

Abstract
Purpose
African Americans have higher incidence and poorer response to lung cancer treatment compared with Caucasians. However, the underlying molecular mechanisms for the significant ethnic difference are not known. The present study examines the ethnic differences in the type and frequency of MET proto-oncogene (MET) mutation in lung cancer and correlated them with other frequently mutated genes such as epidermal growth factor receptor (EGFR), KRAS2, and TP53.
Experimental Design
Using tumor tissue genomic DNA from 141 Asian, 76 Caucasian, and 66 African American lung cancer patients, exons coding for MET and EGFR were PCR amplified, and mutations were detected by sequencing. Mutation carriers were further screened for KRAS2 and TP53 mutations. Functional implications of important MET mutations were explored by molecular modeling and hepatocyte growth factor binding studies.
Results
Unlike the frequently encountered somatic mutations in EGFR, MET mutations in lung tumors were germline. MET-N375S, the most frequent mutation of MET, occurred in 13% of East Asians compared with none in African Americans. The frequency of MET mutations was highest among male smokers and squamous cell carcinoma. The MET-N375S mutation seems to confer resistance to MET inhibition based on hepatocyte growth factor ligand binding, molecular modeling, and apoptotic susceptibility to MET inhibitor studies.
Conclusions
MET in lung cancer tissues contained nonsynonymous mutations in the semaphorin and juxtamembrane domains but not in the tyrosine kinase domain. All the MET mutations were germline. East Asians, African-Americans, and Caucasians had different MET genotypes and haplotypes. MET mutations in the semaphorin domain affected ligand binding.
Lung cancer is a difficult disease to treat and is the most commonly diagnosed form of cancer, and its ravages on human health is underscored by the fact that there are over 213,000 new cases every year in the United States alone. Despite significant advances in treatments over the past two decades, only 15% of lung cancer patients survive for 5 years or longer (1). However, one of the most interesting and intriguing facts that has emerged from recent ethnic studies is that African Americans not only have higher incidence of lung cancer but also suffer from relatively lower survival rate when compared with matched Caucasian population (2). One of the underlying culprits that could partially explain this discrepancy is the tumor suppressor gene TP53. TP53 is mutated in 80% to 100% of small cell lung cancers (SCLC) and 50% to 80% of non-SCLC (NSCLC). There is a strong direct correlation between TP53 polymorphisms (exon 4, intron 3, intron 6) and incidence of lung cancer. The highest frequency of TP53 polymorphisms is, however, seen in African Americans and the least in Mexican Americans (3). Ethnic differences can also play an important role in the design of specific therapeutic approach as shown with studies related to the gain of function somatic mutations in the epidermal growth factor receptor (EGFR) tyrosine kinase in lung cancer. Tumors of Asian origin have relatively high mutation rate of EGFR that results in high basal kinase activity, and the patients respond much better to treatments containing EGFR inhibitor (4–9). In spite of this, the general response to EGFR inhibition is only 15%, and a majority of patients develop resistance. Thus, there are other tyrosine kinases such as MET that play an important role in lung cancer. The focus of this study is, therefore, to map MET mutations in lung tumors collected from patients of different ethnic backgrounds.
The MET receptor tyrosine kinase and its cognitive ligand the hepatocyte growth factor (HGF)/scatter factor play a major role in tumor development and metastasis (10–12). Overexpression of MET and the presence of an autocrine loop of activation have been linked to abnormal cell growth, tumor cell invasion, and subsequent metastasis (12). In a variety of human cancers, MET is also associated with high tumor grade and poor prognosis. Identification of activating germline mutations of MET in hereditary papillary renal carcinoma and subsequent confirmation provided the first direct evidence linking MET to human oncogenesis (13–16). Nonsynonymous mutations of MET have also been reported in a variety of human cancers including head and neck squamous cell carcinoma, small and non–small cell lung cancers, hepatocellular carcinoma, ovarian carcinoma, breast cancer, glioma, and gastric carcinoma (12). A number of MET mutations identified in the tyrosine kinase, juxtamembrane, and the semaphorin domains have been implicated in various cancers (17–21). It is currently not known if these mutations are germline or somatic.
In this study, we screened for mutations in the exons coding for semaphorin, juxtamembrane and tyrosine kinase domains of MET, and tyrosine kinase domain of EGFR in tumor tissues encompassing all non–small cell lung cancer histologic subtypes obtained from patients of East Asian, African-American, and Caucasian ethnicities. The nonsynonymous mutations of MET were further correlated with mutations in other lung cancer biomarkers such as KRAS2 and TP53. In addition, we also screened 74 lung cancer cell lines for MET mutations. The relationship between mutation profiles and patient ethnicity, clinical information, and pathologic characteristics were analyzed and the functional role of important MET mutations was evaluated in cell models.
Translational Relevance
MET receptor kinase is an important therapeutic target in lung cancer as aberrantly expressed or mutated MET serves pleiotropic functions in tumor development, and several germline activating mutations in MET have been implicated in cancers. This is the first study to show that majority of the MET mutations to be germline in lung cancer. This has large implications on hereditary factors and interactions with carcinogens (such as smoke) for lung cancer (which has never been understood previously). The type and frequency MET mutations were different among Caucasians, African Americans, and East Asians. This is the first study to compare the MET and epidermal growth factor receptor mutations in a large set of lung cancers. MET mutation N375S was detected in high proportion of East Asian samples and was correlated to incidence of squamous cell carcinoma. The MET-N375S mutation seems to confer resistance to MET inhibition.
Materials and Methods
Tissue specimen and DNA isolation
The tumor tissue samples were collected at the time of surgery from pathologically documented patients at the University of Chicago Hospitals, Chicago, IL, for whom clinical data were available with informed consent and in conformation with institutional guidelines. Use of human materials in this study was approved by the Institutional Review Board. The corresponding adjacent normal lung tissues obtained at surgery, where available, were also used in sequence analysis. Genomic DNA was isolated from archival formalin-fixed, paraffin-embedded tumor tissues using standard procedures. We also obtained genomic DNA isolated from fresh frozen tumor and adjacent normal tissues as well as from peripheral blood of 141 East Asian lung cancer patients with informed consent and in conformation with institutional guidelines from Taipei Veterans General Hospital, Taipei, Taiwan. Genomic DNA from 74 established lung cancer cell lines was isolated by standard method. Patient clinical information is given in Supplementary Table S1.
Nucleotide sequence analyses of MET, EGFR, KRAS2, and TP53 genes
The individual selected exons of MET (Genbank accession number AC002080), EGFR (X00588), KRAS2 (NG_007524), and TP53 (AF307851) were amplified by multiplex PCR using Qiagen Multiplex PCR reagent, as per manufacturer’s suggested protocol (Qiagen). Primers used in multiplex PCR are listed in Supplementary Table S2.
Recombinant MET ectodomain fused to immunoglobulin Fc region
The ectodomains of MET-WT or MET-N375S were amplified and cloned into pFUSE-mIgG2Aa-Fc1 to generate the Fc fusion construct (Invivogen). Stable transfectants of the wild-type (WT) and mutation-bearing constructs were established in Chinese hamster ovary cells selected in RPMI 1640/FCS-10% containing zeocin (500 μg/mL) and subclones selected on the basis of secreted fusion protein identified by binding of anti–IgG2c-horseradish peroxidase (Jackson Immunoresearch Laboratories, Inc.). Secreted MET-Fc fusion proteins were isolated from conditioned medium by binding to, and elution from, Protein A-sepharose columns (GE Healthcare).
HGF binding to MET-Fc or MET-N375S-Fc by ELISA
Each of the MET proteins [10 μg/mL; 0.05 mol/L carbonate buffer (pH 9.6)] was immobilized on ELISA microplates, followed by blocking overnight with Startingblock T20-TBS (Pierce), and incubation with increasing concentrations of recombinant HGF (Calbiochem) for 2 h; washed thrice in TBS-0.05% Tween 20 (TBS-T), anti-HGF (1 μg/mL in Startingblock; mouse monoclonal IgG1; R&D Systems, Inc.) for 2 h; washed thrice in TBS-T, anti-mouse IgG1 isotype-alkaline phosphatase (1/1,000 in Startingblock; Fisher Biotech; note that the isotype-specific antibody does not detect the γ2c heavy chain of the fusion protein Fc domains); and washed thrice in TBS-T followed by detection of bound alkaline phosphatase by assay at 405 nm of pNPP hydrolysis [1 mg/mL Tris (pH 8.5)].
Functional analysis of MET variants
MET-N375S mutant in pIRES2-EGFP vector was created by site-directed mutagenesis as described previously (19, 21). The recombinant MET mutant and its WT counterpart were transiently expressed in COS-7 cells using Fugene HD transfection reagent. The cells were treated with increasing concentrations of MET-specific inhibitor SU11274 (Pfizer, Inc., R&D) as described previously (19). Apoptosis was measured based on Annexin V–positive staining as determined by fluorescence-activated cell sorter analysis (Annexin–V-Fluos Staining kit; Roche Diagnostics) according to the manufacturer’s directions.
Molecular modeling
Selected MET variants identified in the present study and a few MET variants from our previous study (19) were modeled according to the structural parameters described for MET earlier (22, 23).
Statistical and data analysis
Relationship between mutation carriers and patient clinical and behavioral characteristics were analyzed using Fisher’s exact test or χ2 test using GraphPad Prism version 4.02 and GraphPad InStat version. 3.06 (GraphPad Prism Software). The germline variants were tested for Hardy-Weinberg equilibrium using a χ2 test for goodness-of-fit. P values were calculated using large sample approximations. Haplotypes were inferred using PHASE (24). Pair-wise linkage disequilibrium (LD) across the variants was assessed using r2 values that were calculated and plotted using LD Plotter.11
Results
Type and frequency of MET mutations: ethnic differences
The individual exons of semaphorin, juxtamembrane, and tyrosine kinase domains of MET were amplified using tumor tissue genomic DNA from 141 East Asian, 76 Caucasian, and 66 African American lung cancer patients by PCR, and the mutations were identified by sequencing. In all, we detected nine nucleotide substitutions and six of them involved nonsynonymous amino acid changes (Table 1). Four of the nonsynonymous substitutions were also detected in the corresponding adjacent normal tissues attesting to their germline origin. Corresponding normal tissues were not available for the remaining two mutation carriers. All the nonsynonymous mutations were clustered in the semaphorin domain except R988C of the juxtamembrane domain. Five of the above mutations are listed in the single nucleotide polymorphism database with specific IDs (Supplementary Table S3).
Table 1.
Summary of nonsynonymous MET mutations in lung cancer patients of different ethnicities and lung cancer cell lines
| DNA ID | Amino acid change | Race | Sex | Age(y) | Histology | Stage | Smoker | Adj. normal | Matching lymphocyte | EGFR | kras2 | p53 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| East Asian (n = 141) | ||||||||||||
| T226 | L211W | Asian | M | 74 | Adeno | IB | Y | + | + | - | - | ND |
| T83 | N375S | Asian | M | 79 | Large cell | IB | Y | + | + | - | - | - |
| T85 | N375S | Asian | M | 68 | Adeno | IB | Y | + | + | - | - | - |
| T147 | N375S | Asian | M | 81 | Squamous | IB | NK | + | + | - | - | - |
| T157 | N375S | Asian | M | 85 | Squamous | IB | Y | + | + | - | - | - |
| T208 | N375S | Asian | M | 72 | Adeno | IB | N | + | + | - | - | ND |
| T233 | N375S | Asian | M | 75 | NSCLC | IB | Y | + | + | - | - | ND |
| T297 | N375S | Asian | M | 78 | Adeno | IB | Y | + | + | Del:746-750* | - | ND |
| T30 | N375S | Asian | F | 80 | Adeno | IIB | NK | + | NA | - | G12A | R280K |
| T39 | N375S | Asian | M | 70 | Squamous | IIB | Y | + | NA | - | - | - |
| T104 | N375S | Asian | M | 71 | Adeno | IIB | Y | + | + | Del:746-750* | - | Del:805-814 |
| T372 | N375S | Asian | M | 81 | Squamous | IIB | Y | + | + | - | - | ND |
| T41 | N375S* | Asian | M | 76 | Squamous | IIIA | Y | +* | +* | - | - | - |
| T45 | N375S | Asian | M | 58 | Squamous | IIIA | Y | + | + | - | - | - |
| T256 | N375S | Asian | M | 73 | Adeno | IIIA | NK | + | + | Del:746-750 | - | ND |
| T335 | N375S | Asian | M | 79 | Squamous | IIIA | Y | + | + | - | - | ND |
| T349 | N375S | Asian | M | 50 | Adeno | IIIA | Y | + | + | - | - | ND |
| T139 | N375S | Asian | M | 68 | Squamous | IIIB | Y | + | + | - | - | - |
| T390 | N375S | Asian | M | 80 | Squamous | IIIB | Y | + | + | - | - | ND |
| T165 | N375S | Asian | M | 70 | Adeno | IV | NK | + | + | - | - | - |
| African American and Caucasian (n = 142) | ||||||||||||
| US89 | A347T | Caucasian | M | 73 | Squamous | IB | Y | NA | NA | - | - | ND |
| US58 | E355K | AA | M | 75 | Large cell | IIA | Y | + | NA | - | - | ND |
| US115 | M362T | AA | M | 52 | Adenosquamous | IIIA | Y | NA | NA | - | G12V | ND |
| US79 | N375S | Caucasian | M | 67 | Squamous | IA | Y | + | NA | - | - | ND |
| US88 | N375S | Caucasian | M | 66 | Squamous | IB | Y | + | NA | - | - | ND |
| US114 | R988C | AA | M | 72 | Adenosquamous | IIB | Y | + | NA | - | G12C | ND |
| US60 | R988C | Caucasian | M | 70 | Large cell | IIIA | Y | + | NA | P848L | - | ND |
| Cell lines (n = 74) | ||||||||||||
| H2122 | N375S | Caucasian | F | 46 | Adeno | ND | ND | NA | NA | - | G12C* | - |
| H289 | N375S | Caucasian | F | 45 | Small cell | ND | ND | NA | NA | - | - | - |
| HCC33 | N375S | Caucasian | M | 53 | Small cell | ND | ND | NA | NA | - | - | - |
| H157 | I852F* | Caucasian | M | 59 | Squamous | ND | ND | NA | NA | - | G12R | ND |
| H1437 | R988C | Caucasian | M | 60 | Adeno | ND | ND | NA | NA | - | - | ND |
NOTE: Deletion mutations are represented by nucleotide numbers.
Abbreviations: *, Homozygous; +, mutations present and heterozygous; NK, not known; NA, tissue not available; ND, not determined; AA, African American.
N375S was the most frequent nonsynonymous mutation and occurred at a higher frequency in East Asians compared with Caucasians, and was altogether absent in African Americans. The MET semaphorin domain also contained two synonymous substitutions that occurred with relatively high frequency. 534C > T (S178) was seen mostly in Asians, whereas 1131C > T (I377) was restricted to mainly African Americans. The juxtamembrane domain mutation 2962C > T (R988C) was infrequent and only found in Caucasians and African Americans. In the tyrosine kinase domain, synonymous mutation 3912C > T (D1304) was more commonly found in Caucasians and East Asians than African Americans. No nonsynonymous mutations were detected in the MET kinase domain in any of the tumors.
In addition, 74 lung cancer cell lines were sequenced for all the 21 coding exons of MET gene (Table 1). N375S mutation was detected in two SCLC (H289 and HCC33) and one adenocarcinoma (H2122) cell lines. Juxtamembrane domain mutation R988C was detected in NSCLC cell line H1437. A novel homozygous substitution 2554A > T (I852F) in the IPT-4 domain was detected in NSCLC cell line H157. In addition, we detected seven synonymous substitutions: 144G > A (in 3 cell lines), 534C > T (in 9), 1113C > T (in 1), 1944A > G (in 6), 3912C > T (in 31), 4071G > A (in 31), and 4146G > A (in 31) corresponding to amino acids A48, S178, N371, Q648, D1304, A1357, and P1382, respectively. The three synonymous tyrosine kinase domain substitutions, 3912C > T, 4071G > A, and 4146G > A were linked and occurred together as either homozygous (9/74) or heterozygous (22/74) in cell lines. Interestingly, the synonymous mutations of the tyrosine kinase domain were completely absent in all cell lines derived from African American ethnicity (n = 8). Synonymous mutation 1131C > T, found at high frequency in African American tissues, was not detected in any of the cell lines.
Haplotype analysis
The confirmation of the presence of tumor substitutions in the matching germline DNA gave us the opportunity to describe, for the first time, the haplotype structure of the MET gene in different ethnic groups (Table 2). No deviation from Hardy-Weinberg equilibrium was found (P > 0.05), although we cannot exclude deviations for the less common mutations found in our study. The LD patterns of the identified polymorphisms are shown in Supplementary Fig. S1. Complete LD (r2 = 1) between 534A/G and 1124A/G polymorphisms were observed in both Asian and Caucasian populations, but not in African-Americans, where both variants are monomorphic. Partial LD between 1131 and 2962 in Caucasian (r2 = 0.32) and between 2962 and 3912 in African-American (r2 = 0.24) samples were also observed. Haplotype analysis showed striking differences in the haplotype structure of germline variants among the different ethnic groups as evidenced by differences in the number of variants in each ethnic group and their haplotype composition (Table 2). MET diplotypes were 1/1 (frequency of 0.39), 2/1 (0.21), and 1/2 (0.16) in Caucasians; 2/2 (0.79), 6/2 (0.05), and 2/6 (0.03) in African Americans; and 2/2 (0.44), 1/2 (0.20), 2/1, and (0.18) in East Asians.
Table 2.
Haplotype compositions of MET mutations in different ethnic groups
| East Asian | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Haplotype | 534 | 1124 | 3912 | Number | Frequency | ||||
| 1 | C | A | T | 74 | 0.26 | ||||
| 2 | C | A | C | 189 | 0.67 | ||||
| 3 | T | G | C | 18 | 0.06 | ||||
| 4 | T | G | T | 1 | 0.004 | ||||
|
Caucasian | |||||||||
| Haplotype | 534 | 1039 | 1124 | 1131 | 1216 | 2962 | 3912 | Number | Frequency |
| 1 | C | G | A | C | T | C | C | 95 | 0.63 |
| 2 | C | G | A | C | T | C | T | 46 | 0.30 |
| 3 | C | G | A | C | T | T | T | 1 | 0.006 |
| 4 | T | G | G | C | T | C | C | 1 | 0.006 |
| 5 | C | G | A | C | A | C | T | 3 | 0.02 |
| 6 | C | G | G | C | T | C | C | 1 | 0.006 |
| 7 | C | G | A | T | T | C | C | 2 | 0.01 |
| 8 | C | A | A | C | T | C | C | 1 | 0.006 |
| 9 | C | G | A | C | A | C | C | 2 | 0.01 |
|
African American | |||||||||
| Haplotype | 534 | 1063 | 1085 | 1131 | 1216 | 2962 | 3912 | Number | Frequency |
| 1 | C | G | T | A | T | C | C | 2 | 0.02 |
| 2 | C | G | T | C | T | C | C | 115 | 0.87 |
| 3 | C | A | T | C | T | C | C | 1 | 0.008 |
| 4 | C | G | T | C | T | T | T | 1 | 0.008 |
| 5 | C | G | T | C | T | C | T | 3 | 0.02 |
| 6 | C | G | T | C | A | C | C | 5 | 0.04 |
| 7 | C | G | C | C | T | C | C | 1 | 0.008 |
| 8 | T | G | T | C | T | C | C | 4 | 0.03 |
NOTE: Numbering of nucleotides in the variants is according to c-Met cDNA sequence (Ensembl transcript ID: ENST00000318493).
MET mutations and lung cancer histology, smoking status and gender
The frequency of the MET nonsynonymous mutations in different histologic subtypes of lung cancer was determined (Fig. 1A). Caucasians and East Asians had higher frequency of MET mutations in squamous cell carcinoma than in adenocarcinoma and large cell carcinoma combined [P = 0.0486; 95% confidence interval (95% CI), 1.07–7.8]. In East Asians and Caucasians, the frequency of N375S was higher in squamous cell carcinoma than in adenocarcinoma or in large cell carcinoma (P = 0.05; 95% CI, 1.07–7.8; Fig. 1B). Also, N375S was found at the highest frequency across squamous cell, adenocarcinoma, and large cell carcinoma subtypes in East Asians, when compared with Caucasian (P = 0.0004; 95% CI, 2.004–38.64) or African Americans (P = 0.0006; 95% CI, 1.257–356.5) and none of the 66 tumor samples of African American cohort revealed this mutation.
Fig. 1.

MET mutations in lung cancer. A, frequency of MET mutations based on histologic subtypes of lung cancer and ethnic groups. The percentage frequencies of the occurrence of nonsynonymous mutations are given in parenthesis (blue). Adeno, adenocarcinoma. B, occurrence of MET-N375S mutation among histologic subtypes in different ethnic groups. Carriers of MET-N375S mutation are given as a percentage of total samples. C, frequency distribution of MET-N375S mutation in lung cancer patients of East Asian origin based on gender (i) and smoking history (ii). D, the pie chart shows tumors with nonsynonymous mutations in MET alone, MET and EGFR, and MET and KRAS2 as a percentage of total number of MET mutation carriers.
The frequency of MET-N375S in East Asian males was much higher compared with females (P = 0.0432; 95% CI, 0.8929–54.17; Fig. 1Ci). The most interesting aspect was that a large percentage of N375S mutation carriers (18/19) were smokers (P = 0.0673; 95% CI, 0.7677–48.35; Fig. 1Cii).
EGFR mutations
Gain of function mutations in the kinase domain encoded by exons 18 to 21 of EGFR are known to occur with relatively high frequency in lung cancers of Asian origin (4–9). To understand the relationship between the type and frequency of MET mutations with those of EGFR in lung cancer patients, we also determined EGFR mutations in the same set of lung tumor tissues. The variations in EGFR were mainly clustered in exons 19 and 21 and were most frequent in East Asian samples (Table 3). EGFR mutations were found in 32% (45 of 141) of East Asians compared with only in 3% (4 of 142) of Caucasians and African Americans (P < 0.0001; 95% CI, 5.628–46.47). Heterozygous EGFR-L858R mutation occurred in 13.5% (19 of 141) of the East Asians. Histologically, in East Asians 96% of the EGFR mutations were found in adenocarcinoma and 4% in squamous cell carcinoma, whereas large cell (n = 7) and small cell (n = 2) subtypes showed no mutations. A novel somatic mutation P848L in exon 20 was detected in a large cell lung carcinoma of Caucasian origin. In East Asians, EGFR variations were found preferably in nonsmokers (39% of nonsmokers compared with 24% of smokers had mutations; P < 0.0001; 95% CI, 0.01308–0.1377) and females (34% of females compared with 31% of males had mutations; P = 0.72 by χ2 test). None of the EGFR mutations were present in the adjacent normal or corresponding lymphocyte tissues, indicating that all EGFR mutations were somatic. Out of 283 lung tumors, one adenocarcinoma of East Asian origin and one large cell carcinoma of Caucasian origin had both MET and EGFR mutations (Table 1).
Table 3.
Summary of nonsynonymous EGFR tyrosine kinase domain mutations in lung cancer patients of different ethnicities
| DNA ID | Amino acid change | Race | Sex | Age | Histology | Stage | Smoker | Adj. normal | Matching lymphocyte | KRAS2 | TP53 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| East Asian (n = 141) | |||||||||||
| T176 | E709G | Asian | M | 51 | Adeno | IV | Y | - | NA | - | C176F |
| T126 | S768I | Asian | M | 46 | Adeno | IIIA | Y | - | NA | - | - |
| T206 | L858R | Asian | M | 71 | Adeno | IA | Y | - | NA | - | ND |
| T75 | L858R | Asian | M | 76 | Adeno | IB | Y | - | - | - | - |
| T89 | L858R | Asian | M | 78 | Adeno | IB | Y | - | NA | - | - |
| T223 | L858R | Asian | M | 50 | Adeno | IB | Y | - | NA | - | ND |
| T312 | L858R | Asian | M | 69 | Adeno | IB | N | - | NA | - | ND |
| T132 | L858R | Asian | F | 73 | Adeno | IIA | NK | NA | NA | - | - |
| T249 | L858R | Asian | M | 83 | Adeno | IIA | Y | - | NA | - | ND |
| T317 | L858R | Asian | F | 76 | Adeno | IIA | NK | - | NA | - | ND |
| T68 | L858R | Asian | M | 70 | Adeno | IIB | Y | NA | NA | - | W91Stop |
| T156 | L858R | Asian | M | 54 | Adeno | IIB | Y | - | - | - | - |
| T38 | L858R | Asian | F | 50 | Adeno | IIIA | N | - | NA | - | - |
| T142 | L858R | Asian | F | 71 | Adeno | IIIA | NK | - | - | - | - |
| T232 | L858R | Asian | M | 59 | Adeno | IIIA | Y | - | NA | - | ND |
| T272 | L858R | Asian | M | 63 | Squamous | IIIA | Y | - | NA | - | ND |
| T81 | L858R | Asian | F | 59 | Adeno | IIIB | N | - | - | - | Q32Stop |
| T168 | L858R | Asian | F | 66 | Adeno | IIIB | NK | NA | NA | - | - |
| T121 | L858R | Asian | M | 71 | Adeno | IV | NK | - | - | - | - |
| T176 | L858R | Asian | M | 51 | Adeno | IV | Y | - | NA | - | - |
| T294 | L858R | Asian | M | 58 | Adeno | IV | N | - | NA | - | ND |
| T375 | Del:746-750 | Asian | M | 75 | Adeno | IB | NK | NA | NA | - | ND |
| T210 | Del:746-750 | Asian | M | 50 | Squamous | IB | Y | NA | NA | - | ND |
| T297 | Del:746-750* | Asian | M | 78 | Adeno | IB | Y | NA | NA | - | ND |
| T57 | Del:746-750 | Asian | F | 57 | Adeno | IIB | NK | - | - | - | - |
| T104 | Del:746-750* | Asian | M | 71 | Adeno | IIB | Y | - | - | - | Del:805-814 |
| T66 | Del:746-750 | Asian | M | 70 | Adeno | IIIA | NK | - | - | - | - |
| T43 | Del:746-750 | Asian | M | 58 | Adeno | IIIA | NK | - | - | - | - |
| T80 | Del:746-750 | Asian | M | 71 | Adeno | IIIA | Y | - | - | - | - |
| T256 | Del:746-750 | Asian | M | 73 | Adeno | IIIA | NK | NA | NA | - | ND |
| T113 | Del:746-750 | Asian | M | 80 | Adeno | IIIB | Y | - | - | - | P152L |
| T162 | Del:746-750 | Asian | F | 72 | Adeno | IV | NK | - | - | - | - |
| T44 | Del:746-750 | Asian | M | 62 | Adeno | IV | Y | - | - | - | Q167Stop |
| T62 | Del:746-750 | Asian | M | 92 | Adeno | IV | NK | - | - | - | - |
| T148 | Del:746-750 | Asian | M | 79 | Adeno | IV | NK | - | - | - | - |
| T51 | Del:746-750 Ins A | Asian | F | 68 | Adeno | IB | NK | - | - | - | - |
| T18 | Del:747-753 Ins S | Asian | F | 78 | Adeno | IB | N | - | NA | - | C275F |
| T54 | Del:747-753 Ins S | Asian | M | 49 | Adeno | IB | NK | - | - | - | - |
| T134 | Del:747-752 | Asian | M | 72 | Adeno | IIIA | Y | - | NA | - | - |
| T52 | Del:748-752 | Asian | M | 55 | Adeno | IIIA | NK | NA | NA | - | - |
| T231 | Del:747-752 Ins S | Asian | M | 55 | Adeno | IIIA | N | NA | NA | - | ND |
| T235 | Del:747-752 Ins S | Asian | F | 58 | Adeno | IIIA | N | NA | NA | - | ND |
| T248 | Del:747-751 | Asian | M | 78 | Adeno | IIIA | Y | NA | NA | - | ND |
| T146 | Del:751-758 | Asian | F | 70 | Adeno | IB | N | - | - | - | V272E |
| T391 | Del:710, E709D | Asian | M | 71 | Adeno | IB | N | NA | NA | - | ND |
| African American and Caucasian (n = 142) | |||||||||||
| US94 | P733T | Caucasian | F | 60 | Squamous | IIIA | Y | NA | NA | - | ND |
| US7 | S768I | AA | F | 60 | Adeno | IA | Y | NA | NA | - | ND |
| US60 | P848L | Caucasian | M | 70 | Large cell | IIIA | Y | NA | NA | - | ND |
| US10 | Del:748-753 | AA | F | 59 | Adeno | IIIA | Y | NA | NA | - | ND |
NOTE: Deletion variations are represented by nucleotide numbers.
Abbreviations: AA, African American; *, Homozygous; -, variation not found; NK, not known; ND, not determined; NA, tissue not available.
KRAS2 and TP53 mutations
Screening tissues carrying MET mutations for known KRAS2 somatic mutations in exon 1 (codons 12, 13, and 61) revealed three patients with mutations in both (Table 1). None of the tissues with EGFR mutations had KRAS2 mutations. TP53 mutations were found in 18% (2 of 11) of MET and 29% (8 of 29) of EGFR mutation carriers, respectively (Tables 1 and 3). One tissue with MET-N375S mutation showed KRAS2-G12A and TP53-R280K mutations. Thus, it seems that most frequently occurring multiple gene mutations are in MET or EGFR in combination with TP53 but not with KRAS2; further, the frequency of both MET and EGFR being altered in the same sample is also relatively less (Fig. 1D).
Structure analysis of semaphorin domain mutations by molecular modeling
A summary of all the frequently occurring synonymous as well as nonsynonymous mutations in the MET gene in lung cancer, in relation to its domain structure and ethnic preferences, are summarized in Fig. 2A. The crystal structure of the semaphorin domain in complex with β chain of HGF has been determined earlier (22), providing the opportunity to study the potential effects of variations on ligand binding through comparative molecular modeling. Mutations in the semaphorin domain of MET identified in previous studies by our group (19, 21) were also included in the modeling study. Analysis of the three-dimensional structure of semaphorin domain of MET-HGF complex (1SHY) showed the presence of semaphorin domain residues E168 and L229 in direct contact with HGF (also see Fig. 2B and C), indicating the potential for altering the binding affinity of the complex due to replacement of these residues (Fig. 2B). The three-dimensional model also showed residue E168 in a conserved motif due to the high ω value (>1) of the neighboring residues P169, S170, and P210 (Supplementary Table S4). Replacement of asparagine at 375 by a serine resulted in the loss of a hydrogen bond between the altered residue and the adjacent arginine residue at 280 (Fig. 2C and D), suggesting possible changes to ligand binding. Overall, mutations of semaphorin domain conferred subtle changes to the ligand binding region and also to its physical properties as evidenced from the Root Mean Square Deviation values that were all <1 Å (Supplementary Table S4).
Fig. 2.
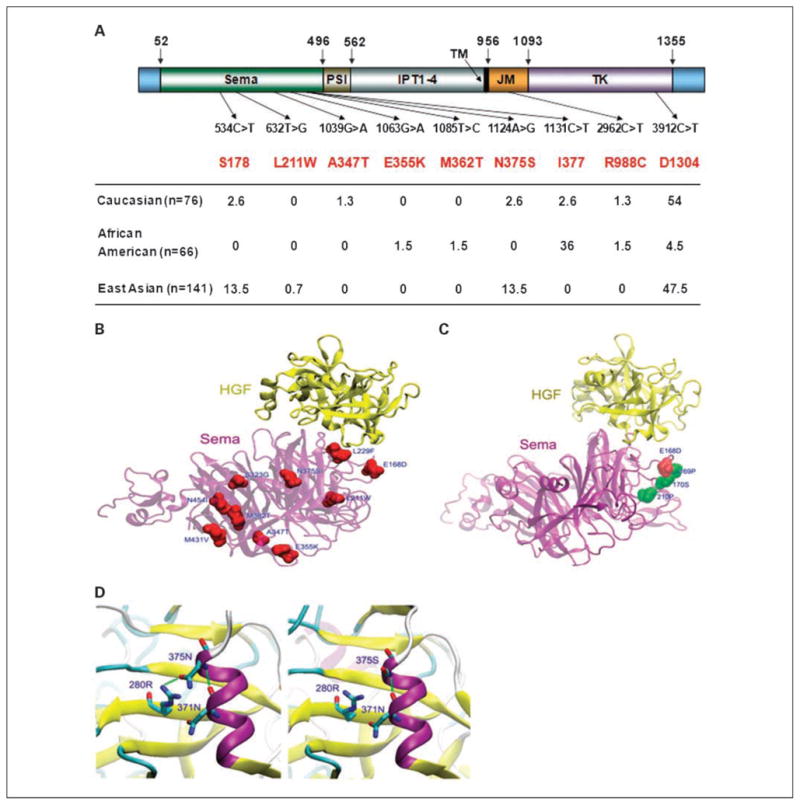
MET mutations: type, frequency, and distribution. Mutations of MET identified in lung tumor tissue samples from different ethnic groups. A, various mutations are illustrated schematically in the context of functional domains of MET. The numbers denote amino acid residues at indicated position in the molecule. Sema, semaphorin domain; PSI, plexins, semaphorins, and integrins domain; IPT1-4, found in immunoglobulin-like regions, plexins, and transcriptional factors; TM, transmembrane region; JM, juxtamembrane domain; TK, catalytic tyrosine kinase domain. Genotype frequencies are represented as a percentage of total number of tumor samples tested in each group. The altered nucleotides as well as the corresponding amino acids (top row) are numbered according to full-length MET (Ensembl transcript ID: ENST00000318493). B, effect of MET mutations on the ligand binding pocket in MET. Structural changes in MET semaphorin domain due to nonsynonymous mutations and their effect on interaction with HGF β-chain are shown by homology modeling: yellow, HGF β-chain; purple, the semaphorin domain of MET as ribbon models. Altered residues are colored red and labeled by residue number; residues 168 and 229 can be seen in direct contact with HGF. C, Van der Waals or space-filling spheres representation of positively selected residues P169, S170, and P210 with high probabilities of ω > 1 are spatially close to mutation E168D, indicating that residue 168 is in the ligand binding region. D, stereo magnification of the mutation N375S; N375 (left) has two potential hydrogen bonds (green dash line), whereas S375 modeling structure (right) shows loss of one hydrogen bond, and therefore likely to weaken ligand binding.
The relative binding between HGF and MET-WT or MET-N375S
Since MET-N375S mutation occurred at highest frequency, we further studied the effect of the amino acid change on ligand binding and susceptibility to the MET inhibitor SU11274. We generated fusion constructs of MET-WT and MET-N375S ectodomains and the Fc portion of the immunoglobulin, expressed them in Chinese hamster ovary cells as described in Materials and Methods, and purified the recombinant proteins. The relative purities of the fusion protein preparations were analyzed by SDS-PAGE and the results are shown in Fig. 3A. The yields as well as the quality of both types of protein preparations obtained from culture supernatants were comparable.
Fig. 3.
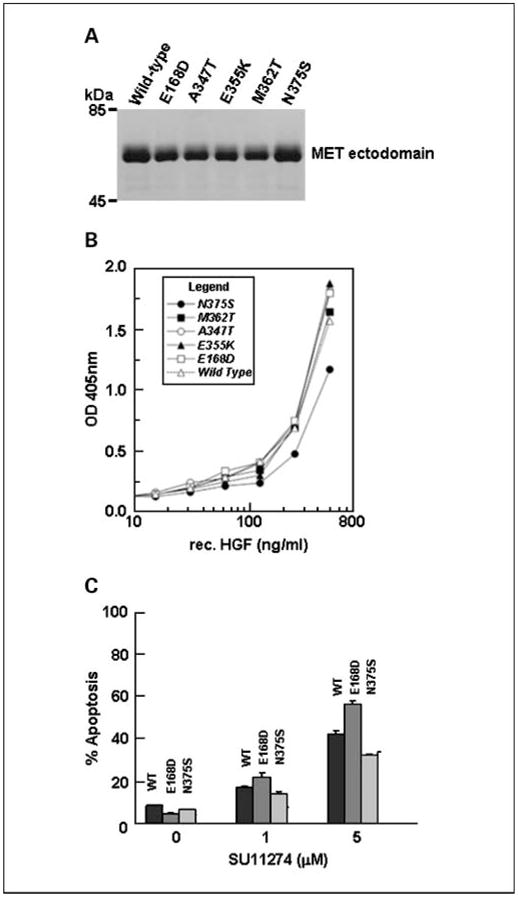
The functional significance of MET-N375S mutation. A, relative purity of MET-WT-Fc and MET-N375S-Fc fusion proteins. B, relative binding of HGF to MET-WT-Fc and MET-N375S-Fc. C, susceptibility of COS-7 cells expressing MET-WT or MET-N375S to SU11274 induced apoptosis.
We next analyzed the binding between HGF and the fusion proteins by an ELISA-based assay as described in Materials and Methods. As shown in Fig. 3B, the amounts of HGF bound to MET-WT were much higher when compared with MET-N375S at every concentration of HGF tested, thereby clearly demonstrating a loss of affinity for HGF in case of the MET-N375S mutant. In contrast, the MET-E168D mutant had relatively higher affinity for HGF. The above finding supports the prediction made by the model presented in Fig. 2B.
MET mutations and their effect on susceptibility to c-Met Inhibitor SU11274
To understand the role of the frequently mutated MET-N375S (and also the E168D for comparison) in modulating sensitivity to MET inhibitor, we did in vitro apoptosis assay in a COS-7 transfection model using the preclinical MET inhibitor SU11274. The recombinant MET-WT, MET-N375S, and MET-E168D constructs were transiently expressed in COS-7 cells and their susceptibility to increasing concentrations of SU11274 was determined by Annexin V–positive staining of cells. Although the relative levels of protein expression for MET-WT and its mutant forms were comparable (data not shown), the MET-N375S cells had less SU11274-induced apoptosis compared with cells that expressed MET-WT, evident at both 1 and 5 μmol/L concentrations of SU11274. In contrast, the cells expressing E168D mutant showed higher susceptibility to the c-Met inhibitor at both concentrations tested (Fig. 3C).
Discussion
Ethnic differences seem to significantly contribute to the incidence and prognosis of lung cancer. One of the underlying molecular differences could be in some of the important receptor tyrosine kinases with major role in lung cancer. The present study aims to understand mutational differences in the receptor tyrosine kinase MET in Caucasians, African Americans, and East Asians. Highest frequency of MET mutations was found in East Asians as was the case with EGFR mutations. However, unlike EGFR mutations that are somatic in nature, majority of the MET mutations were germline. Thus, East Asians seem to show a propensity for mutations in both MET and EGFR RTKs. Incidentally, most of the nonsynonymous mutations were localized to the ligand binding semaphorin domain and the negative regulatory juxtamembrane domain of MET RTK. In our limited previous study, as well as the current multiethnic study, we failed to detect any nonsynonymous mutations (gain of function) in the MET kinase domain, a trait different from that seen in gastric, hereditary papillary renal cell carcinoma, and head and neck cancer (13, 15, 16, 19, 21). In addition, the MET nonsynonymous mutations did not show any significant overlapping or mutually exclusive pattern with mutations in EGFR, KRAS2, or TP53 genes. Although a large number of samples have been screened in this study, due to the low frequency and the germline nature of MET mutations, the results need to be interpreted with caution.
Studies involving specific inhibitors of EGFR clearly showed that there could be other RTKs such as MET that arise in the setting of resistance, and can contribute to tumorogenesis and metastasis (25, 26). This is supported by earlier signaling studies that show interaction between multiple activated RTKs and their downstream signaling target phosphoinositide 3-kinase (27). Earlier studies from our laboratory with specific small inhibitors of MET attest to the above fact and indicate that MET plays a significant role in not only lung tumorogenesis and angiogenesis, but also in its metastasis (19, 28). The present study also unravels a striking difference between the nature of mutations with respect to MET and EGFR. Majority of the nonsynonymous mutations of MET were found to be of germline in origin compared with EGFR that were somatic. In our EGFR mutational analysis, all the adjacent tissue samples were devoid of mutations, whereas the MET mutations were present both in the tumor and the adjacent normal tissues. Interestingly enough, we also had peripheral blood lymphocyte DNA from East Asian subjects, and both L211W and N375S mutations were found in these DNAs. Thus, the mutations seen in the adjacent normal tissues in MET were most likely not due to “field effect.”
A recent report correlated certain African American–specific haplotypes of TP53 to higher incidence and poorer prognosis of lung cancers, and these apparently were absent in Caucasians (3). However, we found that TP53, and not KRAS2, mutations cooccurred with both MET and EGFR tyrosine kinase mutations, and the frequencies were comparable with previous report (29). The higher incidence of the somatic mutations of EGFR as well as the germline mutations of MET in East Asians signifies the underlying hereditary component. As an example, JAK2 mutations are somatic in polycythemia vera, but still, they are controlled by a germline single nucleotide polymorphism identified in the JAK2 gene (30).
The nonsynonymous MET mutations determined in the present study correlated more with squamous cell carcinoma than adenocarcinoma or large cell carcinoma and similarly more with male smokers. A larger study by including detailed quantitative information about the smoking history of lung cancer patients would be useful to determine the true relationship between environmental toxins such as cigarette smoke, MET mutations, and potential risk of developing lung cancer. The epithelial cells that line the air passages and the lung alveoli are maximally exposed to inhaled cigarette smoke (carcinogens) and therefore could develop multiple carcinogenic foci that are commonly seen in patients with squamous cell lung carcinoma. However, such lesions can be diagnosed at preneoplastic stage and therefore can be readily treated with combinatorial chemotherapy that includes specific MET inhibitor. It would be important for the future to determine the stage, metastasis, and other covariables in the context of MET mutations.
The MET semaphorin domain harbors the ligand binding site, and based on results from our laboratory, it seems to be a key region that is targeted for nonsynonymous mutations in lung cancer (19, 21). Our molecular modeling of E168D and N375S mutations of MET revealed subtle changes in the ligand binding region of MET. It is, therefore, likely that the affinity of the above mutant MET variants with HGF is significantly altered, thereby altering MET functionality. This is precisely what we see with MET-E168D and MET-N375S mutants in our functional studies. Our previous study clearly suggested that the lung cancer patients harboring MET mutations could have varying extent of response to specific MET kinase inhibitors such as SU11274 (31). Molecular modeling suggests distancing of the 375 residue from its interacting counterpart in HGF, thereby weakening the ligand and receptor interaction. This is reflected in the lower affinity. It is very likely that ligand binding by the above mutant MET results in less than optimal kinase activation, and therefore, the observed increased resistance to the MET therapeutic inhibitor SU11274. This poses a challenge to the design of patient-directed therapeutic cocktails to achieve the highest efficacy.
In summary, our results identify significant ethnic differences in the type and frequency of MET mutations in lung cancer. Semaphorin domain mutations have an effect on the ligand binding affinity of MET receptor tyrosine kinase. Lung cancer patients do not carry any mutations in the MET kinase domain. The MET mutations, unlike that of EGFR, are germline in nature. This study showed lung cancer patients of East Asian origin as a distinct ethnic group with high incidence of both germline MET-N375S and somatic EGFR mutations compared with Caucasians and African Americans. The germline nature of MET mutations together with their low frequency in lung cancer patients suggests the need for larger number of samples for genotype-phenotype correlational analyses. In light of the reports of amplified MET signaling as the culprit in lung cancer resistance to gefitinib, it is important to determine the role of MET mutations and amplification in lung cancer resistance to therapy.
Supplementary Material
Acknowledgments
Grant support: NIH/National Cancer Institute R01 grants, CA100750-04, CA129501-01A1, and CA-125541-01; V-Foundation/Guy Geleerd Memorial; MARF/Jeffrey P. Hayes Memorial Grant; Cancer Research Foundation/Goldblatt Award (R. Salgia); Lung Cancer Specialized Programs of Research Excellence P50CA70907 (J. Minna); and P01HL058064-13 (J. Garcia).
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Abidoye O, Ferguson MK, Salgia R. Lung carcinoma in African Americans. Nat Clin Pract Oncol. 2007;4:118–29. doi: 10.1038/ncponc0718. [DOI] [PubMed] [Google Scholar]
- 3.Wu X, Zhao H, Amos CI, et al. p53 genotypes and haplotypes associated with lung cancer susceptibility and ethnicity. J Natl Cancer Inst. 2002;94:681–90. doi: 10.1093/jnci/94.9.681. [DOI] [PubMed] [Google Scholar]
- 4.Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–46. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 5.Haber DA, Bell DW, Sordella R, et al. Molecular targeted therapy of lung cancer: EGFR mutations and response to EGFR inhibitors. Cold Spring Harb Symp Quant Biol. 2005;70:419–26. doi: 10.1101/sqb.2005.70.043. [DOI] [PubMed] [Google Scholar]
- 6.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 7.Mitsudomi T, Kosaka T, Yatabe Y. Biological and clinical implications of EGFR mutations in lung cancer. Int J Clin Oncol. 2006;11:190–8. doi: 10.1007/s10147-006-0583-4. [DOI] [PubMed] [Google Scholar]
- 8.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 9.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–7. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 10.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–25. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 11.Birchmeier C, Gherardi E. Developmental roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends Cell Biol. 1998;8:404–10. doi: 10.1016/s0962-8924(98)01359-2. [DOI] [PubMed] [Google Scholar]
- 12.Sattler M, Salgia R. c-Met and hepatocyte growth factor: potential as novel targets in cancer therapy. Curr Oncol Rep. 2007;9:102–8. doi: 10.1007/s11912-007-0005-4. [DOI] [PubMed] [Google Scholar]
- 13.Lee JH, Han SU, Cho H, et al. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene. 2000;19:4947–53. doi: 10.1038/sj.onc.1203874. [DOI] [PubMed] [Google Scholar]
- 14.Liao AT, McMahon M, London CA. Identification of a novel germline MET mutation in dogs. Anim Genet. 2006;37:248–52. doi: 10.1111/j.1365-2052.2006.01415.x. [DOI] [PubMed] [Google Scholar]
- 15.Schmid L, Duh FM, Chen F, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16:68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt L, Junker K, Weirich G, et al. Two North American families with hereditary papillary renal carcinoma and identical novel mutations in the MET proto-oncogene. Cancer Res. 1998;58:1719–22. [PubMed] [Google Scholar]
- 17.Di Renzo MF, Olivero M, Martone T, et al. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene. 2000;19:1547–55. doi: 10.1038/sj.onc.1203455. [DOI] [PubMed] [Google Scholar]
- 18.Jeffers M, Schmidt L, Nakaigawa N, et al. Activating mutations for the met tyrosine kinase receptor in human cancer. Proc Natl Acad Sci U S A. 1997;94:11445–50. doi: 10.1073/pnas.94.21.11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma PC, Jagadeeswaran R, Jagadeesh S, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65:1479–88. doi: 10.1158/0008-5472.CAN-04-2650. [DOI] [PubMed] [Google Scholar]
- 20.Wasenius VM, Hemmer S, Karjalainen-Lindsberg ML, Nupponen NN, Franssila K, Joensuu H. MET receptor tyrosine kinase sequence alterations in differentiated thyroid carcinoma. Am J Surg Pathol. 2005;29:544–9. doi: 10.1097/01.pas.0000156103.37756.e2. [DOI] [PubMed] [Google Scholar]
- 21.Ma PC, Kijima T, Maulik G, et al. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;63:6272–81. [PubMed] [Google Scholar]
- 22.Stamos J, Lazarus RA, Yao X, Kirchhofer D, Wiesmann C. Crystal structure of the HGF β-chain in complex with the Sema domain of the Met receptor. EMBO J. 2004;23:2325–35. doi: 10.1038/sj.emboj.7600243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adamian L, Liang J. Prediction of transmembrane helix orientation in polytopic membrane proteins. BMC Struct Biol. 2006;6:13. doi: 10.1186/1472-6807-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–89. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 26.Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–7. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 28.Puri N, Khramtsov A, Ahmed S, et al. A selective small molecule inhibitor of c-Met, PHA665752, inhibits tumorigenicity and angiogenesis in mouse lung cancer xenografts. Cancer Res. 2007;67:3529–34. doi: 10.1158/0008-5472.CAN-06-4416. [DOI] [PubMed] [Google Scholar]
- 29.Mitsuuchi Y, Testa JR. Cytogenetics and molecular genetics of lung cancer. Am J Med Genet. 2002;115:183–8. doi: 10.1002/ajmg.10692. [DOI] [PubMed] [Google Scholar]
- 30.Kilpivaara O, Mukherjee S, Schram AM, et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat Genet. 2009;41:455–9. doi: 10.1038/ng.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berthou S, Aebersold DM, Schmidt LS, et al. The Met kinase inhibitor SU11274 exhibits a selective inhibition pattern toward different receptor mutated variants. Oncogene. 2004;23:5387–93. doi: 10.1038/sj.onc.1207691. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.