

Abstract
We analyzed MET protein and copy number in NSCLC with or without EGFR mutations untreated with EGFR tyrosine kinase inhibitors (TKIs). MET copy number was examined in 28 NSCLC and 4 human bronchial epithelial cell lines (HBEC) and 100 primary tumors using quantitative real-time PCR. Positive results were confirmed by array comparative genomic hybridization and fluorescence in-situ hybridization. Total and phospho-MET protein expression was determined in 24 NSCLC and 2 HBEC cell lines using Western blot. EGFR mutations were examined for exon 19 deletions, T790M, and L858R. Knockdown of EGFR with siRNA was performed to examine the relation between EGFR and MET activation. High-level MET amplification was observed in 3 of 28 NSCLC cell lines and in 2 of 100 primary lung tumors that had not been treated with EGFR-TKIs. MET protein was highly expressed and phosphorylated in all the 3 cell lines with high MET amplification. In contrast, 6 NSCLC cell lines showed phospho-MET among 21 NSCLC cell lines without MET amplification (p = 0.042). Furthermore, those 6 cell lines harboring phospho-MET expression without MET amplification were all EGFR mutant (p = 0.0039). siRNA-mediated knockdown of EGFR abolished phospho-MET expression in examined 3 EGFR mutant cell lines of which MET gene copy number was not amplified. By contrast, phospho-MET expression in 2 cell lines with amplified MET gene was not down-regulated by knockdown of EGFR. Our results indicated that MET amplification was present in untreated NSCLC and EGFR mutation or MET amplification activated MET protein in NSCLC.
Keywords: MET, amplification, EGFR, gefitinib, lung cancer
Acquired somatic alterations within the cancer genome such as mutations, gene amplification and deletions cause activation of oncogenes or inactivation of tumor suppressor genes, and form the genetic basis of malignancies.1 Cancer-specific somatic mutations have been identified in several protein kinases,2–4 including mutations within the epidermal growth factor receptor (EGFR) gene in non-small cell lung cancer (NSCLC).5–7
The finding of mutations in the tyrosine kinase domain of EGFR in lung adenocarcinoma is of great clinical interest, because many of these tumors are responsive to tyrosine kinase inhibitors (TKIs).5,6,8 Although most EGFR mutant NSCLC initially respond to TKI, the vast majority of these tumors ultimately become resistant to the drug treatment. In approximately half of these cases, resistance is due to the occurrence of a second point mutation in EGFR exon 20 (T790M).9–12 Recently Engelman et al.13 reported that MET proto-oncogene (MET) amplification led to gefitinib resistance in lung cancers lacking the secondary T790M mutation. Bean et al.14 also reported that MET was amplified in lung tumors with acquired resistance more frequently than in untreated lung tumors and accounted for about 20% of cases of acquired resistance to TKIs. MET encodes a heterodimeric transmembrane receptor tyrosine kinase for the hepatocyte growth factor.15–17 Deregulation of MET signaling has been shown to contribute to tumorigenesis in various cancers via activating mutations (e.g. papillary renal carcinomas)18 or via high-level MET amplification (e.g. gastric cancers).19 Although MET amplification in NSCLC may mainly occur after TKI-induced acquired resistance, its status in previously untreated NSCLC has received scant attention. Besides, MET protein status should also be evaluated to understand the functional effect of MET amplification. Furthermore, it is of interest to explore the relation between EGFR alteration and MET protein status because recent reports indicated that mutated or amplified EGFR can drive MET activity.20
In the current study, we investigated the status of MET copy number by quantitative real-time PCR in cell lines and primary lung cancers not previously treated with EGFR-TKIs. We also studied expression of total and phosphorylated MET protein (phospho-MET) in NSCLC cell lines by Western blot and investigated the relation among MET protein expression, MET copy number and EGFR mutational status. Furthermore, we examined the relation between TKI-sensitivity and MET status in NSCLC cell lines. Finally, we performed siRNA-mediated knockdown of EGFR using EGFR mutant or MET amplified NSCLC cell lines to see if EGFR influenced MET protein status.
Material and methods
Cell lines
Most of the human lung cancer cell lines examined in this study were established by the authors (A.F.G and J.D.M)21 at one of 2 locations. The prefix NCI-H- (abbreviated as H-) indicates cell lines established at the National Cancer Institute-Navy Medical Oncology Branch, National Naval Medical Center, Bethesda, MD and the prefix HCC- indicates lines established at the Hamon Center for Therapeutic Oncology Research, the University of Texas Southwestern Medical Center at Dallas, Dallas, TX. A549 was obtained from American Type Culture Collection (Manassas, VA). NCI-H3255 was obtained from Dr. Bruce Johnson (Lowe Center for Thoracic Oncology, Dana-Farber Cancer Institute, Boston, MA).6 PC-9 was obtained from Immuno-Biological Laboratories (Takasaki, Gunma, Japan). All the cancer cell lines except for NCI-H3255 were maintained in RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented with 5 or 10% fetal bovine serum (FBS). NCI-H3255 was maintained in ACL-4.22,23 EGFR mutational status in these cell lines above was available.3 For control non-malignant cell lines, we utilized 4 human bronchial epithelial cell lines (HBECs, HBEC3KT, HBEC5KT, HBEC17KT and HBEC30KT), which were initiated by the authors (J.D.M and A.F.G).24,25 The HBEC cell lines were maintained in Keratinocyte-SFM medium (Invitrogen) with bovine pituitary extract (BPE) and human recombinant epidermal growth factor (EGF). All cell lines were incubated at 37°C in a humidified atmosphere with 5% CO2.
Western blot analysis
Preparation of total cell lysates and Western blot were done as described previously.25 Primary antibodies used were mouse monoclonal anti-Met (25H2, Cell Signaling, Beverly, MA), rabbit monoclonal anti-phospho-Met (3D7, Tyr1234/1235; Cell Signaling), rabbit polyclonal anti-EGFR (Cell Signaling) and mouse monoclonal anti-actin (Sigma-Aldrich, St. Louis, MO) antibodies. Actin levels were used as a control for protein loading. Peroxidase-labeled anti-rabbit or anti-mouse antibodies (Amersham Pharmacia, Piscataway, NJ) were used as the second antibody.
Tumor samples
We analyzed 100 serially collected primary Japanese lung cancers from patients who underwent surgery in Okayama University Hospital (Okayama, Japan) from 2005 to 2007. Resected tumors were frozen at −80°C until DNA was extracted. Corresponding non-malignant peripheral lung tissue was also available. Genomic DNA was obtained from frozen primary lung tumors, corresponding non-malignant peripheral lung tissue and cell lines by standard phenol-chloroform (1:1) extraction followed by ethanol precipitation or by using DNeasy Tissue Kit (Qiagen, Valencia, CA). Institutional Review Board permission and informed consent were obtained at Okayama University Hospital. Clinico-pathological characteristics including age, sex, smoking status (pack-years of exposure), histologic subtype and clinical stage were available.
Quantitative real-time PCR for MET copy number evaluation
Quantitative real-time PCR (qPCR) was performed using Applied Biosystems 7300 Real-Time PCR System (Applied Bio-systems, Foster City, CA). We evaluated MET copy number in each sample by comparing the MET locus to the reference Line-1, a repetitive element for which copy numbers per haploid genome are similar among all of the human normal and neoplastic cells.26,27 We also chose Type III collagen gene (COL8A1, 3q12-q13.1) as another reference gene so as to confirm reliability of this qPCR assay. Primer sequences for COL8A1 were reported previously.28 MET gene copy number in Human Genomic DNA (EMD Biosciences, Darmstadt, Germany) was set as 2 and copy numbers greater than 5 were considered as high-level amplification. Further information including primer sequences for MET and the formula calculating MET copy number is shown in Supporting Data.
Tiling path array comparative genomic hybridization (CGH)
Array hybridization was performed as previously described.29 Detailed information is shown in Supporting Data.
Fluorescence in-situ hybridization (FISH)
To confirm the results of MET copy number obtained by qPCR, FISH was performed using cell lines NCI-H820 and NCI-H3255. Detailed method of FISH is shown in Supporting Data.
EGFR mutational analyses
EGFR mutations in exons 19 (deletions) or 21 (L858R point mutation) were detected by mutant-enriched PCR as described previously.30 The common deletions of exon 19 were distinguished from wild type based on PCR product length polymorphisms using 12% polyacrylamide gel electrophoresis (PAGE). For the point mutation in exon 21 (L858R), Sau96I digestion, which specifically digests the mutant type, was done prior to PAGE analysis.
Secondary EGFR T790M point mutations in exon 20 were detected by direct sequencing as described previously.31 Briefly, PCR products were incubated using ExoSAP-IT (Amersham Biosciences, Piscataway, NJ) and sequenced directly using Applied Biosystems PRISM dye terminator cycle sequencing method (Applied Biosystems). All sequence variants were confirmed by independent PCR amplifications and bidirectional sequencing.
Determining TKI-sensitivity in lung cancer cell lines by MTS assay
The MTS colorimetric assay (Promega, Madison, WI) was performed as per the manufacturer’s instructions. This assay is based on the conversion of MTS into soluble formazan by endogenous dehydrogenase enzymes found in metabolically active cells. Cells were plated at 1 × 103 – 4 × 103 cells/well in tissue culture-treated 96-well plates. The following day, TKI (gefitinib) was added to each plate in a dilution series across the plate such that 8 different concentrations of the drug were tested. On day 5, 20 μl of MTS was added, followed by 1-hr incubation at 37°C and then the absorbance was measured at 490 nm on a plate reader.
96-well plate data were imported into an in-house Database of In VItro drug Sensitivity Assays (DIVISA by Luc Girard, manuscript in preparation) where IC50s are calculated as well as various statistical analyses. To calculate IC50 values, the data was background-subtracted (columns 1 and 12 typically contained media with no cells or drugs and served as background values), and fitted to the DRC model (R package ‘drc’ by Christian Ritz and Jens Streibig, http://www.bioassay.dk) to generate a sigmoidal curve from which the concentration that inhibits 50% of the cells (IC50) was determined.
siRNA-mediated knockdown of EGFR
siRNAs targeting EGFR were designed through the RNAi Co. Ltd website (http://www.rnai.co.jp/e_index.html) based on siDirect online software system,32 which can efficiently select siRNA sequences avoiding off-target effects and were chemically synthesized by RNAi Co., Ltd. (Tokyo, Japan). siRNA sequences targeting 2 different regions of EGFR are as follows (sense and anti-sense, respectively): siEGFR-1 (5′-GCU ACG AAU AUU AAA CAC UUC-3′ and 5′-AGU GUU UAA UAU UCG UAG CAU-3′) and siEGFR-2 (5′-CCG CAA AUU CCG AGA CGA AGC-3′ and 5′-UUC GUC UCG GAA UUU GCG GCA-3′). Cells were transfected with 10 nM of siRNA using Lipofectamine™ RNAiMAX (Invitrogen) according to the manufacturer’s protocol. Control cells were treated with RNAiMAX alone or with scrambled negative control siRNA sequence (RNAi). Cells were grown and harvested 72 hours after the transfection for Western blot analysis.
Statistical analyses
The differences of significance among categorized groups were compared using Fisher’s exact tests. All statistical analyses in this study were performed using Prism 4 software (GraphPad Software, San Diego, CA).
Results
MET gene status in NSCLC cell lines
We investigated 28 NSCLC and 4 HBEC cell lines for MET gene copy number. Ten NSCLC cell lines with EGFR activating mutations were available (all adenocarcinomas from patients untreated with TKIs). Two of these also had the T790M mutation in exon 20 of EGFR (Table I). NCI-H1650 is known to have a homozygous deletion of the PTEN gene and absence of its protein.20,28 MET copy numbers of the 4 HBEC cell lines were near diploid (Fig. 1). Cell lines NCI-H1993 and A549 were used as positive and negative controls for MET copy number evaluation, respectively.13 We also obtained similar results of MET copy numbers in cell lines and tumors using Type III collagen gene (COL8A1, 3q12-q13.1) as another reference gene (representative results are shown in Supporting Table I).
TABLE I.
MET COPY NUMBER STATUS IN EGFR MUTANT OR WILD TYPE NSCLC CELL LINES
| Cell line | Histology | EGFR activating mutation | T790M mutation | MET copy number by qPCR1 | p-MET expression by Western blot2 | IC50s of gefitinib (μM) |
|---|---|---|---|---|---|---|
| NCI-H820 | AD | E746-E749 del | Yes | 6.1 | Yes | 3.0 |
| NCI-H1650 | AD | E746-A750 del | No | 1.9 | No | 11.7 |
| NCI-H1975 | AD | L858R | Yes | 2.7 | Yes | 25 |
| NCI-H3255 | AD | L858R | No | 4.5 | Yes | 0.089 |
| HCC827 | AD | E746-A750 del | No | 2.2 | Yes | 0.04 |
| HCC2279 | AD | E746-A750 del | No | 1.5 | No | 0.048 |
| HCC2935 | AD | E746-S752 del, I ins | No | 3.3 | No | 0.11 |
| HCC4006 | AD | L747-A750 del, P ins | No | 1.3 | Yes | 0.23 |
| HCC4011 | AD | L858R | No | 3.1 | Yes | 0.6 |
| PC-9 | AD | E746-A750 del | No | 2.8 | Yes | 0.031 |
| A549 | AD | WT | No | 2.3 | No | Not done |
| NCI-H226 | SQ | WT | No | 1.5 | Not done | Not done |
| NCI-H522 | AD | WT | No | 3.2 | Not done | Not done |
| NCI-H1299 | LC | WT | No | 2.6 | Not done | 26.4 |
| NCI-H1648 | AD | WT | No | 8.5 | Yes | 36.7 |
| NCI-H1703 | SQ | WT | No | 1.5 | No | Not done |
| NCI-H1819 | AD | WT | No | 3 | No | 19 |
| NCI-H1993 | AD | WT | No | 15.5 | Yes | 17.9 |
| NCI-H2073 | AD | WT | No | 3.8 | No | 0.031 |
| NCI-H2126 | LC | WT | No | 4.7 | No | 21.4 |
| NCI-H2170 | SQ | WT | No | 3.3 | No | 3.2 |
| NCI-H2347 | AD | WT | No | 2.2 | Not done | 60 |
| HCC193 | AD | WT | No | 2.1 | No | 21.1 |
| HCC366 | ADSQ | WT | No | 1.7 | No | 30 |
| HCC1195 | AD | WT | No | 3.9 | No | 27.6 |
| HCC1719 | AD | WT | No | 2.4 | No | Not done |
| HCC2429 | NSCLC | WT | No | 1.9 | No | Not done |
| HCC3051 | LC | WT | No | 2 | No | Not done |
AD, adenocarcinoma; SQ, squamous cell carcinoma; LC, large cell carcinoma; ADSQ, adenosquamous cell carcinoma; NSCLC, non-small cell lung carcinoma; WT, wild type.
Copy numbers greater than 5 were considered as high-level amplification.
See also Figure 4.
Figure 1.
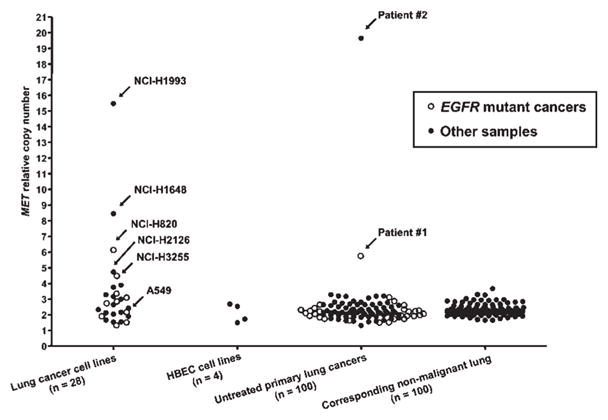
MET gene copy number by quantitative real-time PCR in lung cancer cell lines and untreated primary tumors. MET copy numbers were expressed as relative values compared to pooled human genomic DNA from multiple healthy donors. Twenty-eight lung cancer cell lines (10 EGFR mutant and 18 EGFR wild type cell lines) were analyzed for MET copy number. NCI-H1993 and A549 were used as positive and negative controls, respectively. For primary lung tumors, corresponding adjacent non-malignant tissues (n = 100) were also available. Copy numbers greater than 5 were considered as high-level amplification.
High-level MET amplification was observed in 1 cancer cell line with EGFR mutation and 2 cell lines without EGFR mutation (Fig. 1 and Table I). There was no difference in MET amplification status between EGFR mutant and wild type cell lines. Copy number status was confirmed using CGH in cell lines NCI-H1993 and A549, and one primary tumor (Fig. 2) and using FISH for NCI-H3255 and NCI-H820 (Fig. 3). In addition, we confirmed that samples with copy numbers ranging from 3 to 4 obtained by qPCR were not amplified by CGH (results are shown in Supporting Table II). Furthermore, mean copy number of corresponding normal lung tissue in 100 samples obtained by qPCR was 2.3 and maximal and minimum values were 3.7 and 1.7, respectively. Therefore, we chose copy number more than 5 as high-level amplification. The FISH analyses for NCI-H3255 revealed polysomy and NCI-H820 revealed clustered amplification in addition to polysomy. Summary of MET FISH results on 3 additional cell lines (HCC827, NCI-H1993, NCI-H2073) studied as well as NCI-H3255 and NCI-H820 are shown in Supporting Table III.
Figure 2.
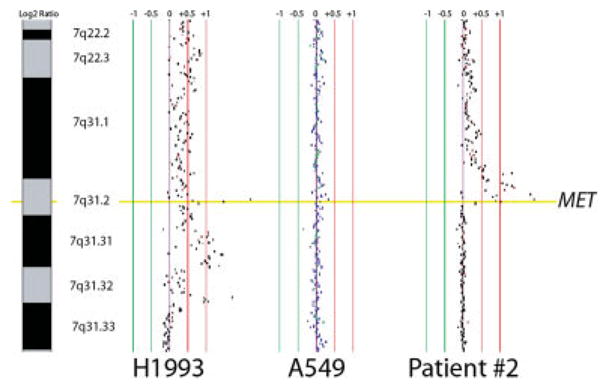
Array CGH. Alignment of representative array CGH profiles at the MET gene locus for lung cancer cell lines and a primary lung tumor. Normalized log 2 signal intensity ratios were plotted using SeeGH software. Vertical lines denote log 2 signal ratios from −1 to 1 with copy number increases to the right (red lines) and decreases to the left (green lines) of 0 (purple line). Each black dot represents a single BAC clone. The region containing MET is shaded yellow. One cell line (NCI-H1993) and one clinical tumor (Patient no. 2) displaying MET gain/amplification are depicted along with cell line A549, which has near diploid copy number for MET. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Figure 3.
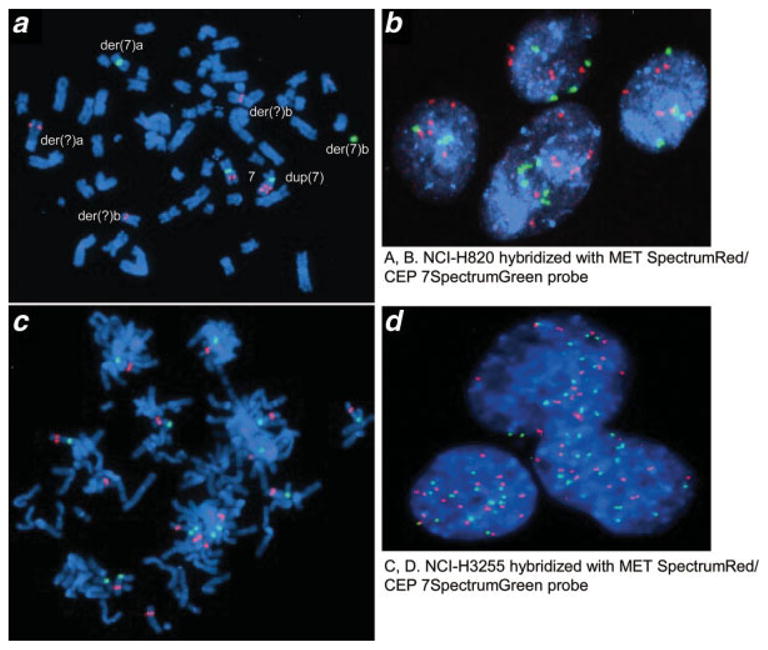
Fluorescence in-situ hybridization (FISH) with MET SpectrumRed/CEP7 Spectrum-Green probe set in EGFR mutant lung cancer cell lines NCI-H820 (a, b) and NCI-H3255 (c, d)., Both cell lines are shown to have high level of polysomy for chromosome 7, in which MET gene is located. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Protein expression status of MET and its association with MET copy number and EGFR mutation
We investigated MET protein expression status in EGFR mutant (n = 10) or wild type (n = 14) cell lines as well as HBEC cell lines (n = 2) and correlated the results with MET copy number and EGFR mutational status.
Total MET protein expression was present in all EGFR mutant cell lines and in 9 of 14 wild type NSCLC cell lines. MET phosphorylation sites at tyrosine residues (Tyr1234/1235) are located in the activation loop of the kinase domain and are indicative of kinase activation. As shown in Fig. 4, 7 (NCI-H1975, HCC4011, NCI-H3255, NCI-H820, PC-9, HCC4006, and HCC827) of 10 EGFR mutant and 2 (NCI-H1993 and NCI-H1648) of 14 wild type cell lines expressed phospho-MET. All the cell lines expressing phospho-MET also expressed total MET protein. All 3 cell lines (NCI-H820, NCI-H1993 and NCI-H1648) with increased MET copy number (1 mutant and 2 wild types), expressed phospho-MET. By contrast, of the 21 cell lines lacking MET increased copy number, 6 cell lines (NCI-H1975, HCC4011, NCI-H3255, PC-9, HCC4006 and HCC827) expressed phospho-MET, indicating that phospho-MET expression was significantly correlated with increased copy number (p = 0.042). Furthermore, those 6 cell lines (NCI-H1975, HCC4011, NCI-H3255, PC-9, HCC4006 and HCC827) expressing phospho-MET without MET amplification were all EGFR mutant, suggesting that EGFR mutation was also associated with phospho-MET expression (p = 0.0039). The HBEC cell lines had total MET but lacked phospho-MET expression.
Figure 4.
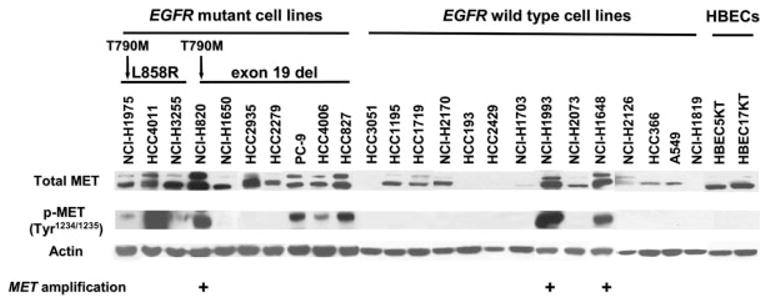
MET protein expression in EGFR mutant or wild type cell lines. Whole cell lysates were subjected to Western blot using the antibodies indicated in this column. MET phosphorylation sites at tyrosine residues (Tyr1234/1235) are located in the activation loop of the kinase domain and are indicative of kinase activation. All the 3 cell lines (NCI-H820, NCI-H1993 and NCI-H1648) with MET amplification had strong expression of phospho-MET (p-MET). Of interest, in 16 NSCLC cell lines with total-MET expression but no MET amplification, while 6 (NCI-H1975, HCC4011, NCI-H3255, PC-9, HCC4006 and HCC827) of 9 EGFR mutant cell lines showed phospho-MET, phosphorylation was not observed among 7 EGFR wild type cell lines.
The effect of EGFR protein for MET phosphorylation
We performed siRNA-mediated knockdown of EGFR to see if activated EGFR affected MET activity. As shown in Figure 5, MET phosphorylation was decreased or abolished after EGFR was knocked down in 3 EGFR mutant cell lines (HCC827, HCC4011 and HCC4006), which had relatively strong phospho-MET expression. By contrast, phospho-MET was retained after knockdown of EGFR in 2 MET amplified cell lines (NCI-H820 and NCI-H1993). NCI-H820 also had exon 19 deletions in EGFR gene.
Figure 5.
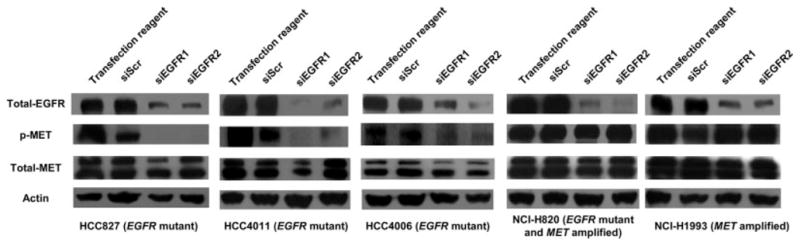
siRNA-mediated knockdown of EGFR. Knockdown of EGFR was performed using 2 different siRNAs (siEGFR1 and siEGFR2). Control cells were treated with transfection reagent alone or scrambled siRNA sequence (siScr). Knockdown of EGFR decreased or abolished phospho-MET expression in EGFR mutant cell lines (HCC827, HCC4011 and HCC4006) whereas phospho-MET expression was retained after knockdown of EGFR in MET amplified cell lines (NCI-H820 and NCI-H1993).
The relation between MET status and TKI sensitivity
We compared the MET status to TKI sensitivity in NSCLC cell lines (Table I). We analyzed the relation between phospho-MET expression and IC50 value of gefitinib in EGFR mutant cell lines without T790M or PTEN loss and in EGFR wild type cell lines. It seems that there is no relation between phospho-MET expression and IC50 value of gefitinib in both groups.
MET amplification in previously untreated lung cancers
Information about 100 primary lung cancers is presented in Table II. EGFR activating mutations were found out in 28 of 100 primary tumors and there were no T790M mutations in this sample set. MET amplification was found in 2 of 100 untreated lung tumors. Of interest, one of them also had a L858R mutation, and was refractory to gefitinib. Detailed information of 2 patients (patient nos. 1 and 2) with MET amplification is provided in Supporting Data.
TABLE II.
CHARACTERISTICS OF PRIMARY LUNG CANCERS (n = 100)
| Characteristics | Number |
|---|---|
| Age | |
| <66 | 48 |
| >=66 | 52 |
| Sex | |
| Male | 69 |
| Female | 31 |
| Smoking History | |
| Never | 32 |
| Ever | 68 |
| Histology | |
| Adenocarcinoma | 75 |
| Squamous cell carcinoma | 16 |
| Adenosquamous cell carcinoma | 1 |
| Large cell carcinoma | 6 |
| Small cell carcinoma | 2 |
| Clinical stage | |
| IA, IB | 74 |
| IIA, IIB | 12 |
| IIIA | 12 |
| IIIB | 2 |
| IV | 0 |
| EGFR activating mutations | |
| Exon 19 deletions | 15 |
| Exon 21 point mutation | 13 |
| Wild type | 72 |
| T790M mutation | 0 |
Discussion
We found that many NSCLC cell lines and bronchial epithelial cells expressed total MET protein. However, a more restricted subset of MET-expressing NSCLC cell lines also expressed the phosphorylated forms. Phospho-MET expression was significantly related to increased MET copy number and was significantly more frequent in EGFR mutant cell lines. As additional evidence for the relation between EGFR mutation and MET activation, recent reports suggested that other tyrosine kinases were also activated in cells with EGFR activation by EGFR mutations,20 supporting our results. Indeed, the results of siRNA experiments confirmed effect of activated EGFR to MET status. Three EGFR mutant cell lines harboring phospho-MET expression without MET amplification lost or decreased phospho-MET expression whereas MET amplified cell lines with or without EGFR mutations retained phospho-MET expression after knockdown of EGFR, indicating that EGFR mutation or MET amplification activate MET protein.
Regarding clinical samples, we found increased MET copy number in 2 of 100 previously untreated primary resected lung cancers. For clinical samples, contamination with non-malignant cells is invariably present (estimated average per tumor = 50% tumor cells, 50% non-malignant cells). Thus cut-off value of amplification may be reduced. One of these 2 MET amplified tumors also had an activating EGFR mutation (L858R), but lacked the resistance associated with T790M mutation. This patient had an advanced tumor and died within 20 days after the start of gefitinib treatment. While he would appear to have been refractory to gefitinib therapy, the short survival duration renders a firm conclusion difficult. We did not examine phospho-MET protein status in primary tumors because formalin fixed materials were unsuitable for such studies.33 We neither had enough frozen clinical samples for Western blot analysis. It is mandatory to confirm the relation among phospho-MET expression, MET copy number and EGFR mutations in primary tumors.
We studied Japanese patients and found the expected high frequency of EGFR mutations in NSCLC arising in patients of East Asian ethnicity.7 The frequency of MET amplification in untreated lung cancers was relatively low. These results were similar with the previous report.14 On the other hand, Beau-Faller et al. recently reported relatively higher frequency (21%) of MET amplification in 106 non-Asian NSCLC.34 Their definition of “amplification” is normalized ratio over mean plus 2 standard deviations of 30 normal lung DNA samples. Even if we use this definition for our sample set, that is, normalized ratio over mean plus 2 standard deviations of 100 normal lung DNA samples, we only have 8 samples with MET amplification out of 100 tumors. The discrepancy of frequency of MET amplification between them may be explained by ethnic difference.
We also examined the IC50 of gefitinib to investigate the impact of phospho-MET on NSCLC cell lines, but the effect of gefitinib did not depend on MET status but depended on EGFR mutation or PTEN status. Our results suggested that although biological significance of MET activation as a result of MET amplification by gefitinib exposure caused acquired resistance to gefitnib,13 MET activation was not solo factor for inherent resistance to gefitinib.
The clinical course of our 2 cases with MET amplification suggests that tumors with MET amplification are very aggressive in nature, that is, MET amplification may have the potential to be involved with development, progression or metastasis of lung cancer, consistent with the known relation of MET expression and tumor invasion and metastasis.35 Lutterbach et al. reported that lung cancer cell lines with MET amplification were dependent on MET for growth and survival.36 Although phospho-MET expression was not analyzed in our clinical samples because of technical issues, further investigation for phospho-MET expression status in primary tumors is important to understand biological significance of MET expression in lung cancer.
In conclusion, MET amplification is present in a subset of untreated lung cancers and cell lines with or without EGFR mutations, and EGFR mutation or MET amplification lead to activation of MET. Further studies clarifying biological significance of MET activation may shed light on the pathogenesis of lung cancer.
Supplementary Material
Acknowledgments
Ministry of Education, Culture, Sports, Science and Technology of Japan; Grant number: 18790993; Grant sponsor: the Specialized Program of Research Excellence in Lung Cancer; Grant numbers: P50CA70907, P01CA58187; Grant sponsor: Genome Canada/British Columbia and Canadian Institutes of Health Research.
This work was supported by the following grants: a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to S.T.); Genome Canada/British Columbia and Canadian Institutes of Health Research (to W.L.L.); the Specialized Program of Research Excellence in Lung Cancer (to A.F.G., J.D.M., and M.V.G.). The authors thank Ms. Fumiko Isobe (Department of Cancer and Thoracic Surgery, Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama University, Okayama, Japan) for her excellent technical support.
Abbreviations
- BAC
bacterial artificial chromosome
- CGH
comparative genomic hybridization
- EGFR
epidermal growth factor receptor
- MET
MET proto-oncogene
- NSCLC
non-small cell lung cancer
- PCR
polymerase chain reaction
- qPCR
quantitative real-time PCR
- SCLC
small cell lung cancer
- siRNA
small interfering RNA
- TKI
tyrosine kinase inhibitor
References
- 1.Weir B, Zhao X, Meyerson M. Somatic alterations in the human cancer genome. Cancer Cell. 2004;6:433–8. doi: 10.1016/j.ccr.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, Stevens C, O’Meara S, Smith R, Parker A, Barthorpe A, Blow M, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431:525–6. doi: 10.1038/431525b. [DOI] [PubMed] [Google Scholar]
- 3.Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, Lee H, Wistuba II, Fong KM, Toyooka S, Shimizu N, Fujisawa T, Minna JD, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005;65:1642–6. doi: 10.1158/0008-5472.CAN-04-4235. [DOI] [PubMed] [Google Scholar]
- 4.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 5.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 6.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 7.Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, Fujisawa T, Feng Z, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–46. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 8.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 10.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, Rabindran SK, McGinnis JP, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA. 2005;102:7665–70. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kosaka T, Yatabe Y, Endoh H, Yoshida K, Hida T, Tsuboi M, Tada H, Kuwano H, Mitsudomi T. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12:5764–9. doi: 10.1158/1078-0432.CCR-06-0714. [DOI] [PubMed] [Google Scholar]
- 13.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 14.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang WC, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA. 2007;104:20932–7. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park M, Dean M, Kaul K, Braun MJ, Gonda MA, Vande Woude G. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc Natl Acad Sci USA. 1987;84:6379–83. doi: 10.1073/pnas.84.18.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giordano S, Ponzetto C, Di Renzo MF, Cooper CS, Comoglio PM. Tyrosine kinase receptor indistinguishable from the c-met protein. Nature. 1989;339:155–6. doi: 10.1038/339155a0. [DOI] [PubMed] [Google Scholar]
- 17.Peruzzi B, Bottaro DP. Targeting the c-Met signaling pathway in cancer. Clin Cancer Res. 2006;12:3657–60. doi: 10.1158/1078-0432.CCR-06-0818. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, Scherer SW, Zhuang Z, Lubensky I, Dean M, Allikmets R, Chidambaram A, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16:68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 19.Smolen GA, Sordella R, Muir B, Mohapatra G, Barmettler A, Archibald H, Kim WJ, Okimoto RA, Bell DW, Sgroi DC, Christensen JG, Settleman J, et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc Natl Acad Sci USA. 2006;103:2316–21. doi: 10.1073/pnas.0508776103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, Rikova K, Possemato A, Nardone J, Innocenti G, Wetzel R, Wang Y, MacNeill J, et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci USA. 2008;105:692–7. doi: 10.1073/pnas.0707270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phelps RM, Johnson BE, Ihde DC, Gazdar AF, Carbone DP, McClintock PR, Linnoila RI, Matthews MJ, Bunn PA, Jr, Carney D, Minna JD, Mulshine JL. NCI-Navy Medical Oncology Branch cell line data base. J Cell Biochem Suppl. 1996;24:32–91. doi: 10.1002/jcb.240630505. [DOI] [PubMed] [Google Scholar]
- 22.Brower M, Carney DN, Oie HK, Gazdar AF, Minna JD. Growth of cell lines and clinical specimens of human non-small cell lung cancer in a serum-free defined medium. Cancer Res. 1986;46:798–806. [PubMed] [Google Scholar]
- 23.Gazdar AF, Oie HK. Re: growth of cell lines and clinical specimens of human non-small cell lung cancer in a serum-free defined medium. Cancer Res. 1986;46:6011–12. [PubMed] [Google Scholar]
- 24.Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, Peyton M, Zou Y, Kurie JM, Dimaio JM, Milchgrub S, Smith AL, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64:9027–34. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- 25.Sato M, Vaughan MB, Girard L, Peyton M, Lee W, Shames DS, Ramirez RD, Sunaga N, Gazdar AF, Shay JW, Minna JD. Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res. 2006;66:2116–28. doi: 10.1158/0008-5472.CAN-05-2521. [DOI] [PubMed] [Google Scholar]
- 26.Wang TL, Maierhofer C, Speicher MR, Lengauer C, Vogelstein B, Kinzler KW, Velculescu VE. Digital karyotyping. Proc Natl Acad Sci USA. 2002;99:16156–61. doi: 10.1073/pnas.202610899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao X, Li C, Paez JG, Chin K, Janne PA, Chen TH, Girard L, Minna J, Christiani D, Leo C, Gray JW, Sellers WR, et al. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res. 2004;64:3060–71. doi: 10.1158/0008-5472.can-03-3308. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto H, Shigematsu H, Nomura M, Lockwood WW, Sato M, Okumura N, Soh J, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 2008;68:6913–21. doi: 10.1158/0008-5472.CAN-07-5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lockwood WW, Coe BP, Williams AC, MacAulay C, Lam WL. Whole genome tiling path array CGH analysis of segmental copy number alterations in cervical cancer cell lines. Int J Cancer. 2007;120:436–43. doi: 10.1002/ijc.22335. [DOI] [PubMed] [Google Scholar]
- 30.Asano H, Toyooka S, Tokumo M, Ichimura K, Aoe K, Ito S, Tsukuda K, Ouchida M, Aoe M, Katayama H, Hiraki A, Sugi K, et al. Detection of EGFR gene mutation in lung cancer by mutant-enriched polymerase chain reaction assay. Clin Cancer Res. 2006;12:43–8. doi: 10.1158/1078-0432.CCR-05-0934. [DOI] [PubMed] [Google Scholar]
- 31.Tokumo M, Toyooka S, Kiura K, Shigematsu H, Tomii K, Aoe M, Ichimura K, Tsuda T, Yano M, Tsukuda K, Tabata M, Ueoka H, et al. The relationship between epidermal growth factor receptor mutations and clinicopathologic features in non-small cell lung cancers. Clin Cancer Res. 2005;11:1167–73. [PubMed] [Google Scholar]
- 32.Naito Y, Yamada T, Ui-Tei K, Morishita S, Saigo K. siDirect: highly effective, target-specific siRNA design software for mammalian RNA interference. Nucleic Acids Res. 2004;32:W124–W129. doi: 10.1093/nar/gkh442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Puri N, Ahmed S, Janamanchi V, Tretiakova M, Zumba O, Krausz T, Jagadeeswaran R, Salgia R. c-Met is a potentially new therapeutic target for treatment of human melanoma. Clin Cancer Res. 2007;13:2246–53. doi: 10.1158/1078-0432.CCR-06-0776. [DOI] [PubMed] [Google Scholar]
- 34.Beau-Faller M, Ruppert AM, Voegeli AC, Neuville A, Meyer N, Guerin E, Legrain M, Mennecier B, Wihlm JM, Massard G, Quoix E, Oudet P, et al. MET gene copy number in non-small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naive cohort. J Thorac Oncol. 2008;3:331–9. doi: 10.1097/JTO.0b013e318168d9d4. [DOI] [PubMed] [Google Scholar]
- 35.Corso S, Migliore C, Ghiso E, De Rosa G, Comoglio PM, Giordano S. Silencing the MET oncogene leads to regression of experimental tumors and metastases. Oncogene. 2008;27:684–93. doi: 10.1038/sj.onc.1210697. [DOI] [PubMed] [Google Scholar]
- 36.Lutterbach B, Zeng Q, Davis LJ, Hatch H, Hang G, Kohl NE, Gibbs JB, Pan BS. Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res. 2007;67:2081–8. doi: 10.1158/0008-5472.CAN-06-3495. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.