarzi MO, Guo X, Taylor KD, Quiñones MJ, Samayoa C, Yang H, Saad MF, Palotie A, Krauss RM, Hsueh WA, et al. Determination and use of haplotypes: ethnic comparison and association of the lipoprotein lipase gene and coronary artery disease in Mexican-Americans. Genet Med. 2003;5:322–327. doi: 10.1097/01.GIM.0000076971.55421.AD. [DOI] [PubMed] [Google Scholar]
- Goodarzi MO, Shah NA, Antoine HJ, Pall M, Guo X, Azziz R. Variants in the 5alpha-reductase type 1 and type 2 genes are associated with polycystic ovary syndrome and the severity of hirsutism in affected women. J Clin Endocrinol Metab. 2006;91:4085–4091. doi: 10.1210/jc.2006-0227. [DOI] [PubMed] [Google Scholar]
- Hatch R, Rosenfield RL, Kim MH, Tredway D. Hirsutism: implications, etiology, and management. Am J Obstet Gynecol. 1981;140:815–830. doi: 10.1016/0002-9378(81)90746-8. [DOI] [PubMed] [Google Scholar]
- Hickey T, Chandy A, Norman RJ. The androgen receptor CAG repeat polymorphism and X-chromosome inactivation in Australian Caucasian women with infertility related to polycystic ovary syndrome. J Clin Endocrinol Metab. 2002;87:161–165. doi: 10.1210/jcem.87.1.8137. [DOI] [PubMed] [Google Scholar]
- Hughes C, Elgasim M, Layfield R, Atiomo W. Genomic and post-genomic approaches to polycystic ovary syndrome--progress so far: Mini Review. Hum Reprod. 2006;21:2766–2775. doi: 10.1093/humrep/del222. [DOI] [PubMed] [Google Scholar]
- Jansen E, Laven JS, Dommerholt HB, Polman J, van Rijt C, van den Hurk C, Westland J, Mosselman S, Fauser BC. Abnormal gene expression profiles in human ovaries from polycystic ovary syndrome patients. Mol Endocrinol. 2004;18:3050–3063. doi: 10.1210/me.2004-0074. [DOI] [PubMed] [Google Scholar]
- Knochenhauer ES, Key TJ, Kahsar-Miller M, Waggoner W, Boots LR, Azziz R. Prevalence of the polycystic ovary syndrome in unselected black and white women of the southeastern United States: a prospective study. J Clin Endocrinol Metab. 1998;83:3078–3082. doi: 10.1210/jcem.83.9.5090. [DOI] [PubMed] [Google Scholar]
- Lamb JR, Tugendreich S, Hieter P. Tetratrico peptide repeat interactions: to TPR or not to TPR? Trends Biochem Sci. 1995;20:257–259. doi: 10.1016/s0968-0004(00)89037-4. [DOI] [PubMed] [Google Scholar]
- Levy JC, Matthews DR, Hermans MP. Correct homeostasis model assessment (HOMA) evaluation uses the computer program. Diabetes Care. 1998;21:2191–2192. doi: 10.2337/diacare.21.12.2191. [DOI] [PubMed] [Google Scholar]
- Liou ST, Wang C. Small glutamine-rich tetratricopeptide repeat-containing protein is composed of three structural units with distinct functions. Arch Biochem Biophys. 2005;435:253–263. doi: 10.1016/j.abb.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Liu FH, Wu SJ, Hu SM, Hsiao CD, Wang C. Specific interaction of the 70-kDa heat shock cognate protein with the tetratricopeptide repeats. J Biol Chem. 1999;274:34425–34432. doi: 10.1074/jbc.274.48.34425. [DOI] [PubMed] [Google Scholar]
- Livak KJ. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet Anal. 1999;14:143–149. doi: 10.1016/s1050-3862(98)00019-9. [DOI] [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- Mazumder B, Seshadri V, Fox PL. Translational control by the 3′-UTR: the ends specify the means. Trends Biochem Sci. 2003;28:91–98. doi: 10.1016/S0968-0004(03)00002-1. [DOI] [PubMed] [Google Scholar]
- Nam Menke M, Strauss JF. Genetics of polycystic ovarian syndrome. Clin Obstet Gynecol. 2007;50:188–204. doi: 10.1097/GRF.0b013e3180305f7c. [DOI] [PubMed] [Google Scholar]
- Pearlman WH, Crepy O, Murphy M. Testosterone-binding levels in the serum of women during the normal menstrual cycle, pregnancy, and the post-partum period. J Clin Endocrinol Metab. 1967;27:1012–1018. doi: 10.1210/jcem-27-7-1012. [DOI] [PubMed] [Google Scholar]
- Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- Prescott J, Coetzee GA. Molecular chaperones throughout the life cycle of the androgen receptor. Cancer Lett. 2006;231:12–19. doi: 10.1016/j.canlet.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Qin ZS, Niu T, Liu JS. Partition-ligation-expectation-maximization algorithm for haplotype inference with single-nucleotide polymorphisms. Am J Hum Genet. 2002;71:1242–1247. doi: 10.1086/344207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfield RL. Ovarian and adrenal function in polycystic ovary syndrome. Endocrinol Metab Clin North Am. 1999;28:265–293. doi: 10.1016/s0889-8529(05)70070-0. [DOI] [PubMed] [Google Scholar]
- Shatkina L, Mink S, Rogatsch H, Klocker H, Langer G, Nestl A, Cato AC. The cochaperone Bag-1L enhances androgen receptor action via interaction with the NH2-terminal region of the receptor. Mol Cell Biol. 2003;23:7189–7197. doi: 10.1128/MCB.23.20.7189-7197.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DF. Tetratricopeptide repeat cochaperones in steroid receptor complexes. Cell Stress Chaperones. 2004;9:109–121. doi: 10.1379/CSC-31.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart DR, Dombroski BA, Urbanek M, Ankener W, Ewens KG, Wood JR, Legro RS, Strauss JF, Dunaif A, Spielman RS. Fine mapping of genetic susceptibility to polycystic ovary syndrome on chromosome 19p13.2 and tests for regulatory activity. J Clin Endocrinol Metab. 2006;91:4112–4117. doi: 10.1210/jc.2006-0951. [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- Tucci S, Futterweit W, Concepcion ES, Greenberg DA, Villanueva R, Davies TF, Tomer Y. Evidence for association of polycystic ovary syndrome in caucasian women with a marker at the insulin receptor gene locus. J Clin Endocrinol Metab. 2001;86:446–449. doi: 10.1210/jcem.86.1.7274. [DOI] [PubMed] [Google Scholar]
- Urbanek M. The genetics of the polycystic ovary syndrome. Nat Clin Pract Endocrinol Metab. 2007;3:103–111. doi: 10.1038/ncpendmet0400. [DOI] [PubMed] [Google Scholar]
- Urbanek M, Legro RS, Driscoll DA, Azziz R, Ehrmann DA, Norman RJ, Strauss JF, Spielman RS, Dunaif A. Thirty-seven candidate genes for polycystic ovary syndrome: strongest evidence for linkage is with follistatin. Proc Natl Acad Sci U S A. 1999;96:8573–8578. doi: 10.1073/pnas.96.15.8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanek M, Woodroffe A, Ewens KG, Diamanti-Kandarakis E, Legro RS, Strauss JF, Dunaif A, Spielman RS. Candidate gene region for polycystic ovary syndrome on chromosome 19p13.2. J Clin Endocrinol Metab. 2005;90:6623–6629. doi: 10.1210/jc.2005-0622. [DOI] [PubMed] [Google Scholar]
- Wallace TM, Levy JC, Matthews DR. Use and abuse of HOMA modeling. Diabetes Care. 2004;27:1487–1495. doi: 10.2337/diacare.27.6.1487. [DOI] [PubMed] [Google Scholar]
- Wang H, Shen H, Wang Y, Li Z, Yin H, Zong H, Jiang J, Gu J. Overexpression of small glutamine-rich TPR-containing protein promotes apoptosis in 7721 cells. FEBS Lett. 2005;579:1279–1284. doi: 10.1016/j.febslet.2004.12.092. [DOI] [PubMed] [Google Scholar]
- Winnefeld M, Grewenig A, Schnölzer M, Spring H, Knoch TA, Gan EC, Rommelaere J, Cziepluch C. Human SGT interacts with Bag-6/Bat-3/Scythe and cells with reduced levels of either protein display persistence of few misaligned chromosomes and mitotic arrest. Exp Cell Res. 2006;312:2500–2514. doi: 10.1016/j.yexcr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- Wood JR, Dumesic DA, Abbott DH, Strauss JF. Molecular abnormalities in oocytes from women with polycystic ovary syndrome revealed by microarray analysis. J Clin Endocrinol Metab. 2007;92:705–713. doi: 10.1210/jc.2006-2123. [DOI] [PubMed] [Google Scholar]
- Yin H, Wang H, Zong H, Chen X, Wang Y, Yun X, Wu Y, Wang J, Gu J. SGT, a Hsp90beta binding partner, is accumulated in the nucleus during cell apoptosis. Biochem Biophys Res Commun. 2006;343:1153–1158. doi: 10.1016/j.bbrc.2006.03.090. [DOI] [PubMed] [Google Scholar]
- Young JC, Barral JM, Ulrich Hartl F. More than folding: localized functions of cytosolic chaperones. Trends Biochem Sci. 2003;28:541–547. doi: 10.1016/j.tibs.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Zawadzki JK, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. In: Dunaif A, Givens JR, Haseltine F, Merriam GR, editors. Polycystic Ovary Syndrome. Blackwell Scientific; Oxford, UK: 1992. pp. 377–384. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix Table I. Clinical Characteristics by rs1640262 genotype in controls.
Appendix Table II. Clinical Characteristics by rs1640262 genotype in PCOS.
Appendix Table III. Clinical Characteristics by haplotype 1 carrier state in controls.
Appendix Table IV. Clinical Characteristics by haplotype 1 carrier state in PCOS.
Appendix Table V. Clinical Characteristics by haplotype 2 carrier state in controls.
Appendix Table VI. Clinical Characteristics by haplotype 2 carrier state in PCOS.