

Abstract
Hepatitis C virus (HCV) infection frequently leads to chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma. There is no effective therapy or vaccine available to HCV-infected patients other than interferon–ribavarin combination, which is effective in a relatively small percentage of infected patients. Our previous results have shown that a synthetic peptide (LAP) corresponding to the N-terminal 18 amino acids of the Lupus autoantigen (La) was a potent inhibitor of HCV IRES-mediated translation. We demonstrate here that LAP efficiently blocks HCV replication of infectious JFH1 virus in cell culture. Our data suggest that LAP forms complexes with IRES-transacting factors (ITAFs) PTB and PCBP2. LAP-mediated inhibition of HCV IRES-mediated translation in vitro could be fully rescued by recombinant PCB and PCBP2. Also transient expression of PTB/PCBP2 combination significantly restores HCV replication in LAP-inhibited cultures. These results suggest that ITAFs could be potential targets to block HCV replication.
Keywords: Hepatitis C virus, JFH-1, IRES-mediated translation, transacting factors, Antivirals, Peptides
Introduction
Hepatitis C virus (HCV) is an enveloped RNA virus belonging to the flaviridae family. HCV infection frequently leads to chronic hepatitis, cirrhosis of the liver, and hepatocellular carcinoma (Saito et al., 1990; Houghton et al., 1991; Choo et al., 1992). There is currently no effective therapy or vaccine available to HCV-infected patients other than interferon–ribavarin combination, which is only effective in a relatively small percentage of infected patients. HCV has been a difficult virus to study due to the lack of an adequate, simple and low cost animal model. Although the RNA transcribed in vitro from a cDNA clone of HCV has been shown to be infectious when injected directly into the livers of chimpanzees (Lolykhalov et al., 1997; Yanagi et al.; 1997), HCV virions have been extremely difficult to propagate in culture. Recently a unique patient isolate called Japanese fulminant hepatitis 1 (JFH1) was found to replicate and produce infectious HCV in cell culture (Wakita et al., 2005). This recent advance has allowed the processes of HCV entry, infectivity and assembly to be investigated for the first time.
While cellular capped mRNAs are translated by a cap-dependent “scanning” mechanism, poliovirus and encepholomyocarditis viral RNAs were first shown to use a cap-independent mechanism, which involves internal entry of ribosome within the 5′ UTR of viral RNA, a process termed internal ribosome entry site (IRES)-mediated translation (Pelletier and Sonenberg, 1988; Jang et al., 1988). Since then, functional IRES elements have been identified in many other RNA viruses including HRV, FMDV, HAV, TMEV, Coxsackie virus, HCV, classical swine fever virus, murine leukemia virus, simian immunodeficiency virus, and cricket paralysis virus (reviewed in Helen and Sarnow, 2001).
The formation of 48S preinitiation complex containing the initiator tRNA, mRNA and 40S ribosome by the HCV IRES element requires only eIF-2 and eIF-3 (Pestova et al., 1998). The 40S ribosome appears to interact with HCV RNA at multiple sites including stems, loops, pseudoknots, as well as the initiator AUG. Ribosomal protein S5/S9 as well as eIF-2B and eIF-2γ have been identified as co-factors of HCV IRES-mediated translation (Fukushi et al., 2001; Kruger et al, 2000). Although these studies have shed light on the minimum required canonical factors for HCV IRES-mediated translation, it is apparent that transacting factors play an important role in modulating IRES activity (Belsham and Sonenberg, 2000). Cellular proteins such as La (lupus autoantigen), PTB (polypyrimidine tract binding protein), PCBP2 (poly rC binding protein 2), C23 (nucleolin), NS1-associated protein 1 (NSAP1) and unr have been shown to interact with viral IRES elements and stimulate IRES-mediated translation (Ali and Siddiqui, 1995 & 1997; Ali et al., 2000; Blyn et al., 1997; Meerovich et al., 1993; Boussadia et al., 2003; Gamarnick and Andino, 1997; Hellen et al., 1993; Hunt et al., 1999; Izumi et al., 2001). It has been hypothesized that the transacting proteins may act as “RNA chaperones” stabilizing IRES secondary and tertiary structures to allow efficient translation to take place (Belsham and Sonenberg, 2000).
Previous results from our laboratory have shown that an 18-amino acid-long sequence from the N-terminus of the La autoantigen was required for efficient interaction of La with viral 5′ UTR elements. A synthetic peptide (called LAP, for La peptide) corresponding to this sequence (amino acids 11 to 28) of La was found to efficiently inhibit viral IRES-mediated translation in vitro (Izumi et al., 2004). The LAP efficiently entered Huh-7 cells and preferentially inhibited HCV IRES-mediated translation programmed by a bicistronic RNA in vivo (Izumi et al., 2004). The LAP did not bind RNA directly. Preliminary competition UV crosslink and translation rescue experiments suggested that LAP inhibits IRES-mediated translation by interacting with proteins rather than RNA. Mutagenesis of LAP demonstrated that single amino acid changes in a highly conserved sequence within LAP are sufficient to eliminate the translation-inhibitory activity of LAP. When one of these mutations (Y23Q) is introduced into full-length La protein, the mutant protein was found to be severely defective in interacting with the viral IRES element and consequently unable to stimulate IRES-mediated translation (Izumi et al., 2004). These results underscored the importance of the La N-terminal amino acids in RNA binding and viral RNA translation.
In this study we have investigated the mechanism by which LAP inhibits HCV IRES-mediated translation. We demonstrate here that LAP, but not the single amino acid LAP mutant Y23Q, efficiently blocks HCV replication as well as formation of infectious JFH1 virus in cell culture. Competition UV crosslink analysis show that LAP is able to block interaction of HCV 5′ UTR with a number of IRES transacting proteins (ITAFs) that include PTB, PCBP2 and La. Gel exclusion chromatography using purified ITAFs and FITC-labeled LAP showed direct interaction of LAP with PTB, PCBP2 and to some extent with La. Consistent with these data, His-tagged, purified PTB and PCBP2 were able to fully rescue HCV 5′ UTR-mediated translation that was inhibited by incubation of Huh-7 translation lysates with LAP. The purified La protein could only partially restore LAP-inhibited translation from the HCV 5′ UTR in vitro. Finally, transient expression of the PTB/PCBP2 combination was able to reverse LAP-mediated inhibition of viral replication in culture. These results underscore the importance of ITAFs in HCV life cycle and demonstrate that these cellular RNA-binding proteins could be targeted to block HCV replication.
Results and Discussion
LAP blocks replication of JFH1 HCV in tissue culture
Previous results from our laboratory have shown that LAP is able to efficiently enter Huh-7 cells and block HCV IRES-mediated translation from a bicistronic RNA without significantly interfering with cap-dependent translation (Izumi et al., 2004). It was, therefore, of considerable interest to determine if LAP could block replication of HCV in tissue culture. We addressed this question by pretreatment of Huh-7.5.1 cells with various concentrations of LAP or the Y23Q LAP mutant for 1–2 hours, followed by removal of excess LAP and washing of cells with PBS. Cells were then infected with the JFH1 virus for 72 hours. Replication of the JFH1 virus was determined by measuring NS5A expression by immunofluorescence using an antibody to NS5A (Zhong et al., 2005). As can be seen in Fig. 1B–1G, LAP inhibited replication of JFH1 virus efficiently at all concentrations tested. This was in contrast to the Y23Q LAP mutant, which was unable to block virus replication significantly even at the highest concentration (Fig. 1H–K). Similar results were obtained when a non-specific peptide with a scrambled LAP sequence was used as a negative control in this experiment (data not shown; see Fig. 1L). To quantify the degree of LAP-mediated inhibition of virus replication, the effect of LAP on virus replication was determined using a monocistronic Renilla luciferase HCV reporter virus, NRFLC (Arumugaswami et al., 2008). The mutant reporter RNA defective in HCV RNA polymerase activity (pol-null) was included as a negative control for genome replication. The genome replication of NRFLC was assessed by measuring the Renilla luciferase activity in the transfected Huh-7.5.1 cells at 6 and 96 hours after transfection (Fig. 1L). At 6h post-transfection the Renilla luciferase activity was mainly due to translation of the input RNA genome. Wt LAP but not the Y23Q or the scrambled NSP inhibited translation of the input RNA significantly at early time points (6h). The significant increase in Renilla luciferase activity at 96h post-transfection is due to replication of the viral RNA genome leading to accumulation of significantly higher levels of translatable viral RNA. The wt LAP significantly inhibited replication of the reporter virus, whereas both the Y23Q LAP and the non-specific peptide (scrambled NSP) had no significant effect on initial translation (6h) or virus replication (96h). While luciferase production from the pol-null mutant RNA at 6h post-transfection was similar to that of the wt RNA, there was no significant increase in luciferase at 96h due to lack of RNA replication by the polymerase mutant. These results suggest LAP is able to block JFH1virus replication significantly in cultured liver cells and are consistent with our previous report that LAP could enter Huh-7 cells efficiently and block HCV IRES-mediated translation (Izumi et al., 2004).
Figure 1. LAP blocks replication of JFH1 HCV in tissue culture.

(A) Schematic representation of wt JFH1 and wt and pol-null luciferase reporter (NRLFC) constructs. (B–K) Huh-7.5.1 cells were pretreated with buffer (panels B and C), various concentrations of wt LAP (panels D–G), or Y23Q mutant LAP (panels H–K) as described in materials and methods. Excess LAP was then removed, and cells were washed with MEM, followed by infection of cells with JFH1 virus (moi=1). Infection was stopped at 72h post-infection, and one half of the cells were stained with DAPI, and the other half was scored for NS5A immunofluorescence using anti-NS5A antibody. (L)10 μg of in vitro transcribed genomic RNA of wild-type NRLFC reporter virus was introduced into Huh-7.5.1 cells (by electroporation) that were pretreated with indicated concentrations of wt LAP, or Y23Q mutant or the non-specific peptide (NSP). The reporter virus lacking polymerase activity (pol-null) is included as negative control. The transfected cells were lysed at indicated time points using Promega passive lysis buffer and the levels of Renilla luciferase were quantified. The experiment was done in triplicate and the mean values with standard deviation of Renilla luciferase (corrected for transfection efficiency and extract protein concentration) values are presented as a bar graph in log10 scale. The lacZ gene (encodes beta-galactosidase protein) under CMV immediate early promoter was used to determine the percentage of transfected cells by comparing the number of cells expressing the reporter protein to the total number of cells in the population.
LAP interferes with binding of ITAFs to the 5′ UTR of HCV RNA
Previous results from our laboratory suggested that LAP-induced inhibition of poliovirus 5′UTR-mediated translation occurred through translation stimulatory proteins rather than through interaction with viral 5′UTR (Izumi et al., 2004). To examine what cellular proteins could be involved in LAP-mediated inhibition of HCV IRES-mediated translation, we examined interaction of cellular proteins (in translation lysates from Huh-7 cells) with 32P-labeled HCV 5′UTR by UV-crosslink assay (Das et al., 1996 & 1998; Dasgupta et al., 2004) in the absence and presence of LAP. As can be seen in Fig. 2A, a number of liver cell-derived proteins having molecular masses between 45 to 100 kDa were found to interact with the HCV 5′UTR (lane 2). Previous studies from our laboratory have shown that interaction of almost all of these polypeptides with the HCV 5′UTR could be specifically competed with the unlabeled 5′UTR but not a non-specific RNA of similar size (Dasgupta et al., 2004). Here we wished to examine if addition of LAP to the reaction would block 5′UTR-protein interaction. Indeed, LAP blocked interaction of these polypeptides with the 5′UTR when used at final concentrations of 40 and 60 μM (lanes 3 and 4). A non-specific scrambled peptide, however, was unable to block RNA-protein interaction significantly (lanes 5 and 6). These results suggest that LAP interferes with cellular protein binding to the HCV 5′UTR.
Figure 2. LAP blocks HCV 5′ UTR-protein interaction of a number of polypeptides from Huh-7.5 cells .
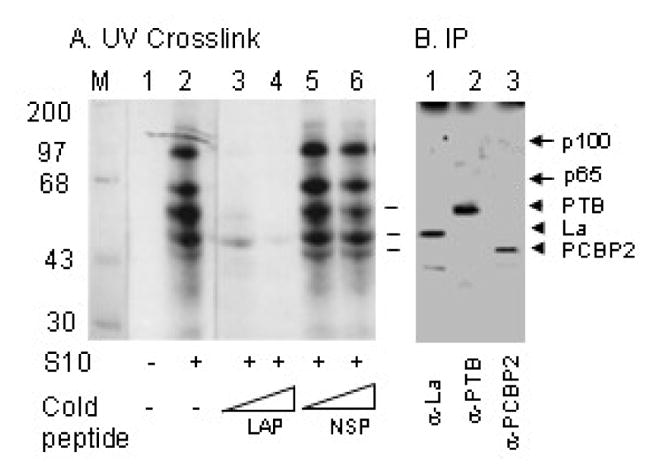
(A) An S10 cell-free translation lysate from Huh-7.5 cells was used for RNA-protein interaction by UV-crosslinking assay using radiolabeled HCV 5′UTR RNA. UV-crosslinked protein-nucleotidyl complexes in the absence (lane1) and presence (lanes 2–6) of S10 containing 40 and 60 μM LAP (lanes 3 and 4) or a non-specific peptide, NSP (lanes 5 and 6) were analyzes by SDS-PAGE. (B) Three pooled reactions as in lane 2 above were used for individual immunoprecipitation of protein-nucleotidyl complexes with anti-La (lane 1), anti-PTB (lane 2), and anti-PCBP2 (lane 3) antibodies.
In order to gain insight into the nature of proteins whose interactions with the HCV 5′UTR are blocked by LAP, we used immunoprecipitation of the UV-crosslinked complexes using antibodies to known IRES-transacting factors (ITAFs). As can be seen in figure 2B, at least three UV-crosslinked protein-nucleotidyl complexes having approximate molecular masses of 57, 52 and 45 kDa could be immunoprecipitated with antibodies to polypyrimidine tract binding protein (PTB), Lupus autoantigen (La), and poly(rC) binding protein 2 (PCBP2), respectively. The identity of the two other major protein-nucleotidyl complexes, p100 and p65 is not known at present. However, we believe that the 65 kDa protein, which is cross-linked to HCV 5′UTR could be the NS1-associated protein 1, which binds to HCV RNA core-encoding sequences downstream of the HCV IRES and stimulates HCV IRES-mediated translation (Kim et al., 2004). A100 kDa protein, which interacts with and stimulates translation programmed by the rhinovirus 5′UTR, was identified to be nucleolin in a previous study (Izumi et al., 2001). It remains to be seen whether the 100kDa protein we detected here (Fig. 2) is actually nucleolin. These results suggest that LAP interferes with binding of ITAFs to the HCV IRES element.
Interaction of LAP with ITAFS in vitro
To determine if LAP directly interacts with PTB, PCBP2 and La, we utilized gel exclusion chromatography capable of separating free LAP from ITAF-bound LAP. For this purpose each recombinant ITAF was expressed in bacteria and the His-tagged proteins were purified by affinity chromatography. Fig. 3B shows two peak fractions from affinity column chromatography containing His-La, His-PTB, and His-PCBP2 proteins. In addition to the full-length wt proteins, we also expressed and purified the N-terminal (amino acids 1–327) and C-terminal (amino acids 328–557) fragments of PTB as well as two La mutants: ΔN28La (N-terminal 28 amino acids deleted) and Y23QLa. We also purified the LAP peptide, which runs on SDS-PAGE as an 8–9 kDa peptide (actual molecular weight 2 kDa). Identity of each recombinant protein was verified by immunoblot analysis using anti-His antibody (data not shown).
Figure 3. Expression and purification of His-tagged recombinant ITAFs.

(A) The schematics of domain structures of wt La, Y23Q La, ΔN28 La, wt PTB, PTB N3, PTB C7 and PCBP2 are shown. The amino acid sequences of wt LAP and y23Q La are shown at the top. (B) Various wt or mutant ITAFs tagged with 6X His as indicated were expressed in bacteria and purified by Ni-affinity column chromatography. The fractions containing recombinant proteins eluting from the final Ni-column were examined by SDS-PAGE followed by coomassie blue staining. The two peak fractions from each preparation used in the following assays are shown. Western blot analysis of FITC-LAP at two concentrations using anti-La antibody is shown.
Interaction of each purified protein with FITC-labeled LAP (Izumi et al., 2004) was examined by gel filtration of the reaction mixture following incubation of the protein with FITC-LAP. Under the assay condition, the majority of free FITC-LAP eluted between fractions 20–33 (Fig. 4B). When incubated with highly purified PTB prior to gel filtration, a significant fraction of the FITC-LAP eluted between fractions 5 through 9 (Fig. 4A). Immunoblot analysis of these fractions using anti-PTB showed co-elution of the PTB with the FITC-LAP (Fig. 4C), suggesting formation of FITC-LAP: PTB complex. Similar results were observed when highly purified PCBP2 was incubated with FITC-LAP followed by gel filtration; the majority of the PCBP2 eluted between fractions 6 through 10, which coincided with the protein bound FITC-LAP elution (Figs. 4A and D). Unlike PTB and PCBP2, significantly smaller quantities of FITC-LAP was found to co-elute from the column with the wt La protein between fractions 6–9 (Figs. 4A and E). In contrast to the ITAFs, ovalbumin, which eluted from the gel filtration column between fractions 7–10, did not bind any FITC-LAP (Figs. 4A and F). These results suggest that LAP interacts with PTB, PCBP2 and to some extent with La. It is interesting to note that another 24 amino acids long synthetic peptide, La R2C, derived from La RRM (amino acids 112–184) was recently reported (Mondal et al., 2008). This peptide also inhibited HCV IRES-mediated translation, but unlike LAP, it retained RNA binding ability and competed with La protein binding to the HCV IRES.
Figure 4. LAP-ITAF interaction.

Various purified His-ITAFs were incubated individually with purified FITC-LAP as described in materials and methods. The final concentrations of ITAFs and FITC-LAP in a reaction volume of 100 μl were 12 μM and 28 μM, respectively. The specific activity of FITC-LAP was 10 fluorescent units per pmole. The reaction mixtures were passed through a Sepahcryl S-200 gel filtration column. Elution profiles of FITC-LAP in the absence (panel B) and in the presence of PTB (PTB: FITC-LAP), PCBP2 (PCBP2: FITC-LAP), La (La: FITC-LAP), and ovalbumin (Ovalbumin: FITC-LAP) (panel A) are shown. Fractions 3–10 from gel filtration columns with individual ITAF were examined by Western blot using anti-His antibody. Panel F shows elution profile of ovalbumin as determined by silver staining of the gel.
PTB is a modular protein, with four noncanonical RNA binding domains (RNA Recognition Motifs, RRM) that are tethered together by conserved linker domains (Fig. 3). The NMR structures of all four RRMs have been resolved (Conte et al., 2000; Simpson et al., 2004). It is not known if the RRMs function independently or synergistically, nor is it understood how they are positioned with respect to each other in the free protein or on an RNA substrate. During poliovirus infection, the viral protease 3CDpro cleaves PTB within the linker between RRM2 and RRM3. This separation of PTB into two fragments suggests that each could be used as independent units by the virus, and perhaps also by the cell. To gain a better understanding of how LAP interacts with PTB that leads to its RNA binding inability, we expressed and purified two fragments of PTB: one having the N-terminal sequences having RRMs 1 and 2 (called N3 PTB), and the other having the C-terminal sequences containing RRMs 3 and 4 (called C7 PTB) (Fig. 3). We then examined interaction of each of these PTB fragments with FITC-LAP by analyzing the protein-FITC-LAP complexes following gel filtration. As can be seen in Fig. 5, although both fragments were defective in interacting with FITC-LAP compared to the wt PTB, small but significant binding of both N3 and C7 PTB was detected. However, the areas under the N3PTB: FITC-LAP and C7PTB: FITC-LAP complexes were significantly reduced compared to that with the wt PTB. The protein profiles of these fractions as determined by immunoblotting using anti-His showed that the high molecular weight fractions containing small but significant amount of FITC-LAP indeed contained the truncated proteins (Fig 5B–D). These results suggest that the entire PTB molecule is required for efficient interaction with LAP.
Figure 5. Interaction of wt PTB, N3PTB and C7 PTB with FITC-LAP.

The elution profiles of PTB, N3 PTB, and C7 PTB in complex with FITC-LAP are shown in panel A. Panels B, C, and D show elution of wt, N3, and C7 PTB, respectively as determined by immunoblot analysis.
PTB and PCBP2 but not La completely rescue LAP-mediated translation inhibition from the HCV IRES
We addressed the functional significance of LAP-ITAF interactions by examining whether the purified ITAFs could rescue LAP-mediated inhibition of translation programmed by the HCV 5′UTR. Cell-free translation lysates prepared from Huh-7 cells were used for in vitro translation of HCV 5′UTR-CAT mRNA in the absence or presence of various His-tagged, purified ITAFs. As can be seen in Fig. 6A, exogenous addition of Huh-7 cell-free extracts to LAP-inhibited translation reactions completely restored translation from the HCV 5′UTR. Purified His-La, on the other hand, was only partially active in rescuing LAP-mediate translation inhibition (Fig. 6B). Addition of even higher concentrations of His-La inhibited translation non-specifically (data not shown). The purified His- La protein, however, was functional since it could restore translation of poliovirus mRNA in rabbit reticulocye lysates, which lacks the La protein (Fig. 6C). These results suggest that a functionally active La is unable to fully restore LAP-induced inhibition of HCV IRES-mediated translation. In contrast to the La protein, both PCBP2 and PTB could fully rescue LAP inhibition of HCV 5′UTR-mediated translation (Fig. 6D and F). Like His-La, addition of higher concentrations of His-PTB and His-PCBP2 also inhibited translation (data not shown). Addition of PCBP2 or PTB to the translation lysates in the absence of LAP only marginally stimulated translation from the HCV 5′UTR (Figs. 6E and G), suggesting that the cell-free lysates were not significantly limiting in these proteins. Ovalbumin, which did not form a detectable complex with FITC-LAP (Fig. 4), was almost totally ineffective in restoring LAP-mediated translation inhibition (Fig. 6H). LAP had no significant effect on cap-dependent translation from a capped-CAT RNA. In fact, marginal stimulation of cap-dependent translation was observed in the presence of LAP (Fig. 6-I, lanes 1 and 2). Under the condition of our assay, addition of La, PTB, or PCBP2 individually to the reaction did not significantly affect cap-dependent translation in the presence or absence of LAP (Fig. 6-I, lanes 3–8). Thus, a direct correlation between translation rescue by the purified ITAFs (Fig. 6) and their ability to form complex with LAP (Fig. 4) was observed. While PTB and PCBP2 were found to completely rescue LAP-induced inhibition of in vitro translation programmed by the HCV 5′UTR, the purified La protein only partially rescued HCV IRES-mediated translation in the presence of LAP, suggesting PTB and PCBP2, but not La, were mainly being sequestered by LAP leading to inhibition of HCV IRES-mediated translation.
Figure 6. PTB and PCBP2 restore LAP-induced inhibition of HCV IRES-mediated translation in vitro.

(A) In vitro translation of HCV 5′UTR-CAT mRNA in translation lysates from Huh-7.5 cells in the absence (lane 1) and presence (lanes 2–5) of 60 μM LAP at 15, 30 and 60 μg of additional lysates (lanes 3, 4 and 5). (B) Translation from HCV 5′UTR-CAT RNA in the absence (lane 1) and presence (lane 2–4) of 60 μM LAP with 500 nM and 1 μM added purified La protein (lanes 3 and 4). (C) Poliovirus IRES (5′UTR-CAT)-mediated translation in reticulocyte lysates was performed in the absence (lane 1) and presence of 250 nM, 500 nM, and 1μM purified La (lanes 2, 3 and 4) and 1 μM purified Y23Q La (lane 5). (D) In vitro translation from HCV 5′UTR-CAT in the absence (lane 1) and presence of 60 μM LAP (lanes 2–4) with exogenously added 275 and 550 nM purified PCBP2 (lanes 3 and 4). (E) Translation from HCV 5′UTR-CAT in the absence (lane 1) and presence of 275 nM (lane 2) and 550 nM (lane 3) PCBP2. (F) In vitro translation from HCV 5′UTR-CAT in the absence (lane 1) and presence of 60 μM LAP (lanes 2–4) with exogenously added 250 and 500 nM purified PTB (lanes 3 and 4). (G) Translation from HCV 5′UTR-CAT in the absence (lane 1) and presence of 250 and 500 nM PTB (lanes 3 and 4) (H) In vitro translation from HCV 5′UTR-CAT in the absence (lane 1) and presence of 60 μM LAP (lanes 2–4) with exogenously added 275 and 550 nM purified ovalbumin (lanes 3 and 4). (I) In vitro cap-dependent translation of capped-CAT RNA in the absence (lanes 1, 3, 5, 7) and presence (lanes 2, 4, 6, 8) of 60 μM LAP with 500 nM PTB (lanes 3 and 4), 550 nM PCBP2 (lanes 5 and 6), and 500 nM La (lanes 7 and 8).
The Y23Q and ΔN28 La mutants sequester LAP efficiently and restore LAP-induced translation inhibition in vitro
We were surprised by the fact that LAP did not interact efficiently with the wt La protein, while it interacted efficiently with two other ITAFs (PTB and PCBP2) (Fig. 4A). A curious finding was that, while the wt La protein bound very little FITC-LAP, both the Y23Q and ΔN28La mutants were as active as PTB (or PCBP2) in sequestering FITC-LAP in the gel filtration assay (Fig. 7). The gel filtration profile of these two La mutants suggested that the single amino acid change in the La protein or the deletion of the N-terminal 28 amino acids altered the structure of these proteins in such a way that they were eluted from the gel filtration column ahead of the wt La protein (Fig 7B). The structural changes induced by these mutations most probably were responsible for renewed LAP binding ability of the mutant La proteins.
Figure 7. The La N-terminal 28 amino acid deletion or Y23Q substitution stimulate interaction with FITC-LAP.

The elution profiles of Wt La: FITC-LAP, ΔN28La: FITC-LAP, and Y23QLa: FITC-LAP are shown in panel A. Panel B shows immunoblot analyses of eluted wt and mutant La proteins in relevant column fractions. Panel C shows rescue of LAP-inhibited HCV 5′UTR-mediated in vitro translation by wt La, ΔN28La, and Y23Q La proteins. All reactions in this experiment contain 60 μM LAP. Averaged results from two separate experiments are shown.
To determine if the renewed ability of the La mutants to bind LAP efficiently could also lead to their ability to restore HCV IRES-mediated translation in LAP-inhibited (translation) extracts, various amounts of the purified, recombinant, His-tagged Y23Q and ΔN28 La mutant polypeptides were added to LAP-inhibited translation extracts. As can be seen in Fig. 7C, LAP-induced inhibition of HCV IRES-mediated translation could be almost fully reversed by both La mutants, but not by the wt La protein. Thus, as found with the wt PTB and PCBP2 proteins, the ability to sequester LAP by the La mutants resulted in rescue of translation in LAP-inhibited translation extracts. These results suggest that deletion of the La N-terminal 28 amino acids or substitution of the La Y23 with Q leads to significant change in protein structure resulting in efficient La-LAP interaction (Fig. 7B) leading to reversal of translation.
PTB and PCBP2 rescues LAP-mediated inhibition of HCV replication in cell culture
To determine whether ITAFs are able to rescue LAP-mediated inhibition of JFH1 replication in cultured Huh-7.5.1 cells, the effects of PTB and PCBP2 expression on NRFLC virus replication were examined in control buffer- and LAP-treated cells. When expressed individually, slight recovery of virus replication in LAP-treated cells, as measured by RLuc production, was observed for both pCPTB and pCPCBP2 compared to the empty vector (pCVector). Inclusion of both pCPTB and pCPCBP2 during transfection, however, resulted in significant recovery of virus replication in LAP-treated cells compared to that in buffer-treated (no LAP) cells. The slight increase in virus replication seen in control buffer-treated cells transfected with both pPTB and pPCBP2 compared to the empty vector-treated cells was possibly due to presence of limiting amounts of these ITAFs in Huh-7.5.1 cells. In contrast to wt NRFLC, the expression of PTB and PCBP2 did not have any significant effect on NRLFC pol-null mutant replication indicating the ability of the ITAFs to reverse LAP-mediated inhibition required active replication of the NRFLC virus. Our cell culture data suggest that PTB and PCBP2 act synergistically in rescuing LAP-mediated inhibition of HCV replication in tissue culture cells (Fig. 8). This is in contrast to our in vitro data where LAP-induced inhibition of HCV IRES-mediated translation could be almost completely rescued by individual ITAFs (Fig. 6). We do not know the precise reason for this discrepancy. One possibility could be that while the in vitro assay measures only translation, the cell based assay measures both translation and multiple cycles of virus replication. How these processes are precisely influenced by LAP as well as ITAFs under these two different assay conditions is not known and could contribute to the observed difference.
Figure 8. Transient expression of PTB and PCBP2 can partially restore LAP-mediated inhibition of JFH1 HCV replication in cell culture.

Ten microgram of in vitro transcribed genomic RNA of wild-type NRLFC reporter virus was co-transfected into control buffer- or LAP-treated Huh-7.5.1 cells with empty mammalian expression vector (pCVector), pCPTB, or pCPCBP2, or both pCPTB and pCPCBP2 as indicated. The mutant reporter virus lacking polymerase activity (pol-null) was included as control. The transfected cells were lysed at 96h post-transfection using Promega passive lysis buffer and the levels of Renilla luciferase were quantified. Three experiments were done in triplicate and the mean values with standard deviation of Renilla luciferase values (RLV) are presented as a bar graph in log10 scale. The data were also corrected with respect to transfection efficiency and extract protein concentration.
The lifecycle of RNA viruses could be potentially interfered with at various stages such as virus-receptor interaction, viral entry, viral protein synthesis, polyprotein processing, RNA synthesis, viral assembly/morphogenesis and release. We have described here the utility of a small, cell-permeable and stable peptide inhibitor, which specifically blocks IRES-mediated translation of RNA viruses that utilize cap-independent translation to synthesize viral proteins. We have shown that the eighteen amino acid long peptide efficiently blocks replication of hepatitis C virus (HCV) in tissue culture. The LAP appears to block viral IRES-mediated protein synthesis by sequestering mainly two IRES-transacting factors: polypyrimidine track binding protein (PTB) and poly(rC) binding protein 2 (PCBP2). These results are consistent with previously described roles of PTB and PCBP2 in HCV RNA replication (Aizaki et al., 2006; Chang and Luo, 2006; Fukushi et al., 2001a; Randall et al., 2007; Zhang et al., 2004). Although our previous data (Izumi et al., 2004) and the results presented here suggest that LAP interferes with HCV IRES-mediated translation possibly through PTB and PCBP2, we cannot rule out the possibility that LAP could also interfere with HCV RNA synthesis. In fact, the roles of both proteins (PTB and PCBP2) in HCV RNA synthesis have been suggested by a number of reports (Aizaki et al., 2006; Chang and Luo, 2006; Domitrovich et al., 2005; Fukushi et al., 2001a). Thus, sequestration of PTB and PCBP2 by LAP could potentially block viral RNA synthesis in addition to viral translation.
Materials and Methods
Cells
The Huh-7.5 and Huh-7.5.1 cell lines (kindly provided by Dr. Francis Chisari, The Scripps Research Institute, La Jolla, and Dr. Charles Rice, Rockefeller University, NY) were cultured in complete DMEM containing 10% fetal bovine serum, 10 mM non-essential amino acids (Invitrogen), 10 mM Hepes, penicillin (100 units/ml), streptomycin (100 mg/ml), and 2 mM L-glutamine at 37°C with 5% CO2.
Peptides and FITC labeling of peptides
LAP peptides were synthesized and purified to >95% homogeneity by Bio-Synthesis, Inc. For fluorescein isothiocyanate (FITC) labeling, the peptides were dissolved in phosphate-buffered saline (PBS; pH 8.0). Peptides were labeled using FluoReporter FITC labeling Kit (F-6434, Molecular Probes), according to manufacturer’s specifications with a slight variation. After labeling, peptides were purified using Quick Spin RNA Purification Column (Roche) (24) Aliquots were stored protected from light at −80°C. For cell treatment, peptides were dissolved in 100mM Tris pH 8.0 to 5mM as previously described (Izumi et al., 2004).
LAP treatment of cells
Before transfection or infection, the cells were pretreated with various concentrations of LAP, Y23Q LAP or a non-specific scrambled peptide (NSP) for 2 h. Cells were then washed three times with PBS to remove excess peptide, and either infected with JFH1 or transfected with appropriate plasmids. At appropriate times post transfection or infection, the cells were examined by confocal microscopy following immunofluorescence using anti-NS5A.
Virus and Plasmid Constructs
The plasmid containing the complete genome of a HCVGT2a strain JFH-1 was kindly provided by Dr. Takaji Wakita, National Institute of Infectious Diseases, Japan (Wakita et al., 2005). An intra-genotype chimeric virus, pJ6/JFH-C, comprising 5′NTR, structural regions and part of non-structural regions (p7 and partial NS2) of the J6CF strain (NCBI accession no. AF177036) and non-structural regions of JFH-1 strain was generated (Arumugaswami et al., 2008). The J6CF genomic region, nt 1 to 2878, was synthesized by PCR based assembly of oligonucleotides (Invitrogen). The T7 promoter sequence (5′-TAATACGACTCACTATAG-3′) and nt 2879 to 2967 of the JFH-1 isolate was engineered at the 5′ and 3′ ends of the J6CF 1–2878 fragment, respectively. The final assembled PCR product (T7-J6CF/JFH) was cloned into pZero-blunt vector (Invitrogen) and sequence verified. The EcoRI-NotI fragment (2.9 kb) of the pJFH-1 was swapped with the assembled T7-J6CF/JFH fragment to obtain pJ6/JFH-C. A monocistronic chimeric reporter virus, pNRLFC based on pJ6/JFH-C parental virus was constructed. A plasmid containing a reporter cassette, T7-5′NTR (388 nucleotides)-Renilla luciferase gene-F2A seqeuce-Core-E1, was constructed. The F2A sequence introduced was 5′-GTGAAACAGACTTTGAATTTTGACCTTCTCAAGTTGGCCGGAGACGTCGAGTC CAACCCTGGGCCC-3′. The EcoRI-BsiSWI fragment containing the reporter cassete was subcloned into pJ6/JFH-C to construct pNRLFC. To construct the envelope-null mutant virus, an in-frame deletion of nt 1040–2215 was engineered in the pJ6/JFH-C genome. This deletion removed most of the E1 and E2 coding regions. An identical E1 and E2 deletion mutant reporter virus pNRLFC was also constructed. An RNA polymerase-null virus with pJ6/JFH-C or pNRLFC background was constructed by mutating the catalytic residues GDD to AAG amino acid residues. The NotI restriction enzyme site present in JFH1 genome at nt 2955 was abolished by PCR-mediated introduction of silent point mutations (CGGC->TGGT). These point mutations did not affect virus infectivity (data not shown)
Virus production and Infection
pJFH-1 plasmid (Wakita et al., 2005) was linearized by XbaI restriction digestion, and treated with MungBean nuclease (New England BioLabs). 1ug linearized DNA was in vitro transcribed using T7 RiboMAX™Large Scale RNA Production System (Promega). 10ug transcribed RNA was electroporated into 106 Huh 7.5.1 cells and virus production monitored. Highest titer virus was used for following infection experiments. Huh 7.5.1 cells were seeded in 48 well plates; when cell confluency reached ~30%, they were treated overnight with LAP peptide in a total volume of 300uL. Cells were infected at an moi of 0.1 to 1 and adsorption allowed for 4hrs, at which time cells were washed and fresh media was added with or without peptides. After 72hrs, samples were collected and assayed for viral RNA and viral protein after cell lysis. Quantification of intracellular virus production and secreted virus were determined as previously described (Arumugaswami et al., 2008). Briefly, cell-free culture supernatants or freeze-thawed lysates were serially diluted and used to infect 3 × 103 naïve Huh 7.5.1 cells in a 96 well plate. After 72 hrs cells were washed with cold PBS, fixed with methanol for 30 min at room temperature and stained for viral protein NS5A for further analysis of foci formation using immunofluorescence.
Determination of virus titer
The virus titer was measured as previously described (Arumugaswami et al/, 2008) by calculating the foci forming unit (ffu) of infectious viral particles per ml of cell-free culture supernatant (Zhong et al., 2005). The infected culture supernatant was 10-fold serially diluted in complete DMEM and inoculated in triplicate onto naïve Huh-7.5.1 cells (3 × 103 cells/well) in 96-well plates. At 72 or 96 hpi, the cells were fixed and immunostained for HCV core antigen. The number of core antigen positive foci were counted at the highest dilution and average foci forming units per ml was calculated.
Renilla Luciferase Reporter Assay for Viral Genome Replication and Infectivity
For viral genome replication assay, the HCV RNA transfected cells were plated in triplicates in 48-well plates. The cells were lysed with passive lysis buffer (Promega) at 6 and 96 hpt. The culture plates were gently rocked at room temperature for 15 min, then stored at −80°C. To determine the supernatant infectivity, 500 μl of cell-free supernatant obtained from HCV RNA transfected cells at 48 and 96 hpt was inoculated in triplicate onto naïve Huh-7.5.1 cells in 48-well plates. At 48 hpi the cells were lysed and stored at −80°C. 10 μl of lysate was used for measuring the Renilla luciferase activity using a Renilla Luciferase Assay System kit (Promega) as previously described (Arumugaswami et al., 2008).
Plasmid construction and protein expression
His-tagged proteins were cloned into the pET-100/D TOPO® Directional Expression System according to manufacturer’s specifications (Invitrogen). Primers (IDT DNA, Coralville, IA) used for cloning of the La proteins are as follows: La/Y23Q fwd (5′c acc atg gct gaa aat ggt gat aat gaa aag atg g 3′), LaΔ28 fwd (5′ c acc aat ttg cca cgg gac aag ttt cta aag 3′) and La/Y23Q/Δ28 rev (5′ cta ctg gtc tcc agc acc att ttc tg 3′). Primers for PTB and truncated proteins: PTB/PTBΔC fwd (5′ c acc atg gac ggc att gtc cc 3′), PTBΔN fwd (5′ c acc aac gtc cac ggc gcc c 3′), PTB rev (5′ cta gat ggt gga ctt gga gaa gga gac c 3′) and PTBΔC rev (5′ cta aac gga aag gcc tgc agc ttg agg 3′). Primers for PCBP2 fwd (5′ c acc atg gac acc ggt gtg att gaa gg 3′) and PCP2 rev (5′ cta gct gct ccc cat gcc acc cg 3′). Proteins were overexpressed in E. coli BL21 star (DE 3) cells (Invitrogen). Briefly, overnight bacterial cultures were inoculated into 1L fresh LB media; when the OD reached 0.6–0.8, protein expression was induced by addition of 1uM IPTG for 4 hrs at 37°C. Cells were collected by centrifugation and stored at −80°C for later protein purification. For expression in tissue culture cells, the cDNAs were subcloned into pCDNA3.1.
Protein purification
Bacterial pellets were lysed in 30mL Resuspension Buffer (25mM Tris, 400mM NaCl, 10% glycerol, pH=7.8, protease inhibitor cocktail) with 75ug lysozyme and incubated for 1hr at room temperature under constant shaking. NP-40 (1%) was added and incubated at 4° for 2hr under constant shaking. The samples were then sonicated and centrifuged at 12K rpm for 30mins. DNA was precipitated with 3% streptomycin sulfate and lysate cleared by centrifugation at 12K rpm for 30 mins. Pre-equilibrated beads (ProBond, Invitrogen) were added to lysate and incubated for 2hrs. Beads were packed into columns (Poly-Prep Chromatography Columns, Bio-rad) and washed with 40mL Wash Buffer (25mM Tris, 15mM Imidazole, 300mM NaCl, 0.05% NP-40, 10% glycerol, pH=6.8). Proteins were eluted using an imidazole gradient: 50–300mM imidazole, and dialyzed overnight against Binding Buffer (25mM Tris, 80mM KCl, 1.5mM Mg(OAc)2, 2.5mM DTT, 5% glycerol, 0.025 NP-40, pH, 7.5). Purified proteins were concentrated, aliquoted and stored at −80°C for further binding assays.
Preparation of cell-free translation extracts and in vitro translation
Trypsinized Huh-7.5 cells as described previously by Svitkin et al. (2005) were collected by centrifugation, washed (three times) and the cell pellet was resuspended in 2 volumes of a buffer containing 25 mM HEPES-KOH, pH 7.3, 50 mM KCl, 1.5 mM MgCl2, 1 mM dithiothreitol (DTT). The cells were allowed to swell on ice for 20 min and then were broken with 20 to 30 strokes of a tight-fitting Dounce homogenizer. One-ninth volume of the buffer containing 25 mM HEPES-KOH, pH 7.3, 1 M K(OAc), 30 mM MgCl2, 30 mM DTT) was added to the homogenate, and the homogenate was centrifuged at 18,000 × g for 20 min at 4°C. The supernatant was frozen in small aliquots and stored at −70°C. Translation reaction mixtures (20 μl) contained 9 μl of S10, 2 μl of a master mix (Svitkin et al., 2005), 2 μl of a salt solution (1 M KCl, 5 mM MgCl2, and 2.5 mM spermidine, unless otherwise specified), 1 μl of [35S]methionine (10 mCi/ml; 1,200 Ci/mmol), and 0.4 μg HCV RNA (unless otherwise specified). Translation in RRL was performed as recommended by the manufacturer (Promega), with the exception that KCl and MgCl2 were included in the reaction cocktail (at 100 mM and 0.75 mM final concentrations, respectively). The reactions were carried out at 32°C for 3 h.
Protein Binding Assay and Gel Filtration
For Binding Assay, 75ug purified protein was incubated with 10uL labeled peptide in Binding Buffer; then, incubated at 37°C for 2hrs. Sample was loaded into a previously equilibrated Gel Filtration Column (Sephacryl S-200 HR, Amersham Biosciences). Fractions were eluted in 200ul fractions and samples analyzed for fluorescence (VersaFluor, Bio-rad) and western blotting.
UV crosslinking assay
Recombinant wt La, PTB, PCBP2 and their mutants were incubated with radiolabeled HCV 5′-UTR (~106 cpm), for 10 min at 30°C. After binding, the reactions were processed as described previously (24).
Western Blotting
Protein samples were ran on Tris-HCl ReadyCast gels (Bio-rad) and transferred to nitrocellulose membranes. Membranes were incubated with primary antibodies in 4% non-fat milk (FITC pAb – Abcam, Penta-His mAb-Qiagen, core pAB, α-tubulin mAb – T5168 Sigma-Aldrich), washed and incubated with secondary antibodies. Proteins were detected using the ImmunStar HRP Detection Kit (Bio-rad).
Immunofluorescence
After methanol fixation, plates were stored at −20°C until further processing. Cells were washed with PBS and blocked overnight at 4°C with Blocking Buffer (3% FBS, 3%BSA, 0.1% Triton X-100 in PBS). Then, cells were incubated with our NS5A polyclonal antibody (1:300) for 1.5hr at room temperature, followed by secondary goat anti-rabbit (#111-116-144, Jackson Immunologicals). After washing with secondary antibody, cell nuclei were stained with DAPI (10ug/mL) for 10 minutes, and samples stored in PBS for examination under fluorescent microscope.
RNA purification and RT-PCR
RNA from infected cells was extracted with TRIzol Reagent (Invitrogen) according to manufacturer’s specifications. Sample was dissolved in nuclease-free water and further used for RT-PCR using SuperScript™III One-Step RT-PCR (Invitrogen). Briefly, 30ng total RNA was used for 25uL reactions containing, 10pmole each NS2 primer, and 1.5pmole each GAPDH primer. RT: 55°C for 40 min; PCR: 28 cycles of 94°C – 30sec, 57°C – 15sec, 68°C 1min. Primers: NS2 fwd (5′gac gca cct gtg cac gga cag 3′); NS2 rev (5′gga gct tcc acc cct tgg agg tg 3′); GAPDH fwd(5′ gag tcc act ggc gtc ttc acc ac 3′) and GAPDH rev (5′cag gtc agg tcc acc act gac ac 3′).
Acknowledgments
This work was supported by the RO1 Grants AI-45733 and AI-38056 to A.D., and the STTR Grant AI-45188 to Virasim Inc. from the National Institutes of Health. The authors are grateful to all the past and present members of the Dasgupta laboratory for their input and help throughout the years.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aizaki H, Choi KS, Liu M, Li YJ, Lai MM. Polypyrimidine tract binding proteinis a component of the HCV RNA replication complex and necessary for RNA synthesis. J Biomed Sci. 2006;13:469–80. doi: 10.1007/s11373-006-9088-4. [DOI] [PubMed] [Google Scholar]
- Ali N, Siddiqui A. Interaction of polypyrimidine tract binding protein with the 5′ non-coding region of the hepatitis C virus RNA genome and its functional requirement in internal initiation of translation. J Virol. 1995;69:6367–6375. doi: 10.1128/jvi.69.10.6367-6375.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali N, Siddiqui A. Molecular mechanisms of translation initiation in eukaryotes. Proc Natl Acad Sci USA. 1997;94:2249–2254. doi: 10.1073/pnas.94.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali N, Prujin GJM, Kenan DJ, Keene JD, Siddiqui A. Human La antigen is required for the hepatitis C virus internal ribosome entry site-mediated translation. J Biol Chem. 2000;275:27531–27540. doi: 10.1074/jbc.M001487200. [DOI] [PubMed] [Google Scholar]
- Arumugaswami V, Remenyi R, Kanagavel V, Yiang Sue E, Ngoc Ho T, Liu C, Fontanes V, Dasgupta A, Sun R. High-Resolution Functional Profiling of Hepatitis C Virus Genome. PLOS Pathogen. 2008;(10):e1000182. doi: 10.1371/journal.ppat.1000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsham GJ, Sonenberg N. Picornavirus RNA translation: roles for cellular proteins. Trends Microbiol. 2000;8:330–335. doi: 10.1016/s0966-842x(00)01788-1. [DOI] [PubMed] [Google Scholar]
- Blyn LB, Towner JS, Semler BL, Ehrenfeld E. Requirement of poly (rC) binding protein 2 for translation of poliovirus RNA. 1997;71:6243–6246. doi: 10.1128/jvi.71.8.6243-6246.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussadia O, Niepmann M, Creancier L, Parts AC, Dautry F, Jacquemin-Sablon H. Unr is required in vivo for efficient initiation of translation from the internal ribosome entry sites of both rhinovirus and poliovirus. J Virol. 2003;77:3353–3359. doi: 10.1128/JVI.77.6.3353-3359.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KS, Luo G. The polypyrimidine tract-binding protein is required for efficient replication of HCV RNA. Virus Research. 2006;115:1–8. doi: 10.1016/j.virusres.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Choo QL, Kuo G, Weiner A, Wang KS, Overby L, Bradley D, Houghton M. Identification of the major, parenteral non-A, non-B hepatitis agent (hepatitis C virus) using a recombinant cDNA approach. Semin Liver Dis. 1992;12:279–288. doi: 10.1055/s-2007-1007399. [DOI] [PubMed] [Google Scholar]
- Conte MR, Grune T, Ghuman J, Kelly G, Ladas A, Matthews S, Curry S. Structure of tandem RNA recognition motifs from polypyrimidine tract binding protein reveals novel features of the RRM fold. EMBO J. 2000;19:3132–3141. doi: 10.1093/emboj/19.12.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Ott M, Yamane A, Tsai W, Gromeier M, Lasher F, Gupta S, Dasgupta A. A small yeast RNA blocks hepatitis C virus internal ribosome entry site (IRES)-mediated translation and inhibits replication of a chimeric poliovirus under translational control of the HCV IRES element. J Virol. 1998;72:5638–5647. doi: 10.1128/jvi.72.7.5638-5647.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Kenan DJ, Bocskai D, Keene JD, Dasgupta A. Sequences within a small yeast RNA required for inhibition of internal initiation of translation: interaction with La and other cellular proteins influences its inhibitory activity. J Virol. 1996;70:1624–1632. doi: 10.1128/jvi.70.3.1624-1632.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta A, Das S, Izumi RE, Venkatesan A, Barat B. Targeting internal ribosome entry site (IRES)-mediated translation to block hepatitis C and other RNA viruses. FEMS Microbiology Letters. 2004;234:189–199. doi: 10.1016/j.femsle.2004.03.045. [DOI] [PubMed] [Google Scholar]
- Domitrovich AM, Diebel KW, Ali N, Sarker S, Siddiqui A. Role of La autoantigen and polypyrimidine tract-binding protein in HCV replication. Virology. 2005;335:72–86. doi: 10.1016/j.virol.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Fukushi S, Okada M, Stahl J, Kagemaya T, Hoshino FB, Katamaya K. Ribosomal protein S5 interacts with the internal ribosome entry site of hepatitis C virus. J Biol Chem. 2001;276:20824–20826. doi: 10.1074/jbc.C100206200. [DOI] [PubMed] [Google Scholar]
- Fukushi S, Okada M, Kageyama T, Hoshino FB, Nagai K, Katayama K. Interaction of poly(rC)-binding protein 2 with the 5′-terminal stem loop of the hepatitis C-virus genome. Virus Res. 2001a;73:67–79. doi: 10.1016/s0168-1702(00)00228-8. [DOI] [PubMed] [Google Scholar]
- Gamarnick AV, Andino R. Two functional complexes formed by KH domain containing proteins with the 5′ non coding region of poliovirus RNA. RNA. 1997;3:882–892. [PMC free article] [PubMed] [Google Scholar]
- Hellen CUT, Witherell GW, Schmid M, Shin SH, Pestova TV, Gil A, Wimmer E. A cytoplasmic 57 kDa protein that is required for translation of picornavirus RNA by internal ribosome entry is identical to the nuclear pyrimidine tract binding protein. Proc Natl Acad Sci USA. 1993;90:7642–7646. doi: 10.1073/pnas.90.16.7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen CUT, Sarnow P. Internal ribosome entry site in eukaryotic mRNA molecules. Genes Dev. 2001;15:1593–1612. doi: 10.1101/gad.891101. [DOI] [PubMed] [Google Scholar]
- Houghton, Weiner MAJ, Han G, Kuo G, Choo QL. Molecular biology of the hepatitis C viruses: implications for diagnosis, development and control of viral disease. Hepatology. 1991;14:381–388. [PubMed] [Google Scholar]
- Hunt HL, Kaminski A, Jackson RJ. unr, a cellular cytoplasmic RNA-binding protein with five cold-shock domains, is required for internal initiation of translation of human rhinovirus RNA. Genes Dev. 1999;13:437–448. doi: 10.1101/gad.13.4.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi RE, Valdez B, Banerjee R, Srivatava M, Dasgupta A. Nucleolin stimulates viral internal ribosome entry site-mediated translation. Virus Res. 2001;76:17–29. doi: 10.1016/s0168-1702(01)00240-4. [DOI] [PubMed] [Google Scholar]
- Izumi RE, Das S, Barat B, Raychaudhuri S, Dasgupta A. A peptide from autoantigen La blocks poliovirus and hepatitis C virus cap-independent translation and reveals a single tyrosine critical for La RNA binding and translation stimulation. J Virol. 2004;78:3763–3776. doi: 10.1128/JVI.78.7.3763-3776.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang SK, Krausslich HG, Nicklin MJH, Duke GM, Palmenberg AC, Wimmer E. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomesduring in vitro translation. J Virol. 1988;62:2636–2643. doi: 10.1128/jvi.62.8.2636-2643.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Paek KY, Ha SH, Cho S, Choi K, Kim CS, Ryu SH, Jang SK. A Cellular RNA-Binding Protein Enhances Internal Ribosomal Entry Site-Dependent Translation through an Interaction Downstreamof the Hepatitis C Virus Polyprotein Initiation Codon. Mol Cell Biol. 2004;24:7878–7890. doi: 10.1128/MCB.24.18.7878-7890.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger M, Berger C, Li QX, Welch PPJ, Tritz R, Levitt M, Barber JR, Wong-Staal F. Identification of eIF-2By and eIF-2y as cofactors for HCV IRES-mediated translation using a functional genomic approach. Proc Natl Acad Sci USA. 2000;97:8566–8571. doi: 10.1073/pnas.97.15.8566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolykhalov A, Agapov EV, Blight KJ, Mihalik K, Feinstone SM, Rice CM. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science. 1997;277:570–574. doi: 10.1126/science.277.5325.570. [DOI] [PubMed] [Google Scholar]
- Meerovich K, Svitkin YV, Lee HS, Lejbkowicz F, Kenan DJ, Chan EK, Agol VI, Keene JD, Sonenberg N. La autoantigen enhances and corrects abberant translation of poliovirus RNA in reticulocyte lysates. J Virol. 1993;67:3798–3807. doi: 10.1128/jvi.67.7.3798-3807.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal T, Ray U, Manna AK, Gupta R, Roy S, Das S. Structural determinant of human La protein critical for IRES translation of hepatitis C virus RNA. J Virol. 2008;82:11927–11938. doi: 10.1128/JVI.00924-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, Sonenberg N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature. 1988;334:320–325. doi: 10.1038/334320a0. [DOI] [PubMed] [Google Scholar]
- Pestova VIN, Shatsky SP, Fletcher RJ, Jackson R, Hellen CUTA. prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal initiation of translation of hepatitis C virus and classical swine fever virus RNAs. Genes Dev. 1998;12:67–83. doi: 10.1101/gad.12.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S, Landthaler M, Landgraf P, Kan S, Lindenbach BD, Chien M, Weir DB, Russo JJ, Ju J, Brownstein MJ, Sheridan R, Sander C, Zavolan M, Tuschl T, Rice CM. Cellular cofactors affecting hepatitis C virus infection and replication. Proc Natl Acad Sci U S A. 2007;104:12884–9. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito I, Miyamura T, Ohbayashi A, Harada H, Katayama T, Kikuchi S, Watanabe TY, Koi S, Onji M, Ohta Y, Choo QL, Houghton M, Kuo G. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma. Proc Natl Acad Sci USA. 1990;87:6547–6549. doi: 10.1073/pnas.87.17.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson PJ, Monie TP, Szendroi A, Davydova N, Tyzack JK, Conte MR, Read CM, Cary PD, Svergun DI, Konarev PV, et al. Structure and RNA interactions of the N-terminal RRM domains of PTB. Structure (Camb) 2004;12:1631–1643. doi: 10.1016/j.str.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Svitkin YV, Pause A, Lopez-Lastra M, Perreault S, Sonenberg N. Complete Translation of the Hepatitis C Virus Genome In Vitro: Membranes Play a Critical Role in the Maturation of All Virus Proteins except for NS3. J Virol. 2005;79:6868–6881. doi: 10.1128/JVI.79.11.6868-6881.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakita, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi M, Purcell RH, Emerson SU, Bukh J. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc Natl Acad Sci USA. 1997;94:8738–8743. doi: 10.1073/pnas.94.16.8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yamada O, Sakamoto T, Yoshida H, Iwai T, Matsushita Y, Shimamura H, Araki H, Shimotohno K. Down-regulation of viral replication by adenoviral-mediated expression of siRNA against cellular cofactors for hepatitis C virus. Virology. 2004;320:135–43. doi: 10.1016/j.virol.2003.11.023. [DOI] [PubMed] [Google Scholar]
- Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci USA. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]