

Abstract
Objectives
To define the role of protein kinase C δ (PKC δ) in acinar cell responses to the hormone cholecystokinin-8 (CCK) using isoform-specific inhibitors and a previously unreported genetic deletion model.
Methods
Pancreatic acinar cells were isolated from 1) rat, and pre-treated with a PKC-δ-specific inhibitor or 2) PKC δ deficient and wild type mice. Isolated cells were stimulated with CCK (0.001-100 nM) and cell responses measured.
Results
The PKC δ inhibitor did not affect stimulated amylase secretion from rat pancreatic acinar cells. CCK stimulation induced a typical biphasic dose response curve for amylase secretion in acinar cells isolated from both PKC δ -/- and wild type mice, with maximal stimulation at 10 pM CCK. CCK (100 nM) induced zymogen and NFkB activation in both PKC δ -/- and wild type mice, although it was up to 50% less in PKC δ -/-.
Conclusions
In contrast to previous studies, this study has used specific and complimentary approaches to examine PKC δ mediated acinar cell responses. We could not confirm that it mediates amylase release, but corroborated its role in the early stages of acute pancreatitis.
Introduction
In recent years, several studies have extensively characterized the roles played by the protein kinase C (PKC) family in both physiological and pathophysiological responses in the exocrine pancreas [1-4]. There are 10-12 isoforms of PKC divided into three classes: conventional (Ca2+ and 1,2 diacylglycerol (DAG)- dependent); novel (Ca2+ independent, DAG- dependent); and atypical (Ca2+ and DAG- independent; activated by phosphatidylinositol-3-kinase (PI3K) and phosphorylation events). In the pancreatic acinar cell conventional PKC α, novel PKC δ and ε, and atypical PKC ζ isoforms have been identified [5,6].
Many studies characterizing the diverse roles of PKC in the acinar cell have used pharmacological inhibitors which may lack specificity. For example, one study which examined inhibition of PKC δ with rottlerin demonstrated an inhibition of cholecystokinin (CCK)-stimulated amylase secretion at physiological concentrations of CCK (0.1nM) [7]. Subsequent studies, however, have suggested that rottlerin may affect non-PKC-δ-dependent mechanisms [8].
Under conditions that induce experimental pancreatitis, such as stimulation of acinar cells with high doses of CCK (100 nM) or its orthologue caerulein, novel PKC isoforms have been shown to mediate both activation of digestive zymogens and the transcription factor NFκB [2-4]. The isoform-specific inhibitors used in these studies prevent PKC δ and ε from translocating to their cellular targets. The specificity of these inhibitors has been extensively characterized although issues of specificity may remain. Li et al. addressed issues of inhibitor specificity by complementing their study with over expression of both PKC δ and a dominant negative form [7]. Over expression of PKC δ increased amylase secretion whereas over expression of the dominant-negative form decreased secretion; the latter finding validating their inhibitor studies. While the over expression approach provides a convincing alternative to pharmacological inhibition, non-specific, off-target effects from protein over expression cannot be ruled out. A feasible means to addressing some of these questions of specificity is to complement studies with a genetic deletion approach. Thus far, the effects of genetic deletion of PKC isoforms in pancreatic acinar cell responses to CCK have not been addressed.
In this study we further characterize the role of PKC δ in the pancreatic acinar cell using 1) PKC δ-isoform-specific translocation inhibitor in isolated acinar cells from rats and 2) PKC δ knockout mice. In both cases we measure the effects of PKC δ inhibition or deletion on stimulated amylase secretion. Further, we assessed the role of PKC δ deletion on zymogen and NFκB activation.
Materials and Methods
Animals
Male Sprague-Dawley rats (50–150 g) were obtained from Charles River Laboratories (Wilmington, MA) and housed at facilities at the Veterans Affairs Connecticut Health Care System, West Haven, CT. Male protein kinase Cδ deficient mice (-/-) and littermate wild type mice (+/+; C57BL/6 background) were bred in pathogen-free facilities at the Veterans Affairs Greater Los Angeles Health Care System (VAGLAHS), Los Angeles, CA. PKCδ deficient mice were generated by Miyamoto et al [9] by replacement of the first and second exons of the PKC gene with a neomycin-resistance cassette as described elsewhere [9]. PKCδ deficient mice were healthy, fertile, and were viable beyond 12 months of age with no signs of cancer or other diseases. Compared to wild type mice, PKCδ deficient mice showed similar levels of other PKC isoforms in pancreas (Figure 3) and other organs such as brain and inflammatory cells [9]. Animal use protocols were approved by the Institutional Animal Care and Use Committees of Yale University and the Veterans Administration in West Haven, CT, and VAGLAHS and the University of California in Los Angeles.
Figure 3. Genetic deletion of PKC δ does not affect expression levels of other PKC isoforms or production of digestive enzymes in mouse acinar cells.

Pancreata from PKC δ -/- and PKC δ +/+ were removed and protein extracts processed for Western blot analysis. Blots were probed with antibodies for A) PKC α, δ, ε and ζ isoforms and B) amylase, trypsinogen and GAPDH; representative blots are shown. These results confirm the absence of PKC δ in the knockout animals and that genetic deletion of PKC δ did not affect the expression of other PKC isoforms or the production of digestive enzymes.
Preparation of isolated pancreatic acini
Acini were isolated from rat or mouse pancreas as previously described [4]. Briefly, animals were euthanized by CO2. Acinar media was prepared as follows (in mM): 10 HEPES (pH 7.4), 95 NaCl, 4.7 KCl, 0.6 MgCl2, 1 NaH2PO4, 10 glucose, 2 glutamine, plus 0.1% BSA, 1× MEM-amino acids (GIBCO-BRL, San Jose, CA) and 1.3mM CaCl2. The pancreas was collected in 15ml of calcium-free acinar media. The pancreas was then minced in a minimal volume of calcium free medium for 5 min, washed 3 times with calcium free medium. The minced tissue was then placed into a 50-ml flask with 12 ml of acinar medium containing 50 U/ml of type-4 collagenase (Worthington, Freehold, NJ) for 60 min at 37°C with shaking (120 rpm). The digest was filtered through a 300-400-μm mesh (Sefar American, Depew, NY) and washed with acinar medium. Isolated acini (groups of 20-100 acinar cells) were distributed among the 24 wells (0.5 ml suspension/well) of a 24-well Falcon tissue culture plate (Becton Dickinson, Franklin Lakes, NJ). All reagents were purchased from Sigma, St. Louis, MO, unless otherwise noted.
Acinar experimental protocol
Acini were recovered for 120 min at 37°C under constant O2 with shaking (90 rpm). Medium was changed at 60 min. At 120 min acini were treated without changing medium. Acini were treated with CCK (0.001-100 nM) or carbachol (0.01-100 μM) for 30 min (unless otherwise noted). Samples were collected, placed in 1.5-ml centrifuge tubes (USA Scientific, Waltham, MA) and centrifuged for 1 min at 30 g. 50μl of the resulting cell-free supernatant was removed to a 0.5-ml microcentrifuge tube to assay for secreted amylase. The remaining 450μl of cells + media was retained for zymogen activation assays and determination of total amylase. All samples were stored at -80°C.
PKC Inhibitors
Rat acinar cells were treated with 10 μM of each of the following: GF109203X; rottlerin; isoform-specific PKC δ translocation inhibitor (δV1-1: S-F-N-S-Y-E-L-G-S-L); and a control scrambled peptide (L-S-E-T-K-P-A-V) for 120min prior to CCK stimulation. Each of these peptides was synthesized as an amino terminal extension to a Drosophila antennapedia peptide (R-QI-K-I-W-F-Q-N-R-R-M-K-W-K-K) to make it cell permeable [4].
Enzymatic activity assays
Enzyme activities were carried out as described [4]. Briefly, samples were thawed, homogenized and centrifuged. To each well of a 24-well plate (Greiner Bio-one Cellstar TC-Plate) the following was added: 100 μl of post-nuclear supernatant, 350 μl of trypsin assay buffer [50 mM Tris (pH 8.1), 150 mM NaCl, 1 mM CaCl2, 0.01% BSA]. The assay was initiated by adding 50 μl of 400 μM enzyme substrate (fluorometric trypsin substrate; Peptides International, Louisville, KY) diluted in trypsin assay buffer (40 μM final). The plate was read using a fluorometric microtiter plate reader (model HTS 7000; Perkin-Elmer Analytical Instruments, Shelton, CT. 380-nm excitation; 440-nm emission; 20 reads /10 min.)
Amylase assay
Amylase activity was determined by using a commercial kit (Phaebadas kit; Pharmacia Diagnostic, Rochester, NY). Amylase release was expressed as % total release [medium/(medium + cells)].
Immunoblot analysis
Western blot analysis was performed to detect PKC isoforms. Briefly, samples were separated on 10% SDS-PAGE gels (Bio- Rad, Hercules, CA) and transferred to Immobilon-P membranes (Millipore, Billerica, MA). Membranes were blocked for 60 min at room temperature with Blotto (TBS, 5% nonfat dry milk, 0.05% Tween-20). Membranes were then probed with primary antibody (rabbit anti-PKC α, δ, ε or ζ, 1:200; Santa Cruz Biotechnology, CA; amylase, Sigma-Aldrich, St. Louis, MO; trypsinogen, Millipore, Temecula, CA) in Blotto for 120 min at room temperature, washed and incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG (Sigma-Adrich, St. Louis, MO) for 60 min at room temperature. Membranes were washed in TBS, and signal was captured on film using a SuperSignal West Pico chemiluminescence kit (Pierce, Rockford, IL).
NFκB activation
Pancreatic acinar cells were isolated from PKC δ -/- and PKC δ +/+ and incubated for 30 min with CCK at a supra-physiologic dose of 100 nM. NF-κB activation was measured using the electromobility shift assay (EMSA) as described [2,3].
Statistical analysis
Data represent means ± SEM of at least 3 individual experiments unless otherwise noted, with each experiment being performed in at least duplicate. A Student's t-test analysis was used to determine statistical significance and P values of < 0.05 were assigned significance.
Results
Rottlerin but not GF109203X affect CCK-induced amylase release
CCK-induced amylase release from rat acini was first assayed in the presence of two PKC inhibitors, GF 109203X and rottlerin (Fig. 1). In contrast to an earlier study using CCK [7], GF109203X did not reduce amylase secretion. However, as shown previously [7], amylase secretion stimulated by both physiologic and supraphysiologic CCK was reduced in the presence of 10 μM rottlerin. Amylase secretion was reduced by approximately 30% with rottlerin compared to that seen with 0.1nM CCK alone. At a higher concentration of CCK (100 nM), rottlerin reduced amylase secretion by approximately 40%.
Figure 1. Rottlerin but not GF109203X affect CCK-induced amylase release.

Isolated acinar cells were pre-incubated with or without broad-spectrum PKC inhibitors GF109203X and rottlerin (10 μM) for 2 hours, followed by 30 min CCK treatment (0.1nM and 100nM). Amylase release is expressed as % total release [medium/(medium + cells)]. N=3; *p<0.05
Isoform-specific PKC δ translocation inhibitor does not affect CCK-induced amylase release
CCK-induced amylase release from acini was next measured in acini in the presence of 10 μM isoform-specific PKC δ translocation inhibitor. We previously demonstrated that this concentration effectively blocks the translocation of PKC δ required for activation without affecting other PKC isoforms (25). As shown in Figure 2a, there was no significant difference in amylase secretion over a concentration range of CCK (0.001-100 nM) in the presence of PKC δ inhibitor. Identical experiments using a scrambled peptide control yielded similar results (data not shown). An earlier study suggested that the effects of PKC δ are time dependent [7]; we observed amylase release over time at a fixed CCK concentration (100 nM) (Figure 2b). Again, no significant difference was observed in the presence of PKC δ inhibitor. Collectively, these findings suggest that PKC δ translocation is not required for CCK-induced amylase secretion in rat acinar cells.
Figure 2. Isoform-specific PKC δ translocation inhibitor does not affect CCK-induced amylase release.


Isolated acinar cells were pre-incubated with or without PKC δ translocation inhibitor (10 μM) for 2 hours, followed by a) 30 min CCK treatment over a concentration range (0.001nM - 100nM); b) 100 nM CCK treatment at set time points (0, 5, 15, 30 min). Amylase release is expressed as % total release [medium/(medium + cells)]. N=3.
CCK-induced amylase release is not reduced in pancreatic acinar cells isolated from PKCδ -/- mice in comparison with wild type
To address issues regarding PKC inhibitor specificity, we next examined responses using mice with genetic deletion of PKC δ -/-. We used two-month old male PKCδ (-/-) and their littermate wild type mice (+/+). Two-month old PKCδ deficient mice had similar body weight compared to littermate wild type mice and displayed normal pancreas morphology (not shown). As illustrated in Figure 3, pancreatic levels of PKC α, ε, and ζ, and amylase and trypsinogen were similar in the PKC δ −/− mice as compared to the wild type (+/+) mice. These results indicate that, at least in physiological conditions, genetic disruption of the PKC δ gene did not affect the expression of other PKC isoforms or the production of digestive enzymes.
Using isolated acini from PKCδ -/- and wild type (PKCδ +/+) mice we measured CCK-stimulated amylase release over a concentration range of CCK (0.001-100 nM) (Fig. 4a). In wild type mice, amylase release peaked at 0.01 nM CCK and subsequently decreased. There was no significant difference between amylase release when comparing PKCδ -/- and wild type (PKC δ +/+) mice although there was a tendency for the wild type to secrete less amylase than the knockout. In addition, acini from PKCδ -/- and wild type (PKCδ +/+) mice were stimulated with the muscarinic agonist carbachol, another activator of PKC, over a concentration range (0.01-100 μM) (Fig 4b). Carbachol-induced amylase release peaked at 1μM, subsequently decreasing. As with CCK-stimulated release, no significant difference was seen in amylase secretion between the two sets of mice. Regardless of how PKC δ is activated, (e.g. CCK vs. carbachol) these results indicate that it does not play a role in stimulated amylase secretion.
Figure 4. CCK- and carbachol-induced amylase release is not reduced in PKCδ -/- mice versus wild type (PKCδ +/+).
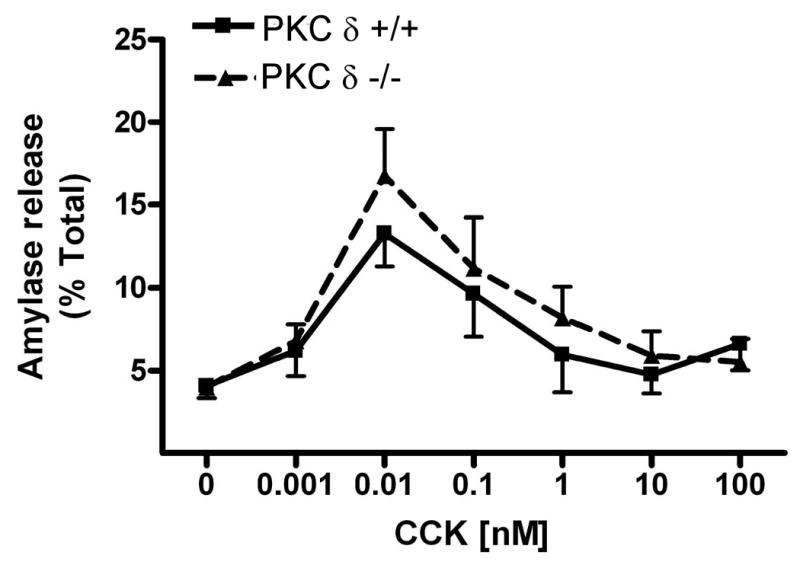

Pancreatic acinar cells were isolated from PKC δ -/- and +/+ mice and then stimulated for 30 min with a) CCK (0.001-100 nM); b) carbachol (0.01-100 μM). Amylase release is expressed as % total release [medium/(medium + cells)]. N=5.
CCK-induced trypsinogen activation is reduced in PKCδ -/- mice versus wild type
Acinar cells isolated from PKCδ -/- and wild type (PKC δ +/+) mice were stimulated with 100 nM CCK for 30 minutes and then assayed for trypsin activity. CCK-induced trypsinogen activation, as measured by trypsin activity, in the PKCδ -/- was reduced 45% compared to wild type (Fig. 5).
Figure 5. CCK-induced trypsinogen activation is reduced in PKCδ -/- mice versus wild type (PKCδ +/+).

Pancreatic acinar cells were isolated from PKC δ -/- and +/+ animals and then stimulated for 30 min with 100 nM CCK. Trypsin activity is normalized to amylase content and expressed as fold vs. control. N=6. A reduction in trypsin activity is seen in the knockout animals approaching significance.
CCK-induced NFκB activation is reduced in PKCδ -/- mice versus wild type
After 30 minutes incubation with a supraphysiologic dose of CCK (100 nM), NF-κB activity in isolated acinar cells was measured by EMSA. A representative EMSA is shown in Fig 6A. CCK stimulated NFκB activity was significantly less in PKC δ -/- acinar cells than in +/+ acinar cells by 50.9%, SEM 16.5% (Fig 6B; p< 0.050; PKC δ +/+ n = 5, PKC δ -/- n = 5).
Figure 6. CCK-induced NFκB activation is reduced in PKCδ -/- mice versus wild type (PKCδ +/+).
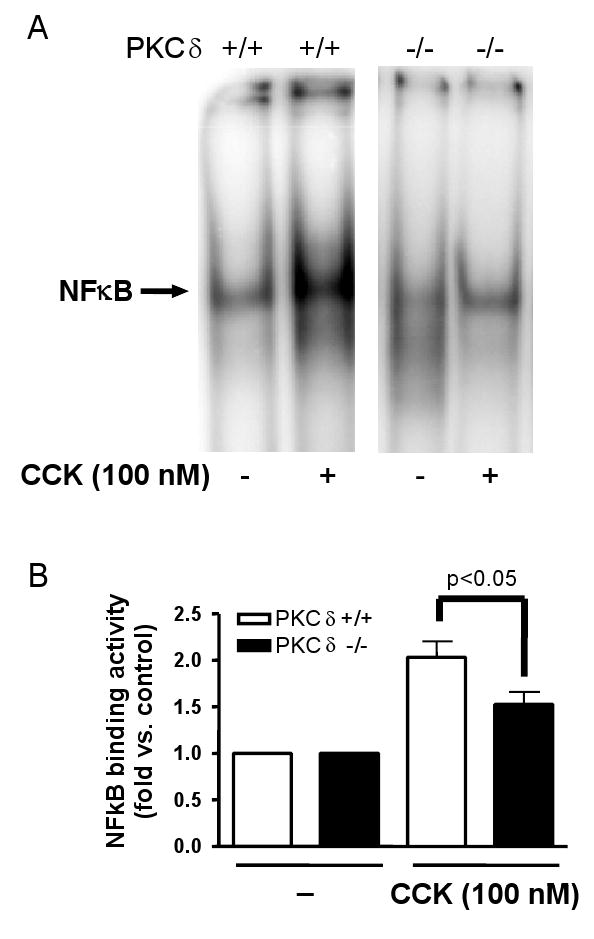
Pancreatic acinar cells were isolated from PKC δ -/- and +/+ animals and then stimulated for 30 min with 100 nM CCK. A) NFκB binding activity was measured in nuclear extracts by EMSA. B) NFκB band intensities were quantified in the PhosphorImager and normalized on the band intensity in the corresponding unstimulated control acini. Values are means +/-SEM (N= 5).
Discussion
PKC δ is the most widely-studied isoform of the PKC family and has a large distribution in a variety of tissues [10,11]. PKC δ participates in many cellular processes including cell growth inhibition and differentiation [12-14], apoptosis [15-19], tumor suppression [20] and motility [21,22]. In the rat pancreatic acinar cell it mediates premature zymogen activation and activation of the transcription factor NFκB during experimental pancreatitis [2,4] as well as mediating the effects of the neuropeptide substance P [23]. In the acinar cell, PKC δ has also been shown to activate a number of cellular targets including the focal adhesion tyrosine kinase PYK2/CAKβ and protein kinase D [24,25]. Lastly, PKC δ has been previously implicated as the key PKC isoform in stimulated secretion from the acinar cell [7].
The role of PKC δ has largely been determined by studies using pharmacological inhibitors believed to be specific for PKC δ [7,10,26-28]. One such inhibitor, the polyphenol rottlerin, was shown to inhibit PKC δ with greater specificity than that seen for other PKC isoforms [29]. However, subsequent studies have shown that this compound also inhibits other PKC isoforms and additional classes of protein kinase [8,30-33]. In a previous publication, amylase secretion induced by physiologic concentrations of CCK in the rat was significantly reduced by rottlerin and the broad spectrum PKC inhibitor GF109203X [7]. The present study has overcome the limitations of past studies by using both a new and specific PKC δ inhibitor and a previously undescribed PKC δ knockout mouse to examine the physiologic and pathologic effects of this enzyme.
In this study, PKC δ functions have been examined in both rat and mouse pancreatic acinar cells. Similar to a previous report, we found that pre-incubation with rottlerin significantly reduced amylase secretion stimulated by physiologic or supraphysiologic CCK. Premature zymogen activation, initiated by supraphysiologic CCK, did not decrease significantly with rottlerin pre-treatment (data not shown). Thus, data obtained using rottlerin as a PKC δ inhibitor is consistent with a role for PKC δ in stimulated amylase secretion, but not zymogen activation. However, in contrast to earlier studies, we observed no effect of broad spectrum PKC inhibitor GF109203X on CCK-stimulated amylase secretion at both physiologic and supraphysiologic concentrations (Fig. 1). Further, in earlier studies we have shown a significant reduction in CCK-induced activation of zymogens and NFκB after pre-treatment with the broad spectrum PKC inhibitor, GF [2,4]. Hence, data obtained from GF studies contradicts studies with rottlerin and suggests that PKC δ does not affect secretion, but can mediate activation of zymogens and NFκB.
To reconcile these conflicting findings, we employed an isoform-specific peptide inhibitor and a genetic deletion approach. We demonstrated that CCK-stimulated amylase secretion from rat pancreatic acinar cells, over a range of concentrations and time points, was unaffected by PKC δ inhibition using the isoform-specific inhibitor (Fig 2). Further, we demonstrated that genetic deletion of PKC δ in mice did not affect CCK- or carbachol-stimulated pancreatic amylase secretion over a range of concentrations (Fig 4). However, trypsinogen activation and NFkB activation were reduced in PKC δ -/- mice. This result is consistent with previous studies in rats during which PKC δ was inhibited using the isoform-specific translocation inhibitor (Fig 5 and 6) [2,4]. The data obtained from this approach would indicate that PKC δ does not play a major role in amylase secretion, but does participate in the early stages of acute pancreatitis.
Together our data also suggests that the effects we observed with rottlerin are through a PKC δ-independent mechanism and this reflects a non-specific effect of the inhibitor. Indeed, one study showed that effects of rottlerin could be mimicked by a classical mitochondrial uncoupler, FCCP indicating that ATP depletion maybe affecting acinar cell secretion, rather than direct PKC δ inhibition [8]. Our studies failed to confirm a role for PKC δ in stimulated secretion as shown by another study [7]. Though their initial assessments included the use of rottlerin, they substantiated their findings with over expression of PKC δ and a dominant negative PKC δ in cultured primary acinar cells. Overexpression of PKC δ enhanced CCK-stimulated amylase release and over expression of dominant negative PKC δ decreased it in isolated rat pancreatic acinar cells [7]. They further found that over expression of wild type PKC ε also enhanced amylase release but expression of dominant-negative PKC ε had no inhibitory effect. The authors suggested that PKC ε, when over expressed, may be substituting for PKC δ. Although the authors conclude that PKC δ is the dominant isoform in secretion, we cannot rule out that the over expression of PKC isoforms and their dominant-negative counterparts are having non-specific, off-target effects, as seen with over expression of PKC ε. Furthermore, the amylase release properties of cultured acinar cells can change over time. For example, in overnight cultured mouse and rat acini amylase release is reduced and higher concentrations of CCK are required to invoke maximal amylase release [7,34]. Although amylase release was detectable in these studies and effects of genetically manipulating PKC δ were observed, changes in the secretory behavior of the cultured acini or off target effects of the genetic manipulations may ultimately account for the findings.
In conclusion, by using recently developed PKC isoform specific inhibitors and a previously unreported PKC δ genetic deletion approach, this study provides a new perspective on the role of this enzyme in regulating acinar cell responses. Thus, we have used new approaches and find that PKC δ does not mediate amylase release, but have confirmed that it does modulate some of the key early acinar cell responses in acute pancreatitis. These observations might also have therapeutic implications. Thus, the initiation of acute pancreatitis appears to require both zymogen activation and the retention of active enzymes in the acinar cell. Together with this study, this suggests a testable hypothesis: that PKC δ inhibition would reduce zymogen activation, but not affect secretion in acute pancreatitis and thus might be useful if employed very early in the course of disease.
Acknowledgments
This work was supported by a National Institutes of Health Grant R21 (DK69702 to E.C.T.), National Institutes of Health Grants RO1 (DK33850 to JRR, Jr.), (DK54021 to F.S.G), USC-UCLA Research Center for Alcoholic Liver and Pancreatic Injury Grant (5 P60 AA11999 from NIAAA; Tsukamoto; to S.J.P.) and a Veterans Administration Merit and Senior Career Development Award (to F.S.G.).
References
- 1.Cosen-Binker LI, Lam PP, Binker MG, et al. Alcohol/cholecystokinin-evoked pancreatic acinar basolateral exocytosis is mediated by protein kinase C alpha phosphorylation of Munc18c. J Biol Chem. 2007;282:13047–13058. doi: 10.1074/jbc.M611132200. [DOI] [PubMed] [Google Scholar]
- 2.Satoh A, Gukovskaya AS, Nieto JM, et al. PKC-delta and -epsilon regulate NF-kappaB activation induced by cholecystokinin and TNF-alpha in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G582–591. doi: 10.1152/ajpgi.00087.2004. [DOI] [PubMed] [Google Scholar]
- 3.Satoh A, Gukovskaya AS, Reeve JR, Jr, et al. Ethanol Sensitizes NF-{kappa}B Activation in Pancreatic Acinar Cells through effects on Protein Kinase C Epsilon. Am J Physiol Gastrointest Liver Physiol. 2006;291:G432–G438. doi: 10.1152/ajpgi.00579.2005. [DOI] [PubMed] [Google Scholar]
- 4.Thrower EC, Osgood S, Shugrue CA, et al. The novel protein kinase C isoforms -delta and -epsilon modulate caerulein-induced zymogen activation in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1344–1353. doi: 10.1152/ajpgi.00020.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bastani B, Yang L, Baldassare JJ, et al. Cellular distribution of isoforms of protein kinase C (PKC) in pancreatic acini. Biochim Biophys Acta. 1995;1269:307–315. doi: 10.1016/0167-4889(95)00120-0. [DOI] [PubMed] [Google Scholar]
- 6.Pollo DA, Baldassare JJ, Honda T, et al. Effects of cholecystokinin (CCK) and other secretagogues on isoforms of protein kinase C (PKC) in pancreatic acini. Biochim Biophys Acta. 1994;1224:127–138. doi: 10.1016/0167-4889(94)90120-1. [DOI] [PubMed] [Google Scholar]
- 7.Li C, Chen X, Williams JA. Regulation of CCK-induced amylase release by PKC-delta in rat pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G764–771. doi: 10.1152/ajpgi.00111.2004. [DOI] [PubMed] [Google Scholar]
- 8.Tapia JA, Jensen RT, Garcia-Marin LJ. Rottlerin inhibits stimulated enzymatic secretion and several intracellular signaling transduction pathways in pancreatic acinar cells by a non-PKC-delta-dependent mechanism. Biochim Biophys Acta. 2006;1763:25–38. doi: 10.1016/j.bbamcr.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Miyamoto A, Nakayama K, Imaki H, et al. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cdelta. Nature. 2002;416:865–869. doi: 10.1038/416865a. [DOI] [PubMed] [Google Scholar]
- 10.Gschwendt M. Protein kinase C delta. Eur J Biochem. 1999;259:555–564. doi: 10.1046/j.1432-1327.1999.00120.x. [DOI] [PubMed] [Google Scholar]
- 11.Liu WS, Heckman CA. The sevenfold way of PKC regulation. Cell Signal. 1998;10:529–542. doi: 10.1016/s0898-6568(98)00012-6. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe T, Ono Y, Taniyama Y, et al. Cell division arrest induced by phorbol ester in CHO cells overexpressing protein kinase C-delta subspecies. Proc Natl Acad Sci U S A. 1992;89:10159–10163. doi: 10.1073/pnas.89.21.10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mischak H, Goodnight JA, Kolch W, et al. Overexpression of protein kinase C-delta and -epsilon in NIH 3T3 cells induces opposite effects on growth, morphology, anchorage dependence, and tumorigenicity. J Biol Chem. 1993;268:6090–6096. [PubMed] [Google Scholar]
- 14.Gargiulo BJ, Cragg P, Richardson JB, et al. Phenotypic modulation of human articular chondrocytes by bistratene. A Eur Cell Mater. 2002;3:9–18. doi: 10.22203/ecm.v003a02. [DOI] [PubMed] [Google Scholar]
- 15.Blass M, Kronfeld I, Kazimirsky G, et al. Tyrosine phosphorylation of protein kinase Cdelta is essential for its apoptotic effect in response to etoposide. Mol Cell Biol. 2002;22:182–195. doi: 10.1128/MCB.22.1.182-195.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Basu A. Involvement of protein kinase C-delta in DNA damage-induced apoptosis. J Cell Mol Med. 2003;7:341–350. doi: 10.1111/j.1582-4934.2003.tb00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basu A, Woolard MD, Johnson CL. Involvement of protein kinase C-delta in DNA damage-induced apoptosis. Cell Death Differ. 2001;8:899–908. doi: 10.1038/sj.cdd.4400885. [DOI] [PubMed] [Google Scholar]
- 18.Yoshida K, Wang HG, Miki Y, et al. Protein kinase Cdelta is responsible for constitutive and DNA damage-induced phosphorylation of Rad9. Embo J. 2003;22:1431–1441. doi: 10.1093/emboj/cdg134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eitel K, Staiger H, Rieger J, et al. Protein kinase C delta activation and translocation to the nucleus are required for fatty acid-induced apoptosis of insulin-secreting cells. Diabetes. 2003;52:991–997. doi: 10.2337/diabetes.52.4.991. [DOI] [PubMed] [Google Scholar]
- 20.Lu Z, Hornia A, Jiang YW, et al. Tumor promotion by depleting cells of protein kinase C delta. Mol Cell Biol. 1997;17:3418–3428. doi: 10.1128/mcb.17.6.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwabu A, Smith K, Allen FD, et al. Epidermal growth factor induces fibroblast contractility and motility via a protein kinase C delta-dependent pathway. J Biol Chem. 2004;279:14551–14560. doi: 10.1074/jbc.M311981200. [DOI] [PubMed] [Google Scholar]
- 22.Frank DE, Carter WG. Laminin 5 deposition regulates keratinocyte polarization and persistent migration. J Cell Sci. 2004;117:1351–1363. doi: 10.1242/jcs.01003. [DOI] [PubMed] [Google Scholar]
- 23.Ramnath RD, Sun J, Adhikari S, et al. Role of PKC-delta on substance P-induced chemokine synthesis in pancreatic acinar cells. Am J Physiol Cell Physiol. 2008;294:C683–692. doi: 10.1152/ajpcell.00360.2007. [DOI] [PubMed] [Google Scholar]
- 24.Satoh A, Gukovskaya AS, Edderkaoui M, et al. Tumor necrosis factor-alpha mediates pancreatitis responses in acinar cells via protein kinase C and proline-rich tyrosine kinase 2. Gastroenterology. 2005;129:639–651. doi: 10.1016/j.gastro.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 25.Berna MJ, Hoffmann KM, Tapia JA, et al. CCK causes PKD1 activation in pancreatic acini by signaling through PKC-delta and PKC-independent pathways. Biochim Biophys Acta. 2007;1773:483–501. doi: 10.1016/j.bbamcr.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kikkawa U, Matsuzaki H, Yamamoto T. Protein kinase C delta (PKC delta): activation mechanisms and functions. J Biochem. 2002;132:831–839. doi: 10.1093/oxfordjournals.jbchem.a003294. [DOI] [PubMed] [Google Scholar]
- 27.Yokoyama G, Fujii T, Tayama K, et al. PKCdelta and MAPK mediate G(1) arrest induced by PMA in SKBR-3 breast cancer cells. Biochem Biophys Res Commun. 2005;327:720–726. doi: 10.1016/j.bbrc.2004.12.070. [DOI] [PubMed] [Google Scholar]
- 28.Kim MJ, Moon CH, Kim MY, et al. Role of PKC-delta during hypoxia in heart-derived H9c2 cells. Jpn J Physiol. 2004;54:405–414. doi: 10.2170/jjphysiol.54.405. [DOI] [PubMed] [Google Scholar]
- 29.Gschwendt M, Muller HJ, Kielbassa K, et al. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–98. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- 30.Davies SP, Reddy H, Caivano M, et al. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Susarla BT, Robinson MB. Rottlerin, an inhibitor of protein kinase Cdelta (PKCdelta), inhibits astrocytic glutamate transport activity and reduces GLAST immunoreactivity by a mechanism that appears to be PKCdelta-independent. J Neurochem. 2003;86:635–645. doi: 10.1046/j.1471-4159.2003.01886.x. [DOI] [PubMed] [Google Scholar]
- 32.Leitges M, Elis W, Gimborn K, et al. Rottlerin-independent attenuation of pervanadate-induced tyrosine phosphorylation events by protein kinase C-delta in hemopoietic cells. Lab Invest. 2001;81:1087–1095. doi: 10.1038/labinvest.3780321. [DOI] [PubMed] [Google Scholar]
- 33.Zhao H, Tian W, Cohen DM. Rottlerin inhibits tonicity-dependent expression and action of TonEBP in a PKCdelta-independent fashion. Am J Physiol Renal Physiol. 2002;282:F710–717. doi: 10.1152/ajprenal.00303.2001. [DOI] [PubMed] [Google Scholar]
- 34.Chen X, Edwards JA, Logsdon CD, et al. Dominant negative Rab3D inhibits amylase release from mouse pancreatic acini. J Biol Chem. 2002;277:18002–18009. doi: 10.1074/jbc.M201248200. [DOI] [PubMed] [Google Scholar]