

Abstract
To ensure safety, regulatory agencies recommend elimination of antibiotic resistance markers from therapeutic and vaccine plasmid DNA vectors. Here, we describe the development and application of a novel antibiotic-free selection system. Vectors incorporate and express a 150 bp RNA-OUT antisense RNA. RNA-OUT represses expression of a chromosomally integrated constitutively expressed counter-selectable marker (sacB), allowing plasmid selection on sucrose. Sucrose selectable DNA vaccine vectors combine antibiotic-free selection with highly productive fermentation manufacturing (>1 gm/L plasmid DNA yields), while improving in vivo expression of encoded proteins and increasing immune responses to target antigens. These vectors are safer, more potent, alternatives for DNA therapy or vaccination.
Keywords: DNA vaccine, plasmid, antibiotic-free
1) Introduction
Plasmid based DNA vaccines and therapeutics are in development for a variety of human, animal, bird and fish applications. Antibiotic resistance markers, typically kanamycin resistance (kanR), allow selective retention of plasmid DNA during bacterial fermentation and are the most commonly utilized selectable markers. The presence of an antibiotic resistance gene in the plasmid backbone is considered undesirable by regulatory agencies, due to: 1) the potential transfer of antibiotic resistance to endogenous microbial fauna; and 2) the potential activation and transcription of the genes from mammalian promoters after cellular incorporation into the genome [Reviewed in 1,2]. For example, a regulatory guidance with regard to DNA vaccine plasmids states: “The use of certain selection markers, such as resistance to antibiotics, which may adversely impact on other clinical therapies in the target population, should be avoided”[3]. Further, the use of antibiotics in fermentation culture requires expensive process validation of antibiotic removal during plasmid purification, to prevent contamination of the final product with residual antibiotics. Ideally, the plasmid would not contain any protein coding regions other than the gene of interest, since these could potentially be expressed in mammalian cells. Alternative selection strategies to address these concerns are described below.
1.1. Balanced lethals
An essential gene is maintained on the plasmid, with a corresponding chromosomal deletion or suppressible mutation. These systems eliminate the need for antibiotic resistance markers on the plasmid. For example, the pCOR vector contains a tRNA selectable marker to suppress a host chromosomal arg gene mutation, allowing minimal media growth [4].
1.2. Repressor titration
In this case, an operator sequence, placed on a multicopy plasmid, derepresses a chromosomal gene. For example, the lac operator or tet operator may be on the plasmid, and an antibiotic gene, regulated by the relevant operator is on the chromosome. Titration of the repressor by the operator leads to expression of the chromosomal gene, and antibiotic resistance. Therapeutic plasmids incorporating this design have been constructed (e.g. pGM509 [5]). An alternative system [6] has the dapD essential chromosomal gene under the control of the lac operator/promoter. Three copies of the operator on the plasmid titrate the lac repressor, allowing expression of the essential chromosomal gene. In the absence of the multicopy plasmid, dap expression is repressed, and the cell dies. The advantage of such systems is small size and elimination of antibiotic resistance (in the case of dapD). However, high yield fermentation with repressor titration plasmids has not been reported.
1.3. Antidote/poison selection schemes
A plasmid stabilization system wherein the F-plasmid ccd antidote-poison operon was modified for plasmid selection has been reported [7]. The antidote protein gene (ccdA) was incorporated onto the plasmid, and the ccdB poison gene was expressed from the chromosome; this scheme allowed plasmid stabilization. This system does contain a protein based selection marker (ccdA) and has not been evaluated in high yield plasmid fermentation.
1.4. RNA based selectable markers
Two groups have developed RNA based selection systems that utilize the endogenous RNAI/RNAII antisense regulators of the pMB1 origin. In both examples, chromosomal genes were engineered to contain an RNAII sequence within the untranslated region of the mRNA. In the presence of a ColE1 type plasmid, the plasmid encoded RNAI repressor binds the RNAII sequence and represses translation of the chromosomal gene. This gene can be an antibiotic resistance marker, transcriptional repressor (controlling expression of a second gene) or a lethal gene [8,9,10]. These systems are limited to existing ColE1 type origin containing plasmid vectors only.
We report herein novel vectors and host strains for application of a new, generally applicable, RNA based system for plasmid selection, maintenance, and high yield plasmid production.
2) Materials and Method
2.1. Phage λ attachment site integration vectors
Replication incompetent plasmids can be site specifically integrated into the genome at bacteriophage attachment sites utilizing bacteriophage recombinase expressing plasmids. Such systems, to allow integration at various bacteriophage attachment sites have been developed [11]. In brief, integration of the sacB counter-selectable marker (Bacillus subtilis levansucrase) at the Phage lambda attachment site required cloning the gene into the modified integration plasmid pCAH63-CAT. The chloramphenicol resistant (chlorR) pCAH63-CAT vector was constructed by ligating the bacteriophage lambda attachment site-ori-R-chlorR-rgnB containing DNA fragment of pCAH63 (2.2 kb NheI/PstI DNA fragment) with a 350 bp NheI/PstI tL3 containing DNA fragment from pAH81 (both vectors are described in Haldiman and Wanner, 2001 [11] and were obtained from the E. coli stock center, Yale NH). The resultant pCAH63-CAT plasmid contains the R6K conditional replication origin (which requires engineered pir+ E. coli host cells such as BW23474 for propagation; the R6K origin is non functional in DH5α) a multiple cloning site, a chloramphenicol resistance marker (chlorR) and the lambda attachment site. The plasmid is integrated into the phage lambda attachment site using the pINT-ts helper plasmid (ampicillin resistant, temperature sensitive R plasmid replicon; this plasmid is lost at 42°C) that encodes a heat inducible phage lambda integrase. Transformation of DH5α containing the pINT helper with the pCAH63-CAT derivative plasmid to be integrated results in integration of the pCAH63-CAT plasmid into the genome at the phage lambda attachment site; recombinants are selected with chlorR and integration verified using PCR as described [11].
2.2. pCAH63-CAT RNA-IN-SacB (p5/6 5/6)
All cloning was performed in BW23474 with chloramphenicol selection. All clones were sequence verified. The pCAH63-CAT vector was digested with PstI/BamHI and the linear fragment purified (F1). The sacB gene was PCR amplified from the pSIREN-DNR vector using following primers:
SacBF01: 5′ ctccagcacctgcctatCAAGatgaacatcaaaaagtttgcaaaacaagc 3′ (5′ end of gene) and;
SacBR01: 5′ cgtgagcacctgcaacgGATCcttatttgttaactgttaattgtccttg 3′ (3′ end of gene)
The 1.4 kb PCR product was digested with AarI (underlined, with sticky ends bolded in primer sequences above) and the linear fragment purified (F2).
A constitutive promoter P5/6 5/6 –RNA-IN leader sequence was made using synthetic primers as follows. The following primer pairs were annealed to generate F3 (P5/6 5/6 promoter = 5/6 identity to consensus for both −35 and −10 promoter boxes):
RNA-INF02: 5′ ggtagacacacatcttgtcatatgatagaatggtttcgccaaaaatcaataatcagacaa 3′
RNA-INR02: 5′ cttgttgtctgattattgatttttggcgaaaccattctatcatatgacaagatgtgtgtctacctgca 3′
The P5/6 5/6 promoter −35 (double underlined) and −10 (double underlined, bold) regions are indicated in the RNA-INF02 primer. The 3 fragments (F1, F2 and F3) were ligated, and transformed into the BW23474 cell line. Recombinants [pCAH63-CAT RNA-IN-SacB (p5/6 5/6)] were selected on choramphenicol, correct clones identified by restriction digestion, and verified by sequencing with tL3-r and rgnB-f sequencing primers [11].
2.3. pCAH63-CAT RNA-IN-SacB (P5/6 6/6)
To increase expression of RNA-IN from the above P5/6 5/6 construct, a promoter mutation was introduced that increased promoter activity (P5/6 6/6 has a single base change from P5/6 5/6 that increases promoter strength to 6/6 match with −10 Pribnow box, by changing the TAGAAT sequence to TATAAT). The pCAH63-CAT RNA-IN-SacB (p5/6 5/6) plasmid from above was PCR amplified using the following primers:
6/6-10F01: 5′ ctccagcacctgcctatTGGTttcgccaaaaatcaataatcagacaac 3′
6/6-10R01: 5′ cgtgagcacctgcaacgACCATTATATCATATGACAAGATGTGTGTCTAC 3′
The single base change from C to A is double underlined in the 6/6-10R01 primer (encodes G to T change in −10 forward sequence). The 4 kb PCR product was digested with AarI (underlined in primers, bold bases are complementary sticky ends) and the linear fragment purified and self-ligated. The BW23474 cell line was transformed and choramphenicol resistant colonies selected and correct clones [pCAH63-CAT RNA-IN-SacB (p5/6 6/6)] identified by restriction digestion. The recombinant plasmid (Fig. 4) was sequenced with rgnB-f to confirm the promoter change.
Fig. 4.

Restriction map of the integration plasmid pCAH63-Cat, containing P5/6 6/6 RNA-IN controlled SacB counter-selectable marker
2.4. pCAH63-CAT RNA-IN-SacB (P5/6 6/6) integrated cell lines
RNA-IN-SacB
The pCAH63-CAT RNA-IN-SacB (P5/6 6/6) plasmid was integrated into DH5α and the resultant cell line (RNA-IN-SacB) confirmed by PCR using P1–P4 primers [11]. RNA-IN-SacB did not grow on LB (no NaCl) +6% sucrose plates. This demonstrates that a strain containing a single chromosomal copy of constitutive promoter expressed RNAI-IN-SacB produces sufficient sacB to prevent growth on sucrose media.
NTC3016
The pCAH63-CAT RNA-IN-SacB (P5/6 6/6) plasmid (Fig. 4) was PCR amplified to delete the R6K replication origin using the following primers:
R6KdR01: 5′ ctccagcacctgcttttACACaggaacacttaacggctgacatg 3′
R6KdF01: 5′ cgtgagcacctgcaactGTGTtgaactgctgatcttcagatcctctac 3′
The 3.5 kb linear product was DpnI digested (to eliminate parent plasmid), AarI digested (underlined in primers) to create compatible sticky ends (bolded in primer), purified, and self-ligated (to make exonuclease resistant circles for integration). The ligation was electroporated into pINTts lambda integration plasmid-containing NTC3012 cell line (autolytic production host [12]) and colonies were selected with chloramphenicol. The cell line (NTC3016) was confirmed with PCR using P1–P4 primers [11].
2.5. pDNAVACCUltra- P5/6,4/6-RBS-EGFP (RNA-OUT)
A fluorescent RNA-OUT plasmid was created. The pDNAVACCUltra- P5/6,5/6-RBS-EGFP plasmid (expresses EGFP constitutively) was digested with DraIII/KpnI and the linear vector (2029, 1015) purified. The following 2 RNA-OUT primers were annealed. These produce an annealed RNA-OUT fragment that is compatible with DraIII and KpnI (Fig. 3).
Fig. 3.

a) the IS10 functional map, with locations of RNA IN and RNA-OUT regulatory RNAs is shown. b) the RNA-OUT sequence as adapted for cloning into DNA vaccine plasmid is shown. Flanking DraIII (cacgttgtg) and KpnI (ggtacc) sites are underlined.
RNA-OUTF01: 5′ gtggtagaattggtaaagagagtcgtgtaaaatatcgagttcgcacatcttgttgtctgattattgatttttggcgaaaccatttgatcatatg acaagatgtgtatctaccttaacttaatgattttgataaaaatcattaggtac 3′
RNA-OUTR01: 5′ ctaatgatttttatcaaaatcattaagttaaggtagatacacatcttgtcatatgatcaaatggtttcgccaaaaatcaataatcagacaacaa gatgtgcgaactcgatattttacacgactctctttaccaattctaccacaac 3′
The annealed primers were ligated into the vector, and transformed into the RNA-IN-sacB integrated cell line from step 2.3 above. Colonies were selected on LB (no NaCl) +6% sucrose and plasmid from two fluorescent colonies were sequence verified as correct. Transformation of sequence-verified pDNAVACCUltra-P5/6,4/6-RBS-EGFP (RNA-OUT) plasmid into RNA-IN –SacB competent cells was performed; all recovered colonies after sucrose selection were fluorescent (i.e. 100% of recovered cells contained the plasmid).
2.6. Cell culture, SEAP, ELISA, and Western Blot analysis
HEK293 (human embryonic kidney) cells were propagated on DMEMF12 media supplemented with 10% fetal bovine serum. For transfections, cells were plated on 24 well tissue culture plates. The EGFP or influenza H5 hemagglutinin gene (HA) RNA-OUT or kanR plasmids (0.4 ug each well) were transfected into HEK293 using lipofectamine 2000 following the manufacturers instructions (Invitrogen, Carlsbad, CA) and total cellular protein 48 hrs post transfection assayed by FLX800 microplate fluorescence reader (EGFP) or Western blot analysis (HA). Western blot analysis was performed on protein extracts using rabbit anti-HA H5N1 rabbit IgG (eEnzyme, Gaithersburg, MD) primary antibody, and the amplified alkaline phosphatase immuno-blot assay kit and AP conjugate substrate kit (Biorad, Hercules, CA).
SEAP levels were determined using the Phospha-light SEAP Reporter Gene Assay System from Applied Biosystems (Foster City, CA) according to the manufacturer’s instructions. Anti-HA specific total IgG, IgG2a, and IgG1 levels were determined by standard ELISA assay using plates coated with Nature Technology Corporation produced recombinant E. coli produced A/Vietnam/1203/2004 HA HA2 domain, Sigma (St Louis, Mo) detection antibodies, and the BD Biosciences OptEIA kit (BD Biosciences Pharmingen, San Diego CA) block, wash and detection reagents. Briefly, coated wells were blocked and incubated 2 hrs with diluted serum samples, the wells washed and signal detected by subsequent sequential incubation and washing with:
total IgG: goat anti mouse IgG (Fc)-biotin, then Streptavidin-horse radish peroxidase conjugate
IgG1: goat anti-mouse IgG1, then anti-goat IgG-biotin, then Streptavidin-horse radish peroxidase conjugate
IgG2a: goat anti-mouse IgG2a, then anti-goat IgG-biotin, then Streptavidin-horse radish peroxidase conjugate (IgG1).
2.7. Murine studies
NTC7382 41H-HA and NTC8382-41H-HA (Fig. 1) influenza H5 Hemagglutinin DNA vaccine plasmids were tested for immunogenicity versus control NTC7382-SEAP vector. Groups of 8 mice were immunized in an IACUC approved study performed at OLAW certified Explora Laboratories animal facility, San Diego CA. Immunogenicity was compared to expression levels using the corresponding vectors containing SEAP rather than HA in a second study. For expression, groups of 3 mice were injected with plasmid in an IACUC approved study performed at OLAW certified Aldevron animal facility, Fargo ND. In both studies, 3 μg HA or SEAP plasmid was injected intra-muscular (IM, quadriceps) of female BALB/c mice (6–8 weeks old) in a single dose (day 0) prior to electroporation (EP) using the MedPulser® system (Inovio Biomedical, San Diego CA) using a 2 needle array (27 G), 4 mm apart, 2×60 ms pulses at 50 mA. Serum samples were taken and tested for SEAP or anti-HA IgG response.
Fig. 1.

Immunostimulatory influenza H5 HA ‘bird flu’ pDNAVACCultra plasmids (NTC8382-41H-HA) are shown. The RNA-OUT selectable marker in NTC8382-41H-HA is cloned as a 140 bp DraIII-KpnI fragment, replacing the kanR gene in the otherwise equivalent NTC7382-41H HA vector. The immunostimulatory eRNA41H RNA element (41H) is cloned between the Eukaryotic terminator and the prokaryotic replication origin in this case.
3) Results and Discussion
The NTC7382 and NTC8382 vectors (Fig. 1) are kanamycin resistant (kanR, NTC7382) or antibiotic free (NTC8382) DNA vaccine vectors incorporating the chimeric NTC7382 promoter. Both vectors facilitate cloning of genes or epitopes seamlessly into the desired intracellular targeting gene leader cassettes as described for the original CMV promoter based pDNAVACCUltra vectors [13]. All plasmid elements have been optimized and minimized to comply with FDA guidelines [14] regarding content and elimination of extraneous materials. The vector can simultaneously produce DNA polymerase III (pol III) driven ssRNA, dsRNA or shRNA (e.g. eRNA41H; Fig. 1) that are utilized to customize the immune response [15].
3.1. NTC7382 promoter
The NTC7382 promoter is shown in Fig. 2. The NTC7382 chimeric promoter is composed of the novel combination of; 1) CMV promoter and start of exon 1; 2) the HTLV-I R-U5 sequence which contains the 5′ splice acceptor site; 3) a synthetic 3′ acceptor site based on the rabbit β globin intron; 4) Exon 2 splicing enhancer comprised of SR protein binding site (3 copies of GAAGAAGAC) to improve RNA export [16], prior to 5) exon 2 kozak sequence upstream of the start codon for the gene of interest. Incorporation of the HTLV-1 R-U5 region downstream of the CMV promoter has been demonstrated to improve expression and cellular immune responses to HIV DNA vaccines in mice and nonhuman primates [17] and improve humoral responses to an influenza pDNAVACCUltra based DNA vaccine in mice (compared to CMV promoter based vectors; J. Williams, unpublished observations). Expression levels (of EGFP) from plasmids incorporating the NTC7382 promoter, after transfection of HEK293, are dramatically increased versus CMV based promoter plasmids (Table 1).
Fig. 2.

An annotated map of the NTC7382 promoter is shown
Table 1.
Expression levels (EGFP Fluorescence units) from plasmids incorporating the NTC7382, or CMV based promoter
| Vector name† | Promoter | Selection | EGFP (fluorescence units) |
|---|---|---|---|
| pDNAVACCUltra-EGFP | CMV | Kan | 6,668 |
| NTC7382-EGFP (UP) | NTC7382 | Kan | 20,074 |
| NTC8382-EGFP (UP) | NTC7382 | RNA-OUT | 31,788 |
(UP) is a modified backbone wherein the SV40 enhancer and ISS sequences (see Fig. 1) are removed
3.2. RNA-OUT based antibiotic-free selection system
In the insertion sequence 10 (IS10) system, RNA-OUT RNA hybridizes to the RNA-IN RNA, and reduces translation of the downstream IS10 transposase gene (Fig. 3). Jaenecke and colleagues reported engineering a pLac-expressed RNA-OUT from a plasmid that weakly repressed expression of a single copy integrated pR-RNA-IN-lacZ construct [18]. While this demonstrates that the RNA-IN, RNA-OUT system can be used to obtain some level of regulation of a heterologous chromosomal gene, it does not demonstrate sufficient repression for use of this system for plasmid selection and maintenance. We report herein adaptation of RNA-OUT, RNA-IN regulation to plasmid selection as follows.
A counter-selectable marker (sacB; Bacillus subtilis levansucrase [19]) expressed under the control of the RNA-IN promoter and leader (Fig. 4) was integrated into the chromosome of DH5α (RNA-IN-SacB cell line). Expression of sacB was determined to be constitutive since cells containing sacB encoded levansucrase were killed in the presence of sucrose (see Materials and Methods). Translation of SacB from this RNA was designed to be repressed in the presence of the antisense regulator RNA-OUT (Fig. 3). RNA-OUT was cloned into the pDNAVACCUltra-P5/6,4/6-RBS-EGFP vector, in place of the kanR gene, by replacement of the DraIII-KpnI cassette (Fig. 1 and 3; see Materials and Methods). pDNAVACCUltra- P5/6,4/6-RBS-EGFP (RNA-OUT) repressed expression of the RNA-IN regulated chromosomally integrated counter-selectable marker SacB. This allowed antibiotic free plasmid selection in the presence of sucrose on solid and liquid media (see Materials and Methods).
3.3. RNA-OUT plasmid construction efficiency
The RNA-OUT selectable marker was substituted for the kanR gene in a DNA vaccine plasmid as follows. The RNA-OUT cassette was excised from pDNAVACCUltra- P5/6,4/6-RBS-EGFP (RNA-OUT) using DraIII/XbaI and the 154 bp restriction fragment cloned in a 3 fragment ligation with DraIII-SalI and SalI/XbaI fragments from a target DNA vaccine plasmid. Correct recombinants have the three fragments ligated together. The ligation was transformed into serial diluted Z-competent cells (Zymo Research, Orange, CA) of the RNA-IN-SacB cell line and the transformations plated on LB (no NaCl) +6% sucrose plates. Colonies from each transformation were screened for insert. 8/10 recovered colonies from the undiluted Z competent cells were correct, as were 7/9 from the 1:10 diluted competent cells, and 2/2 from the 1:100 diluted competent cells. Similar results were obtained with a second independent ligation reaction for a different construct. Collectively, these results demonstrate that RNA-IN containing plasmids can be easily selected from ligation reactions on LB Sucrose plates, and that the selection is robust.
A construct was made with the RNA-OUT cassette in the opposite orientation in the vector. Correct colonies were recovered as described above. This demonstrates that the RNA-OUT cassette can function for sucrose selection in either orientation in the vector.
Over 50 different plasmid vectors have been constructed using RNA-OUT selection. This demonstrates the robustness and general utility of the RNA-OUT sucrose selection methodology.
3.4. RNA-OUT plasmid fermentation
The RNA-OUT sequence has no repetitive sequences that may be toxic or unstable in E. coli, so undesirable effects on quality in fermentation production were not expected. To verify this, an RNA-OUT vector was tested for fermentation productivity. An inducible fed-batch fermentation was performed using Nature Technology Corporation’s (NTC) high yield plasmid production process [20,21] with an RNA-OUT plasmid in DH5α derived E. coli strain RNA-IN-SacB. The fermentation media contained 0.5% sucrose for plasmid selection since it was determined that ≥0.5% sucrose in fermentation media is growth inhibitory to the parent plasmid-less strain. The yield from this fermentation was 1213 mg plasmid/L (Fig. 5) and the plasmid was of a high quality (supercoiled monomer) with no detected replication intermediates or aberrant bands. This yield is typical for kanR based pDNAVACCUltra derived vectors. This demonstrates that replacement of kanR with RNA-OUT in the vector backbone is not detrimental to plasmid production yield or quality.
Fig. 5.
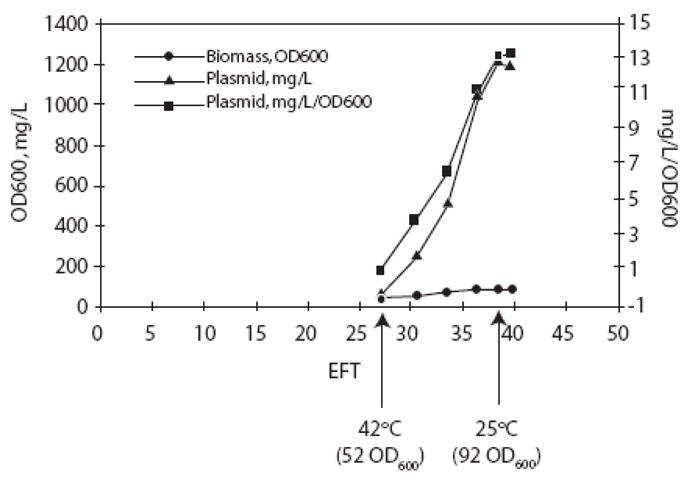
Analytical data including production yield, specific yield and biomass (OD600) from an RNA-OUT influenza DNA vaccine plasmid fermentation (RNA-OUT-41H-HA).
3.5. RNA-OUT Autolytic plasmid production cell line (NTC3016)
For general application to production of a variety of plasmids, it is preferable for the production cell line to not have plasmid replication origins (e.g. R6K replication origin) in the chromosome. The R6K origin from the pCAH63-CAT RNA-IN-SacB (P5/6 6/6) plasmid was deleted, and the insert integrated into the NTC3012 cell line (autolytic plasmid production host [12]) to create NTC3016 (see Materials and Methods).
Several RNA-OUT plasmids have been successfully transformed and propagated in the NTC3016 cell line, without toxicity or interference with autolytic plasmid DNA processing. A fermentation yield of 1150 mg plasmid/liter culture was obtained with the RNA-OUT NTC8382-41H-HA plasmid (Fig. 1) in the NTC3016 cell line (data not shown) demonstrating RNA-OUT compatibility with autolytic host strains and the high yield NTC fermentation process.
3.6. NTC8382 DNA vaccine vectors
NTC8382 contains the 150 bp DraIII-KpnI RNA-OUT based selectable marker in place of the kanR marker in NTC7382 (Fig. 1). Derivatives were constructed with either the influenza H5 hemagglutinin gene (HA), SEAP or EGFP reporter genes, with or without various pol III transcribed immunostimulatory RNAs (eRNAs) that activate the RIG-I and MDA5 receptor (e.g. eRNA41H [15]). These vectors were made by swapping of: 1) the eRNA region into the plasmid backbone using flanking sites common to all the vectors, such as NotI-AflIII or NheI-ApaLI fragment swaps; and 2) the gene of interest such as HA or EGFP using SalI-BglII fragment swaps.
3.7. NTC8382 expression
The RNA-OUT NTC8382-41H-HA vector (Fig. 1; RNA-OUT equivalent of kanR NTC7382 41H-HA) and derivatives were tested for expression and immunogenicity compared to parent kanR vectors.
Antigen expression after Lipofectamine™ 2000 transfection of HEK293 was determined for RNA-OUT plasmids, compared to control kanR NTC7382 plasmids (see Materials and Methods). NTC8382 plasmids were higher expressing than the corresponding kanR plasmid (otherwise same backbones) for EGFP (Table 1) and HA (data not shown) expression. Expression levels of EGFP and HA from NTC7382 promoter containing vectors (kanR or RNA-OUT) were higher than the corresponding CMV promoter vectors. This demonstrates that the presence of RNA-OUT in the vector backbone improves NTC7382 promoter-driven antigen expression.
3.8. NTC8382 in vivo expression and immunogenicity
NTC7382 41H-HA and NTC8382-41H-HA (Fig. 1) influenza H5 hemagglutinin DNA vaccine plasmids were tested for immunogenicity. Expression levels for each vector were determined, using SEAP expressing versions of the vectors. Serum samples were taken and tested for SEAP or anti-HA IgG response (see Materials and Methods).
The results are summarized in Table 2 and demonstrate the antibiotic free NTC8382 backbone is equivalent or superior to the kanR NTC7382 backbone for expression and immunogenicity.
Table 2.
NTC7382 41H-HA and NTC8382-41H-HA in vivo expression and immunogenicity++
| Backbone | 4 day SEAP (ng/mL) | 7 day SEAP (ng/mL) | 14 Day SEAP (ng/mL) | 21 day anti-HA2 IgG (total) (Abs 1/12,500) | 21 day anti- HA2 IgG2a (Abs 1/500) | 21 day anti-HA2 IgG1 (Abs 1/500) | % seroconverted (1/2500) |
|---|---|---|---|---|---|---|---|
| NTC7382-41H | 216 ± 147 | 300 ± 121 | 158 ± 43 | 0.173 ± 0.101 | 0.318 ± 0.229 | 0.140 ± 0.069 | 6/8 |
| NTC8382-41H | 241 ± 71 | 305 ± 83 | 243 ± 43 | 0.302 ± 0.134 | 0.483 ± 0.236 | 0.377 ± 0.384 | 7/8 |
| Control† | 0.066 ± 0.010 | 0.079 ± 0.023 | 0.086 ± 0.020 | 1/8 |
All day 0 SEAP undetected. For HA, NTC7382-SEAP was the negative control plasmid
Results presented as average ± standard deviation
In conclusion, we report the development of antibiotic-free DNA Vaccine vectors, that combine high (>1 gm/L) fermentation yield and plasmid quality with improved in vivo expression and immunogenicity. The RNA based selectable marker is not restricted to ColE1 vectors, and has general utility for retrofitting antibiotic-containing vectors.
Acknowledgments
We thank Sheryl Anderson for her tireless efforts cleaning, batching and operating fermentors, Marni England-Hill for oversight of the murine SEAP study at Aldevron, and Jonas Soderholm for oversight of the murine immunogenicity study at Explora Laboratories. This paper described work supported by NIH grants R43GM073394, R44GM072141 and R43A1071660.
Footnotes
Conflict of Interest Statement
JL, AEC, CPH and JAW have an equity interest in Nature Technology Corporation
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carnes AE, Williams JA. Plasmid DNA manufacturing. Technology Recent Patents on Biotechnology. 2007;1:151–166. doi: 10.2174/187220807780809436. [DOI] [PubMed] [Google Scholar]
- 2.Glenting J, Wessels S. Ensuring safety of DNA vaccines. Microbial Cell Factories. 205(4):26–30. doi: 10.1186/1475-2859-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.European Medicines Agency. Note for guidance on the quality, preclinical and clinical aspects of gene transfer medicinal products. 2001 CPMP/BWP/3088/99. [Google Scholar]
- 4.Soubrier F, Cameron B, Manse B, Somarriba S, Dubertret C, Jaslin G, Jung G, Caer CL, Dang D, Mouvault JM, Scherman D, Mayaux JF, Crouzet J. pCOR: a new design of plasmid vectors for nonviral gene therapy. Gene Ther. 1999;6:1482–1488. doi: 10.1038/sj.gt.3300968. [DOI] [PubMed] [Google Scholar]
- 5.Eastman EM, Durland RH. Plasmid for delivery of nucleic acid to cells and method of use. 2003 US Patent No 6103470. [Google Scholar]
- 6.Cranenburgh RM, Hanak JA, Williams SG, Sherratt DJ. Escherichia coli strains that allow antibiotic-free plasmid selection and maintenance by repressor titration. Nucleic Acids Res. 2001;29:E26. doi: 10.1093/nar/29.5.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szpirer CY, Milinkovitch MC. Separate-component-stabilization system for protein and DNA production without the use of antibiotics. Biotechniques. 2005;38:775–781. doi: 10.2144/05385RR02. [DOI] [PubMed] [Google Scholar]
- 8.Mairhofer J, Pfaffenzeller I, Merz D, Grabherr R. A novel antibiotic free plasmid selection system: Advances in safe and efficient DNA therapy. Biotechnol J. 2008;3:83–89. doi: 10.1002/biot.200700141. [DOI] [PubMed] [Google Scholar]
- 9.Grabherr R, Pfaffenzeller I. Host-Vector system for antibiotic-free ColE1 Plasmid Propagation. 2006 World Patent Application WO2006029985. [Google Scholar]
- 10.Cranenburgh RM. Plasmid Maintenance. 2005 World Patent Application WO2005052167. [Google Scholar]
- 11.Haldimann A, Wanner BL. Conditional-Replication, Integration, Excision, and Retrieval Plasmid-Host Systems for Gene Structure-Function Studies of Bacteria. J Bacteriol. 2001;183:6384–6393. doi: 10.1128/JB.183.21.6384-6393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carnes AE, Williams JA, Hodgson CP. Improved E. coli plasmid DNA production. 2008 US patent application US60931465. [Google Scholar]
- 13.Williams JA, Luke J, Johnson L, Hodgson C. pDNAVACCultra vector family: high throughput intracellular targeting DNA vaccine plasmids. Vaccine. 2006;24:4671–4676. doi: 10.1016/j.vaccine.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 14.CBER. Guidance for industry: Considerations for Plasmid DNA Vaccines for Infectious Disease Indications. 2005 [Google Scholar]
- 15.Williams JA. Vectors and methods for genetic immunization. 2008 US Patent Application 60932160. [Google Scholar]
- 16.Lavigueur A, La Branche H, AR Kornblihtt, Chabot B. A splicing enhancer in the human fibronectin alternate ED1 exon interacts with SR proteins and stimulates U2 snRNP binding. Genes Develop. 1993;7:2405. doi: 10.1101/gad.7.12a.2405. [DOI] [PubMed] [Google Scholar]
- 17.Barouch DH, Yang ZY, Kong WP, Korioth-Schmitz B, Sumida SM, Truitt DM, Kishko MG, Arthur JC, Miura A, Mascola JR, Letvin NL, Nabel GJ. A human T-cell leukemia virus type 1 regulatory element enhances the immunogenicity of human immunodeficiency virus type 1 DNA vaccines in mice and nonhuman primates. J Virol. 2005;79:8828–8834. doi: 10.1128/JVI.79.14.8828-8834.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaenecke S, de Lorenzo V, Timmis KN, Diaz E. Molec. Microbiol. 1996;21:293–300. doi: 10.1046/j.1365-2958.1996.6411358.x. [DOI] [PubMed] [Google Scholar]
- 19.Reyrat JM, Pelicic V, Gicquel B, Rappuoli R. Counterselectable Markers: Untapped Tools for Bacterial Genetics and Pathogenesis. Infection Immun. 1998;66:4011–4017. doi: 10.1128/iai.66.9.4011-4017.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carnes AE, Hodgson CP, Williams JA. Inducible Escherichia coli fermentation for increased plasmid DNA production. Biotechnol Appl Biochem. 2006;45:155–66. doi: 10.1042/BA20050223. [DOI] [PubMed] [Google Scholar]
- 21.Carnes AE, Williams JA. Process for plasmid DNA fermentation. 2006 World Patent Application WO2006023546. [Google Scholar]