

Abstract
AIM
Experimental pain models may help to evaluate the mechanisms of action of analgesics and target the clinical indications for their use. This review addresses how the efficacy of opioids can be assessed in human volunteers using experimental pain models. The drawback with the different study designs is also discussed.
METHOD
A literature search was completed for randomized controlled studies which included human experimental pain models, healthy volunteers and opioids.
RESULTS
Opioids with a strong affinity for the µ-opioid receptor decreased the sensation in a variety of experimental pain modalities, but strong tonic pain was attenuated more than short lasting pain and non-painful sensations. The effects of opioids with weaker affinity for the µ-opioid receptor were detected by a more narrow range of pain models, and the assessment methods needed to be more sensitive.
CONCLUSION
The way the pain is induced, assessed and summarized is very important for the sensitivity of the pain models. This review gives an overview of how different opioids perform in experimental pain models. Generally experimental pain models need to be designed with careful consideration of pharmacological mechanisms and pharmacokinetics of analgesics. This knowledge can aid the decisions needed to be taken when designing experimental pain studies for compounds entering phase 1 clinical trials.
Keywords: clinical trials, experimental pain, opioids
Introduction
Pain is a very prevalent symptom in medicine, and characterization of pain is of major importance in the diagnosis and choice of treatment [1]. The use of various opioids is the prevailing treatment of moderate to severe pain. The clinical effects typically guide the selection of the analgesics and titration of the dose. However, when treating clinical pain analgesic effects are difficult to evaluate due to a number of factors other than the pain intensity. These modifiers of the effect may include complaints relating to psychological, cognitive and social aspects of the illness, as well as systemic reactions such as fever and general malaise [2]. Hence, any change in these factors will invariably also interfere with pain intensity and pain quality and bias the assessment of analgesics in clinical trials.
Because of the confounders, experimental pain models are often advantageous for characterizing analgesics [3]. Using such models, the investigator can control the experimentally induced pain (including the nature, localization, intensity, frequency and duration of the stimulus), and provide quantitative measures of the psychophysical, behavioural or neurophysiologic responses [2, 4]. Discrete changes in pain intensity can be detected and the analgesic effect can be evaluated. Application of these experimental pain models offers a unique opportunity to investigate analgesic effects on different pain modalities arising from different tissues as well as peripheral and/or central pain mechanisms [5].
Reproducibility is an important factor in the testing of analgesics where it is necessary to repeat the pain stimulation several times during active and placebo treatments. If the reproducibility is low, then the change in the pain measure needs to be large for the model to detect it.
This review, as the first in a series of two, deals with opioids tested in human pain models. The next review will describe non-opioid analgesics tested in human pain models [6]. To be able to illustrate the importance of various experimental designs, only drugs that have been tested in at least five different trials each including at least seven volunteers were included. These limits ensured that a sufficient number of trials could be included and that these trials were of a quality ensuring a meaningful discussion. Data from patient studies have not been included. Generally these are more difficult to interpret due to the heterogeneity of the patients and the confounders associated with the illness mentioned above.
Hence, the aim of this review was to characterize how various experimental models of acute pain and evoked hyperalgesia detect analgesia of clinically used analgesics. This was divided into the following: i) to investigate the sensitivity of various experimental models to test clinically used analgesics and ii) to investigate how the dose and dosing regimen can affect the findings. As physiology of the deep (viscera or muscle) and superficial (skin) pain differs the results were furthermore divided into the tissue (skin, muscle or viscera) in which the pain was induced, and into modalities used for pain stimulation.
Methods
PubMed searches were conducted for articles and abstracts. MeSH and free-text terms for opioids were combined with the terms ‘experimental pain’, ‘human’, and ‘randomized’. Only manuscripts published in English were included. There was no limit for the time of publication. Furthermore we did not feel that the level of evidence could be graded due to the exploratory nature of many of the studies. Some trials test combinations of analgesics but to avoid too complex results we only included trials where the analgesic in question was tested alone in one of the treatment arms.
To be able to illustrate the importance of various experimental designs we only included drugs that had been tested in at least five different trials. Trials involving experimental pain often use very small sample sizes because the variation of the outcome measures are less variable than in traditional clinical trials. Trials with less than 10–12 subjects are hard to test statistically and the findings therefore questionable. However it has been shown that in experimental models with a high reproducibility sample sizes below 10 are powered to show the effect of analgesics [7]. Accordingly we found a well designed study with a sample size of seven, and this was the minimum sample size for the studies included in this review [8].
Results are summed up and discussed on a pharmacological mechanistic basis at the end of each drug class section.
Short introduction to experimental pain models
Several in-depth reviews exist in this area [2, 9–11]. Clinical pain is the net effect of peripheral activation and sensitization of afferent nerves, followed by complex multidimensional mechanisms that involve most parts of the CNS [12]. The nociceptive impulses are transmitted by thin myelinated (Aδ) or non-myelinated (C) fibres [13]. Modifications of the CNS follow long-lasting or strong pain, and may result in sensitization of the nociceptive system [14].
Fundamentally pain models can be divided into acute models and models inducing hyperalgesia. Acute models activate normal physiological mechanisms by activation of peripheral nociceptors by, for example, heat stimulation of the skin [10]. Traditionally such pain mechanisms are believed to be less relevant to mimic pathological pain. Models such as intradermally injected capsaicin induce hyperalgesia and allodynia [15, 16]. Such models alter the peripheral and central pain system and are thought to reflect chronic pain processes to a greater extent than the acute pain models [17]. It is important to realize that experimental pain only activates part of the multidimensional mechanisms involved in pathological pain and this limits the translation of analgesic effects in experimental pain into clinical analgesic effects. An overview of frequently used models where clinical correlates exist is given in Table 1.
Table 1.
Frequently used experimental pain models
| Reference | Model | Proposed mechanism | Clinical correlate |
|---|---|---|---|
| [8, 59, 102] | Pressure algometry | Activation of group III and IV afferents | Palpation |
| [8, 52, 108] | Cold pressor test | Activation of DNIC | DNIC is impaired in many functional pain syndromes |
| [8, 102] | Ischaemic muscle pain | Release of various transmitters involved in pain such as adenosine, serotonin and prostaglandins. Probably also activation of DNIC | Chronic musculo-skeletal pain, intermittent claudication |
| [8, 18, 53, 57, 100, 102] | Temporal summation of electrical, thermal or mechanical stimuli (applied in all tissue) | Activation of the NMDA receptor | The NMDA receptor is known to be activated in most types of clinical pain |
| Capsaicin applied in the skin | Activation of the TRPV1 receptor and C fibre mediated pain. Central and peripheral sensitisation evoking Aβ mediated allodynia and Aδ mediated hyperalgesia | Exhibits features seen in neuropathic pain | |
| [32, 33] | Continuous electrical stimulation | Central and peripheral sensitization | Not known |
DNIC: diffuse noxious inhibitory control, NMDA: N-methyl D-aspartate, TRPV1: transient receptor potential cation channel, subfamily V, member 1.
Acute models
Models applied in the skin
When determining the heat pain thresholds, rapid skin heating (faster than 1°C s−1) activates first Aδ-fibres, where the evoked sensation corresponds to the ‘first pain’ felt within 0.4 s after the heat stimulus [18]. Slow heating (1°C s−1 or less) gives a preferential activation of the C-fibres (thought to be most important for peripheral opioid receptors, see below) and the best evaluation of the ‘second pain’[10]. It is generally believed that lasers stimulate Aδ fibres giving a pricking pain followed by C-fibre mediated second pain [19]. The neuronal activation after cold pain is less well described than for heat pain, but probably involves a mosaic of primary afferent input with a definite involvement of C-fibres [20, 21]. Electrical stimulation excites the nerve directly and bypasses the nociceptors and therefore pharmacological effects on peripheral nociceptors (e.g. activation of peripheral opioid receptors) cannot be elucidated by this method. Electrical stimulation of the tooth pulp is considered to be a C-fibre selective stimulation [22].
Models applied in the muscle
Pressure algometry (deep pressure) is the most frequently applied technique for quantification of pain. The method is an experimental parallel to palpation in clinical practice [23]. The pain mainly originates from deep tissue group III and IV afferents [9]. When pressure algometry is performed at the tibia, the pain probably contains elements of pain originating from the bone as well as the skin. The mechanism of pain evoked in the cold pressor test is not well described, but it is known that the immersion of the hand in ice water activates the sympathetic nervous system innervating muscle and nerve fascicles [24, 25]. The cold pressor test is known to activate the ‘diffuse noxious inhibitory control system’, which is a system of descending neuronal pathways arising in the brain stem that exerts negative feed-back control of the incoming activity to the spinal cord.
Models evoking central integration of pain and hyperalgesia
By evoking different central phenomena like allodynia, hyperalgesia, referred pain or temporal summation in the experimental situation, central pain mechanisms can be studied in humans [26]. This is of major importance since abnormal central processing of pain characterizes many disorders associated with pain [14]. The central phenomena can be evoked by stimulation of all tissues, but have been investigated most thoroughly in the skin, where, for example, repeated electrical or thermal stimulation can induce temporal summation mechanisms [26, 27]. The drawback with models evoking hyperalgesia is that they are more difficult to control regarding reproducibility compared with the acute stimulations (Figure 1).
Figure 1.
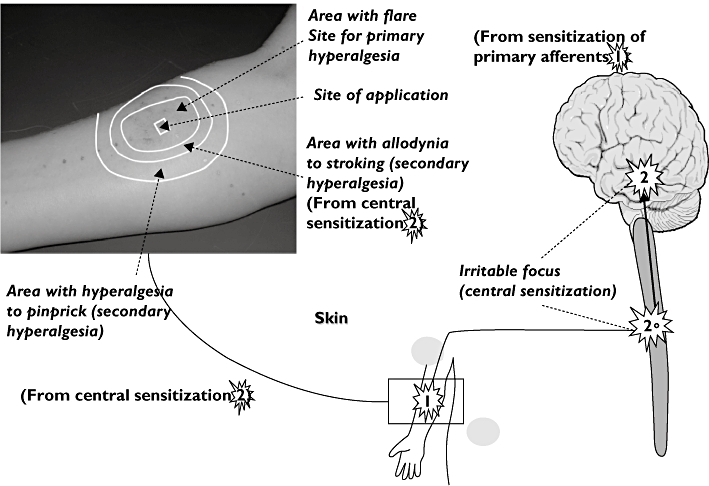
Experimental hyperalgesia exemplified by capsaicin-evoked hyperalgesia in the skin. The stars illustrate how sensitization of the pain system is evoked at peripheral (arm) and central levels (spinal and supraspinal). The picture illustrates (top left) how hyperalgesia is manifested locally (peripheral sensitisation in the area of flare formation) and by central sensitization (allodynia and hyperalgesia to pinprick)
Models applied in the skin
Allodynia and hyperalgesia can be evoked at an injury site (primary hyperalgesia) or at sites adjacent to or remote from an injury site (secondary hyperalgesia). It is well established that primary hyperalgesia is the sensory consequence of the sensitization of peripheral nociceptors. The mechanisms of secondary hyperalgesia are far from clear. It is generally accepted that these mechanisms are based on alterations of sensory processing in the CNS [28].
The burn injury model produces a first degree burn injury, where primary and secondary hyperalgesia exist. Hyperalgesia to heat in the burned area (primary hyperalgesia) is probably mediated by both central and peripheral sensitization of C- and Aδ-fibers. The model also produces hyperalgesia to heat in normal skin surrounding the burn (secondary hyperalgesia) [29]. Intradermal injection or topical application of capsaicin evokes C-fibre mediated pain in the skin [16, 30]. This model produces allodynia and primary and secondary hyperalgesia which are prominent symptoms in, for example, neuropathic pain. Induction of a freeze lesion provides a model of hyperalgesia, which predominantly is caused by peripheral mechanisms [31].
The electrical hyperalgesia model offers stable long-lasting hyperalgesia demonstrating a central mechanism of sensitization. This model provides an index for the hyperalgesic vs the analgesic properties of a drug [32]. The model is well tested and has proven both positive and negative predictive value of the clinical effects of drugs [33, 34].
Models applied in the muscle
Eccentric contractions induce a delayed onset (24–48 h) of muscle pain or soreness. The long-lasting nature of the pain evoked in the ‘delayed-onset muscle soreness model’ mimics clinical pain [35].
In the viscera, central modifications manifested by hyperalgesia/allodynia, can also be induced by application of strong or long lasting noxious stimuli [36].
Pain assessment
Another important factor is the assessment method of the induced pain. It can be quantified by psychophysical, neurophysiologic or imaging methods [12]. Psychophysical methods are based on the subjective experience of pain, measured on standard scales or as pain thresholds. It should be emphasized that pain models which evoke higher pain intensities recruit more C-fibres than pain intensities close to or under the pain detection threshold. Accordingly suprathreshold pain measures are traditionally thought to be more sensitive in drug research than the pain detection threshold [37]. Part of the pain system can also be evaluated objectively with neurophysiologic techniques. Examples of such methods are measuring of nociceptive withdrawal reflexes or evoked brain potentials. The evoked potentials result from summation of a series of time-locked electro encephalogram (EEG) responses to a stimulus. Potentials elicited by mechanical and electrical stimulation are mainly the result of a cortical response to Aδ fibres, whereas laser stimulation mainly elicits potentials resulting from cortical responses to both Aδ and C fibres [38]. Furthermore the central processing of pain can be assessed by imaging techniques like functional magnetic resonance imaging (fMRI) and positron emission tomography (PET). Such methods can assert the role of the cortex in pain perception and may subdivide different cortical and sub-cortical areas as to their specific role in pain perception and modulation [39].
In models evoking hyperalgesia, the sensitization of the pain system can be assessed in different ways. Hyperalgesia can be seen as an increased pain response to painful stimulation or a lowering of the pain detection threshold [40, 41]. Often such models also evoke allodynia, for example, to gently stroking of the skin by a cotton wool tip [42, 43].
Opioids in experimental pain
Overviews of various opioids and the sensitivity of various experimental pain models are given in Tables 1–7.
Table 7.
Schematic overview of studies involving tramadol in human experimental pain models
| Reference | Dose | Model | Main findings | |
|---|---|---|---|---|
| Acute models | [117] (n= 15) | 50 mg orally. three times daily for 3 days | Ischaemic pain performance and PTT | No parameters were affected. |
| [111] (n= 27) | 100 mg iv | Nociceptive reflex to electrical sural nerve stimulation (single and repeated) PDT, PTT Cold pressor test peak pain, discomfort (VAS) | Extensive metabolizers: Nociceptive reflexes and pain thresholds ↔ Cold pressor test: discomfort ↓, peak pain ↔ Poor metabolizers: Single stimulation PTT (single electrical) ↓ Cold pressor test: discomfort ↓ | |
| [114] (n= 12) | 50 mg orally | Electrical tooth pulp stimulation sensation threshold, subjective pain rating, evoked brain potentials | No parameters were affected | |
| [113] (n= 20) | 100 and 200 mg sustained release orally | Stimulation of nasal mucosa by CO2 and dry air (tonic pain) VAS, evoked brain potentials (to CO2 stimulation) EEG (frequency analysis) | VAS to tonic pain ↓ VAS to CO2 stimulation ↔ Amplitudes of evoked potentials ↓ Latencies of evoked potentials ↔ EEG frequency spectrum was changed | |
| [121] (n= 27) | 2 mg kg−1 orally | Pressure algometry PDT, PTT (phalanx) Nocicpetive reflex to electrical sural nerve stimulation (single and repeated) stimulus response curve to electrical stimulation Cold pressor test AUCVAS peak pain, discomfort (VAS) | Extensive metabolizers: Pressure PDT and PTT ↑ Thresholds to nociceptive reflex ↑, stimulus response curve ↔ Cold pressor test: peak pain and AUCVAS↓, discomfort ↔ Poor metabolizers: Only significant effect on pressure PTT and single nociceptive reflex | |
| [112] (n= 16) | 150 mg orally | Stimulation of nasal mucosa by CO2VAS, evoked brain potentials (to CO2 stimulation), EEG (frequency analysis) | VAS and amplitudes of evoked potentials ↓ Latencies of evoked potentials ↔ EEG frequency spectrum ↔ | |
| [116] (n= 10) | 50 and 100 mg orally | Electrical tooth pulp stimulation sensation threshold, subjective pain rating, evoked brain potentials | All parameters were affected (dose-response manner) | |
| Models inducing hyperalgesia | [118] (n= 17) | 75 mg orally | Continuous electrical skin stimulation Ongoing pain (numeric rating scale) Area of secondary HA to pinprick (von Frey) | Ongoing pain ↓ Area of HA ↔ |
| [117] (n= 15) | 50 mg three times daily for 3 days | Delayed onset muscle sorenss Pressure algometry PDT (thigh, knee joint) | No parameters were affected |
In the column ‘model’ the method for pain assessment is normal font, and the method for pain induction is bolded. Abbreviations: pain detection threshold (PDT), pain tolerance threshold (PTT), area under curve (AUC), visual analogue scale score (VAS), hyperalgesia (HA).
Short acting opioids with strong affinity for the µ-opioid receptor
Alfentanil (Table 2)
Table 2.
Schematic overview of studies involving alfentanil in human experimental pain models
| Reference | Dose | Model (pain assessments) | Main findings | |
|---|---|---|---|---|
| Acute models | [45] (n= 36) | 50–250 ng ml−1 iv for 30 min | Electrical skin stimulation, PTT Heat skin stimulation VAS (0.5°C/s rise to set temperatures) | Electrical PTT↓ Heat pain ↓ |
| [44] (n= 12) | 13.4–126.1 ng ml−1 iv | Electrical skin stimulation, PDT, PTT Heat skin stimulation PDT, PTT(1°C s−1) | Electrical and heat PDT, PTT ↓ | |
| [49] (n= 11) | 25–75 ng ml−1 iv | Warmth and cool (sensory threshold) Heat and cold skin stimulation PDT (1–1.5°C/s) Touch sensory threshold (von Frey) | Sensory threshold ↑ Hot and cold PDT ↔ Sensation from touch ↔ | |
| [48] (n= 16) | 25 and 75 ng ml−1 iv | IM injection of hypertonic saline, AUCVASTranscutaneous and intramuscular electrical stimulation PDT | AUC ↓ Electrical PDT ↓ But only for the high dose | |
| [53] (n= 15) | 60 ng ml−1 iv | Skin and intramuscular single and repeated electrical stimulation, PDT, tonic pain to intramuscular electrical stimulation (pain to 1.5 × PDT for 10 s) Im injection of hypertonic saline, AUCVAS | Electrical PDT ↓ VAS to electrical stim. and hypertonic saline ↓ | |
| [8] (n= 7) | 50–200 ng ml−1 iv | Electrical skin stimulation Heat skin stimulation PTT (2°C s−1), Deep pressure, PTT Cold pressor test, AUCVASIschemic pain AUCVAS | Electrical PTT ↑ Cold pressor test, AUC ↓ Deep pressure PTT ↑ Heat PTT↔ Ischaemic pain AUC ↔ | |
| [52] (n= 10) | 16–65 ng ml−1 iv | Cold pressor test, VAS, McGill pain questionnaire | VAS and bothersomeness (McGill pain questionnaire) ↓ | |
| [47] (n= 12) | 7.5 µg kg−1 followed by 0.1 µg kg−1 min−1 and 15 µg kg−1 followed by 0.3 µg kg−1 min−1 iv | Nocicpetive reflex to sural nerve stimulation Deep pressure, PDT, PTT Argon laser pain Intracutaneous electrical stimulation, VAS Evoked brain potentials (electrical and laser) | Reflex threshold ↑ (dose-response relation) Pressure PDT and PTT ↑ (high dose) Evoked potentials amplitude ↓ Argon laser pain ↓ (high dose) | |
| [46] (n= 8) | 30 µg kg−1 im | Nocicpetive reflex to electrical sural nerve stimulation Heat skin stimulation PTT (laser) Pressure algometry (deep pressure), PDT, PTT Cold pressor test AUCVAS | Reflex threshold ↑ (dose-response relation) Pressure PDT ↔ and PTT ↑ Heat PTT ↓ AUCVAS↓ | |
| [51] (n= 10) | 15 µg kg−1 iv | Electrical stimulation of the teeth Evoked brain potentials Subjective pain report (6 point scale) | Evoked potentials amplitude ↓ Pain was reduced | |
| [50] (n= 25) | 19.6–76.6 ng ml−1 iv | Stimulation of nasal mucosa by CO2VAS, functional magnetic resonance imaging | PDT↓ dose dependently Pain associated brain activation ↓ | |
| Models inducing hyperalgesia | [57] (n= 16) | 70 ng ml−1 iv | Burn injury PDT to pinprick in injured skin, area of secondary HA to pinprick Repeated pinprick AUCVASContinuous electrical skin stimulation PDT to pinprick, area of secondary HA to pinprick, repeated pinprick (AUCVAS), | Pain and HA was reduced for all parameters |
| [49] (n= 11) | 25, 50 and 75 ng ml−1 iv | Intradermal capsaicin, evoked pain (VAS) Area of secondary HA to pinprick (von Frey), heat and allodynia to stroking | Pain from capsaicin↓ Area of secondary HA to pinprick & allodynia to stroking↓, HA to heat ↔ | |
| [32] (n= 12) | 100 ng ml−1 iv | Continuous electrical skin stimulation Evoked pain (threshold to VAS5/10) Area of secondary HA to pinprick (von Frey) and allodynia to stroking | Electrically evoked pain ↓ Area of secondary HA to pinprick ↓ (during infusion) | |
| [55] (n= 12) | 28 µg kg−1 and 7 µg kg−1, iv | Intradermal capsaicin, evoked pain (VAS) Pinprick hyperalgesia and allodynia to stroking | No significant dose-response could be demonstrated | |
| [54] (n= 46) | 200 ng ml−1 iv | Intradermal capsaicin, evoked pain (VAS) Area of secondary HA to pinprick (von Frey) and allodynia to stroking | Pain from capsaicin↓ Area of secondary HA to pinprick & allodynia to stroking↓ |
In the column ‘model’ the method for pain assessment is normal font, and the method for pain induction is bolded. Abbreviations: pain detection threshold (PDT), pain tolerance threshold (PTT), area under curve (AUC), visual analogue scale score (VAS), hyperalgesia (HA).
Acute models
Skin/teeth:
Alfentanil has been tested in several acute models in the skin using heat, cold and electrical stimulation [8, 44–49]. The tested models were generally all sensitive to alfentanil.
Short pulses of gaseous CO2 applied to the nasal mucosa evoked pain that was dose dependently decreased by alfentanil. More interestingly this study applied functional magnetic resonance imaging to investigate the opioid effects and found this assessment method sufficiently sensitive to see differential effects on the affective and sensory components of the pain [50]. Furthermore this study, as one of few, was able to demonstrate how carriers of different genetic variants of the µ-opioid receptor responded differently to alfentanil.
Alfentanil has furthermore provided robust analgesia in electrically evoked pain in the teeth [51].
Muscle:
Alfentanil is well characterized in experimental muscular pain such as deep pressure, intramuscular electrical stimulation, cold pressor test, and intramuscular injection of hypertonic saline and ischaemic pain. In the study by Black et al. both pain intensity and the affective component ‘bothersomeness’ was scored and alfentanil reduced both parameters [8, 52].
Alfentanil showed analgesic effects in tests involving electrical stimulation (both repeated and single stimulation), pressure, cold pressor test and injection of hypertonic saline [8, 44, 45, 48, 53]. The tourniquet model (evoking muscle ischaemia mainly), with continuous pain assessment for 120 s and area under the visual analogue scale (VAS) curve used as a pain measure, was not sensitive to alfentanil [8].
Models of hyperalgesia
Skin:
Alfentanil has been tested in a model that evokes hyperalgesia by intradermal injection of capsaicin [49, 54–56]. Two out of three studies found an effect on the evoked pain, hyperalgesia and allodynia [49, 54, 56].
Furthermore alfentanil has been tested against electrically evoked secondary hyperalgesia and in this model hyperalgesia and allodynia was reduced [32, 57].
In the burn injury model alfentanil reduced secondary hyperalgesia to pinprick suggesting a central effect of this opioid [57].
Dose:
Alfentanil is a very potent opioid showing convincing analgesia in experimental pain over a broad dose range. However, in the study by Schulte et al. a dose–response relationship was seen and the pain parameters were mainly affected at the high dose [48].
Fentanyl (Table 3)
Table 3.
Schematic overview of studies involving fentanyl in human experimental pain models
| Reference | Dose | Model | Findings | |
|---|---|---|---|---|
| Acute models | [58] (n= 20) | Transdermal 12.5 or 25 µg h−1 | Electrical and heat (1.4°°C s−1) skin stimulation PTT cold pressor test AUC, peak pain, mean pain intensity | Electrical pain ↔ Heat pain: ↓dose dependently Cold pressor test: AUC ↓ (both doses), peak pain and mean pain intensity (only high dose) ↓ |
| [59] (n= 10) | Targeted iv infusion 0.2 to 1.20 ng ml−1 | Electrical and heat (1°C s−1) skin stimulation PDT Deep pressure, PDT | PDT for electrical and pressure pain ↑ in a dose dependent manner Heat PDT ↔ | |
| [60] (n= 10) | Epidural, 0.03 mg followed by 0.1 mg | Electrical and heat (1°C s−1) skin stimulation, PTT | PTT for both parameters ↑ | |
| [62] (n= 14) | 0.75 and 1.5 µg kg−1 iv | Heat skin stimulation (52°°C, applied as single and repeated taps) VAS Cold skin stimulation (0.3–1°°C applied as repeated taps) VAS | Heat pain (single and repeated) ↔ Cold repeated pain ↓ | |
| [51] (n= 10) | 2 µg kg−1 iv | Dental electrical stimulation VAS, evoked brain potentials | Amplitude (brain potentials) ↓ VAS ↓ |
In the column ‘model’ the method for pain assessment is normal font, and the method for pain induction is bolded. Abbreviations: pain detection threshold (PDT), pain tolerance threshold (PTT), area under curve (AUC), visual analogue scale score (VAS), hyperalgesia (HA).
Acute models
Skin/dental:
Fentanyl has been tested against electrical and thermal (heat and cold) skin pain. Electrical pain was unaffected in one study, whereas two studies found an effect on this pain modality [58–60]. Furthermore electrical dental pain has been tested and found to be attenuated by fentanyl [51, 61]. Heat pain has been tested through various stimulation paradigms and conflicting results exist. The studies by Koltzenburg et al. and Ginosar et al. found that fentanyl attenuated this parameter, which was contradictory to the finding of Tucker et al[58–60]. Repetitive heat pain was unaffected by fentanyl where repetitive cold pain was attenuated by fentanyl [62].
Muscle:
Fentanyl has been tested in the cold pressor test and this model was sensitive to the analgesia induced by this opioid. The analgesic effect was shown more robustly when assessed as the area under the VAS curve compared with the peak pain intensity and the mean pain intensity [58].
Dose:
The studies involving the same pain models (heat pain with ascending ramp) did not use comparable methods of drug administration. Most likely transdermal dosing (as in the study by Koltzenburg et al.) with slow ascending plasma concentrations will produce a different analgesic profile from iv dosing with fast ascending and high peak plasma concentrations [58–60]. However, in the study applying transdermal drug administration, for less sensitive pain measures (peak pain and mean pain intensity in the cold pressor test) only the highest dose (25 µg h−1 transdermally) produced analgesia [58].
Remifentanil (Table 4)
Table 4.
Schematic overview of studies involving remifentanil in human experimental pain models
| Reference | Dose | Model (pain assessment) | Main findings | |
|---|---|---|---|---|
| Acute models | [68] (n= 7) | 0.05 and 0.15 µg min−1 kg−1 iv | Heat skin stimulation (pulses 0.3°°C under −1°C above PDT) evaluated by positron emission tomography | Remifentanil decreased the pain induced brain activation |
| [65] (n= 14) | 1 and 2 ng ml−1 iv | Single and repeated skin and intramuscular electrical stimulation PDT Deep pressure PTT | Pain from all modalities was decreased. Remifentanil induced HA was detected by pressure pain | |
| [69] (n= 10) | 0.025, 0.05 and 0.1 µg min−1 kg−1 iv | Tibial pressure evaluated by cerebral blood flow | Pain induced increase in cerebral blood flow was attenuated | |
| [66] (n= 18/14) | 0.1 µg min−1 kg−1 iv | Heat skin stimulation PDT and V s−1 to 1 min of 45°°C | PDT ↑ VAS ↓ | |
| [64] (n= 20) | 0.01 increasing to 0.17 µg min−1 kg−1 iv | Heat skin stimulation PDT (0.5°C s−1) | PDT ↑ in a dose-related manner | |
| [63] (n= 14) | 1 and 2 ng ml−1 iv (target control) | Skin and intramuscular repeated electrical stimulation PDT | PDT ↑ in both tissues, but mostly in the muscles | |
| Models inducing hyperalgesia | [71] (n= 15) | 0.05 µg min−1 kg−1 iv | Continuous electrical skin stimulation Ongoing pain (numeric rating scale) Area of secondary HA to pinprick (von Frey) | Electrically evoked pain ↓ Area of secondary HA to pinprick ↓ (during infusion). Post infusion HA was detected |
| [72] (n= 15) | 0.1 µg min −1kg−1 iv | Continuous electrical skin stimulation Ongoing pain (numeric rating scale) Area of secondary HA to pinprick (von Frey) | Electrically evoked pain ↓ Area of secondary HA to pinprick ↓ (during infusion). Post infusion HA was detected | |
| [33] (n= 13) | 0.05–0.1 µg min−1 kg−1 iv | Continuous electrical skin stimulation Ongoing pain (numeric rating scale) Area of secondary HA to pinprick (von Frey) | Dose-dependent reduction of evoked pain and HA. Post infusion HA was detected | |
| [31] (n= 12) | 0–6 ng ml−1 iv (target control) | Freeze lesion. Primary HA to electrical stimulation, stroking, pinprick and blunt pressure | No HA to electrical stimulation was seen. Electrical pain ↓ HA to blunt pressure ↓ twice as much as to pinprick. Stroking did not evoke pain | |
| [73] (n= 10) | 3.1 ng ml−1 iv (target control) | Heat-capsaicin sensitisation area of allodynia/secondary HA to brushing/pinprick von Frey Heat skin stimulation in inflamed skin (three different temperatures for 5 min.) numeric raring scale | Area of allodynia and HA ↓ Heat pain ↓ Post infusion HA was detected | |
| [66] (n= 18/14) | 0.1 µg min−1 kg−1 iv | Heat/capsaicin secondary area of HA to pinprick stimulation and brushing | The area of HA for both modalities was reduced |
In the column ‘model’ the method for pain assessment is normal font, and the method for pain induction is bolded. Abbreviations: pain detection threshold (PDT), pain tolerance threshold (PTT), area under curve (AUC), visual analogue scale score (VAS), hyperalgesia (HA).
Acute models
Skin:
Remifentanil has been tested against heat and electrical stimulation [63–67]. The studies showed a reduction in pain to heat stimuli as well as to single and repeated electrical stimulation. The effect of remifentanil on heat pain was further evaluated by positron emission tomography. The study showed a decrease of the pain induced brain activation and increased brain activity in the cingulofrontal cortex and periaqueductal gray [68].
Muscle:
Pressure pain has been tested against remifentanil and here it was found that hyperalgesia was induced by this opioid [65].
Furthermore, the effect of remifentanil against pain from pressure applied to the tibia has been tested via the cerebral blood flow, where the drug decreased the pain induced increase in the cerebral blood flow [69]. Two studies investigated and found effect of remifentanil to single and repeated electrical stimulation of the muscle [63, 65] (Figure 2).
Figure 2.
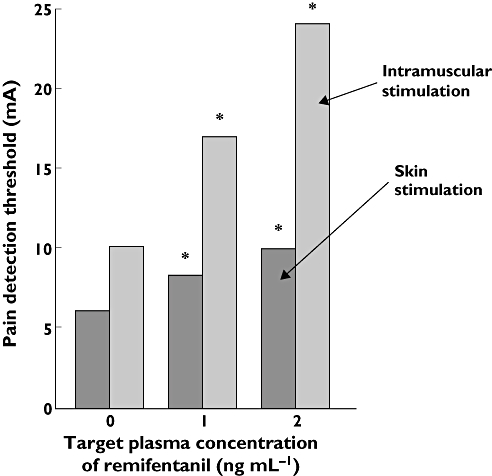
Example of tissue differences in opioid analgesia (remifentanil). The pain detection threshold to electrical intramuscular stimulation is attenuated more than the pain detection threshold to electrical stimulation in the skin [63]
Models of hyperalgesia
Skin:
Four studies induced hyperalgesia by continuous electrical stimulation to the skin, showing a reduction in ongoing pain and hyperalgesia [33, 70–72]. Furthermore, three of the studies found hyperalgesia after withdrawal of remifentanil [33, 71, 72]. Lötsch et al. found an effect of remifentanil on hyperalgesia to mechanical (brush, punctuated and blunt) and electrical stimulation before and after induction of hyperalgesia with a freeze lesion [31]. Remifentanil has also been tested in the capsaicin model, where two studies demonstrated that the area of secondary hyperalgesia obtained with heat/capsaicin stimulation was reduced for both pinprick and brush [66, 67, 73].
Dose:
Seven of the 11 studies were comparable in dosing [64, 66–69, 71, 72]. Except for the studies by Gustorff et al. and Wagner et al. these studies applied doses in the therapeutic range, where four of the remaining studies used a dose at the upper end of the therapeutic range (0.1 µg min−1 kg−1). The studies generally showed robust analgesia in both acute and hyperalgesic pain models and for this drug doses throughout the therapeutic interval seems to work in experimental pain models.
Mechanistic aspects:
The traditional opinion that opioids attenuate mainly C-fibre mediated pain is not always correct. An example of this is seen when alfentanil is applied in experimental pain where two studies could not detect analgesia towards heat pain and one from cold pain [8, 49]. This cannot be explained by an insufficient dose since heat pain was affected by alfentanil in a study using a lower dose than in the study by Luginbuhl et al. [8, 44]. Generally it would be expected that a µ-opioid agonist would affect pain conveyed through C-fibres and heat pain is traditionally believed to be conveyed through these fibres [74, 75]. However, nociception to a fast increase in temperature, which is associated with Aδ fibre stimulation can be less sensitive to opioids. Hence it could be argued that the increase in temperature (1.5–2°°C s−1) was too fast in the non-sensitive studies [8, 49, 76]. On the other hand, another study using 200 ms argon laser stimulation showed analgesia to alfentanil although the heating rate was higher [8, 47, 49]. However the heating rate for laser stimulation is measured in Joules and cannot readily converted into °C s−1 making a direct comparison difficult.
The cold pressor test has been shown to induce descending noxious inhibitory control and since opioids reinforce this mechanism it is not surprising that opioids work well in the cold pressor test [8, 52, 77].
The capsaicin model evokes intense and tonic pain and therefore it could be expected that opioid analgesia would be shown in this model. However, conflicting findings exist and one study did not find any effects of alfentanil which could be related to the problematic repeatability of the evoked secondary hyperalgesia [55]. Theoretically an analgesic can inhibit secondary hyperalgesia by lowering the nociceptive barrage from the periphery to the spinal synapse. Furthermore hyperalgesia can be prevented by inhibition of central mechanisms such as wind-up [78]. Since alfentanil decreased the immediate pain response to capsaicin the incoming nociceptive barrage is probably lowered. Hence it is difficult to conclude whether the peripheral effect is the cause for the subsequent decrease in the secondary hyperalgesic area, or if alfentanil by a direct spinal/supraspinal mechanism prevent the development of hyperalgesia.
The model by Koppert at al. where hyperalgesia is evoked from continuous intradermal electrical stimulation illustrates how alfentanil affects both peripheral and central pain mechanisms. Here it was found that both peripheral (shown by flare reduction) and central inhibition (shown by reduction of secondary hyperalgesic area) of the hyperalgesia, but the effect was short lasting and opioid-induced hyperalgesia appeared after termination of the drug infusion [32]. This model has furthermore been used to mimic opioid induced hyperalgesia after withdrawal of remifentanil and fentanyl. This is an unwanted effect that can occur with several types of opioids and in other settings than withdrawal [79]. However, hyperalgesia has repeatedly been shown in the experimental setting after withdrawal of short acting opioids like fentanyl and remifentanil [33, 70, 73, 80]
Longer acting opioids with strong affinity for the µ-opioid receptor
Morphine (Table 5)
Table 5.
Schematic overview of studies involving morphine in human experimental pain models
| Reference | Dose | Model | Findings | |
|---|---|---|---|---|
| Acute models | [96] (n= 13) | 0.02 mg kg−1 iv | Heat skin stimulation VAS (affective and sensory) (37, 49 and 51°C) | Affective or sensory responses ↔ |
| [102] (n= 10) | 0.08 mg kg−1 iv | Heat skin stimulation PDT, PTT (0.5°°C s−1) Deep pressure PDT Ischemic pain PDT, PTT | All thresholds were increased | |
| [81] (n= 12) | 30 mg p.o. | Heat skin stimulation PDT, PTT (2°C s−1), cold, Pressure algometry PTT (finger pulp) Electrical skin stimulation (single and repeated) Cold pressor test VAS, peak pain, mean pain, AUCVAS | Pain to pressure, cold pressor test, single and repeated electrical stimulation were decreased Heat pain ↔ | |
| [37] (n= 10) | 4 mg epidural | Heat and cold skin stimulation detection threshold, PDT, PTT (1 and 2°C s−1) Electrical skin stimulation PDT, VAS to 1 ms stimulation Pressure algometry PDT, PTT, moderate pain to 20 ms stimulation (toe nail) Short lasting radiant heat VAS (argon laser, 200 ms) | Cold detection threshold ↔ Warmth detection threshold, PDT, PTT to heat, pressure, and electrical stimulation ↑ (PTT>PDT) VAS after short lasting radiant pain ↓ Short lasting pain (1 and 20 ms) to mechanical and electrical stimulation ↔ | |
| [83] (n= 30) | 0.15 mg kg−1 iv | Short lasting radiant heat warmth detection threshold, pinprick PDT (argon laser, 200 ms) | Warmth detection or pinprick PDT ↔ | |
| [86] (n= 7) | 10 mg iv | Intra cutaneous electrical stimulation VAS, evoked brain potentials and frequency analysis | Amplitude of evoked potentials and VAS↓ EEG frequency spectrum ↔ | |
| [51] (n= 10) | 0.142 mg kg−1 iv | Electrical tooth stimulation evoked brain potential, subjective pain report (6 point scale) | Amplitude of evoked potentials and pain score ↓ | |
| [48] (n= 16) | 0.14 and 0.28 mg kg−1 iv | Intramuscular injection of hypertonic saline AUCVASIntramuscular electrical stimulation PDT, AUCVAS to suprathreshold stimulation for 10 s | High dose: All parameters affected Low dose: All parameters ↔ | |
| [53] (n= 15) | 0.1 mg kg−1 iv | Cutaneous and intramuscular electrical stimulation PDT, AUCVAS to suprathreshold stimulation for 10 s Intramuscular injection of hypertonic saline AUCVAS | PDT to intramuscular electrical stimulation ↑ PDT to electrical skin stimulation, AUCVAS after suprathreshold stimulation and hypertonic saline ↔ | |
| [86] (n= 12) | 60 mg extended release orally | Cold pressor test PTT | PTT ↑ | |
| [87] | 0.5 mg kg−1 orally | Cold pressor test PDT, PTT, VAS | Latency to onset of pain and PTT ↑ | |
| [90] (n= 34) | 0.5 mg kg−1 orally | Cold pressor test PDT, PTT, VAS | PDT and PTT ↑ VAS ↓ | |
| [89] (n= 19) | 0.1 mg kg−1 iv | Ischemic pain VAS (summated) | VAS ↓ | |
| [5] (n= 24) | 30 mg orally | Heat skin stimulation PTT (2°C/s) Deep pressure PTT Pinching PTT Cutaneous and intramuscular electrical stimulation PTT Oesophageal distension and electrical pain PTT Oesophageal heat pain PDT | Oesophageal heat pain ↔ The remaining pain thresholds parameters ↓ | |
| [95] (n= 10) | 100 ng ml−1 iv | Electrical skin stimulation PDT, PTT | PDT and PTT ↑ | |
| [106] (n= 28) | 20 mg or 30 mg single dose orally | Cold pressor test AUCVAS, peak pain, discomfort (VAS) Heat skin stimulation and deep pressure PDT | Peak pain and discomfort in cold pressor test ↓ AUCVAS and heat PDT and PTT ↔ Pressure PTT ↓ whereas PDT ↔ | |
| [91] (n= 9) | 0.1 mg kg−1 iv | Ischaemic pain AUCVAS | AUCVAS↓ | |
| [97] (n= 47) | 0.04–0.08 mg kg−1 iv | Heat skin stimulation VAS (sensory and affective dimension) to graded temperatures and VAS to brief pulses (first and second pain) | Dose < 0.06: VAS (affective) ↓, VAS (sensory) ↔ Dose > 0.06: VAS (affective and sensory and first and second pain) ↓ | |
| [92] (n= 45) | 10 mg 70 kg−1 iv | Ischaemic pain PTT | PTT ↑ | |
| [84] (n= 10) | 4 mg injected perineurally to ulnar nerve and epidurally | Short lasting radiant heat PDT and warmth detection threshold), evoked brain potentials (argon laser, 200 ms) | PDT and warmth detection threshold ↑ Amplitude of evoked brain potentials ↓ Latencies of evoked brain potentials↑ after perineural administration | |
| Models inducing hyperalgesia | [57] (n= 16) | 15 and 30 ng ml−1 measured at steady state | Burn injury PDT to pinprick in injured skin, area of secondary HA to pinprick Repeated pinprick AUCVASContinuous electrical skin stimulation PDT to pinprick, area of secondary HA to pinprick, repeated pinprick (AUCVAS) | Only the high dose had significant effect. Continuous electrical skin stimulation: PDT ↑ and area of HA↓ Burn injury and repeated pinprick: no parameters were significantly affected |
| [94] (n= 11) | 10 µg kg−1 min−1 for 45 min iv | Burn injury PDT to pinprick in injured skin, area of secondary HA to pinprick Repeated pinprick AUCVAS | No parameters were affected | |
| [41] (n= 12) | 0.15 mg kg−1 iv | Burn injury heat and cold detection threshold and PDT in primary and secondary HA area (1°C/s), area of secondary HA to pinprick, detection threshold to pinprick, appearance of wind-up like pain to repeated pinprick stimulation | No parameters were affected | |
| [93] (n= 12) | 2 mg sub-cutaneously | Burn injury, PDT to heat (1°C s−1) and deep pressure | Heat PDT ↑ Mechanical PDT ↑ | |
| [42] (n= 12) | 40 ml 0.01% iv | Ultraviolet (UV-B) radiation PDT to heat VAS to mechanical impact | Heat PDT ↑ VAS to mechanical impact ↔ | |
| [95] (n= 10) | 100 ng ml−1 iv | Freeze lesion PDT to pinprick Concentric and eccentric muscle contraction VAS | PDT to prinptick ↓ Muscle pain intensity ↔ |
In the column ‘model’ the method for pain assessment is normal font, and the method for pain induction is bolded. Abbreviations: pain detection threshold (PDT), pain tolerance threshold (PTT), area under curve (AUC), visual analogue scale score (VAS), hyperalgesia (HA).
Morphine is a widely used analgesic and it has been tested extensively in experimental pain (Table 4).
Acute models
Skin/teeth:
Morphine has been tested against cutaneous heat and cold pain, mechanical (pinching) and electrical pain [5, 37, 53, 81]. For heat pain to be sensitive to morphine it has been argued that it needs to be applied with slow temperature rises (<1°C s−1) [82, 83]. However, morphine has also shown effect on pain from rapid increases in temperature [37, 81, 83–85]. Three studies found sensitivity of electrical pain for morphine, whereas another did not [5, 37, 53]. Pain evoked by pinching of the skin was sensitive to morphine [5]. Two studies showed an effect of morphine on the warmth detection threshold and pain detection threshold to heat, pressure and electrical stimulation [37, 84, 85]. Electrically-induced pain in the teeth and skin has been assessed by electroencephalogram (EEG) recordings. This type of pain assessment showed opioid analgesia in accordance with the psychophysical pain scoring [51, 86].
Muscle:
Morphine has been tested against pain from deep pressure algometry, electrical stimulation, cold pressor test, injection of hypertonic saline and ischaemic pain [5, 53, 87–90]. Ischaemic pain, cold pressor pain and pain to electrical stimulations were decreased by morphine [87, 89–92]. Pain to hypertonic saline was sensitive to modulation from morphine when a high dose was administered [48, 53].
Viscera:
Morphine analgesia was significantly better than placebo in attenuating mechanical and electrical oesophageal pain, but not in thermal oesophageal pain [5] (Figure 3).
Figure 3.
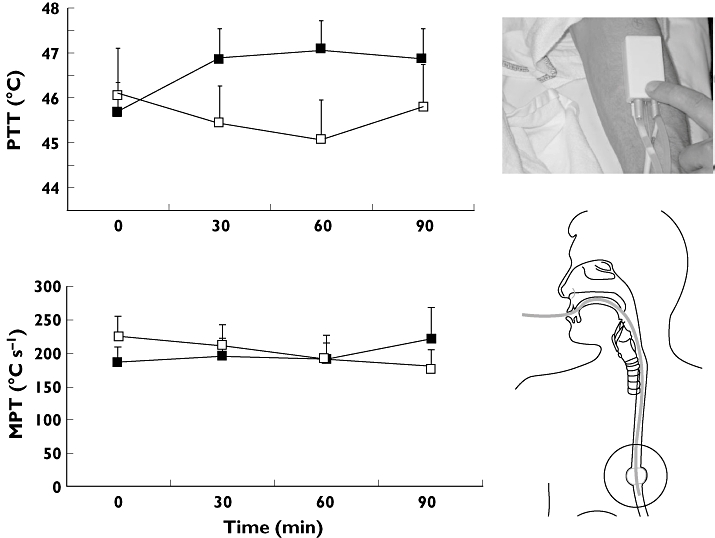
Tissue differences in morphine analgesia. Top graph: Compared with placebo, morphine significantly raises the pain tolerance threshold (PTT) to heat pain in the skin. Bottom graph: morphine has no effect on the moderate pain tolerance (MPT, measured as the area under the temperature curve) threshold to oesophageal heat pain [5]. Error bars represent SE. Placebo (□); Morphine ( )
)
Models of hyperalgesia
Skin:
Morphine has been tested in various models involving hyperalgesia such as burn injuries, freeze lesions, continuous electrical stimulation and radiation with ultraviolet light [41, 42, 93–95].
Hyperalgesia and allodynia from burn injuries were unaffected in two studies [41, 57]. However, when Schulte et al. applied a higher dose of morphine (0.2 mg kg−1 for 15 min and 0.66 mg kg−1 for 110 min) reduction of the area of secondary hyperalgesia was seen as the only modulation [57]. However, peripheral effects of morphine were detected by the burn injury model in the study by Moiniche et al. [93]. The study by Koppert et al. investigated the peripheral effects of morphine (applied as iv regional anaesthesia) in the UV-B induced hyperalgesia model and found that morphine attenuated primary hyperalgesia to heat pain [42].
Muscle:
Hyperalgesia produced by eccentric muscle contraction was decreased by morphine [91, 92].
Dose:
In the study by Roberts et al. it would be expected that the applied heat pain would be sensitive to morphine. The lack of effect could be explained by the low dose used in this study, which was designed to determine the synergistic effect for morphine in combination with tetrahydrocannabinol (0.0.2 mg kg−1 iv) [96]. Schulte et al. did a dose–response study of morphine concentration against pain to the injection of hypertonic saline and found that a dose above 0.14 mg kg−1 iv is necessary to show an effect in this model [48, 53]. However, other studies which used other pain models applied doses under 0.14 mg kg−1 iv and found an effect of morphine, illustrating how the pain models differ in sensitivity to a given dose [89, 95, 97]. Accordingly the study by Brennum et al. had a good sensitivity of almost all sensory tests towards morphine. Here 4 mg morphine was administered in the epidural space. This dose is at upper end of the therapeutic range [37]. In the study by Staahl et al. analgesia was seen for several pain parameters in various tissues and this group also applied a high dose, compared with the normal dose used in the clinic, although in the therapeutic range [5].
Morphine is generally effective towards pain from many different stimulus modalities [37, 51, 81, 94, 95]. However, the results are not as clear-cut as seen with alfentanil and this could be due to the complex pharmacokinetic profile of morphine. The amount of morphine absorbed is very individual and this opioid enters the main effect site (the CNS), by crossing the blood brain barrier slowly [98]. All this causes increased variability of the individual subject's response to morphine, blurring the findings in experimental pain research [51, 99].
Mechanistic aspects:
As stated previously opioids mainly attenuate pain intensities above the pain detection threshold [100, 101]. However two studies showed an effect of morphine on the warmth detection threshold and pain detection threshold to heat, pressure and electrical stimulation [37, 84, 85]. Warmth sensations are conveyed by C-fibres and hence there is a neurophysiologic explanation for morphine modulating the sensation of warmth [76]. Since C-fibres are pain selective and morphine mainly affects dorsal horn activity produced from tonic C-fibre activation, it is most likely that morphine will produce a significant effect on a pain tolerance threshold evoked by a tonic type of pain [5, 83, 102]. Accordingly the study by Brennum et al. did see a more pronounced effect on the longer lasting stimulations (>2000 ms) and on the pain tolerance threshold [37]. However, exceptions exist and the study by Roberts et al. did not find any effect of morphine on 5 s stimulation at 51°°C, a stimulus intensity normally considered well above the pain detection threshold.
Compared with models where the painful stimulus is applied to the skin it appears that morphine analgesia is more robust in deep pain. The reason for this could be that deep pain is often considered more unpleasant than skin pain and the muscular models often apply a more tonic type of pain (hypertonic saline, cold pressor test etc.). The unpleasantness of pain is associated with the limbic structures in the brain, an area where opioids traditionally are known to modulate the pain response [39, 103]. As one of few opioids, morphine has been evaluated in visceral pain [5]. This study revealed important tissue differences in opioid analgesia, particularly when comparing somatic and visceral pain. This study applied thermal pain to the skin and viscera at the same heating rate and morphine analgesia was prominent only in the skin (Figure 2) [5]. This reflects the clinical situation where visceral pain, in contrast to somatic pain, can be difficult to treat with traditional µ opioid agonists [104].
Opioids with weak affinity for the µ-opioid receptor
Codeine (Table 6)
Table 6.
Schematic overview of studies involving codeine in human experimental pain models
| Reference | Dose | Model | Findings | |
|---|---|---|---|---|
| Acute models | [99] (n= 18) | 125 mg orally | Pressure algometry (phalanx), PTT, single and repeated electrical sural nerve stimulation PDT, PTT Cold pressor test peak pain AUCVAS, discomfort | Pain and discomfort for all stimulations was decreased |
| [104] (n= 27) | 100 mg orally | Heat skin stimulation and deep pressure PDT, PTT Cold pressor test VAS, peak pain, AUCVASSural nerve electrical stimulation, PDT, PTT stimulus response curve | Heat from electrical, thermal and pressure ↔, cold pressor, peak pain ↓ | |
| [108] (n= 12) | 60/120 mg orally | Cold pressor test VAS, level of ‘bothersomeness’ | VAS ↓ (not dose-related), level of bothersomeness ↔ | |
| [107] (n= 14) | 75 mg or 100 mg orally | Cold pressor test AUCVAS, peak pain, discomfort (VAS) Heat skin stimulation and deep pressure PDT, PTT | Peak pain and discomfort in cold pressor test ↓ (only extensive metabolizers) AUCVAS and heat and pressure (PDT and PTT) ↔ | |
| [108] (n= 48/32) | 60 mg orally | Electrical and heat skin stimulation (PDT, PTT (electrical) | Pain from both modalities was decreased |
In the column ‘model’ the method for pain assessment is normal font, and the method for pain induction is bolded. Abbreviations: pain detection threshold (PDT), pain tolerance threshold (PTT), area under curve (AUC), visual analogue scale score (VAS), hyperalgesia (HA).
Acute models
Skin:
Codeine worked against acute experimental pain to heat, pressure, single/repeated electrically stimulation and in the cold pressor test [100, 105–108]. Two studies applied both the cold pressor test and more phasic pain tests, like heat, electrical and pressure pain. These studies did only find effect in the more tonic pain from the cold pressor test [105, 106].
Dose:
Codeine has been applied in supratherapeutic doses in all studies and this could be the explanation for the effects of this weak analgesic in a variety of acute and short lasting pain models as well as more tonic pain models. However the application of supratherapeutic doses has probably given a significant plasma concentration of morphine/morphine-6-glucuronide and this could explain the convincing effect seen in the more phasic pain models that traditionally are thought to be less sensitive to opioid analgesia.
Mechanistic aspects:
Codeine is a weak opioid, which is metabolized in the liver to morphine. The main effect of codeine is thought to be mediated via µ-receptors mainly through the main metabolites, morphine and morphine-6 glucoronide [109, 110]. Seven percent of Caucasians lack the ability to metabolize codeine whereas 25% of Ethiopians are ultra-rapid metabolizers, due to a polymorphism of the enzyme responsible for this metabolism (P4502D6) [106]. In experimental pain the analgesic effect of codeine seems to depend on the conversion of codeine to morphine since subjects who are slow metabolizers do not have any analgesic effect of codeine [106]
Opioids with mixed binding profiles
Tramadol (Table 7)
Acute models
Skin/nasal mucosa:
Tramadol has shown an effect in experimental pain from pressure stimulation, electrical stimulation of the sural nerve (nociceptive reflex) and the cold pressor test [106, 111].
Phasic pain from stimulating the nasal mucosa by gaseous carbon dioxide was attenuated by tramadol [112, 113]. Here the pain was assessed by subjective pain ratings, but also by electrophysiological assessment of pain using evoked brain potentials. In the study by Thurauf et al. an effect was found only on evoked brain potentials and not on pain ratings [113]. Furthermore, tonic pain from stimulating the nasal mucosa with dry air is sensitive to tramadol [37, 113]. Pain from electrical stimulation of the tooth pulp was also sensitive to tramadol, but mainly with doses over 50 mg (100 mg) [114–116].
Muscle:
Ischaemic pain was unaffected by tramadol [117].
Models of hyperalgesia
Skin:
Tramadol has been tested against continuous electrically evoked secondary hyperalgesia and in this model, the ongoing pain intensity was reduced, but the hyperalgesia and allodynia was not affected significantly [118].
Muscle:
The delayed-onset muscle soreness was unaffected by tramadol in the study by Loram et al. (see discussion below) [117].
Dose:
Hummel et al. used doses above the therapeutic range (150 mg) of tramadol and this could be a cofactor explaining the robust effect seen in this study [112]. Ischaemic pain and delayed onset muscle soreness was unaffected in a study engaging a therapeutic like dosing regime, and the use of doses in the lower end of the therapeutic range in the study by Loram et al. may explain the lack of analgesia in this otherwise well-designed study, mimicking the clinical situation with multiple dosing [117]. Similarly tramadol mainly affected pain from electrical tooth pulp stimulation when doses in the range of 100 mg were applied [114, 116]. These findings indicate that doses above or at the upper end of the therapeutic range are necessary to show an effect under experimental conditions. When a drug with an active metabolite is being tested it can also be crucial to locate poor/extensive metabolizers since such individuals can add increased variation. Such variation would decrease the statistical power of the trial [106].
Mechanistic aspects:
Besides effects on the opioid system tramadol exerts analgesia through actions on the noradrenergic and serotonergic systems [119]. Tramadol exerts its opioid action through a metabolite (O-desmethyl-tramadol), which has an affinity for the µ opioid receptor approximately 10 times lower than that of morphine [120]. As for codeine, polymorphism of the enzyme (P4502D6) responsible for this metabolism exists in a significant proportion of the population [106]. This genetic factor is reflected in the findings in experimental pain studies, where only extensive metabolizers exhibit convincing analgesia in experimental pain from pressure stimulation, electrical stimulation of the sural nerve (nociceptive reflex) and the cold pressor test [106, 111].
Discussion
Opioids in experimental pain
Strong opioids are potent analgesics and suitable models for detecting opioid analgesia are found amongst both acute models and models evoking hyperalgesia [46, 53, 81, 90]. Assessment of the analgesic effects of opioids has most frequently been done by skin stimulation [6, 32, 45, 49, 54, 86, 97, 105]. Fast acting opioids like alfentanil and remifentanil have been tested extensively and these pure µ-receptor agonists have a short half-life and straightforward kinetics. Accordingly these opioids have consistently shown robust analgesia in a variety of pain models. However several examples can be found where opioids preferentially modulate the higher pain intensities (see also Figure 4) [37, 46, 106].
Figure 4.
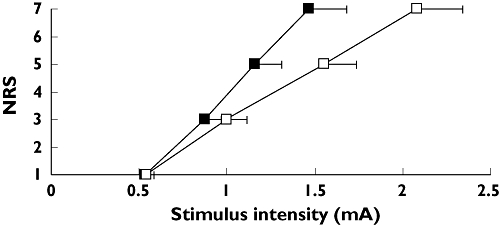
Stimulus–response curve for pain after intramuscular electrical stimulation before ( ) and 90 min after administration of 30 mg morphine orally. A score of 5 on the numeric rating scale (NRS) is the pain detection threshold. NRS = 7 corresponds to moderate pain [5]. Error bars represent SE. Baseline (
) and 90 min after administration of 30 mg morphine orally. A score of 5 on the numeric rating scale (NRS) is the pain detection threshold. NRS = 7 corresponds to moderate pain [5]. Error bars represent SE. Baseline ( ); 90 min after morphine administration (□)
); 90 min after morphine administration (□)
The situation for the weak opioids is more complex and trials with tramadol and codeine, drugs which have active metabolites, showed how genetic factors influencing the drug metabolism, can affect the results [111, 121].
A model that has been used extensively for the testing of opioids is the cold pressor test, and this model is sensitive to opioid analgesia, possibly due to the strong intensity and tonic nature of the pain induced by this model.
Designing of experimental studies involving opioids (Figure 5)
Figure 5.

Suggestions of important topics to consider when designing trials with analgesics and experimental pain models. To meet these criteria it is advantageous to include several (yet a feasible number) pain models and methods of pain assessment. Regarding point 1f, this is particularly relevant for visceral pain, where this can be a problem in many studies, where the tissue impact is hard to determine and often not reproducible [2]
To obtain a good trial design, at least three factors need to be considered: i) a model (including an appropriate induction and assessment method) that activates mechanisms and pain pathways sensitive to the analgesic in question, ii) correct dose, which ensures sufficient efficacy combined with a limited amount of side effects, iii) correct dosing regime (single dose/multiple dose) and time points of testing for analgesia and iv) methodology and trial design ensuring appropriate signal detection.
Choosing the right models
Since the opioidergic system is universal for pain modulation most types of experimental pain are affected by the administration of exogenous opioids. However the most sensitive models include tonic pain, with stimulus intensity evoking pain above the pain detection threshold [37, 58, 83]. As opioids mainly affect dorsal horn activity produced from tonic C-fibre activation, it is most likely that the analgesic effect will be on a pain tolerance threshold evoked by a tonic type of pain [82]. However exceptions exist and ischaemic muscle pain has been tested with alfentanil, morphine and tramadol, where only morphine decreased ischaemic pain [8, 89, 117].
Some assessment methods summarize the pain over time, whereas others register the peak pain. Normally summarized pain measures are more robust giving a high signal to noise ratio, which determines the sensitivity of the pain model [122]. The ‘method of levels’ is a stimulation paradigm where certain levels of stimulus intensities are presented to the subject in preferentially random order and the subject then scores the level of pain. The ‘method of limits’ is a stimulation paradigm where the level of stimulus intensity is gradually increased until, for example, the pain detection threshold is reached. This method often gives a higher variability than the ‘method of levels’ probably because reaction time is an important factor in the ‘method of limits’[123].
In the study by Thurauf et al. the value of objective pain assessment was shown since an effect of tramadol was found only on evoked brain potentials and not on pain ratings [113]. It is however important to note that, although evoked potentials can be a sensitive measure of nociceptive processes, they only measure a single dimension of pain. Pain is a multidimensional sensation and this is reflected better in the subjective pain measure. This limits the translation of analgesic effect on evoked brain potentials into effect on clinical pain measures.
Revealing detailed human cerebral opioid pharmacology is possible in experimental pain. For this purpose various imaging techniques have been used. Accordingly it was shown in a positron emission tomography study that remifentanil decreased the pain induced brain activation in the cingulofrontal cortex and peri-aqueductal gray, indicating that opioidergic activation modulates activity in pain inhibitory circuitries [68]. Furthermore in a functional magnetic resonance imaging study it has been shown that alfentanil decreased both sensory and affective brain processing of pain and that genotypes of the µ-opioid receptor determined the degree of opioid activity in the sensory brain processing of pain [50].
It is important to note that opioid analgesia exhibits tissue differences and therefore the inclusion of models in more types of tissue than just skin is optimal. Accordingly Curatolo et al. showed a more pronounced effect of remifentanil in inhibiting muscular pain than cutaneous pain [124]. Visceral tissue seemed to differ a lot from somatic tissue in the analgesic opioid response as shown with morphine on oesophageal pain [5].
Choosing the right dosing regime and time points for testing the analgesia
The kinetic profile is necessary to determine when it is optimal to perform the pain tests. For opioids it is particularly important to remember that they often need to cross the blood-brain-barrier and enter the CNS to have analgesic effect. This causes a lag-time to the onset of analgesia. The study design should consider these different lag-times for different opioids.
Choosing the right dose
Most opioids are powerful analgesics and doses in the therapeutic range are generally sufficient for detectable analgesia in experimental pain models [5, 58, 87, 88]. However to increase the validity of the findings in experimental pain the finding of dose-response curves with significant slopes supports the idea of an analgesic effect that could be translatable into clinical pain [8, 125].
Genetics can alter the metabolism and hence exposures to the active metabolite for opioids like tramadol and codeine. Similarly genetics can affect the structure and activity of opioid receptors altering the response to a given drug concentration. This has been shown for alfentanil and the same is true for morphine, although this has not yet been shown in experimental pain [50, 126]. Hence genetic profiling of the healthy volunteers entering a study can be helpful in eliminating large variation in the response to a given opioid.
Methodology and trial design
The sensitivity of a given experimental model for detecting opioid analgesia is affected by the method used to measure this pain. Hence good sensitivity of a model is obtained by combining a pain mechanism potently affected by opioids (large effect size or signal), and using a pain assessment that is reliable producing data with modest variance (noise). In general parallel studies give a weaker statistical power than a cross-over design, demanding larger sample sizes [127]. Furthermore, pain measures that only have a small dynamic range allow only a limited sensitivity for detecting analgesic effects.
Pain measures that are summated give a more robust pain measure, and are therefore more sensitive to modulation. This is exemplified by the findings in the cold pressor model. Pain in the cold pressor model is often assessed by the area under the VAS curve and accordingly opioid analgesia can be shown for most opioids [8, 58, 81, 107, 125].
Another way of reducing the variance of the outcome is to maintain control with psychophysiological factors, including personality factors and anxiety induced by application of the model [7, 128].
When opioids are applied in experimental pain blinding can be troublesome due to the side effects. Sedation is particularly troublesome since it can affect the pain scoring. For controlling this EEG can be useful via spectral analysis [86].
The role of experimental pain in drug testing
There is a need for translational studies between animal studies and complex phase III studies in patients. Part of the complexity seen in clinical trials with analgesics for the treatment of pathological (neuropathic) pain is that there are many confounding factors. For example, the best predictors of the chronicity of back pain are not pathology or genetic, but rather psychosocial [129]. The experimental human pain models can provide additional information about drugs overcoming species differences and partly avoiding the bias seen in clinical trials involving analgesics. Despite the fact that experimental pain models only explore a limited and differential part of the cascade of processes involved in clinical pain some models predict how the analgesic will behave in the clinic [124]. An example of this is the model of ischaemic muscle pain. This model is thought to mimic clinical inflammatory musculoskeletal pain and for morphine there is consensus between the findings in the model and the clinical situation [91, 92]. Another example, showing the limit of experimental pain, is the capsaicin model, thought to mimic neuropathic pain because the evoked hyperalgesia has features (allodynia) that is seen also in the clinic. Opioids attenuate both hyperalgesia and allodynia in many cases but have a more limited effect in clinical neuropathic pain.
Because only single or a few pain mechanisms are activated it is possible to investigate on a mechanistic basis how analgesics work [32, 130]. This may give the possibility of investigating, for example, tissue differentiated effects of morphine, where visceral and somatic pain responds differentially to morphine and oxycodone [5]. These opioids have, in clinical trials, also shown subtle differences and this illustrates the link between experimental pain and the clinical situation [131].
There are still major problems in the exact determination of the activated pathways and pain mechanisms in human experimental pain [14]. Nevertheless, the experimental human models give the possibility to obtain reproducible results in test-retest experiments and hence be useful for drug screening [10].
It should be noted that there is still a need for basic investigations of opioids in well designed human experimental pain models. Such trials furthermore may give valuable knowledge about the human opioidergic system. However, experimental pain models have so far produced many contradictive findings even in studies using the same opioid and pain stimulus [5, 37, 53]. Differences are often caused by different pain assessment methods (where the intensity and modality are often poorly controlled), different populations of volunteers and/or dosing regimes, and the use of more homogenous trial designs in this respect, would make study comparisons more useful. However trial designs can be difficult to export to other laboratories, and often it can be seen that two laboratories cannot reproduce the results of a specific pain model [112, 113].
Competing interests
None declared.
REFERENCES
- 1.Konig HH, Bernert S, Angermeyer MC. Health Status of the German Population: results of a Representative Survey Using the euroqol questionnaire. Gesundheitswesen. 2005;67:173–82. doi: 10.1055/s-2005-857991. [DOI] [PubMed] [Google Scholar]
- 2.Drewes AM, Gregersen H, Arendt-Nielsen L. Experimental pain in gastroenterology: a reappraisal of human studies. Scand J Gastroenterol. 2003;38:1115–30. doi: 10.1080/00365520310004399. [DOI] [PubMed] [Google Scholar]
- 3.Arendt-Nielsen L, Curatolo M, Drewes A. Human experimental pain models in drug development: translational pain research. Curr Opin Investig Drugs. 2007;8:41–53. [PubMed] [Google Scholar]
- 4.Graven-Nielsen T, Mense S. The peripheral apparatus of muscle pain: evidence from animal and human studies. Clin J Pain. 2001;17:2–10. doi: 10.1097/00002508-200103000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Staahl C, Christrup LL, Andersen SD, Arendt-Nielsen L, Drewes AM. A comparative study of oxycodone and morphine in a multi-modal, tissue-differentiated experimental pain model. Pain. 2006;123:28–36. doi: 10.1016/j.pain.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 6.Staahl C, Olesen AE, Andresen T, Arendt-Nielsen L, Drewes AM. Assessing analgesic actions of non-opioid analgesics by experimental pain models in healthy volunteers – an updated review. Br J Clin Pharmacol. 2009 doi: 10.1111/j.1365-2125.2009.03433.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Staahl C, Reddy H, Andersen SD, Arendt-Nielsen L, Drewes AM. Multi-modal and tissue-differentiated experimental pain assessment: reproducibility of a new concept for assessment of analgesics. Basic Clin Pharmacol Toxicol. 2006;98:201–11. doi: 10.1111/j.1742-7843.2006.pto_211.x. [DOI] [PubMed] [Google Scholar]
- 8.Luginbuhl M, Schnider TW, Petersen-Felix S, Arendt-Nielsen L, Zbinden AM. Comparison of five experimental pain tests to measure analgesic effects of alfentanil. Anesthesiology. 2001;95:22–9. doi: 10.1097/00000542-200107000-00009. [DOI] [PubMed] [Google Scholar]
- 9.Graven-Nielsen T, Mense S, Arendt-Nielsen L. Painful and non-painful pressure sensations from human skeletal muscle. Exp Brain Res. 2004;159:273–83. doi: 10.1007/s00221-004-1937-7. [DOI] [PubMed] [Google Scholar]
- 10.Handwerker HO, Kobal G. Psychophysiology of experimentally induced pain. Physiol Rev. 1993;73:639–71. doi: 10.1152/physrev.1993.73.3.639. [DOI] [PubMed] [Google Scholar]
- 11.Staahl C, Drewes AM. Experimental human pain models: a review of standardised methods for preclinical testing of analgesics. Basic Clin Pharmacol Toxicol. 2004;95:97–111. doi: 10.1111/j.1742-7843.2004.950301.x. [DOI] [PubMed] [Google Scholar]
- 12.Gracely RH. Pain measurement. Acta Anaesthesiol Scand. 1999;43:897–908. doi: 10.1034/j.1399-6576.1999.430907.x. [DOI] [PubMed] [Google Scholar]
- 13.Besson JM. The neurobiology of pain. Lancet. 1999;353:1610–5. doi: 10.1016/s0140-6736(99)01313-6. [DOI] [PubMed] [Google Scholar]
- 14.Woolf CJ, Max MB. Mechanism-based pain diagnosis: issues for analgesic drug development. Anesthesiology. 2001;95:241–9. doi: 10.1097/00000542-200107000-00034. [DOI] [PubMed] [Google Scholar]
- 15.Dirks J, Petersen KL, Rowbotham MC, Dahl JB. Gabapentin suppresses cutaneous hyperalgesia following heat-capsaicin sensitization. Anesthesiology. 2002;97:102–7. doi: 10.1097/00000542-200207000-00015. [DOI] [PubMed] [Google Scholar]
- 16.Wallace MS, Barger D, Schulteis G. The effect of chronic oral desipramine on capsaicin-induced allodynia and hyperalgesia: a double-blinded, placebo-controlled, crossover study. Anesth Analg. 2002;95:973–8. doi: 10.1097/00000539-200210000-00034. table. [DOI] [PubMed] [Google Scholar]
- 17.Negus SS, Vanderah TW, Brandt MR, Bilsky EJ, Becerra L, Borsook D. Preclinical assessment of candidate analgesic drugs: recent advances and future challenges. J Pharmacol Exp.Ther. 2006;319:507–14. doi: 10.1124/jpet.106.106377. [DOI] [PubMed] [Google Scholar]
- 18.Hughes AM, Rhodes J, Fisher G, Sellers M, Growcott JW. Assessment of the effect of dextromethorphan and ketamine on the acute nociceptive threshold and wind-up of the second pain response in healthy male volunteers. Br J Clin Pharmacol. 2002;53:604–12. doi: 10.1046/j.1365-2125.2002.01602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arendt-Nielsen L, Chen AC. Lasers and other thermal stimulators for activation of skin nociceptors in humans. Neurophysiol Clin. 2003;33:259–68. doi: 10.1016/j.neucli.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 20.Harrison JL, Davis KD. Cold-evoked pain varies with skin type and cooling rate: a psychophysical study in humans. Pain. 1999;83:123–35. doi: 10.1016/s0304-3959(99)00099-8. [DOI] [PubMed] [Google Scholar]
- 21.Davis KD. Cold-induced pain and prickle in the glabrous and hairy skin. Pain. 1998;75:47–57. doi: 10.1016/S0304-3959(97)00203-0. [DOI] [PubMed] [Google Scholar]
- 22.Narhi M, Jyvasjarvi E, Virtanen A, Huopaniemi T, Ngassapa D, Hirvonen T. Role of intradental A- and C-type nerve fibres in dental pain mechanisms. Proc Finn Dent Soc. 1992;88(Suppl. 1):507–16. [PubMed] [Google Scholar]
- 23.Chaves TC, Nagamine HM, de Sousa LM, de Oliveira AS, Grossi DB. Intra- and interrater agreement of pressure pain threshold for masticatory structures in children reporting orofacial pain related to temporomandibular disorders and symptom-free children. J Orofac Pain. 2007;21:133–42. [PubMed] [Google Scholar]
- 24.Pasini FL, Capecchi PL, Colafati M, Randisi P, Puccetti L, Di PT. Systemic adenosine increase during cold pressor test is dependent on sympathetic activation. Clin Exp Pharmacol Physiol. 1999;26:774–8. doi: 10.1046/j.1440-1681.1999.03126.x. [DOI] [PubMed] [Google Scholar]
- 25.Fagius J, Karhuvaara S, Sundlof G. The cold pressor test: effects on sympathetic nerve activity in human muscle and skin nerve fascicles. Acta Physiol Scand. 1989;137:325–34. doi: 10.1111/j.1748-1716.1989.tb08760.x. [DOI] [PubMed] [Google Scholar]
- 26.Arendt-Nielsen L, Graven-Nielsen T, Svensson P, Jensen TS. Temporal summation in muscles and referred pain areas: an experimental human study. Muscle Nerve. 1997;20:1311–3. doi: 10.1002/(sici)1097-4598(199710)20:10<1311::aid-mus15>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 27.Arendt-Nielsen L, Petersen-Felix S, Fischer M, Bak P, Bjerring P, Zbinden AM. The effect of N-methyl-D-aspartate antagonist (ketamine) on single and repeated nociceptive stimuli: a placebo-controlled experimental human study. Anesth Analg. 1995;81:63–8. doi: 10.1097/00000539-199507000-00013. [DOI] [PubMed] [Google Scholar]
- 28.Cervero F, Laird JM, Garcia-Nicas E. Secondary hyperalgesia and presynaptic inhibition: an update. Eur J Pain. 2003;7:345–51. doi: 10.1016/s1090-3801(03)00047-8. [DOI] [PubMed] [Google Scholar]
- 29.Pedersen JL. Inflammatory pain in experimental burns in man. Dan Med Bull. 2000;47:168–95. [PubMed] [Google Scholar]
- 30.LaMotte RH, Lundberg LE, Torebjork HE. Pain, hyperalgesia and activity in nociceptive C units in humans after intradermal injection of capsaicin. J Physiol. 1992;448:749–64. doi: 10.1113/jphysiol.1992.sp019068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lotsch J, Angst MS. The mu-opioid agonist remifentanil attenuates hyperalgesia evoked by blunt and punctuated stimuli with different potency: a pharmacological evaluation of the freeze lesion in humans. Pain. 2003;102:151–61. doi: 10.1016/s0304-3959(02)00349-4. [DOI] [PubMed] [Google Scholar]
- 32.Koppert W, Dern SK, Sittl R, Albrecht S, Schuttler J, Schmelz M. A new model of electrically evoked pain and hyperalgesia in human skin: the effects of intravenous alfentanil, S(+)-ketamine, and lidocaine. Anesthesiology. 2001;95:395–402. doi: 10.1097/00000542-200108000-00022. [DOI] [PubMed] [Google Scholar]
- 33.Koppert W, Sittl R, Scheuber K, Alsheimer M, Schmelz M, Schuttler J. Differential modulation of remifentanil-induced analgesia and postinfusion hyperalgesia by S-ketamine and clonidine in humans. Anesthesiology. 2003;99:152–9. doi: 10.1097/00000542-200307000-00025. [DOI] [PubMed] [Google Scholar]
- 34.Segerdahl M. Multiple dose gabapentin attenuates cutaneous pain and central sensitisation but not muscle pain in healthy volunteers. Pain. 2006;125:158–64. doi: 10.1016/j.pain.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 35.Slater H, Arendt-Nielsen L, Wright A, Graven-Nielsen T. Experimental deep tissue pain in wrist extensors – a model of lateral epicondylalgia. Eur J Pain. 2003;7:277–88. doi: 10.1016/S1090-3801(02)00141-6. [DOI] [PubMed] [Google Scholar]
- 36.Sarkar S, Aziz Q, Woolf CJ, Hobson AR, Thompson DG. Contribution of central sensitisation to the development of non-cardiac chest pain. Lancet. 2000;356:1154–9. doi: 10.1016/S0140-6736(00)02758-6. [DOI] [PubMed] [Google Scholar]
- 37.Brennum J, Arendt-Nielsen L, Horn A, Secher NH, Jensen TS. Quantitative sensory examination during epidural anaesthesia and analgesia in man: effects of morphine. Pain. 1993;52:75–83. doi: 10.1016/0304-3959(93)90117-8. [DOI] [PubMed] [Google Scholar]
- 38.Bromm B, Treede RD. Laser-evoked cerebral potentials in the assessment of cutaneous pain sensitivity in normal subjects and patients. Rev Neurol (Paris) 1991;147:625–43. [PubMed] [Google Scholar]
- 39.Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain. 2005;9:463–84. doi: 10.1016/j.ejpain.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 40.Andersen OK, Felsby S, Nicolaisen L, Bjerring P, Jensen TS, rendt-Nielsen L. The effect of ketamine on stimulation of primary and secondary hyperalgesic areas induced by capsaicin – a double-blind, placebo-controlled, human experimental study. Pain. 1996;66:51–62. doi: 10.1016/0304-3959(96)02995-8. [DOI] [PubMed] [Google Scholar]
- 41.Warncke T, Stubhaug A, Jorum E. Ketamine, an NMDA receptor antagonist, suppresses spatial and temporal properties of burn-induced secondary hyperalgesia in man: a double-blind, cross-over comparison with morphine and placebo. Pain. 1997;72:99–106. doi: 10.1016/s0304-3959(97)00006-7. [DOI] [PubMed] [Google Scholar]
- 42.Koppert W, Likar R, Geisslinger G, Zeck S, Schmelz M, Sittl R. Peripheral antihyperalgesic effect of morphine to heat, but not mechanical, stimulation in healthy volunteers after ultraviolet-B irradiation. Anesth Analg. 1999;88:117–22. [PubMed] [Google Scholar]
- 43.Poyhia R, Vainio A. Topically administered ketamine reduces capsaicin-evoked mechanical hyperalgesia. Clin J Pain. 2006;22:32–6. doi: 10.1097/01.ajp.0000149800.39240.95. [DOI] [PubMed] [Google Scholar]
- 44.Angst MS, Ramaswamy B, Davies MF, Maze M. Comparative analgesic and mental effects of increasing plasma concentrations of dexmedetomidine and alfentanil in humans. Anesthesiology. 2004;101:744–52. doi: 10.1097/00000542-200409000-00024. [DOI] [PubMed] [Google Scholar]
- 45.Olofsen E, Romberg R, Bijl H, Mooren R, Engbers F, Kest B, Dahan A. Alfentanil and placebo analgesia: no sex differences detected in models of experimental pain. Anesthesiology. 2005;103:130–9. doi: 10.1097/00000542-200507000-00020. [DOI] [PubMed] [Google Scholar]
- 46.Petersen-Felix S, Arendt-Nielsen L, Bak P, Bjerring P, Breivik H, Svensson P, Zbinden AM. Ondansetron does not inhibit the analgesic effect of alfentanil. Br J Anaesth. 1994;73:326–30. doi: 10.1093/bja/73.3.326. [DOI] [PubMed] [Google Scholar]
- 47.Petersen-Felix S, Arendt-Nielsen L, Bak P, Fischer M, Zbinden AM. Psychophysical and electrophysiological responses to experimental pain may be influenced by sedation: comparison of the effects of a hypnotic (propofol) and an analgesic (alfentanil) Br J Anaesth. 1996;77:165–71. doi: 10.1093/bja/77.2.165. [DOI] [PubMed] [Google Scholar]
- 48.Schulte H, Segerdahl M, Graven-Nielsen T, Grass S. Reduction of human experimental muscle pain by alfentanil and morphine. Eur J Pain. 2006;10:733–41. doi: 10.1016/j.ejpain.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 49.Wallace MS, Ridgeway B, III, Leung A, Schulteis G, Yaksh TL. Concentration-effect relationships for intravenous alfentanil and ketamine infusions in human volunteers: effects on acute thresholds and capsaicin-evoked hyperpathia. J Clin Pharmacol. 2002;42:70–80. doi: 10.1177/0091270002042001008. [DOI] [PubMed] [Google Scholar]
- 50.Oertel BG, Preibisch C, Wallenhorst T, Hummel T, Geisslinger G, Lanfermann H, Lotsch J. Differential opioid action on sensory and affective cerebral pain processing. Clin Pharmacol Ther. 2008;83:577–88. doi: 10.1038/sj.clpt.6100441. [DOI] [PubMed] [Google Scholar]
- 51.Chapman CR, Hill HF, Saeger L, Gavrin J. Profiles of opioid analgesia in humans after intravenous bolus administration: alfentanil, fentanyl and morphine compared on experimental pain. Pain. 1990;43:47–55. doi: 10.1016/0304-3959(90)90049-J. [DOI] [PubMed] [Google Scholar]
- 52.Black ML, Hill JL, Zacny JP. Behavioral and physiological effects of remifentanil and alfentanil in healthy volunteers. Anesthesiology. 1999;90:718–26. doi: 10.1097/00000542-199903000-00013. [DOI] [PubMed] [Google Scholar]
- 53.Schulte H, Graven-Nielsen T, Sollevi A, Jansson Y, Arendt-Nielsen L, Segerdahl M. Pharmacological modulation of experimental phasic and tonic muscle pain by morphine, alfentanil and ketamine in healthy volunteers. Acta Anaesthesiol Scand. 2003;47:1020–30. doi: 10.1034/j.1399-6576.2003.00204.x. [DOI] [PubMed] [Google Scholar]
- 54.Eisenach JC, Hood DD, Curry R, Tong C. Alfentanil, but not amitriptyline, reduces pain, hyperalgesia, and allodynia from intradermal injection of capsaicin in humans. Anesthesiology. 1997;86:1279–87. doi: 10.1097/00000542-199706000-00008. [DOI] [PubMed] [Google Scholar]
- 55.Sethna NF, Liu M, Gracely R, Bennett GJ, Max MB. Analgesic and cognitive effects of intravenous ketamine-alfentanil combinations versus either drug alone after intradermal capsaicin in normal subjects. Anesth Analg. 1998;86:1250–6. doi: 10.1097/00000539-199806000-00022. [DOI] [PubMed] [Google Scholar]
- 56.Wallace MS, Braun J, Schulteis G. Postdelivery of alfentanil and ketamine has no effect on intradermal capsaicin-induced pain and hyperalgesia. Clin J Pain. 2002;18:373–9. doi: 10.1097/00002508-200211000-00005. [DOI] [PubMed] [Google Scholar]
- 57.Schulte H, Sollevi A, Segerdahl M. Dose-dependent effects of morphine on experimentally induced cutaneous pain in healthy volunteers. Pain. 2005;116:366–74. doi: 10.1016/j.pain.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 58.Koltzenburg M, Pokorny R, Gasser UE, Richarz U. Differential sensitivity of three experimental pain models in detecting the analgesic effects of transdermal fentanyl and buprenorphine. Pain. 2006;126:165–74. doi: 10.1016/j.pain.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 59.Tucker AP, Kim YI, Nadeson R, Goodchild CS. Investigation of the potentiation of the analgesic effects of fentanyl by ketamine in humans: a double-blinded, randomised, placebo controlled, crossover study of experimental pain [ISRCTN83088383. BMC Anesthesiol. 2005;5:2. doi: 10.1186/1471-2253-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ginosar Y, Riley ET, Angst MS. The site of action of epidural fentanyl in humans: the difference between infusion and bolus administration. Anesth Analg. 2003;97:1428–38. doi: 10.1213/01.ANE.0000081793.60059.10. [DOI] [PubMed] [Google Scholar]
- 61.Hill HF, Chapman CR, Saeger LS, Bjurstrom R, Walter MH, Schoene RB, Kippes M. Steady-state infusions of opioids in human. II. Concentration-effect relationships and therapeutic margins. Pain. 1990;43:69–79. doi: 10.1016/0304-3959(90)90051-E. [DOI] [PubMed] [Google Scholar]
- 62.Price DD, Staud R, Robinson ME, Mauderli AP, Cannon R, Vierck CJ. Enhanced temporal summation of second pain and its central modulation in fibromyalgia patients. Pain. 2002;99:49–59. doi: 10.1016/s0304-3959(02)00053-2. [DOI] [PubMed] [Google Scholar]
- 63.Curatolo M, Petersen-Felix S, Gerber A, rendt-Nielsen L. Remifentanil inhibits muscular more than cutaneous pain in humans. Br J Anaesth. 2000;85:529–32. doi: 10.1093/bja/85.4.529. [DOI] [PubMed] [Google Scholar]
- 64.Gustorff B, Felleiter P, Nahlik G, Brannath W, Hoerauf KH, Spacek A, Kress HG. The effect of remifentanil on the heat pain threshold in volunteers. Anesth Analg. 2001;92:369–74. doi: 10.1097/00000539-200102000-00017. [DOI] [PubMed] [Google Scholar]
- 65.Luginbuhl M, Gerber A, Schnider TW, Petersen-Felix S, rendt-Nielsen L, Curatolo M. Modulation of remifentanil-induced analgesia, hyperalgesia, and tolerance by small-dose ketamine in humans. Anesth Analg. 2003;96:726–32. doi: 10.1213/01.ANE.0000048086.58161.18. [DOI] [PubMed] [Google Scholar]
- 66.Petersen KL, Jones B, Segredo V, Dahl JB, Rowbotham MC. Effect of remifentanil on pain and secondary hyperalgesia associated with the heat – capsaicin sensitization model in healthy volunteers. Anesthesiology. 2001;94:15–20. doi: 10.1097/00000542-200101000-00008. [DOI] [PubMed] [Google Scholar]
- 67.Petersen KL, Maloney A, Hoke F, Dahl JB, Rowbotham MC. A randomized study of the effect of oral lamotrigine and hydromorphone on pain and hyperalgesia following heat/capsaicin sensitization. J Pain. 2003;4:400–6. doi: 10.1016/s1526-5900(03)00718-1. [DOI] [PubMed] [Google Scholar]
- 68.Wagner KJ, Sprenger T, Kochs EF, Tolle TR, Valet M, Willoch F. Imaging human cerebral pain modulation by dose-dependent opioid analgesia: a positron emission tomography activation study using remifentanil. Anesthesiology. 2007;106:548–56. doi: 10.1097/00000542-200703000-00020. [DOI] [PubMed] [Google Scholar]
- 69.Lorenz IH, Kolbitsch C, Hinteregger M, Bauer P, Spiegel M, Luger TJ, Schmidauer C, Streif W, Pfeiffer KP, Benzer A. Remifentanil and nitrous oxide reduce changes in cerebral blood flow velocity in the middle cerebral artery caused by pain. Br J Anaesth. 2003;90:296–9. doi: 10.1093/bja/aeg055. [DOI] [PubMed] [Google Scholar]
- 70.Koppert W, Angst M, Alsheimer M, Sittl R, Albrecht S, Schuttler J, Schmelz M. Naloxone provokes similar pain facilitation as observed after short-term infusion of remifentanil in humans. Pain. 2003;106:91–9. doi: 10.1016/s0304-3959(03)00294-x. [DOI] [PubMed] [Google Scholar]
- 71.Singler B, Troster A, Manering N, Schuttler J, Koppert W. Modulation of remifentanil-induced postinfusion hyperalgesia by propofol. Anesth Analg. 2007;104:1397–403. doi: 10.1213/01.ane.0000261305.22324.f3. table. [DOI] [PubMed] [Google Scholar]
- 72.Troster A, Sittl R, Singler B, Schmelz M, Schuttler J, Koppert W. Modulation of remifentanil-induced analgesia and postinfusion hyperalgesia by parecoxib in humans. Anesthesiology. 2006;105:1016–23. doi: 10.1097/00000542-200611000-00024. [DOI] [PubMed] [Google Scholar]
- 73.Hood DD, Curry R, Eisenach JC. Intravenous remifentanil produces withdrawal hyperalgesia in volunteers with capsaicin-induced hyperalgesia. Anesth Analg. 2003;97:810–5. doi: 10.1213/01.ANE.0000078811.80093.88. [DOI] [PubMed] [Google Scholar]
- 74.Tian YL, Guo Y, Cao DY, Zhang Q, Wang HS, Zhao Y. Local application of morphine suppresses glutamate-evoked activities of C and Adelta afferent fibers in rat hairy skin. Brain Res. 2005;1059:28–34. doi: 10.1016/j.brainres.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 75.Hallin RG, Torebjork HE, Wiesenfeld Z. Nociceptors and warm receptors innervated by C fibres in human skin. J Neurol Neurosurg Psychiatry. 1982;45:313–9. doi: 10.1136/jnnp.45.4.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Le Bars D, Gozariu M, Cadden SW. Animal models of nociception. Pharmacol Rev. 2001;53:597–652. [PubMed] [Google Scholar]
- 77.Willer JC, Le Bars D, De Broucker T. Diffuse noxious inhibitory controls in man: involvement of an opioidergic link. Eur J Pharmacol. 1990;182:347–55. doi: 10.1016/0014-2999(90)90293-f. [DOI] [PubMed] [Google Scholar]
- 78.Dickenson AH. Spinal cord pharmacology of pain. Br J Anaesth. 1995;75:193–200. doi: 10.1093/bja/75.2.193. [DOI] [PubMed] [Google Scholar]
- 79.Cohen SP, Christo PJ, Wang S, Chen L, Stojanovic MP, Shields CH, Brummett C, Mao J. The effect of opioid dose and treatment duration on the perception of a painful standardized clinical stimulus. Reg Anesth Pain Med. 2008;33:199–206. doi: 10.1016/j.rapm.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 80.Joly V, Richebe P, Guignard B, Fletcher D, Maurette P, Sessler DI, Chauvin M. Remifentanil-induced postoperative hyperalgesia and its prevention with small-dose ketamine. Anesthesiology. 2005;103:147–55. doi: 10.1097/00000542-200507000-00022. [DOI] [PubMed] [Google Scholar]
- 81.Naef M, Curatolo M, Petersen-Felix S, Arendt-Nielsen L, Zbinden A, Brenneisen R. The analgesic effect of oral delta-9-tetrahydrocannabinol (THC), morphine, and a THC-morphine combination in healthy subjects under experimental pain conditions. Pain. 2003;105:79–88. doi: 10.1016/s0304-3959(03)00163-5. [DOI] [PubMed] [Google Scholar]
- 82.Le Bars D, Guilbaud G, Jurna I, Besson JM. Differential effects of morphine on responses of dorsal horn lamina V type cells elicited by A and C fibre stimulation in the spinal cat. Brain Res. 1976;115:518–24. doi: 10.1016/0006-8993(76)90370-x. [DOI] [PubMed] [Google Scholar]
- 83.van der Burght M, Rasmussen SE, Arendt-Nielsen L, Bjerring P. Morphine does not affect laser induced warmth and pin prick pain thresholds. Acta Anaesthesiol Scand. 1994;38:161–4. doi: 10.1111/j.1399-6576.1994.tb03859.x. [DOI] [PubMed] [Google Scholar]
- 84.Arendt-Nielsen L, Bjerring P, Dahl JB. A quantitative double-blind evaluation of the antinociceptive effects of perineurally administered morphine compared with lidocaine. Acta Anaesthesiol Scand. 1991;35:24–9. doi: 10.1111/j.1399-6576.1991.tb03236.x. [DOI] [PubMed] [Google Scholar]
- 85.Arendt-Nielsen L, Oberg B, Bjerring P. Hypoalgesia following epidural morphine: a controlled quantitative experimental study. Acta Anaesthesiol Scand. 1991;35:430–5. doi: 10.1111/j.1399-6576.1991.tb03323.x. [DOI] [PubMed] [Google Scholar]
- 86.Quante M, Scharein E, Zimmermann R, Langer-Brauburger B, Bromm B. Dissociation of morphine analgesia and sedation evaluated by EEG measures in healthy volunteers. Arzneimittelforschung. 2004;54:143–51. doi: 10.1055/s-0031-1296951. [DOI] [PubMed] [Google Scholar]
- 87.Eckhardt K, Ammon S, Hofmann U, Riebe A, Gugeler N, Mikus G. Gabapentin enhances the analgesic effect of morphine in healthy volunteers. Anesth Analg. 2000;91:185–91. doi: 10.1097/00000539-200007000-00035. [DOI] [PubMed] [Google Scholar]
- 88.Grach M, Massalha W, Pud D, Adler R, Eisenberg E. Can co-administration of oxycodone and morphine produce analgesic synergy in humans? An experimental cold pain study. Br J Clin Pharmacol. 2004;58:235–42. doi: 10.1111/j.1365-2125.2004.02141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Plesan A, Sollevi A, Segerdahl M. The N-methyl-D-aspartate-receptor antagonist dextromethorphan lacks analgesic effect in a human experimental ischemic pain model. Acta Anaesthesiol Scand. 2000;44:924–8. doi: 10.1034/j.1399-6576.2000.440805.x. [DOI] [PubMed] [Google Scholar]
- 90.Pud D, Yarnitsky D, Sprecher E, Rogowski Z, Adler R, Eisenberg E. Can personality traits and gender predict the response to morphine? An experimental cold pain study. Eur J Pain. 2006;10:103–12. doi: 10.1016/j.ejpain.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 91.Segerdahl M, Ekblom A, Sollevi A. The influence of adenosine, ketamine, and morphine on experimentally induced ischemic pain in healthy volunteers. Anesth Analg. 1994;79:787–91. doi: 10.1213/00000539-199410000-00029. [DOI] [PubMed] [Google Scholar]
- 92.Smith GM, Egbert LD, Markowitz RA, Mosteller F, Beecher HK. An experimental pain method sensitive to morphine in man: the submaximum effort tourniquet technique. J Pharmacol Exp Ther. 1966;154:324–32. [PubMed] [Google Scholar]
- 93.Moiniche S, Dahl JB, Kehlet H. Peripheral antinociceptive effects of morphine after burn injury. Acta Anaesthesiol Scand. 1993;37:710–2. doi: 10.1111/j.1399-6576.1993.tb03795.x. [DOI] [PubMed] [Google Scholar]
- 94.Schulte H, Sollevi A, Segerdahl M. The synergistic effect of combined treatment with systemic ketamine and morphine on experimentally induced windup-like pain in humans. Anesth Analg. 2004;98:1574–80. doi: 10.1213/01.ANE.0000113237.89875.5D. [DOI] [PubMed] [Google Scholar]
- 95.Tegeder I, Meier S, Burian M, Schmidt H, Geisslinger G, Lotsch J. Peripheral opioid analgesia in experimental human pain models. Brain. 2003;126:1092–102. doi: 10.1093/brain/awg115. [DOI] [PubMed] [Google Scholar]
- 96.Roberts JD, Gennings C, Shih M. Synergistic affective analgesic interaction between delta-9-tetrahydrocannabinol and morphine. Eur J Pharmacol. 2006;530:54–8. doi: 10.1016/j.ejphar.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 97.Price DD, Von der GA, Miller J, Rafii A, Price C. A psychophysical analysis of morphine analgesia. Pain. 1985;22:261–9. doi: 10.1016/0304-3959(85)90026-0. [DOI] [PubMed] [Google Scholar]
- 98.D’Honneur G, Gilton A, Sandouk P, Scherrmann JM, Duvaldestin P. Plasma and cerebrospinal fluid concentrations of morphine and morphine glucuronides after oral morphine. The influence of renal failure. Anesthesiology. 1994;81:87–93. doi: 10.1097/00000542-199407000-00013. [DOI] [PubMed] [Google Scholar]
- 99.Lotsch J. Pharmacokinetic-pharmacodynamic modeling of opioids. J Pain Symptom Manage. 2005;29:S90–103. doi: 10.1016/j.jpainsymman.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 100.Enggaard TP, Poulsen L, Arendt-Nielsen L, Hansen SH, Bjornsdottir I, Gram LF, Sindrup SH. The analgesic effect of codeine as compared to imipramine in different human experimental pain models. Pain. 2001;92:277–82. doi: 10.1016/s0304-3959(01)00267-6. [DOI] [PubMed] [Google Scholar]
- 101.Poulsen L, Rendt-Nielsen L, Brosen K, Nielsen KK, Gram LF, Sindrup SH. The hypoalgesic effect of imipramine in different human experimental pain models. Pain. 1995;60:287–93. doi: 10.1016/0304-3959(94)00142-2. [DOI] [PubMed] [Google Scholar]
- 102.Fillingim RB, Ness TJ, Glover TL, Campbell CM, Hastie BA, Price DD, Staud R. Morphine responses and experimental pain: sex differences in side effects and cardiovascular responses but not analgesia. J. Pain. 2005;6:116–24. doi: 10.1016/j.jpain.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 103.Sprenger T, Valet M, Boecker H, Henriksen G, Spilker ME, Willoch F, Wagner KJ, Wester HJ, Tolle TR. Opioidergic activation in the medial pain system after heat pain. Pain. 2006;122:63–7. doi: 10.1016/j.pain.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 104.De Schepper HU, Cremonini F, Park MI, Camilleri M. Opioids and the gut: pharmacology and current clinical experience. Neurogastroenterol Motil. 2004;16:383–394. doi: 10.1111/j.1365-2982.2004.00513.x. [DOI] [PubMed] [Google Scholar]
- 105.Arendt-Nielsen L, Pedersen P, Poulsen L, Andersen OK, Bjerring P, Coste I, Insuasty J, Sindrup SH, Drewes AM. A 5-HT antagonist (UP 26-91) versus codeine and placebo in a human experimental pain study. Pain Res Manage. 2000;4:135–140. [Google Scholar]
- 106.Poulsen L, Brosen K, Arendt-Nielsen L, Gram LF, Elbaek K, Sindrup SH. Codeine and morphine in extensive and poor metabolizers of sparteine: pharmacokinetics, analgesic effect and side effects. Eur J Clin Pharmacol. 1996;51:289–95. doi: 10.1007/s002280050200. [DOI] [PubMed] [Google Scholar]
- 107.Stacher G, Bauer P, Schneider C, Winklehner S, Schmierer G. Effects of a combination of oral naproxen sodium and codeine on experimentally induced pain. Eur J Clin Pharmacol. 1982;21:485–90. doi: 10.1007/BF00542043. [DOI] [PubMed] [Google Scholar]
- 108.Walker DJ, Zacny JP. Subjective, psychomotor, and analgesic effects of oral codeine and morphine in healthy volunteers. Psychopharmacology (Berl) 1998;140:191–201. doi: 10.1007/s002130050757. [DOI] [PubMed] [Google Scholar]
- 109.Guay DR, Awni WM, Halstenson CE, Findlay JW, Opsahl JA, Abraham PA, Jones EC, Matzke GR. Pharmacokinetics of codeine after single- and multiple-oral-dose administration to normal volunteers. J Clin Pharmacol. 1987;27:983–7. doi: 10.1002/j.1552-4604.1987.tb05601.x. [DOI] [PubMed] [Google Scholar]
- 110.Srinivasan V, Wielbo D, Tebbett IR. Analgesic effects of codeine-6-glucuronide after intravenous administration. Eur J Pain. 1997;1:185–90. doi: 10.1016/s1090-3801(97)90103-8. [DOI] [PubMed] [Google Scholar]
- 111.Enggaard TP, Poulsen L, Arendt-Nielsen L, Brosen K, Ossig J, Sindrup SH. The analgesic effect of tramadol after intravenous injection in healthy volunteers in relation to CYP2D6. Anesth Analg. 2006;102:146–50. doi: 10.1213/01.ane.0000189613.61910.32. [DOI] [PubMed] [Google Scholar]
- 112.Hummel T, Hummel C, Friedel I, Pauli E, Kobal G. A comparison of the antinociceptive effects of imipramine, tramadol and anpirtoline. Br J Clin Pharmacol. 1994;37:325–33. doi: 10.1111/j.1365-2125.1994.tb04285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Thurauf N, Fleischer WK, Liefhold J, Schmid O, Kobal G. Dose dependent time course of the analgesic effect of a sustained-release preparation of tramadol on experimental phasic and tonic pain. Br J Clin Pharmacol. 1996;41:115–23. doi: 10.1111/j.1365-2125.1996.tb00168.x. [DOI] [PubMed] [Google Scholar]
- 114.Hogger P, Rohdewald P. Comparison of tilidine/naloxone, tramadol and bromfenac in experimental pain: a double-blind randomized crossover study in healthy human volunteers. Int J Clin Pharmacol Ther. 1999;37:377–85. [PubMed] [Google Scholar]
- 115.Narhi M, Jyvasjarvi E, Virtanen A, Huopaniemi T, Ngassapa D, Hirvonen T. Role of intradental A- and C-type nerve fibres in dental pain mechanisms. Proc Finn Dent Soc. 1992;88(Suppl. 1):507–16. [PubMed] [Google Scholar]
- 116.Rohdewald P, Granitzki HW, Neddermann E. Comparison of the analgesic efficacy of metamizole and tramadol in experimental pain. Pharmacology. 1988;37:209–17. doi: 10.1159/000138468. [DOI] [PubMed] [Google Scholar]
- 117.Loram LC, Mitchell D, Fuller A. Rofecoxib and tramadol do not attenuate delayed-onset muscle soreness or ischaemic pain in human volunteers. Can J Physiol Pharmacol. 2005;83:1137–45. doi: 10.1139/y05-113. [DOI] [PubMed] [Google Scholar]
- 118.Filitz J, Ihmsen H, Gunther W, Troster A, Schwilden H, Schuttler J, Koppert W. Supra-additive effects of tramadol and acetaminophen in a human pain model. Pain. 2008;136:262–70. doi: 10.1016/j.pain.2007.06.036. [DOI] [PubMed] [Google Scholar]
- 119.Minami K, Uezono Y, Ueta Y. Pharmacological aspects of the effects of tramadol on G-protein coupled receptors. J Pharmacol Sci. 2007;103:253–60. doi: 10.1254/jphs.cr0060032. [DOI] [PubMed] [Google Scholar]
- 120.Gillen C, Haurand M, Kobelt DJ, Wnendt S. Affinity, potency and efficacy of tramadol and its metabolites at the cloned human mu-opioid receptor. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:116–21. doi: 10.1007/s002100000266. [DOI] [PubMed] [Google Scholar]
- 121.Poulsen L, Arendt-Nielsen L, Brosen K, Sindrup SH. The hypoalgesic effect of tramadol in relation to CYP2D6. Clin Pharmacol Ther. 1996;60:636–44. doi: 10.1016/S0009-9236(96)90211-8. [DOI] [PubMed] [Google Scholar]
- 122.Angst MS, Clark JD, et al. Comment on Koltzenburg. differential sensitivity of three experimental pain models in detecting the analgesic effects of transdermal fentanyl and buprenorphine. Pain. 2006;126:165–74. doi: 10.1016/j.pain.2006.06.028. Pain 2007; 128: 292–94. [DOI] [PubMed] [Google Scholar]
- 123.Yarnitsky D, Sprecher E, Zaslansky R, Hemli JA. Multiple session experimental pain measurement. Pain. 1996;67:327–33. doi: 10.1016/0304-3959(96)03110-7. [DOI] [PubMed] [Google Scholar]
- 124.Petersen-Felix S, Arendt-Nielsen L. From pain research to pain treatment: the role of human experimental pain models. Best Pract Res Clin Anaesthesiol. 2002;16:667–80. doi: 10.1053/bean.2002.0258. [DOI] [PubMed] [Google Scholar]
- 125.Chizh BA, Priestley T, Rowbotham M, Schaffler K. Predicting therapeutic efficacy – Experimental pain in human subjects. Brain Res Rev. 2009;60:243–54. doi: 10.1016/j.brainresrev.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 126.Campa D, Gioia A, Tomei A, Poli P, Barale R. Association of ABCB1/MDR1 and OPRM1 gene polymorphisms with morphine pain relief. Clin Pharmacol Ther. 2008;83:559–66. doi: 10.1038/sj.clpt.6100385. [DOI] [PubMed] [Google Scholar]
- 127.Garcia R, Benet M, Arnau C, Cobo E. Efficiency of the cross-over design: an empirical estimation. Stat Med. 2004;23:3773–80. doi: 10.1002/sim.2072. [DOI] [PubMed] [Google Scholar]
- 128.Keogh E, Barlow C, Mounce C, Bond FW. Assessing the relationship between cold pressor pain responses and dimensions of the anxiety sensitivity profile in healthy men and women. Cogn Behav Ther. 2006;35:198–206. doi: 10.1080/16506070600898330. [DOI] [PubMed] [Google Scholar]
- 129.Pincus T, Burton AK, Vogel S, Field AP. A systematic review of psychological factors as predictors of chronicity/disability in prospective cohorts of low back pain. Spine. 2002;27:E109–120. doi: 10.1097/00007632-200203010-00017. [DOI] [PubMed] [Google Scholar]
- 130.Arendt-Nielsen L, Nielsen J, Petersen-Felix S, Schnider TW, Zbinden AM. Effect of racemic mixture and the (S+)-isomer of ketamine on temporal and spatial summation of pain. Br J Anaesth. 1996;77:625–31. doi: 10.1093/bja/77.5.625. [DOI] [PubMed] [Google Scholar]
- 131.Kalso E, Poyhia R, Onnela P, Linko K, Tigerstedt I, Tammisto T. Intravenous morphine and oxycodone for pain after abdominal surgery. Acta Anaesthesiol Scand. 1991;35:642–6. doi: 10.1111/j.1399-6576.1991.tb03364.x. [DOI] [PubMed] [Google Scholar]