

Abstract
AIMS
Whether glucocorticoids induce gastric mucosal injury remains uncertain. We investigated whether very high-dose steroids caused gastric mucosal injury in systemic lupus erythematous (SLE) patients and evaluated the possible risk factors for mucosal injury.
METHODS
In this prospective paired study, 67 SLE patients who had received pulse methylprednisolone therapy were enrolled. Each patient underwent endoscopic examination and tissue and blood sampling before and after pulse steroid therapy. Mucosal injury was diagnosed if the follow-up injury scale was higher than the initial scale. Examined parameters included Helicobacter pylori infection, cyclooxygenase (COX)-1 and COX-2 activity, and current nonsteroidal anti-inflammatory drug (NSAID) usage including aspirin.
RESULTS
Eleven (16.4%) of 67 cases who developed gastric mucosal injury after pulse therapy had significantly higher rates of peptic ulcer history, NSAID/aspirin use, lower gastric thromboxane B2 and prostaglandin E2 levels when compared with cases without gastric mucosal injury (P < 0.05). Infection by H. pylori was not a risk factor for gastric mucosal injury. Multivariate logistic regression analysis showed that NSAID/aspirin use was the only risk factor for gastric mucosal injury in these patients (odds ratio 26.99, 95% confidence interval 4.91, 148.57, P < 0.0001). Pulse steroid therapy alone did not induce gastric mucosal injury in fifty SLE patients without taking any NSAID/aspirin.
CONCLUSIONS
Use of NSAIDs/aspirin, but not H. pylori infection, increases gastric mucosal injury in SLE patients receiving pulse methylprednisolone therapy. Very high-dose steroids de novo seem not to induce gastric mucosal injury in these patients. A larger case-controlled study enrolling a heterogeneous population is needed to clarify the role of glucocorticoids in gastric mucosal injury.
Keywords: gastric mucosal injury, Helicobacter pylori, nonsteroidal anti-inflammatory drugs, pulse methylprednisolone therapy, systemic lupus erythematosus
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Whether glucocorticoids induce gastric mucosal injury remains uncertain, and Helicobacter pylori infection in steroid users has not been well evaluated in the past.
Pulse methylprednisolone therapy with the very high dose of steroid that is 100 times larger than the physiological dose will be a special model to evaluate whether glucocorticoids induce gastric mucosal injury.
WHAT THIS STUDY ADDS
Use of nonsteroidal anti-inflammatory drugs/aspirin, but not H. pylori infection, increases gastric mucosal injury in systemic lupus erythematosus patients receiving pulse methylprednisolone therapy.
Very high-dose steroids de novo seem not to induce gastric mucosal injury in these patients.
However, a larger, case-controlled study enrolling a heterogeneous population is needed to clarify the role of glucocorticoids in gastric mucosal injury.
Introduction
Corticosteroids are a well-known treatment for many diseases for more than five decades via their anti-inflammatory and immunosuppressive properties. Although long-term application of corticosteroids can clearly cause various adverse effects, the association between corticosteroids and gastric mucosal injury remains controversial [1]. An early meta-analysis of 26 placebo-controlled, double-blinded studies of corticosteroid therapies revealed no association between corticosteroid usage and peptic ulcer disease (PUD) [2]. However, subsequent meta-analysis of 71 clinical trials showed that systemic corticosteroids or adrenocorticotropic hormone doubled the risk of PUD [3]. Recently, a large epidemiological study in the UK showed that steroid use increased ulcer complications from gastric ulcer rather than from duodenal ulcer [4]. It has been suggested that the mechanisms responsible for gastric mucosal damage induced by corticosteroids include inhibition of gastric mucus synthesis, enhancement of gastrin and parietal cell hyperplasia with augmented acid secretion, and suppression of arachidonic acid metabolism and prostaglandin (PG) synthesis [5, 6]. Our previous study revealed that a 0.2 mg kg−1 day−1 dose of dexamethasone was non-ulcergenic to rat gastric mucosa and only inhibited cyclooxygenase (COX)-2 expression, but did not influence COX-1 expression and its activity [7]. However, dexamethasone delayed ulcer healing [7, 8].
Nonsteroidal anti-inflammatory drugs (NSAIDs) are ulcerogenic agents and cause mucosal damage by inhibiting COX-1 and COX-2 activity with decreasing PG synthesis, increasing expression of intercellular cell adhesion molecule-1, inducing neutrophil infiltration with oxygen-derived free radical formation, and so on [9]. Further studies have demonstrated that inhibition of both COX-1 and COX-2 is required for NSAID-induced gastric injury in rat [10]. NSAIDs also delay ulcer healing via decreasing cell proliferation, angiogenesis of granulation tissue and cell migration [11].
Whether corticosteroids are ulcerogenic to human gastric mucosa is unclear. In studies of rats given high-dose NSAIDs, every rat developed gastric mucosal injury within hours [11, 12]. To the best of our knowledge, there are no data demonstrating whether and to what extent very high-dose corticosteroids alone, or in combination with NSAIDs, will induce gastric mucosal injury. Patients with systemic lupus erythematosus (SLE) often receive pulse methylprednisolone therapy (1000 mg day−1 for 2–3 days) for their critical disease manifestations [13, 14]. The corticosteroid dose received by these patients may be as high as 100 times the physiological corticosteroid dose. SLE patients also took NSAIDs for treating their inflammatory arthritis and took aspirin for preventing thrombosis if they showed positive antiphospholipid antibodies [15, 16]. This prospective paired study evaluated whether gastric mucosa injury occurred in SLE patients who received pulse methylprednisolone therapy and identified possible risk factors, including NSAIDs/aspirin, for gastric mucosal injury.
Methods
Participants
SLE patients who received intravenous pulse methylprednisolone therapy (methylprednisolone sodium succinate, Sterile Soul-Medrol; Pharmacia/Pfizer, Puurs, Belgium) 1000 mg day−1 for 2 days with 2-week intervals were enrolled. SLE was diagnosed according to the American College of Rheumatology 1997 Revised Criteria for SLE [17]. Referral SLE patients who had taken acid-reducing agents or mucosal protective agents such as proton pump inhibitor, histamine 2 receptor antagonists or misoprostol, who had taken selective serotonin reuptake inhibitor, had current PUD or history of surgery for PUD complications, had thrombocytopenia (platelets <80 000 mm−3), bleeding tendency or poor general condition were excluded from this study. The study was approved by the Institutional Review Board of Taipei Veterans General Hospital (VGHIRB No: 93-09-08A) and was conducted in accordance with Declaration of Helsinki guidelines. All patients provided written informed consent before participating.
Procedures and measurement
Within 1 week before methylprednisolone pulse therapy, upper gastrointestinal symptoms including epigastralgia, epigastric distension, heartburn, and acid regurgitation were recorded in all patients. Habitual consumption of cigarette tobacco (≥ half pack daily), alcohol (≥80 g weekly), tea (≥ three cups daily) or coffee (≥ three cups daily), continuous, current NSAID medications (nonselective or selective COX-2 inhibitor) including aspirin, and peptic ulcer history were reviewed. Use of NSAIDs, aspirin and steroids was defined as receiving these medication during all the course of the study. After overnight fasting and before taking any medication on the day of endoscopy, each subject received pharyngeal anaesthesia with 10% xylocaine (AstraZeneca AB, Sodertalje, Sweden) and then underwent video oesophagogastroduodenoscopy (EGD) (Olympus GIF-XQ260 gastrointestinal videoscope; Aizo Olympus, Fukushima, Japan). All endoscopies were performed and recorded on a digital file system by a senior gastroenterologist (J-C.L. or F-Y.C.) so that mucosal lesions could be subsequently scored by two gastroenterologists while blinded to the treatment status of each patient. Gastric and duodenal mucosal injuries were scored using the scale developed by Lanza et al., with some modifications (Table 1) [18, 19]. Erosions were defined as mucosal breaks smaller than 3 mm. Oesophageal mucosal injury was defined and graded using the Los Angeles system (grade A as scale 1; grade B as scale 2; grade C as scale 3; and grade D as scale 4). If the scale for the same patient differed between the two gastroenterologists, the digital file was reviewed and a consensus was reached.
Table 1.
Rating scale for assessment of gastric and duodenal mucosal injury by video oesophagogastroduodenoscopy
| Scale | Definition |
|---|---|
| 0 | Normal or erythema |
| 1 | 1–3 submucosal haemorrhage or oedema without erosions |
| 2 | More than 3 submucosal haemorrhages or oedema without erosions |
| 3 | One erosion ± submucosal haemorrhage or oedema |
| 4 | 2–4 erosions ± submucosal haemorrhage or oedema |
| 5 | 5 or more erosions and/or a single ulcer ± submucosal haemorrhage or oedema |
| 6 | Multiple ulcers ± submucosal haemorrhage or oedema |
Erosions were defined as mucosal breaks <3 mm.
Helicobacter pylori infection was diagnosed based on positive rapid urease test (CLO test; Ballard Medical Products, Draper, UT, USA) plus histological observation showing H. pylori in the stomach specimens (Giemsa stain)[1]. Gastric juice was also collected for pH assessment (Microcomputer pH-vision 6071; JENCO Electronics Ltd, Taipei, Taiwan). Antral gastric mucosa (2–4 cm proximal to the pylorus) was collected to measure thromboxane (TX) B2 and PGE2 levels. Serum was collected before EGD to measure serum albumin, creatinine, haemoglobin, platelet count, gastrin, pepsinogen I, TXB2 and PGE2 levels.
Blood COX-2 activity was measured as lipopolysaccharide (LPS)-induced PGE2, and blood COX-1 activity was measured as serum TXB2[20]. LPS-induced PGE2 in whole blood, a marker of monocyte COX-2 activity, and serum-generated TXB2 in whole blood, a marker of platelet COX-1 activity, were measured as in a previous study [20]. PGE2 and TXB2 were measured in duplicate using PGE2 and TXB2 enzyme-linked immunosorbent assay (ELISA) kit, respectively (Quantikine; R&D Systems Inc., Minneapolis, MN, USA). Plasma gastrin (Incstar Corp., Stillwater, MN, USA) and pepsinogen I (Alpco Diagnostics, Windham, NH, USA) levels were measured in duplicate by radioimmunoassay and ELISA kit, respectively. Blood count, including haemoglobin and platelet count, were checked by autoanalyser and technicians. Serum albumin and creatinine were measured by an autoanalyser (Hitachi Model 736 automatic analyser; Tokyo, Japan).
Gastric tissues were homogenized with homogenizing buffer (0.05 m Tris–HCl at pH 7.4, 0.1 m NaCl, 0.001 m CaCl2, 1 mg ml−1 D-glucose, 28 µm indomethacin to inhibit further PGE2 formation) for 30 s, then centrifuged at 24 148 g for 15 min at 4 oC. Supernatants were assayed in duplicate using a commercial available TXB2 and PGE2 ELISA kit, respectively (Quantikine; R&D systems Inc.). Optical densities were determined by the MRX microplate reader (Dynex Technologies, Chantilly, VA, USA). Protein levels in each sample were determined by protein assay kit, and mucosal TXB2 and PGE2 levels were expressed as pg mg−1 protein [1, 21].
Within 48 h, patients received pulse methylprednisolone therapy after initial EGD. To avoid the previous biopsy lesion from interfering with the endoscopic assessment of gastric mucosal injury, all enrolled patients received follow-up EGD after at least the second course of pulse methylprednisolone therapy with at least a 2-week interval after the initial endoscopic evaluation and tissue sampling (Figure 1). To detect any newly developed mucosal lesion probably induced by pulse therapy, all enrolled patients received follow-up EGD within 24 h after the last dose of pulse methylprednisolone therapy. Mucosal injury was diagnosed if the follow-up injury scale exceeded the initial scale. Gastric juice pH, TXB2 and PGE2 levels of the gastric mucosa as well as serum gastrin, pepsinogen I, TXB2 and PGE2 were again measured during follow-up EGD.
Figure 1.
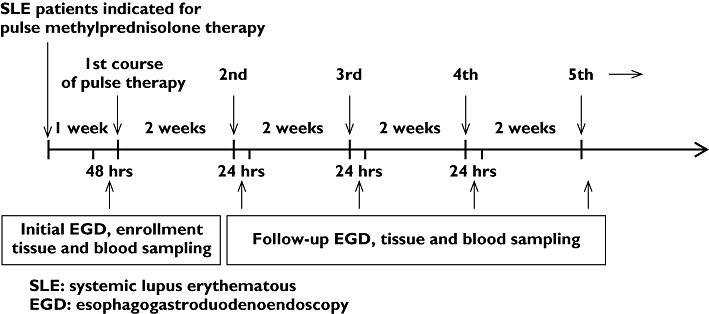
Scheme of gastric mucosal injury in SLE patients receiving pulse methylprednisolone therapy
Statistical analysis
We intended to enrol 62 eligible patients, based upon 80% power to detect newly developed mucosal lesions after pulse methylprednisolone therapy in SLE patients, assuming that 15% of subjects developed a new mucosal lesion after therapy using two-sided testing at the 5% significance level. All data were expressed as means ± SD. Because the endoscopic scale, TXB2 and PGE2 were not normally distributed, nonparametric analysis was used. Results were compared between groups depending on the type of data analysed using either χ2 test, Fisher's exact test, Student's t-test or the nonparametric Mann–Whitney U-test, where appropriate. Paired t-test or nonparametric Wilcoxon signed rank test were used to compare continuous data for the same patient before and after methylprednisolone therapy, McNemar test was used to compare categorical data for the same patient before and after therapy. Continuous variables were transformed to categorical variables with the cut-off points determined by the median of each variable in logistic regression analysis. Univariate and multivariate logistic regressions were performed to evaluate risk factors for mucosal injury in SLE patients receiving pulse methylprednisolone therapy. All statistical analyses were performed using SamplePower release 2.0 and SPSS for Windows version 14.0 (both by SPSS Inc., Chicago, IL, USA). All P-values were two-tailed, and a P-value <0.05 was considered statistically significant.
Results
From March 2005 to June 2007, 78 SLE patients who were to receive intravenous pulse methylprednisolone therapy were considered for enrolment. Three subjects were excluded due to PUD identified by EGD during screening test, eight subjects did not receive follow-up EGD after pulse therapy. A final population of 67 eligible SLE patients remained for data analysis. The mean cumulative dose of methylprednisolone was 4300 ± 2200 mg per subject (range 2000–12 000 mg). The mean course was 2.7 ± 1.2 sessions of pulse therapy per subject (range two to right sessions). Average age was 29 ± 7 years (range 19–50 years). Five (7%) patients were male, 62 (93%) were female, two (3%) had smoked, one (1%) had drunk, 23 (34%) had H. pylori infection, and 17 (25%) used NSAIDs/aspirin (six used nonselective NSAIDs, including five with meloxicam, one with naproxen, six used the COX-2 inhibitor celecoxib, one used the nonselective NSAIDs melocixam plus aspirin, one used the COX-2 inhibitor celecoxib plus aspirin, and three used aspirin alone). Three (4%) had a past history of PUD. Three reported frequent acid regurgitation, and four had occasional epigastralgia. After pulse therapy, one developed a new onset of acid regurgitation, and another developed a new onset of epigastric pain. Eleven (16.4%) patients developed gastric mucosal injury (three from scale 0 to 1, two from scale 0 to 2, one from scale 0 to 3, three from scale 1 to 2, two from scale 2 to 3), whereas three patients had a decreased gastric injury scale after pulse therapy (from scale 1 to 0). None developed oesophageal or duodenal mucosal injury. Among these 17 NSAID/aspirin users, two of five who took meloxicam, one of one who took naproxen, two of six who took celecoxib, one of one who took meloxicam with aspirin, one of one who took celecoxib with aspirin, and two of three who took aspirin developed gastric mucosal injury.
Data comparison between patients with and without gastric mucosal injury
The incidence of past history of PUD and NSAID/aspirin use was significantly higher in the gastric mucosal injury group (n= 11) than in the 56 patients without mucosal injury (Table 2). The patients with mucosal injury also had significantly lower gastric TXB2 and PGE2 before pulse therapy as well as after pulse therapy than those without gastric mucosal injury (Table 3). All other measured variables were not significantly different between these two groups (Tables 2 and 3).
Table 2.
Comparison of the demographic and laboratory data between SLE patients with gastric mucosal injury and those without gastric mucosal injury
| Gastric mucosal injury (+) N= 11 | Gastric mucosal injury (−) N= 56 | P-value | |
|---|---|---|---|
| Age | 27 ± 4 | 30 ± 8 | 0.259 |
| Sex (M/F) | 2/9 | 3/53 | 0.186 |
| Smoking +/− | 1/10 | 1/55 | 0.303 |
| Alcohol +/− | 1/10 | 0/56 | 0.164 |
| Past PUD history +/− | 3/8 | 0/56 | 0.003 |
| Use of NSAIDs/ASA +/− | 9/2 | 8/48 | <0.001 |
| Helicobacter pylori infection +/− | 3/8 | 20/36 | 0.434 |
| Pre-therapy mucosal injury (scale ≥1) +/− | 5/6 | 22/34 | 0.476 |
| Serum albumin (g dl−1) | 3.1 ± 0.3 | 2.9 ± 0.4 | 0.166 |
| Serum creatinine (mg dl−1) | 1.6 ± 0.4 | 1.4 ± 0.6 | 0.536 |
| Haemoglobin (g dl−1) | 10.4 ± 1.4 | 10.5 ± 1.5 | 0.731 |
| Platelets count (mm−3) | 209 500 ± 69 800 | 210 600 ± 65 900 | 0.962 |
| MTP cumulative dose (mg) | 4 000 ± 1 800 | 4 400 ± 2 200 | 0.574 |
| Therapy course (session) | 2.45 ± 0.69 | 2.70 ± 1.31 | 0.554 |
SLE, systemic lupus erythematous; PUD, peptic ulcer disease; NSAIDs, nonsteroidal anti-inflammatory drugs; ASA, aspirin; MPT, methylprednisolone. Data are expressed as means ± SD.
Table 3.
Comparison of the laboratory data (pre- and post therapy) between SLE patients with gastric mucosal injury and those without gastric mucosal injury
| Gastric mucosal injury (+) N = 11 | Gastric mucosal injury (−) N = 56 | p value | |
|---|---|---|---|
| Pre-therapy data | |||
| Gastric pH | 3.2 ± 1.7 | 3.1 ± 1.5 | 0.962 |
| Serum gastrin (pg ml−1) | 69.4 ± 30.3 | 88.5 ± 51.4 | 0.237 |
| Serum pepsinogen I (ng ml−1) | 110.9 ± 32.8 | 118.3 ± 50.3 | 0.639 |
| Serum TXB2 (pg ml−1) (COX-1 activity) | 20 195 ± 32 555 | 33 291 ± 42 143 | 0.334 |
| Serum PGE2 (pg ml−1) (COX-2 activity) | 385.3 ± 183.9 | 450.8 ± 210.6 | 0.498 |
| Gastric TXB2 (ng mg−1 protein) | 637.6 ± 643.5 | 1 525.8 ± 1 223.2 | 0.006 |
| Gastric PGE2 (ng mg−1 protein) | 288.3 ± 258.5 | 660.9 ± 551.2 | 0.004 |
| Post-therapy data | |||
| Gastric pH | 3.8 ± 2.1 | 3.0 ± 1.6 | 0.273 |
| Serum gastrin (pg ml−1) | 69.6 ± 36.0 | 89.9 ± 59.9 | 0.284 |
| Serum pepsinogen I (ng ml−1) | 113.8 ± 32.4 | 121.0 ± 52.8 | 0.662 |
| Serum TXB2 (pg ml−1) (COX-1 activity) | 18 169 ± 34 073 | 34 644 ± 41 877 | 0.178 |
| Serum PGE2 (pg ml−1) (COX-2 activity) | 356.6 ± 201.4 | 420.7 ± 209.8 | 0.310 |
| Gastric TXB2 (ng mg−1 protein) | 506.2 ± 591.8 | 1 670.0 ± 1 329.6 | <0.001 |
| Gastric PGE2 (ng mg−1 protein) | 253.5 ± 281.8 | 766.7 ± 574.2 | 0.001 |
SLE, systemic lupus erythematous; TXB2, thromboxane B2; COX, cyclooxygenase; PGE2, prostaglandin E2. Data are expressed as means ± SD.
Data comparison between patients taking and not taking NSAIDs/aspirin
Laboratory findings before therapy were also compared between the 17 patients who took NSAIDs/aspirin and the 50 patients who did not. Patients who took NSAIDs/aspirin had significantly lower serum gastrin (70.0 ± 24.7 vs. 90.6 ± 53.9 pg ml−1, P= 0.039), lower serum TXB2 (COX-1 activity) (9002 ± 10 000 vs. 38 668 ± 44 514 pg ml−1, P= 0.032), lower serum PGE2 (COX-2 activity) (356.3 ± 228.0 vs. 468.5 ± 193.1 pg ml−1, P= 0.020), lower gastric TXB2 (540.1 ± 380.7 vs. 1665.5 ± 1239.6 ng mg−1 protein, P < 0.001) and lower gastric PGE2 (247.3 ± 136.8 vs. 719.6 ± 562.8 ng mg−1 protein, P < 0.001). However, the two groups did not significantly differ in H. pylori infection rate, serum albumin, serum creatinine, haemoglobin, platelet count, gastric pH, or serum pepsinogen I.
Risk factor analysis in patients with gastric mucosal injury
Univariate logistic regressions analysis showed that NSAID/aspirin usage, past history of PUD, low gastric TXB2 (<1000 ng mg−1 protein) and low gastric PGE2 (<400 ng mg−1 protein) were the risk factors for gastric mucosal injury in SLE patients receiving pulse methylprednisolone therapy. Multivariate logistic regression analysis showed NSAID/aspirin usage was the only risk factor for gastric mucosal damage in the 67 SLE patients who received pulse therapy (odds ratio 26.99, 95% confidence interval 4.91, 148.57, P < 0.0001).
Data comparison in 67 SLE patients before and after pulse therapy
Comparison of pre-therapy and post-therapy parameters in these 67 subjects revealed no significant difference in gastric pH (3.1 ± 1.5 vs. 3.1 ± 1.7, P= 0.858), serum gastrin (85.4 ± 48.8 vs. 86.6 ± 57.0 pg ml−1, P= 0.778), pepsinogen I (117.1 ± 47.7 vs. 119.8 ± 49.9 ng ml−1, P= 0.559), serum TXB2 (COX-1 activity) (31 141 ± 40 798 vs. 31 939 ± 41 013 pg ml−1, P= 0.644), gastric TXB2 (1380.0 ± 1191.4 vs. 1478.9 ± 1309.6 ng mg−1 protein, P= 0.930) or gastric PGE2 (599.8 ± 531.6 vs. 682.5 ± 568.8 ng mg−1 protein, P= 0.060). However, there was a trend to increased gastric injury scale after pulse therapy (0.67 ± 1.05 vs. 0.82 ± 1.15, P= 0.058), and serum PGE2 (COX-2 activity) (440.0± 206.6 vs. 410.2 ± 208.3 pg ml−1, P= 0.043) dropped significantly after pulse therapy.
Very high-dose steroids de novo in 50 SLE patients
To evaluate the effect of very high-dose steroids alone on gastric mucosal damage, 50 patients who had not taken NSAIDs/aspirin were further analysed. The mean cumulative dose of methylprednisolone was 4100 ± 2000 mg per person (range 2000–12 000 mg). The mean course of pulse therapy was 2.5± 1.0 sessions (range two to six sessions per person). Mean age was 30 ± 8 years (range 19–50 years) in the 50 patients. Among them, four (8%) were male, 46 (92%) were female, two (4%) were smokers, one (2%) regularly consumed alcohol, 17 (34%) had H. pylori infection and none had a history of PUD. No significant differences in mucosal injury scale (oesophagus, stomach and duodenum) were observed between pre- and post therapy (Table 4). Although one (2%) patient developed gastric mucosal injury after pulse therapy with initial scale of 2 to final scale of 3, three patients had a decreased gastric injury scale after pulse therapy. No significant differences were noted in gastric pH, serum gastrin, pepsinogen I, serum TXB2 (COX-1 activity), gastric TXB4 or PGE2 between pre- and post therapy except that serum PGE2 (COX-2 activity) significantly dropped after pulse therapy (Table 4).
Table 4.
Comparison of pre-therapy and post-therapy laboratory data in 50 SLE patients receiving pulse therapy of methylprednisolone without taking NSAIDs/aspirin
| Pre-therapy | Post therapy | P-value | |
|---|---|---|---|
| Gastric injury scale median (range) | 0.70 ± 1.15 | 0.64 ± 1.16 | 0.180 |
| 0 (0–4) | 0 (0–4) | ||
| Gastric pH | 3.2 ± 1.4 | 3.0 ± 1.5 | 0.460 |
| Serum gastrin (pg ml−1) | 90.6 ± 53.9 | 91.4 ± 63.6 | 0.893 |
| Serum pepsinogen I (ng ml−1) | 120.5 ± 53.2 | 119.5 ± 52.7 | 0.864 |
| Serum TXB2 (pg ml−1) (COX-1 activity) | 38 668 ± 44 514 | 40 060 ± 44 619 | 0.532 |
| Serum PGE2 (pg ml−1) (COX-2 activity) | 468.5 ± 193.0 | 388.2 ± 143.1 | 0.001 |
| Gastric TXB2 (ng mg−1 protein) | 1 665.5 ± 1 239.6 | 1 821.1 ± 1 343.4 | 0.732 |
| Gastric PGE2 (ng mg−1 protein) | 719.6 ± 562.8 | 828.3 ± 582.7 | 0.171 |
SLE, systemic lupus erythematous; NSAIDs, nonsteroidal anti-inflammatory drugs; TXB2, thromboxane B2; COX, cyclooxygenase; PGE2, prostaglandin E2. Data are expressed as means ± SD.
Discussion
This study has demonstrated that NSAID/aspirin use, but not H. pylori infection, increased the incidence of gastric mucosal injury in SLE patients who had received pulse methylprednisolone therapy; very high-dose steroid therapy de novo did not induce gastric mucosal injury in these patients. The study appears to be the first prospective study revealing that very high-dose steroids alone do not induce mucosa injury of the upper gastrointestinal tract in humans, at least in a young SLE population group.
Aspirin, which effectively inhibits COX-1 function, is a NSAID and causes gastric erosions and ulcers. This study has also shown that SLE patients who had received pulse methylprednisolone therapy and developed gastric mucosal damage had a significantly higher rate of NSAID/aspirin use, lower gastric TXB2 and lower gastric PGE2 than those without gastric mucosa damage. This study also showed that patients who took NSAIDs/aspirin had significantly lower serum TXB2, lower serum PGE2, lower gastric TXB2, and lower gastric PGE2, which is consistent with other studies [18, 22]. Taken together, these observations suggest that lower gastric TXB2 and lower gastric PGE2 in these SLE patients with gastric mucosal injury may be due to NSAID/aspirin effects rather than the methylprednisolone effect. This suggestion was supported by logistic regression analysis showing that NSAID/aspirin usage was the only risk factor for gastric mucosal injury in those SLE patients who had received pulse therapy. It is a limitation of this study that we did not divide nonselective NSAIDs, COX-2 inhibitors and aspirin as independent risk factors for further analysis for gastric mucosal injury due to the limited number of cases taking NSAIDs/aspirin, although they may exhibit some differences in inhibiting TXB2 and PGE2 formation.
Pulse methylprednisolone therapy was administered for urgent critical conditions such as proliferative lupus nephritis, acute rejection after organ transplantation, and multiple sclerosis [14]. Methylprednisolone is an intermediate-acting steroid with a serum half-life of approximately 1–4 h and a tissue biological half-life of 24 h or more [13, 14, 23]. In our investigation we found that 50 SLE patients have significantly reduced serum PGE2 (COX-2 activity) after methylprednisolone therapy alone, but that no mucosal injury developed. Furthermore, serum TXB2 (COX-1 activity) as well as gastric TXB2 and PGE2 did not significantly differ between the pre-therapy and post-therapy condition of the 50 SLE patients who received blood and tissue sampling within 24 h after the last dose of pulse therapy. These findings support that pulse methylprednisolone therapy does not inhibit COX-1 function of serum and gastric mucosa and does not cause gastric mucosal injury, although it decreases serum COX-2 activity, which is consistent with previous findings that inhibition of both COX-1 and COX-2 function is required for rat gastric mucosa injury [10]. Although the intra- and interassay variability of ELISA for PGE2 and TXB2 is reportedly high [18, 22], Wilcoxon signed rank test was used in this inquiry to compare individual eicosonoids in the same patients before and after pulse therapy. In addition, all the eicosonoids were prepared and measured simultaneously to decrease intra- and interassay variability.
Ulcer formation is an imbalance between aggressive factors and defensive mucosal factors. When the function of defence and repairing factors is less than that of aggressive factors, mucosal injury worsens, and finally ulcer formation develops. A study by Weusten has demonstrated that seven out of 50 (14%) refractory rheumatoid arthritis patients developed endoscopically proven peptic ulcer during an average 3.8-year follow-up and after receiving an average of 1.7 courses of pulse methylprednisolone therapy [24]. However, in this retrospective study, some cases had received NSAID therapy and no pre-therapy scopy procedures were evaluated. As we know, combined use of NSAIDs and steroids increase PUD risk [1, 25], and previous animal studies also have demonstrated that a non-ulcerogenic dose of steroid delayed gastric ulcer healing [7, 8].
Infection with H. pylori is a risk factor for PUD in aspirin users and naive NSAID users [26]. However, no currently available data indicate whether H pylori infection is a risk factor for PUD in steroid users other than that in our previous study showing H. pylori infection is not a risk factor for PUD in autoimmune disease patients receiving low-dose oral steroid therapy [1]. The current study found that H. pylori infection is also not a risk factor for gastric mucosal damage in SLE patients receiving pulse methylprednisolone therapy.
The uncontrolled nature of this study dose not allow the conclusion that a very high dose is safe for gastric mucosa of humans due to numerous limitations in our study design. Limited case number (only 50 subjects after excluding confounding by NSAID/aspirin use) without the control group are the major limitation of this study. In addition, these 50 SLE patients were relatively low-risk patients, since they were young, had not been previously diagnosed with PUD and had no PUD at initial screening for this study. Furthermore, gastric adaptation has been reported in animal and human studies. Adaptation is the process by which visible gastric mucosal injury is improved despite continued administration of an injurious substance such as aspirin or NSAIDs [27]. All SLE patients in this study were treated with various doses of oral prednisolone daily before pulse methylprednisolone therapy. Whether steroid usage causes a gastric adaptation phenomenon similar to that of NSAIDs requires further investigation. It is too early to say that very high-dose steroid therapy is safe for gastric mucosa until we design and complete a case-controlled study enrolling a larger population of healthy subjects, elderly patients, high-risk patients receiving pulse therapy and enrolling a control group to clarify the issue.
In conclusion, NSAID/aspirin use, but not H. pylori infection, increases gastric mucosal injury in SLE patients receiving pulse methylprednisolone therapy. Very high-dose steroid therapy de novo seems not to induce gastric mucosal injury in these low-risk SLE patients. A larger case-controlled study enrolling a heterogeneous population is needed to clarify the role of glucocorticoids in gastric mucosal injury.
Competing interests
None to declare.
This work was supported by a grant from Taipei Veteran General Hospital (V96C1-017) and National Health Research Institutes of Taiwan (90P001).
REFERENCES
- 1.Luo JC, Chang FY, Lin HY, Lu RH, Lu CL, Chen CY, Lu RH, Lee SD. The potential risk factors leading to peptic ulcer formation in autoimmune disease patients receiving corticosteroid treatment. Aliment Pharmacol Ther. 2002;16:1241–8. doi: 10.1046/j.1365-2036.2002.01279.x. [DOI] [PubMed] [Google Scholar]
- 2.Conn HO, Blitzer BL. Nonassociation of adrenocorticosteroid therapy and peptic ulcer. N Engl J Med. 1976;294:473–9. doi: 10.1056/NEJM197602262940905. [DOI] [PubMed] [Google Scholar]
- 3.Messer J, Reitman D, Sacks HS, Smith H, Jr, Chalmers TC. Association of adrenocorticosteroid therapy and peptic-ulcer disease. N Engl J Med. 1983;309:21–4. doi: 10.1056/NEJM198307073090105. [DOI] [PubMed] [Google Scholar]
- 4.Hernandez-Diaz S, Rodriguez LAG. Steroid and risk of upper gastrointestinal complications. Am J Epidemiol. 2001;153:1089–93. doi: 10.1093/aje/153.11.1089. [DOI] [PubMed] [Google Scholar]
- 5.Bandyopadhyay U, Biswas K, Bandyopadhyay D, Ganguly CK, Banerjee RK. Dexamethasone makes the gastric mucosa susceptible to ulceration by inhibiting prostaglandin synthetase and peroxidase – two important gastroprotective enzymes. Mol Cell Biochem. 1999;202:31–6. doi: 10.1023/a:1007018212822. [DOI] [PubMed] [Google Scholar]
- 6.Okazaki K, Chiba T, Hajiro K. Downregulation of gastric mucin gene expression and its biosynthesis by dexamethasone in the human. J Clin Gastroenterol. 1998;27(Suppl. 1):S91–6. doi: 10.1097/00004836-199800001-00015. [DOI] [PubMed] [Google Scholar]
- 7.Luo JC, Shin VY, Liu ESL, So WHL, Ye YN, Chang FY, Cho CH. Non-ulcerogenic dose of dexamethasone delays gastric ulcer healing in rats. J Pharmacol Exp Ther. 2003;307:692–8. doi: 10.1124/jpet.103.055202. [DOI] [PubMed] [Google Scholar]
- 8.Luo JC, Shin VY, Liu ESL, Ye YN, Wu WKK, So WHL, Chang FY, Cho CH. Dexamethasone delays ulcer healing by inhibition of angiogenesis in rat stomachs. Eur J Pharmacol. 2004;485:275–81. doi: 10.1016/j.ejphar.2003.11.038. [DOI] [PubMed] [Google Scholar]
- 9.Hawkey CJ. Nonsteroidal anti-inflammatory drugs gastropathy. Gastroenterology. 2000;119:521–35. doi: 10.1053/gast.2000.9561. [DOI] [PubMed] [Google Scholar]
- 10.Wallace JL, McKnight W, Reuter BK, Vergnolle N. NSAID-induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology. 2000;119:706–14. doi: 10.1053/gast.2000.16510. [DOI] [PubMed] [Google Scholar]
- 11.Schmassmann A. Mechanisms of ulcer healing and effects of nonsteroidal anti-inflammatory drugs. Am J Med. 1998;104:43S–53S. doi: 10.1016/s0002-9343(97)00211-8. [DOI] [PubMed] [Google Scholar]
- 12.Gretzer B, Maricic N, Respondek M, Schuligoi R, Peskar BM. Effects of specific inhibition of cyclooxygenase-1 and cyclooxygenase-2 in the rat stomach with normal mucosa and after acid challenge. Br J Pharmacol. 2001;132:1565–73. doi: 10.1038/sj.bjp.0703955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Badsha H, Edwards CJ. Intravenous pulse of methylprednisolone for systemic lupus erythematous. Semin Arthritis Rheum. 2003;32:370–7. doi: 10.1053/sarh.2002.50003. [DOI] [PubMed] [Google Scholar]
- 14.Czock D, Keller F, Rasche FM, Haussler U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin Pharmacokinet. 2005;44:61–98. doi: 10.2165/00003088-200544010-00003. [DOI] [PubMed] [Google Scholar]
- 15.Hereng T, Lambert M, Hachulla E, Samor M, Dubucquoi S, Caron C, Launay D, Morell-Dubois S, Queyrel V, Hatron PY. Influence of aspirin on the clinical outcome of 103 anti-phospholipid antibodies-positive patients. Lupus. 2008;17:11–5. doi: 10.1177/0961203307084724. [DOI] [PubMed] [Google Scholar]
- 16.Tektonidou MG, Laskari K, Panagiotakos DB, Moutsopoulos HM. Risk factors for thrombosis and primary thrombosis prevention in patients with systemic lupus erythematous with or without antiphospholipid antibodies. Arthritis Rheum. 2009;61:29–36. doi: 10.1002/art.24232. [DOI] [PubMed] [Google Scholar]
- 17.Hochberg MC. Updating the American College of Rheumatology Revised Criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 18.Cryer B, Feldman M. Effects of very low dose daily, long-term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury in healthy humans. Gastroenterology. 1999;117:17–25. doi: 10.1016/s0016-5085(99)70545-7. [DOI] [PubMed] [Google Scholar]
- 19.Lanza F, Royer GL, Nelson RS, Chen TT, Seckman CE, Rack MF. A comparative endoscopic evaluation of the damaging effects of nonsteroidal anti-inflammatory agents on the gastric and duodenal mucosa. Am J Gastroenterol. 1981;75:17–21. [PubMed] [Google Scholar]
- 20.Wight NJ, Gottesdiener K, Garlick NM, Atherton CT, Novak S, Gertz BJ, Calder NA, Cote J, Wong P, Dallob A, Hawkey CJ. Rofecoxib, a COX-2 inhibitor, does not inhibit human gastric mucosal prostaglandin production. Gastroenterology. 2001;120:867–73. doi: 10.1053/gast.2001.22432. [DOI] [PubMed] [Google Scholar]
- 21.Wallace JL. Glucocorticoid-induced gastric mucosal damage: inhibition of leukotriene, but not prostaglandin biosynthesis. Prostaglandins. 1987;34:311–23. doi: 10.1016/0090-6980(87)90252-8. [DOI] [PubMed] [Google Scholar]
- 22.Treiber G, Wex T, Link A, Vieth M, Laufer S, Malfertheiner P. The effect of single-dose naproxen on eicosanoid formation in human gastroduodenal mucosa. Aliment Pharmacol Ther. 2006;23:155–67. doi: 10.1111/j.1365-2036.2006.02725.x. [DOI] [PubMed] [Google Scholar]
- 23.Tornatore KM, Reed KA, Venuto RC. Repeated assessment of methylprednisolone pharmacokinetics during chronic immunosuppression in renal transplant recipients. Ann Pharmacol. 1995;29:120–4. doi: 10.1177/106002809502900202. [DOI] [PubMed] [Google Scholar]
- 24.Weusten BLMA, Jacobs JWG, Bijlsma JWJ. Corticosteroid pulse therapy in active rheumatoid arthritis. Semin Arthritis Rheum. 1993;23:183–92. doi: 10.1016/s0049-0172(05)80039-3. [DOI] [PubMed] [Google Scholar]
- 25.Piper JM, Ray WA, Daugherty JR, Griffin MR. Corticoid use and peptic ulcer disease: role of nonsteroidal anti-inflammatory drugs. Ann Intern Med. 1991;114:735–40. doi: 10.7326/0003-4819-114-9-735. [DOI] [PubMed] [Google Scholar]
- 26.Huang JQ, Sridhar S, Hunt RH. Role of Helicobacter pylori and non-steroidal anti-inflammatory drugs in peptic ulcer disease: a meta-analysis. Lancet. 2002;359:14–22. doi: 10.1016/S0140-6736(02)07273-2. [DOI] [PubMed] [Google Scholar]
- 27.Graham DY, Smith JL, Spjut HJ, Torres E. Gastric adaptation. Studies in humans during continuous aspirin administration. Gastroenterology. 1988;85:327–33. [PubMed] [Google Scholar]