

Abstract
The endogenous opioid peptide dynorphin A is distinct from other endogenous opioid peptides in having significant neuronal excitatory and neurotoxic effects that are not mediated by opioid receptors. Some of these non-opioid actions of dynorphin contribute to the development of abnormal pain resulting from a number of pathological conditions. Identifying the mechanisms and the sites of action of dynorphin is essential for understanding the pathophysiology of dynorphin and for exploring novel therapeutic targets for pain. This review will discuss the mechanisms that have been proposed and the recent finding that spinal dynorphin may be an endogenous ligand of bradykinin receptors under pathological conditions to promote pain.
Keywords: Neuropathic pain, inflammatory pain, G protein coupled receptor, opioid, dorsal root ganglion, sensory neuron, voltage gated calcium channels, kinins, kininogen
Introduction
Endogenous opioid neuropeptides are classified into the enkephalins, the endorphins and the dynorphins [30]. The peptides are characterized by their high affinity for opioid mu, delta and kappa receptors and the ability of the opioid receptor antagonist, naloxone, to block or reverse agonist actions of these substances. The physiological actions of enkephalins and endorphins are the result of inhibitory effects, predominately on neuronal cells, to elicit analgesia, to alter motor and secretory functions of the gastrointestinal tract, as well as other functions including respiration, cardiovascular activity, hormonal balance, temperature and responses to stress. The physiological role of the dynorphins, on the other hand, remains somewhat more obscure.
The dynorphins are all C terminal extensions of leu-enkephalin [16]. Dynorphin A(1–17) is one of the major proteolytic fragments of prodynorphin [8] [Figure 1] and is widely distributed in the CNS [8, 53]. Dynorphin A(1–17) has been suggested to inhibit neuronal activity through opioid kappa receptors[5]. In vivo, dynorphin A is converted rapidly to the des-tyrosyl fragment by aminopeptidase activity, which is considered to be the major inactivation mechanism of its opioid actions upon release [58] because these des-tyrosyl fragments of dynorphin A (e.g., dynorphin A(2–13)) have very low affinity for opioid receptors [49]. However, subsequent analyses showed that dynorphin A is distinct from other prodynorphin-derived peptides by its neuronal excitatory actions and excitotoxicity that are not mediated by opioid receptors. Dynorphin A given intracerebroventricularly [50] or intrathecally [12, 37] produces little or no antinociception. Relatively low doses of dynorphin A induce pronociceptive behaviors including biting, licking and scratching [41], or transient hypersensitivity to innocuous touch and noxious heat stimuli to the hind paw [19]. At non-paralytic doses, dynorphin A elicits long-lasting tactile hypersensitivity and thermal hyperalgesia [45]. At higher doses, intrathecal dynorphin A produces paralysis which is indicative of severe motor effects [12, 37]. At these doses, dynorphin A is neurotoxic, depleting sensory neurons, motor neurons, and interneurons in the spinal cord [22], and potentiating excitatory neurotransmitter release [10, 34]. The pronociceptive and excitotoxic effects of dynorphin A are not reversed by naloxone, and can be elicited by the des-tyrosyl fragments of dynorphin. Other dynorphin fragments, namely dynorphin B and neo-endorphin, do not produce these effects [40].
Figure 1.
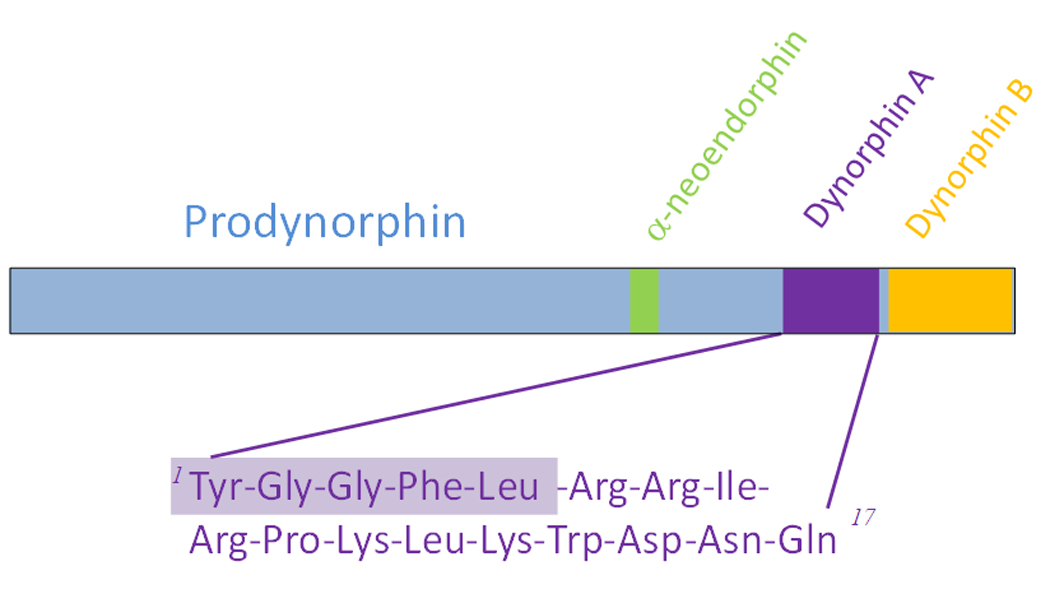
Dynorphin A(1–17) is a major proteolytic fragment from the precursor peptide, prodynorphin.
The level of spinal dynorphin expression appears to be easily perturbed [7]. Many experimental models of pathological pain show significant regional elevations of dynorphin A in the spinal cord. These include inflammatory pain [31], neuropathic pain [17, 27, 48], bone cancer pain [29], chronic pancreatitis[46], abnormal pain (hyperalgesia) following sustained exposure to morphine [44] or nicotine[25], spinal cord trauma [1, 13, 39], and arthritis [54]. Elevated levels of dynorphin A are critical for the expression of chronic pain because approaches that inhibit dynorphin A activity consistently normalize the enhanced sensory responses (i.e., diminish hyperalgesia). Spinal administration of an anti-dynorphin A antiserum reduces neurological impairment after nerve injury [11], blocks the hypersensitivity sensory input and attenuates trauma induced changes in the spinal cord [55], but does not alter normal sensory thresholds in non-injured rats [27, 47]. Transgenic mice that carry a null mutation in the prodynorphin gene do not exhibit persistent pain states after peripheral nerve injury when compared with their wild type littermates [52]. These observations suggest that approaches that prevent or reverse the excitatory actions of dynorphin A in the spinal cord are likely to have potential therapeutic relevance.
The NMDA receptor as a non-opioid site of action of dynorphin A
The underlying mechanism(s) of this pronociceptive and neuroexcitatory actions of dynorphin A has been proposed to be a direct agonist action of dynorphin A at the NMDA receptor based on the following evidence: First, dynorphin A-induced neurological damage, including hind limb paralysis, loss of tail-flick reflex, and loss of neuronal cell bodies, can be prevented by antagonists and modulators of the NMDA receptor [3, 23, 38]. Second, the long-lasting hyperresponsiveness to sensory stimuli by intrathecal dynorphin A is prevented by pretreatment with the NMDA receptor antagonist MK-801[45].Biting and scratching precipitated by intrathecal dynorphin likewise is blocked by antagonists of the NMDA receptor [40]. More recently, we reported that spinal infusion of dynorphin A(2–13) stimulated the release of excitatory amino acids and PGE2, and the effect could be blocked by the NMDA receptor antagonist, AP5 [18]. Although the evidence suggests that NMDA receptor may ultimately mediate the neuroexcitatory effects of dynorphin A, it is not sufficient to differentiate between direct versus indirect actions of dynorphin A at the NMDA receptor.
The evidence that implicates a direct interaction of dynorphin A at the NMDA receptor came from radioligand binding studies showing that dynorhpin A competes for the binding of radioligands that are selective for the NMDA receptor, e.g., [3H]MK-801 or [3H]CGP39,653 [9, 28, 32]. The inhibition constants based on the indirect binding analysis suggest moderate to low affinity. Attempts to define the affinity of dynorphin at the NMDA receptor using [125I]dynorphin A(2–17) suggest the existence of a low capacity, high affinity binding site (~10 nM) [42]. The most direct functional electrophysiological study used recombinant, heteromeric NMDA receptors expressed in Xenopus oocytes. Data showed that dynorphin inhibited NMDA-induced currents [4] except under conditions of low extracellular concentrations of glycine (<0.1 µM), where dynorphin potentiated the NMDA-induced currents probably by mimicking the action of glycine at the NMDA receptor [59]. The excitatory effects of dynorphin seen in vivo may not be a consequence of this interaction. Studies using primary neuronal cultures further illustrate the complex effects of dynorphin A on NMDA currents [6, 20]. Overall, the in vitro evidence did not satisfactorily validate a direct agonist action at the NMDA receptor as the primary mechanism for the excitatory actions of dynorphin in vivo.
In cultured cortical neurons, dynorphin A(2–17) stimulates a transient increase in intracellular calcium in response to dynorphin A [43]. Similar response is seen in embryonic spinal cord neurons, while prolonged exposure of these cells to dynorphin A results in a significant loss of neurons [15]. Thus dynorphin A may exert its excitatory actions on neurons through a calcium dependent mechanism, which could account for both the acute neuroexcitatory effect as well as the excitotoxic actions upon prolonged exposure to dynorphin. In cortical neurons, the acute effect of dynorphin A on the intracellular calcium persists despite the blockade of NMDA receptors by MK-801, suggesting that the acute effect of dynorphin A(2–17) is not mediated by the NMDA receptor [43]. In spinal cord neurons, the neuronal loss due to prolonged treatment with dynorphin A(1–13) was blocked by MK-801; thus the neurotoxic effect of dynorphin is contributed at least in part through the NMDA receptors [15]. Sustained exposure to dynorphin A(1–17) induces caspase-mediated apoptotic cell death via AMPA/kainate receptor activation, presumably due to glutamate release from the cultured cells[33]. Dynorphin A(2–13) also potentiates the release of CGRP evoked by capsaicin from spinal cord tissues [14]. Taken together, these findings link the excitatory actions of dynorphin A to transmitter release through a non-opioid, calcium dependent mechanism, but the question remains as to the primary site(s) of action of dynorphin that mediate the increase in intracellular calcium and excitatory transmitter release.
The bradykinin receptor as a non-opioid site of action of dynorphin A
We recently reported that dynorphin A and its des-tyrosyl fragment, dynorphin A(2–13), stimulate calcium influx via L-type and P/Q type voltage sensitive calcium channels through interaction with bradykinin receptors in a dorsal root ganglion X neuroblastoma hybrid cell line, F-11[19]. This novel, non-opioid agonist action of dynorphin was not predicted by any structural similarity either between dynorphin and bradykinin, or between the opioid receptor and the bradykinin receptor. Dynorphin A displaced the binding of [3H]bradykinin and [3H]kallidin in brain tissues as well as cell lines that express either endogenous or heterologous bradykinin B1 and B2 receptors with apparent moderate affinity (~ 1 µM). Despite the moderate affinity, the interaction between dynorphin and bradykinin receptors appears to have physiological relevance because intrathecal injection of a B2 selective bradykinin receptor antagonist, HOE 140, blocks the hyperalgesia induced by intrathecal injection of dynorphin A(2–13), suggesting that spinal bradykinin receptors ultimately mediate the pronociceptive actions of spinal dynorphin. In addition, intrathecal dynorphin does not induce hyperalgesia in transgenic mice that lack B2 receptors, further supporting bradykinin receptors as a site of action of spinal dynorphin. Our observation that dynorphin A may activate bradykinin receptors identifies an unexpected putative direct neuroexcitatory target of dynorphin.
Bradykinin receptors in the spinal cord are associated with the central terminals of the primary afferent projecting to the superficial laminae of the dorsal horn [36, 56] as well as spinal cord neurons [24]. The contribution of these receptors to pain and hyperalgesia has been implicated by the effects of bradykinin receptor antagonists injected intrathecally. For example, both the B2 antagonist HOE140, and the B1 antagonist des-Arg9-Leu8-bradykinin (DALBK) given intrathecally attenuate thermal hyperalgesia in rats with peripheral nerve injury [19]. The antihyperalgesic effect of DALBK is delayed, which is consistent with the upregulation of the B1 receptor at 2 weeks but not at 48 hr after injury. The source of kinins in the spinal cord and their prospective role as transmitters is subject to debate. Kinins are extremely labile, with a half life of around 30 seconds. This makes kinin levels very difficult to measure in any tissue. Measurements of kininogen, the precursor of bradykinin in the spinal cord by quantitative RT-PCR showed no appreciable expression in either naïve rats, rats that had been treated with the inflammatory agent complete Freud’s adjuvant [26], or nerve injured rats [19]. Hence, there is a lack of evidence of de novo action of kinins in the spinal cord. It is also difficult to evaluate the possible actions of kinins in the spinal cord by intrathecal administration because the peptides would have been degraded before they can reach the site of action. Prior reports on intrathecal injection of bradykinin have yielded conflicting nociceptive behavior, ranging from transient hyperalgesia followed by increased thermal latency in the tail flick test attributed to the release of norepinephrine [21], to long lasting hyperalgesia that peaks at 75 min and slowly recovers over 4 hr [51]. The effect of bradykinin also appears to vary with dose, which was found to be hyperalgesic at low dose, but antinociceptive at high dose [35].
In our hands, several doses of bradykinin ranging from 0.15 to 10 µg given i.t. to male, Sprague Dawley rats elicited immediate but transient bouts of hyperactivity and escape behaviors that lasted less than 30 sec, and normal behavior returned within one min (Chen and Porreca, unpublished observations). Spinal bradykinin did not alter behavioral responses to either tactile stimuli elicited by von Frey filaments, or to noxious thermal stimuli using noxious radiant heat or hot-plate test. Therefore, whether spinal bradykinin receptors contribute to pain and hyperalgesia due to their activation by kinins remains to be clarified.
Both bradykinin B1 and B2 receptors are primarily coupled to the phospholipase C (PLC) pathway via Gq/11 resulting in the mobilization of intracellular calcium by inositol trisphosphate. The activation of calcium influx by dynorphin via the bradykinin receptors as seen in the F-11 cells is not due to the PLC pathway and is not sensitive to PKC inhibitors [19], suggesting that dynorphin and bradykinin induce different coupling mechanisms via the bradykinin receptor. Our observation that dynorphin A activation of calcium influx via bradykinin receptors is blocked by the PKA inhibitor, H89 [19], links this function to the cAMP pathway, where the downstream effectors may be the phosphorylation activation of the L-type and P/Q type channels.
The pathophysiological relevance of the agonist action of dynorphin A at the bradykinin receptors
The agonist activity of dynorphin A at bradykinin receptors is a possible mechanism for the pronociceptive actions of dynorphin A based on the following considerations. First, prodynorphin and bradykinin receptors are both localized to the superficial laminae of the dorsal horn of the spinal cord making potential interactions between the two anatomically feasible. Second, bradykinin receptors are localized to small diameter DRG neurons that express substance P or CGRP; the receptors’ distribution is relevant to dynorphin A’s enhancing effect on neuropeptide release from the central terminals of the primary afferent in spinal cord tissues[14]. Third, activation of bradykinin receptors activates sensory neurons. Fourth, the calcium channels that are modulated by dynorphin A via the bradykinin receptors are associated with neuronal excitability and transmitter release.
Indeed, transient hyperalgesia induced by the intrathecal administration of the non-opioid fragment, dynorphin A(2–13), was blocked by intrathecal HOE140 [19] suggesting that spinal B2 receptors are essential for the pronociceptive actions of spinal dynorphin. In neuropathic rats, both HOE140 and a B1 receptor selective antagonist, des-Arg9Leu-bradykinin (DALBK), acutely reversed neuropathic pain states when given intrathecally, seen about a week after nerve injury. At the earlier time points (first 4 to 6 days), however, the antagonists had no significant effect. This delayed effect of bradykinin receptor antagonists correlated with the time course of the upregulation of spinal dynorphin but not with changes in expression of bradykinin receptors. An important consideration is that there is no evidence for de novo synthesis of bradykinin precursors in the spinal cord either in control rats or nerve injured rats. The data support the postulation that activation of spinal bradykinin receptors promotes pain, and they are likely activated by dynorphin when the latter is present in elevated concentration. Elevated concentrations of spinal dynorphin may also enhance its opioid activity at the kappa opioid receptors as proposed by Xu et al[57], showing that transgenic mice that lack kappa opioid receptors exhibited enhanced hyperalgesia upon injury when compared to that of injured wild-type mice. The enhanced opioid activity, however, is insufficient to counter the peptide’s non-opioid, pronociceptive actions.
Future directions
Despite the in vitro and in vivo evidence stated above, many questions remain regarding this putative binding site for dynorphin at the bradykinin receptors. First and foremost is the apparent moderate affinity of dynorphin for the bradykinin receptor based on indirect competitive binding analysis, which raises the issue about the specificity of this interaction in vivo, and whether a low affinity agonist has physiological relevance. In principle, having a moderate affinity for the bradykinin receptor is consistent with the pronociceptive action of dynorphin only upon its elevated production in the spinal cord upon an injurious insult, such that dynorphin’s action at spinal bradykinin receptors does not occur under normal, physiological conditions but is promoted by an overabundance of dynorphin. Thus, dynorphin may be considered a “pathogenic” endogenous ligand for bradykinin receptors [2]. This concept, however, raises a second question of whether the extent of upregulation of spinal dynorphin in vivo is sufficient to directly activate bradykinin receptors. Third, our data show that dynorphin activates the bradykinin receptor to stimulate voltage gated calcium channels; this is different from the action of bradykinin, which primarily stimulates the coupling of the B2 bradykinin receptor to phospholipase Cβ via Gαq/11. The differential coupling induced by dynorphin and bradykinin suggests that the two peptides may occupy the receptor in different manner to induce two distinct functional receptor conformations.
The distinct signaling pathways activated by bradykinin and dynorphin, perhaps indicative of their structure-activity relationship, raise the possibility that dynorphin may bind to a distinct or allosteric site from that for bradykinin. Strategies that give a direct measure of dynorphin’s affinity are necessary to address the issue of its affinity and specificity at the bradykinin receptors, as well as a comparative analysis of the binding of dynorphin versus bradykinin at the same receptor. Ultimately, the pathophysiological relevance of this interaction between dynorphin and bradykinin receptors will be ascertained by identifying bradykinin receptor antagonists that are designed based on the structure of dynorphin and can block the actions of dynorphin at the bradykinin receptors in vitro and in vivo. Their effect on the pronociceptive actions of spinal dynorphin, either by intrathecal administration of dynorphin, or by elevated spinal dynorphin after injury, will establish whether this binding site of dynorphin at the bradykinin receptor is necessary for abnormal pain. The validation of bradykinin receptors as a non-opioid site of action of dynorphin will represent an important breakthrough in elucidating the mechanisms of the excitatory and pronociceptive actions of dynorphin in pathological pain states. Furthermore, antagonists of bradykinin receptors that selectively block the action of dynorphin but not that of endogenous bradykinin may be effective against abnormal pain states without disrupting the normal physiological actions (e.g., cardiovascular) of bradykinin.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abraham KE, McGinty JF, Brewer KL. The role of kainic acid/AMPA and metabotropic glutamate receptors in the regulation of opioid mRNA expression and the onset of pain-related behavior following excitotoxic spinal cord injury. Neuroscience. 2001;104:863–874. doi: 10.1016/s0306-4522(01)00134-8. [DOI] [PubMed] [Google Scholar]
- 2.Altier C, Zamponi GW. Opioid, cheating on its receptors, exacerbates pain. Nat Neurosci. 2006;9:1465–1467. doi: 10.1038/nn1206-1465b. [DOI] [PubMed] [Google Scholar]
- 3.Bakshi R, Faden AI. Blockade of the glycine modulatory site of NMDA receptors modifies dynorphin-induced behavioral effects. Neurosci Lett. 1990;110:113–117. doi: 10.1016/0304-3940(90)90797-d. [DOI] [PubMed] [Google Scholar]
- 4.Brauneis U, Oz M, Peoples RW, Weight FF, Zhang L. Differential sensitivity of recombinant N-methyl-D-aspartate receptor subunits to inhibition by dynorphin. J Pharmacol Exp Ther. 1996;279:1063–1068. [PubMed] [Google Scholar]
- 5.Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- 6.Chen L, Gu Y, Huang LY. The opioid peptide dynorphin directly blocks NMDA receptor channels in the rat. J Physiol. 1995;482(Pt 3):575–581. doi: 10.1113/jphysiol.1995.sp020541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho HJ, Basbaum AI. Increased staining of immunoreactive dynorphin cell bodies in the deafferented spinal cord of the rat. Neurosci Lett. 1988;84:125–130. doi: 10.1016/0304-3940(88)90395-3. [DOI] [PubMed] [Google Scholar]
- 8.Civelli O, Douglass J, Goldstein A, Herbert E. Sequence and expression of the rat prodynorphin gene. Proc Natl Acad Sci U S A. 1985;82:4291–4295. doi: 10.1073/pnas.82.12.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dumont M, Lemaire S. Dynorphin potentiation of [3H]CGP-39653 binding to rat brain membranes. Eur J Pharmacol. 1994;271:241–244. doi: 10.1016/0014-2999(94)90288-7. [DOI] [PubMed] [Google Scholar]
- 10.Faden AI. Dynorphin increases extracellular levels of excitatory amino acids in the brain through a non-opioid mechanism. J Neurosci. 1992;12:425–429. doi: 10.1523/JNEUROSCI.12-02-00425.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faden AI. Opioid and nonopioid mechanisms may contribute to dynorphin's pathophysiological actions in spinal cord injury. Ann Neurol. 1990;27:67–74. doi: 10.1002/ana.410270111. [DOI] [PubMed] [Google Scholar]
- 12.Faden AI, Jacobs TP. Dynorphin-related peptides cause motor dysfunction in the rat through a non-opiate action. Br J Pharmacol. 1984;81:271–276. doi: 10.1111/j.1476-5381.1984.tb10074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faden AI, Molineaux CJ, Rosenberger JG, Jacobs TP, Cox BM. Increased dynorphin immunoreactivity in spinal cord after traumatic injury. Regul Pept. 1985;11:35–41. doi: 10.1016/0167-0115(85)90029-1. [DOI] [PubMed] [Google Scholar]
- 14.Gardell LR, Wang R, Burgess SE, Ossipov MH, Vanderah TW, Malan TP, Jr, Lai J, Porreca F. Sustained morphine exposure induces a spinal dynorphin-dependent enhancement of excitatory transmitter release from primary afferent fibers. J Neurosci. 2002;22:6747–6755. doi: 10.1523/JNEUROSCI.22-15-06747.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hauser KF, Foldes JK, Turbek CS. Dynorphin A (1–13) neurotoxicity in vitro: opioid and non-opioid mechanisms in mouse spinal cord neurons. Exp Neurol. 1999;160:361–375. doi: 10.1006/exnr.1999.7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hughes J, Smith TW, Kosterlitz HW, Fothergill LA, Morgan BA, Morris HR. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature. 1975;258:577–580. doi: 10.1038/258577a0. [DOI] [PubMed] [Google Scholar]
- 17.Kajander KC, Sahara Y, Iadarola MJ, Bennett GJ. Dynorphin increases in the dorsal spinal cord in rats with a painful peripheral neuropathy. Peptides. 1990;11:719–728. doi: 10.1016/0196-9781(90)90187-a. [DOI] [PubMed] [Google Scholar]
- 18.Koetzner L, Hua XY, Lai J, Porreca F, Yaksh T. Nonopioid actions of intrathecal dynorphin evoke spinal excitatory amino acid and prostaglandin E2 release mediated by cyclooxygenase-1 and -2. J Neurosci. 2004;24:1451–1458. doi: 10.1523/JNEUROSCI.1517-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai J, Luo MC, Chen Q, Ma S, Gardell LR, Ossipov MH, Porreca F. Dynorphin A activates bradykinin receptors to maintain neuropathic pain. Nat Neurosci. 2006;9:1534–1540. doi: 10.1038/nn1804. [DOI] [PubMed] [Google Scholar]
- 20.Lai SL, Gu Y, Huang LY. Dynorphin uses a non-opioid mechanism to potentiate N-methyl-Daspartate currents in single rat periaqueductal gray neurons. Neurosci Lett. 1998;247:115–118. doi: 10.1016/s0304-3940(98)00293-6. [DOI] [PubMed] [Google Scholar]
- 21.Laneuville O, Reader TA, Couture R. Intrathecal bradykinin acts presynaptically on spinal noradrenergic terminals to produce antinociception in the rat. Eur J Pharmacol. 1989;159:273–283. doi: 10.1016/0014-2999(89)90158-1. [DOI] [PubMed] [Google Scholar]
- 22.Long JB, Petras JM, Mobley WC, Holaday JW. Neurological dysfunction after intrathecal injection of dynorphin A (1–13) in the rat. II. Nonopioid mechanisms mediate loss of motor, sensory and autonomic function. J Pharmacol Exp Ther. 1988;246:1167–1174. [PubMed] [Google Scholar]
- 23.Long JB, Rigamonti DD, Oleshansky MA, Wingfield CP, Martinez-Arizala A. Dynorphin A-induced rat spinal cord injury: evidence for excitatory amino acid involvement in a pharmacological model of ischemic spinal cord injury. J Pharmacol Exp Ther. 1994;269:358–366. [PubMed] [Google Scholar]
- 24.Lopes P, Kar S, Tousignant C, Regoli D, Quirion R, Couture R. Autoradiographic localization of [125I-Tyr8]-bradykinin receptor binding sites in the guinea pig spinal cord. Synapse. 1993;15:48–57. doi: 10.1002/syn.890150106. [DOI] [PubMed] [Google Scholar]
- 25.Lough C, Young T, Parker R, Wittenauer S, Vincler M. Increased spinal dynorphin contributes to chronic nicotine-induced mechanical hypersensitivity in the rat. Neurosci Lett. 2007;422:54–58. doi: 10.1016/j.neulet.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo MC, Chen Q, Porreca F, Lai J. Dynorphin Maintains Inflammatory Hyperalgesia by the Activation of Spinal Bradykinin Receptors. Journal of Pain. in press. [Google Scholar]
- 27.Malan TP, Ossipov MH, Gardell LR, Ibrahim M, Bian D, Lai J, Porreca F. Extraterritorial neuropathic pain correlates with multisegmental elevation of spinal dynorphin in nerve-injured rats. Pain. 2000;86:185–194. doi: 10.1016/s0304-3959(00)00243-8. [DOI] [PubMed] [Google Scholar]
- 28.Massardier D, Hunt PF. A direct non-opiate interaction of dynorphin-(1–13) with the N-methyl- D-aspartate (NMDA) receptor. Eur J Pharmacol. 1989;170:125–126. doi: 10.1016/0014-2999(89)90149-0. [DOI] [PubMed] [Google Scholar]
- 29.Peters CM, Lindsay TH, Pomonis JD, Luger NM, Ghilardi JR, Sevcik MA, Mantyh PW. Endothelin and the tumorigenic component of bone cancer pain. Neuroscience. 2004;126:1043–1052. doi: 10.1016/j.neuroscience.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 30.Rossier J. Opioid peptides have found their roots. Nature. 1982;298:221–222. doi: 10.1038/298221a0. [DOI] [PubMed] [Google Scholar]
- 31.Ruda MA, Iadarola MJ, Cohen LV, Young WS., 3rd In situ hybridization histochemistry and immunocytochemistry reveal an increase in spinal dynorphin biosynthesis in a rat model of peripheral inflammation and hyperalgesia. Proc Natl Acad Sci U S A. 1988;85:622–626. doi: 10.1073/pnas.85.2.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shukla VK, Bansinath M, Dumont M, Lemaire S. Selective involvement of kappa opioid and phencyclidine receptors in the analgesic and motor effects of dynorphin-A-(1–13)-Tyr-Leu-Phe- Asn-Gly-Pro. Brain Res. 1992;591:176–180. doi: 10.1016/0006-8993(92)90994-k. [DOI] [PubMed] [Google Scholar]
- 33.Singh IN, Goody RJ, Goebel SM, Martin KM, Knapp PE, Marinova Z, Hirschberg D, Yakovleva T, Bergman T, Bakalkin G, Hauser KF. Dynorphin A (1–17) induces apoptosis in striatal neurons in vitro through alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate/kainate receptor-mediated cytochrome c release and caspase-3 activation. Neuroscience. 2003;122:1013–1023. doi: 10.1016/j.neuroscience.2003.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skilling SR, Harkness DH, Larson AA. Experimental peripheral neuropathy decreases the dose of substance P required to increase excitatory amino acid release in the CSF of the rat spinal cord. Neurosci Lett. 1992;139:92–96. doi: 10.1016/0304-3940(92)90865-5. [DOI] [PubMed] [Google Scholar]
- 35.Sot U, Misterek K, Gumulka SW, Dorociak A. Intrathecal bradykinin administration: opposite effects on nociceptive transmission. Pharmacology. 2002;66:76–80. doi: 10.1159/000065629. [DOI] [PubMed] [Google Scholar]
- 36.Steranka LR, Manning DC, DeHaas CJ, Ferkany JW, Borosky SA, Connor JR, Vavrek RJ, Stewart JM, Snyder SH. Bradykinin as a pain mediator: receptors are localized to sensory neurons, and antagonists have analgesic actions. Proc Natl Acad Sci U S A. 1988;85:3245–3249. doi: 10.1073/pnas.85.9.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stevens CW, Yaksh TL. Dynorphin A and related peptides administered intrathecally in the rat: a search for putative kappa opiate receptor activity. J Pharmacol Exp Ther. 1986;238:833–838. [PubMed] [Google Scholar]
- 38.Stewart P, Isaac L. A strychnine-sensitive site is involved in dynorphin-induced paralysis and loss of the tail-flick reflex. Brain Res. 1991;543:322–326. doi: 10.1016/0006-8993(91)90044-v. [DOI] [PubMed] [Google Scholar]
- 39.Tachibana T, Miki K, Fukuoka T, Arakawa A, Taniguchi M, Maruo S, Noguchi K. Dynorphin mRNA expression in dorsal horn neurons after traumatic spinal cord injury: temporal and spatial analysis using in situ hybridization. J Neurotrauma. 1998;15:485–494. doi: 10.1089/neu.1998.15.485. [DOI] [PubMed] [Google Scholar]
- 40.Tan-No K, Esashi A, Nakagawasai O, Niijima F, Tadano T, Sakurada C, Sakurada T, Bakalkin G, Terenius L, Kisara K. Intrathecally administered big dynorphin, a prodynorphin-derived peptide, produces nociceptive behavior through an N-methyl-D-aspartate receptor mechanism. Brain Res. 2002;952:7–14. doi: 10.1016/s0006-8993(02)03180-3. [DOI] [PubMed] [Google Scholar]
- 41.Tan-No K, Takahashi H, Nakagawasai O, Niijima F, Sato T, Satoh S, Sakurada S, Marinova Z, Yakovleva T, Bakalkin G, Terenius L, Tadano T. Pronociceptive role of dynorphins in uninjured animals: N-ethylmaleimide-induced nociceptive behavior mediated through inhibition of dynorphin degradation. Pain. 2005;113:301–309. doi: 10.1016/j.pain.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 42.Tang Q, Gandhoke R, Burritt A, Hruby VJ, Porreca F, Lai J. High-affinity interaction of (des-Tyrosyl)dynorphin A(2–17) with NMDA receptors. J Pharmacol Exp Ther. 1999;291:760–765. [PubMed] [Google Scholar]
- 43.Tang Q, Lynch RM, Porreca F, Lai J. Dynorphin A elicits an increase in intracellular calcium in cultured neurons via a non-opioid, non-NMDA mechanism. J Neurophysiol. 2000;83:2610–2615. doi: 10.1152/jn.2000.83.5.2610. [DOI] [PubMed] [Google Scholar]
- 44.Vanderah TW, Gardell LR, Burgess SE, Ibrahim M, Dogrul A, Zhong CM, Zhang ET, Malan TP, Jr, Ossipov MH, Lai J, Porreca F. Dynorphin promotes abnormal pain and spinal opioid antinociceptive tolerance. J Neurosci. 2000;20:7074–7079. doi: 10.1523/JNEUROSCI.20-18-07074.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vanderah TW, Laughlin T, Lashbrook JM, Nichols ML, Wilcox GL, Ossipov MH, Malan TP, Jr, Porreca F. Single intrathecal injections of dynorphin A or des-Tyr-dynorphins produce long-lasting allodynia in rats: blockade by MK-801 but not naloxone. Pain. 1996;68:275–281. doi: 10.1016/s0304-3959(96)03225-3. [DOI] [PubMed] [Google Scholar]
- 46.Vera-Portocarrero LP, Xie JY, Kowal J, Ossipov MH, King T, Porreca F. Descending facilitation from the rostral ventromedial medulla maintains visceral pain in rats with experimental pancreatitis. Gastroenterology. 2006;130:2155–2164. doi: 10.1053/j.gastro.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 47.Wagner R, Deleo JA. Pre-emptive dynorphin and N-methyl-D-aspartate glutamate receptor antagonism alters spinal immunocytochemistry but not allodynia following complete peripheral nerve injury. Neuroscience. 1996;72:527–534. doi: 10.1016/0306-4522(95)00495-5. [DOI] [PubMed] [Google Scholar]
- 48.Wagner R, DeLeo JA, Coombs DW, Willenbring S, Fromm C. Spinal dynorphin immunoreactivity increases bilaterally in a neuropathic pain model. Brain Res. 1993;629:323–326. doi: 10.1016/0006-8993(93)91339-t. [DOI] [PubMed] [Google Scholar]
- 49.Walker JM, Moises HC, Coy DH, Baldrighi G, Akil H. Nonopiate effects of dynorphin and des-Tyr-dynorphin. Science. 1982;218:1136–1138. doi: 10.1126/science.6128791. [DOI] [PubMed] [Google Scholar]
- 50.Walker JM, Moises HC, Coy DH, Young EA, Watson SJ, Akil H. Dynorphin (1–17): lack of analgesia but evidence for non-opiate electrophysiological and motor effects. Life Sci. 1982;31:1821–1824. doi: 10.1016/0024-3205(82)90219-3. [DOI] [PubMed] [Google Scholar]
- 51.Wang H, Kohno T, Amaya F, Brenner GJ, Ito N, Allchorne A, Ji RR, Woolf CJ. Bradykinin produces pain hypersensitivity by potentiating spinal cord glutamatergic synaptic transmission. J Neurosci. 2005;25:7986–7992. doi: 10.1523/JNEUROSCI.2393-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Z, Gardell LR, Ossipov MH, Vanderah TW, Brennan MB, Hochgeschwender U, Hruby VJ, Malan TP, Jr, Lai J, Porreca F. Pronociceptive actions of dynorphin maintain chronic neuropathic pain. J Neurosci. 2001;21:1779–1786. doi: 10.1523/JNEUROSCI.21-05-01779.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watson SJ, Khachaturian H, Akil H, Coy DH, Goldstein A. Comparison of the distribution of dynorphin systems and enkephalin systems in brain. Science. 1982;218:1134–1136. doi: 10.1126/science.6128790. [DOI] [PubMed] [Google Scholar]
- 54.Weihe E, Millan MJ, Hollt V, Nohr D, Herz A. Induction of the gene encoding pro-dynorphin by experimentally induced arthritis enhances staining for dynorphin in the spinal cord of rats. Neuroscience. 1989;31:77–95. doi: 10.1016/0306-4522(89)90031-6. [DOI] [PubMed] [Google Scholar]
- 55.Winkler T, Sharma HS, Gordh T, Badgaiyan RD, Stalberg E, Westman J. Topical application of dynorphin A (1–17) antiserum attenuates trauma induced alterations in spinal cord evoked potentials, microvascular permeability disturbances, edema formation and cell injury: an experimental study in the rat using electrophysiological and morphological approaches. Amino Acids. 2002;23:273–281. doi: 10.1007/s00726-001-0138-y. [DOI] [PubMed] [Google Scholar]
- 56.Wotherspoon G, Winter J. Bradykinin B1 receptor is constitutively expressed in the rat sensory nervous system. Neurosci Lett. 2000;294:175–178. doi: 10.1016/s0304-3940(00)01561-5. [DOI] [PubMed] [Google Scholar]
- 57.Xu M, Petraschka M, McLaughlin JP, Westenbroek RE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Terman GW, Chavkin C. Neuropathic pain activates the endogenous kappa opioid system in mouse spinal cord and induces opioid receptor tolerance. J Neurosci. 2004;24:4576–4584. doi: 10.1523/JNEUROSCI.5552-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Young EA, Walker JM, Houghten R, Akil H. The degradation of dynorphin A in brain tissue in vivo and in vitro. Peptides. 1987;8:701–707. doi: 10.1016/0196-9781(87)90046-5. [DOI] [PubMed] [Google Scholar]
- 59.Zhang L, Peoples RW, Oz M, Harvey-White J, Weight FF, Brauneis U. Potentiation of NMDA receptor-mediated responses by dynorphin at low extracellular glycine concentrations. J Neurophysiol. 1997;78:582–590. doi: 10.1152/jn.1997.78.2.582. [DOI] [PubMed] [Google Scholar]