

Abstract
Aim
Increased production of cytokines and chemokines in serum and tissues upon oxidative stress caused by severe systemic infections are the major cause of sepsis. Aldose reductase (AR) known to mediate oxidative stress- induced NF-κB activation and transcription of cytokines and chemokines are the main mediator of bacterial endotoxin - and lipopolysaccharide -induced inflammatory cytokines. Our aim is to investigate the effect of AR inhibitors on the prevention of inflammatory cytokines in the cecal ligation and puncture (CLP) model of polymicrobial sepsis which closely mimics the sepsis syndrome in humans.
Results
Mice were rendered septic by CLP in the absence and presence of AR inhibitor, sorbinil. The levels of cytokines, chemokines and other inflammatory markers in the plasma, peritoneal fluid and heart of mice were significantly inhibited by sorbinil. Inhibition of AR also prevented CLP-induced COX-2, iNOS and HMGB-1 in heart, kidney and spleen.
Conclusions
Our results showed that the inhibition of AR significantly prevented the polymicrobial sepsis-induced increase in inflammatory markers and thus indicate the use of AR inhibitors as anti-inflammatory agents.
Keywords: Sepsis, Cecum Ligation and Puncture, Cytokines, Inflammation, Aldose Reductase
1 INTRODUCTION
Sepsis is a serious medical condition that results in increased fatalities in elderly, immunocompromised and critically ill intubated patients [1]. This disease is characterized by whole-body inflammation due to severe systemic infection [2]. Severely burnt patients usually develop sepsis which is the major cause of death if not aggressively and timely treated[3]. In the United States alone, sepsis is the second leading cause of death in non-coronary intensive care unit patients [4], which account for 20 to 50% deaths per year out of 660,000 affected cases [5]. Despite significant advances in the understanding of pathogenesis of sepsis and its complications, barely a small number of restorative strategies have been employed that could decrease mortality associated with septic shock. Although sepsis is generally initiated by a bacterial infection, the pathogenesis of sepsis is characterized by increased levels of inflammatory markers such as cytokines and chemokines that could lead to severe complications such as multi-organ failure and death [6]. Eventhough the treatment with broad range antibiotics is generally effective in killing the replicating bacteria, the dead bacterial cell wall components act as bacterial toxins which cause overwhelming systemic inflammatory response [7]. Thus controlling bacterial infection is not sufficient to treat sepsis. It is rather important to neutralize the immunogenic signals that activate transcription factors that transcribe inflammatory markers. Although several new therapeutic strategies have been described in the literature for the treatment of sepsis to date, various anti-inflammatory strategies have produced only modest therapeutic effects in critically ill sepsis patients [8].
Recently, we have shown that the treatment with AR inhibitors results in profoundly improved survival in the lipopolysaccharide (LPS) -induced endotoximia in mice[9]. The observed improvement in survival after treatment with AR inhibitors or AR-siRNA was associated with restoration of cardiac muscle contractility and cardiac functions. We have demonstrated that a significant increase in the levels of serum as well as heart cytokines, chemokines and other inflammatory markers such as COX-2, iNOS and PGE2 in LPS-treated mice which significantly contributes to pathophysiology of sepsis, was markedly prevented by AR inhibition [9]. Also inhibition of AR prevented the endotoxin, high glucose and TNF-α induced increase in the redox sensitive transcription factors, NF-κB and AP-1, which are known to transcribe various inflammatory markers [9], [10], [11], [12], [13], [14] and [15]. These results suggest that AR inhibitors, developed as anti-diabetic drugs, could be used as therapeutic intervention to prevent sepsis [16]. AR inhibitors such as zopolrestat, fidarestat have been found to be safe and passed in FDA’s Phase-I and clinical trials for diabetic neuropathy but failed in Phase-III clinical trials as they have been shown to be not as effective. There are several AR inhibitors such as raneristat that are still undergoing phase-III trails for diabetic neuropathy. An AR inhibitor, epalrestat is available in Japan mainly to treat patients with diabetic neuropathy [16] and [17]. Our recent results demonstrate that AR inhibitors could be used therapeutically for inflammatory disorders other than diabetic complications [15] and [18]. For such use a careful examination of the effect of AR inhibition in clinically relevant animal models is mandatory. In the present study, we investigated whether treatment with AR inhibitors could prevent alterations in the cytokine and chemokine levels in a clinically more relevant model that mimics sepsis conditions in humans. Our results indicate that AR inhibitor is a powerful repressor of the expression of major cytokines and chemokines in a mouse model of polymicrobial sepsis indicating its use as potential anti-inflammatory agent in sepsis and associated complications.
2 MATERIALS AND METHODS
2.1. Materials
Antibodies against COX-2, iNOS p65-NF-κB and HMGB-1 were purchased from Santa Cruz Biotech, Inc. (Santa Cruz, CA). Antibodies against AR were custom raised and tested for specificity. Mouse specific ELISA kits for cytokines were purchased from BD biosciences (Franklin Lakes, NJ). Cytokine antibody array was purchased from RayBiotech, Inc. (Norcross, GA). Sorbinil was gift from Pfizer (Groton, CT). ACTICHROME® Tissue factor activity assay kit was purchased from American diagnostic Inc (Stamford, CT), Tramadol Hydrochloride from Mallinckrodt Inc. (St. Louis, MO). All other reagents were of the highest purity available.
2.2. CLP procedure
BALB/c female mice (19-23 g) were obtained from Jackson laboratories (Bar Harbor, ME) and housed with regular diet and ambience for 48 hrs before recruiting to the study. The animals were maintained in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health and in accordance with the Institute’s “Guideline of the Animal Care and Use Committee”. Using previously described method [19] CLP procedure was performed by anesthetizing the mice with ketamine (60 μg/g) and xylazine (10 μg/g) and a 2-cm midline incision was made through the linea alba. The cecum was located, ligated with sterile 3-0 silk, and perforated with double puncture using a 18-gauge needle. A small amount of stool was extruded to ensure wound potency. Sham-treated mice also had surgery done along with cecal manipulations but without ligation and puncture. The cecum was then replaced in its original position within the abdomen, and incision was immediately closed. Soon after surgery, each mouse received a subcutaneous injection of 1 ml of warm (37°C) normal saline with traMADOL hydrochloride (20 μg/g body wt). To inhibit AR, mice received 25 mg/kg body wt sorbinil 2 h after surgery and then 6 h after surgery. All mice (5 in each group) were kept at 22°C and killed after 14 h of surgery. Blood was collected from the heart in EDTA-coated tubes. Plasma was separated from cellular components by centrifugation at 600x g for 5 min and stored at -20°C. For peritoneal lavage, 2 cm skin was removed leaving the peritoneal membrane intact. Then 1 ml of ice-cold 1× Hanks balanced salt solution (without CaCl2, MgCl2, Mg2SO4, or phenol red; GIBCO) was injected using 26 gauge needle. After injection, peritoneum was gently palpated for 30 s, and peritoneal fluid was aspirated out using a 20 gauge needle. The fluid was centrifuged (600 × g, 5 min), and the supernatant was stored at -20°C. Heart and aorta tissues were collected and stored at -70°C for tissue homogenates.
2.3. Measurement of Cytokines in plasma
Mice after 14 h of surgery were euthanized and blood was collected in to EDTA-coated vacutainers by heart puncture. plasma was separated by centrifugation at 600 g for 5 min. Levels of IL-1β, IL-6, TNF-α and MCP-1 in the plasma were measured by using respective mouse specific ELISA kits according to the manufacturer’s instructions. Known concentrations of standards were used to generate the standard curves. The reaction was initiated by the addition of 0.1 ml of the serum, stopped and read at 450 nm on an ELISA plate reader. The results obtained are expressed as pg/ml of the plasma.
2.4. Measurement of cytokines and chemokines in heart
To measure the expression of cytokines and chemokines in heart tissue homogenates, we used commercially available RayBio Mouse cytokine antibody array system that determines the expression of 64 inflammatory markers from a single sample by following manufacturer’s instructions. In brief, 100 μg heart tissue homogenates were incubated with array support membrane where the membrane was specifically coated with antigens to capture an array of cytokines. The tagged proteins were detected by conjugating with biotinylated antobodies and streptavidin system. Fold change was calculated from the measured intensities of the individual spots signal developed on X-ray film using Kodak-densitometry software.
2.5. Western blot analysis
To examine the effect of AR inhibition on CLP-induced expression of COX-2, iNOS, AR, NF-κB and HMGB-1 we homogenized heart, spleen and kidney tissues obtained from euthanized mice after 14 h of CLP procedure. Equal amount of protein from homogenates were subjected to SDS-PAGE and Western blotting and membranes were probed for against specific antibodies AR, iNOS, pP65-NFκB, COX-2, HMG-1 (HMGB1) and GAPDH. The antigen-antibody complexes were detected by enhanced chemiluminescence (Pierce, Rockford, IL). All blots were probed with GAPDH as a loading control and densitometric analysis was carried out by using Kodak Image station.
2.6. Tissue Factor Assay
Tissue factor was measured in mice plasma by using ACTICHROME® Tissue factor activity assay kit from American diagnostic Inc by following manufacturer’s protocol. In-brief After the experimental procedure mice plasma were collected as described in methods and the reaction was initiated by adding equal amount of plasma to reaction mixture containing lapidated tissue factor, factor VIII and factor X, allowing to form complex with fVIIa to generate TF/fVIIa complexes and convert fX into fXa. fXa cleaves the chromogenic substrate and the formation of color is measured by reading absorbance at 409 nm using micro well plate reader. Amount of tissue factor in the sample were calculated using the standards. The results obtained were measured in pMoles.
2.7. Statistical analysis
Data are presented as mean ± SEM and the p values were determined using the one-way ANOVA and unpaired Student’s t-test.
3 RESULTS
3.1. Prevention of CLP-induced increase in plasma and peritoneal cytokines by AR inhibition
To investigate the effect of AR inhibition on polymicrobial infection -induced inflammatory response, we performed CLP-surgery on mice injected without or with AR inhibitor, sorbinil. Increased redness around the cecum caused by increased blood flow due to dilatory expansion of microcirculatory blood vessels observed in the CLP operated mice was prevented by AR inhibitor (Fig. 1). The plasma and peritoneal fluids were collected 14 h after CLP procedure and analyzed for cytokines and chemokines. The plasma and peritoneal fluid levels of IL-1β, IL-6, TNF-α and MCP-1 proteins in sham-operated controls were low but detectable (Fig. 1A-D, left panel). However in the CLP mice the plasma levels of TNF-α, IL-6, IL-1β and MCP-1 increased by approximately ∼0.8, 37, 11 and 10 folds, respectively (Fig. 1A-D, left panel). On the other hand, administration of sorbinil to the CLP mice significantly (∼45-80%) prevented the increase in the plasma levels of cytokines and chemokines. Similarly, in CLP mice the peritoneal fluid levels of TNF-α, IL-6, IL-1β and MCP-1 increased by 5.2, 31, 10 and 5 folds, respectively (Fig. 2A-D, right panel) and administration of sorbinil to the CLP mice significantly (∼40-70%) prevented the increase in cytokine levels. AR inhibitors alone had no effect on the basal levels of these cytokines and chemokines in plasma or peritoneal fluid. Further, the measurement of inflammatory cytokines (such as TNF-α, IL-1, IL-6) in the serum at 3 h after CLP surgery did not cause any increase in the cytokine levels as compared to controls and ARI treated mice (data not shown). These results suggest that AR inhibition could prevent polymicrobial infection-induced systemic production of inflammatory cytokines and chemokines in mice.
Figure-1. AR inhibition prevents CLP-induced redness around the wound.

After 14 h of CLP, mice were killed and wound was accessed for severity of the inflammation. Digital pictures were taken immediately after the opening of wound. The region showing redness was circled. A) Sham B) Sham+ ARI C) CLP and D) CLP+ARI.
Figure-2. AR inhibition prevents CLP-induced cytokine secretion in mouse plasma and peritoneal fluid.

After 14 h of CLP, mice were killed and plasma was separated from blood. Peritoneal fluid was collected by injecting 1 ml of ice-cold 1× Hanks balanced salt solution in to peritoneum as described in methods. A) IL-1β B) IL-6, C) TNF-α and D) MCP-1 were measured in both plasma and in peritoneal fluid using mouse specific ELISA kits as described in Methods. Data are expressed as Mean ± SEM (n = 4). *P<0.001 Vs Control, #P<0.001 Vs. CLP, **P<0.05 Vs Control, ## P<0.05 Vs. CLP
3.2. Prevention of CLP-induced increase in tissue cytokines by AR inhibition
Since increased cardiac dysfunction and cardiomyopathy due to augmented local production of inflammatory cytokines and chemokines are the major cause of mortality and morbidity observed in patients with sepsis [20], we specifically examined the effect of AR inhibition on CLP-induced cytokines and chemokines in the mice hearts. We used an antibody array that determines the expression of 64 cytokines and chemokines and other inflammatory markers from a single sample (Fig. 3A). As shown in Fig. 3, the levels of MIP-2, MIP-1 and lymphotoxin increased by ∼6 fold in CLP mice group whereas the increase was only ∼3 fold in case of CLP+ sorbinil group. Similarly the levels of other cytokines and chemokines were significantly elevated in CLP group of mice and administration of AR inhibitor significantly prevented these cytokines and chemokines (Fig. 3B).
Figure-3. Inhibition of AR prevents inflammatory cytokines in mice hearts.
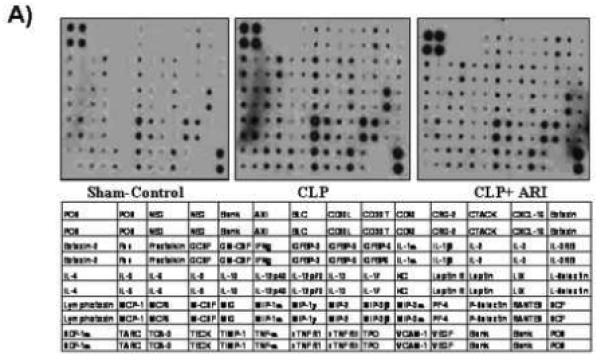
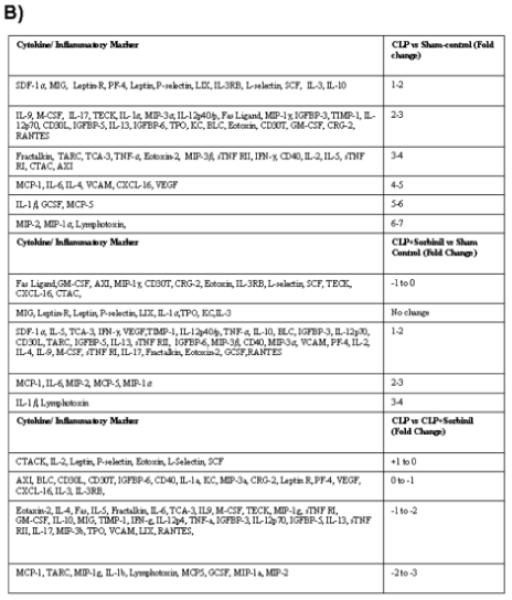
After 14 h of CLP, mice hearts were dissected out and homogenates were made. The levels of various (as indicated in table under the figure) inflammatory markers were measured by using Ray Bio Mouse cytokine antibody array. The intensity of the spots was measured by densitometry and compared with positive and negative controls. (A) Representative picture is shown. (B) Table showing the fold change was calculated by measuring the intensities of individual array spots and normalized with controls and compared with CLP group.
We next determined the effect of AR inhibition on the CLP-induced levels of inflammatory markers COX-2, iNOS and HMGB-1 in heart, spleen and kidney homogenates by Western blot analysis (Fig.4). As shown in Fig. 4A and B, polymicrobial sepsis caused ∼3.5, ∼2.5 fold induction of COX-2 and iNOS respectively which was significantly (∼90%) prevented by the administration of AR inhibitor. Similarly in CLP mice tissue homogenates, a significant increase in AR protein was observed and inhibitors of AR prevented CLP-induced increase in the expression of AR protein (Fig. 4C). Since the expression of cytokines and chemokines, COX-2, iNOS and AR depends on the activation of NF-κB, we next measured the activated form of NF-κB in mice heart, spleen and kidney homogenates. As shown in Fig. 4D the CLP-induced activation of NF-κB in mice tissue homogenates was significantly prevented by AR inhibition. Since activated macrophages and monocytes secrete HMGB-1 protein which acts as a mediator of inflammation [21], we next measured the effect of AR inhibition on the expression of HMGB-1. As shown in Fig. 4E, increased expression of HMGB-1 protein was observed in CLP tissues as compared to sham controls and ARI treatment significantly prevented CLP-induced HMGB-1. These results suggested that AR inhibition could prevent CLP-induced inflammation by inhibiting redox-sensitive transcription factor, NF-κB - dependent expression of various inflammatory markers such as cytokines and chemokines.
Figure-4. AR inhibition prevents CLP-induced expression of COX-2, iNOS and HMGB-1 in mice heart, spleen and kidneys.
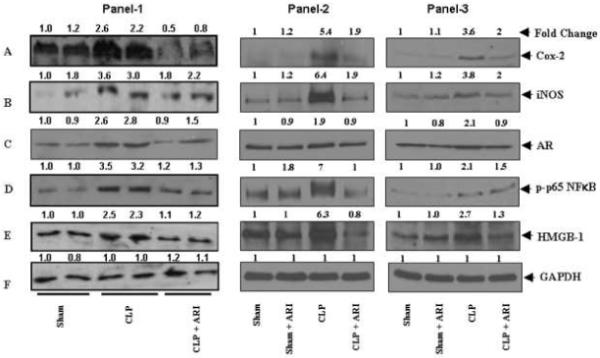
After 14 h of CLP, mice hearts, spleen and kidneys were dissected out and homogenates were made. Equal amounts of homogenates were subjected to Western Blot analysis using antibodies against A) COX-2, B) iNOS, C) AR, D) p-p65 (NF-κB), E) HMGB-1 and F)GAPDH. Panel 1: heart, Panel 2: Spleen and Panel 3: Kidney. The antibody binding was detected by enhanced pico chemiluminescence (Pierce). The fold change was determined by densitometry scanning, as indicated at the top of the Western blot.
3.3. Prevention of CLP-induced activation of plasma Tissue Factor by AR inhibition
Previous studies have shown that the use of anti-coagulants would be beneficial in preserving platelets and organ functions in endotoxemia-model via mechanisms involving inactivation of tissue factor, which plays a critical role in the initiation of complex mechanisms in the process of blood clot which causes organ dysfunction [22]. We therefore, measured the activation of tissue factor in CLP-mice plasma. Our results (Fig. 5) show that tissue factor activity was much higher in CLP group as compared to CLP+ARI group of mice as well as sham-operated control mice indicating that inhibition of AR could prevent secretion of tissue factor.
Figure-5. AR inhibition prevents tissue factor activity.
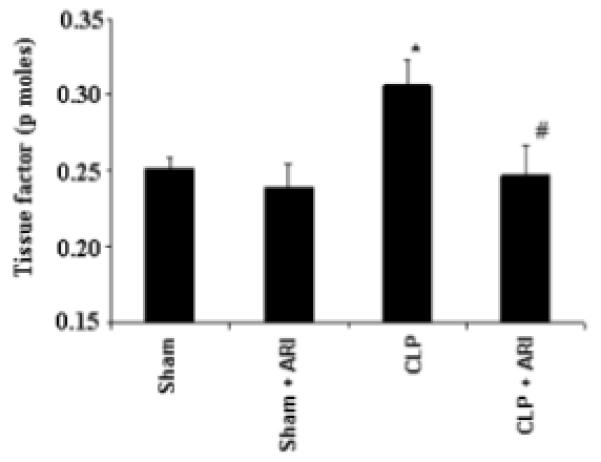
After 14 h of CLP, the mice plasma was collected. The tissue factor activity was measured by using ACTICHROME® Tissue factor activity assay kit from American diagnostic inc. as described in methods. Data are expressed as Mean ± SEM (n = 4). *P<0.001 Vs. Control and #P<0.001 vs CLP.
4 Discussion
Polymicrobial sepsis is a serious medical condition characterized by shock, hypoxemia, and organ dysfunction and disseminated intravascular coagulation [1] and [23]. Although subjects of all age groups upon severe bacterial infections can get sepsis, higher mortality has been observed in overweight, older age, intubated and immunocompromised patients[5]. Severe sepsis results from the body’s systemic over-response to infection that disrupts homeostasis through an uncontrolled cascade of inflammation [24], blood coagulation and impaired fibriolysis [21]. Even though controlling bacterial infections by aggressive antibiotics therapy and maintaining homeostasis in immune response saves a number of lives, 20-25% patients die because of multiorgan failure [4], [5] and [25]. Although precise mechanism(s) of multiorgan failure is not clearly understood, several clinical models have been developed to investigate the mechanism(s) involved in sepsis pathogenesis [26]. Among them, CLP-model has been shown to closely replicate the nature and course of clinical sepsis. This model is highly reproducible and widely used to study the mechaninsm(s) of multiorgan failure as well as for testing the efficacy of drugs [27].
Uncontrolled generation of proinflammatory cytokines and chemokines has been noted in experimental models of sepsis as well as in clinical settings. Early release of macrophage-derived proinflammatory cytokines, such as TNF-α, IL-6, IL-1βand MCP-1 have been shown to be important in the pathogenesis of septic shock and multiorgan failure [28], [29], [30] and [31]. In our study, AR inhibitor administration resulted in a significant decrease in these proinflammatory cytokines not only in the plasma and peritoneal fluids but in the heart as well. Thus the ability of AR inhibited mice to produce less inflammatory cytokines in response to CLP-induced sepsis could indicate the potential use of AR inhibitors in prevention of sepsis and its associated complications. It is well known that under oxidative stress induced by infections, bacterial toxins and during various pathological conditions NF-κB is significantly activated which regulates the expression of genes involved in inflammatory responses [10] and [18]. The mechanisms by which AR inhibition exerts an inhibitory effect on proinflammatory cytokine levels may involve the suppression of NF-κB activity [11] and [29]. In fact, several reports suggest that inhibition of NF-κB could prevent inflammatory response observed in sepsis. For example, antioxidants such as N-acetylcystene, ascorbic acid, and α-tocopherol [32], [33], [34] and [35] which prevent the activation of ROS-induced NF-κB have been shown to protect animals and humans from septic shock. We have recently shown that AR inhibitors which act like anti-oxidants and prevent bacterial endotoxin, LPS-induced generation of ROS prevent the activation of NF-κB and the expression of inflammatory markers in macrophages as well as in mice serum [9], [12], [13] and [14]. We have shown that AR-catalyzed reduced products of lipid aldehyde glutathione conjugates such as GS-DHN could mediate the signaling pathway that eventually increases transcription of inflammatory markers in vascular smooth muscle cells and other cells [14]. The increased cytokines, chemokines and growth factors subsequently initiate the signal transduction pathways that result in increased secretion of inflammatory markers which cause inflammation and multi organ failure. Our cellular studies using vascular smooth muscle cells, vascular endothelial cells and macrophages have demonstrated that ROS-induced NF-κB activation and transcription of inflammatory markers such as TNF-α, IL-1β, COX-2 and iNOS can be prevented by inhibiting AR [10], [11], [12], [13], [14] and [15]. In the present study also our results indicate that the release of bacteria into the peritoneum by CLP-procedure causes a significant increase in various inflammatory markers in the peritoneal fluid which is significantly prevented by AR inhibitor. Since several of the AR inhibitors have been found to be safe and have already been studied for phase-1 and phase-2 clinical trails for diabetic neuropathy [16] and [17], the use of AR inhibitors as anti-inflammatory drug would be less time consuming and if found as effective in human as in experimental model of sepsis, they could be used therapeutically in the inflammatory disorder patients. In summary, our results suggest that the AR inhibition has anti-inflammatory effects in murine model of sepsis. Further studies are required to use AR inhibition as an excellent therapeutic approach to treat sepsis especially those patients in which antibiotics though kill the bacteria, the circulating bacterial toxins including bacterial wall that takes several days to clear from the tissues cause fatal inflammatory disorders.
ACKNOWLEDGMENTS
This work was supported by NIH grant GM71036. We are thankful to Dr. E.R Sherwood, Department of Anesthesiology for help in performing CLP.
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Hotchkiss RS, Karl IE. The Pathophysiology and Treatment of Sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- [2].Brown K, Brain S, Pearson J, Edgeworth J, Lewis S, Treacher D. Neutrophils in development of multiple organ failure in sepsis. The Lancet. 2006;368:157–169. doi: 10.1016/S0140-6736(06)69005-3. [DOI] [PubMed] [Google Scholar]
- [3].Cho S, Minn Y, Kwon K. Stroke after Burn. Cerebrovasc Dis. 2007;24:261–263. doi: 10.1159/000104488. [DOI] [PubMed] [Google Scholar]
- [4].Parrillo JE, Parker MM, Natanson C, Suffredini AF, Danner RL, Cunnion RE, Ognibene FP. Septic shock in humans: advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann Intern Med. 1990;113:227–242. doi: 10.7326/0003-4819-113-3-227. [DOI] [PubMed] [Google Scholar]
- [5].Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- [6].Parrillo JE. Pathogenetic Mechanisms of Septic Shock. N Engl J Med. 1993;328:1471–1478. doi: 10.1056/NEJM199305203282008. [DOI] [PubMed] [Google Scholar]
- [7].Kurahashi K, Kajikawa O, Sawa T, Ohra M, Gropper MA, Frank DW, Martin TR, Wiener-kronish JP. Pathogenesis of septic shock in Pseudomonas aeruginosa pneumonia. J Clin Invest. 1999;104:743–750. doi: 10.1172/JCI7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Eichacker PQ, Parent C, Kalil A, Esposito C, Cui X, Banks SM, Gerstenberger EP, Fitz Y, Danner RL, Natanson C. Risk and the efficacy of antiinflammatory agents: retrospective and confirmatory studies of sepsis. Am J Respir Crit Care Med. 2002;166:1197–1205. doi: 10.1164/rccm.200204-302OC. [DOI] [PubMed] [Google Scholar]
- [9].Ramana KV, Willis MS, White MD, Horton JW, DiMaio JM, Srivastava D, Bhatnagar A, Srivastava SK. Endotoxin-induced cardiomyopathy and systemic inflammation in mice is prevented by aldose reductase inhibition. Circulation. 2006;24:1838–1846. doi: 10.1161/CIRCULATIONAHA.106.630830. [DOI] [PubMed] [Google Scholar]
- [10].Ramana KV, Chandra D, Srivastava S, Bhatnagar A, Aggarwal BB, Srivastava SK. Aldose reductase mediates mitogenic signaling in vascular smooth muscle cells. J Biol Chem. 2002;277:32063–32070. doi: 10.1074/jbc.M202126200. [DOI] [PubMed] [Google Scholar]
- [11].Ramana KV, Friedrich B, Srivastava S, Bhatnagar A, Srivastava SK. Activation of nuclear factor-kappaB by hyperglycemia in vascular smooth muscle cells is regulated by aldose reductase. Diabetes. 2004;53:910–920. doi: 10.2337/diabetes.53.11.2910. [DOI] [PubMed] [Google Scholar]
- [12].Ramana KV, Fadl AA, Tammali R, Reddy AB, Chopra AK, Srivastava SK. Aldose reductase mediates the lipopolysaccharide-induced release of inflammatory mediators in RAW264.7 murine macrophages. J Biol Chem. 2006;281:33019–33029. doi: 10.1074/jbc.M603819200. [DOI] [PubMed] [Google Scholar]
- [13].Ramana KV, Srivastava SK. Mediation of aldose reductase in lipopolysaccharide-induced inflammatory signals in mouse peritoneal macrophages. Cytokine. 2006;36:115–122. doi: 10.1016/j.cyto.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ramana KV, Bhatnagar A, Srivastava S, Yadav UC, Awasthi S, Awasthi YC, Srivastava SK. Mitogenic responses of vascular smooth muscle cells to lipid peroxidation-derived aldehyde 4-hydroxy-trans-2-nonenal (HNE): role of aldose reductase-catalyzed reduction of the HNE-glutathione conjugates in regulating cell growth. J Biol Chem. 2006;281:17652–17660. doi: 10.1074/jbc.M600270200. [DOI] [PubMed] [Google Scholar]
- [15].Ramana KV, Reddy AB, Tammali R, Srivastava SK. Aldose reductase mediates endotoxin-induced production of nitric oxide and cytotoxicity in murine macrophages. Free Radic Biol Med. 2007;42:1290–1302. doi: 10.1016/j.freeradbiomed.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bruno S, Cattaneo D, Perico N, Remuzzi G. Emerging drugs for diabetic nephropathy. Expert Opin Emerging Drugs. 2005;10:747–771. doi: 10.1517/14728214.10.4.747. [DOI] [PubMed] [Google Scholar]
- [17].Hamada Y, Nakamura J. Clinical potential of Aldose reductase inhibitors in diabetic neuropathy. Treat Endocrinol. 2004;3:245–255. doi: 10.2165/00024677-200403040-00006. [DOI] [PubMed] [Google Scholar]
- [18].Srivastava SK, Ramana KV, Bhatnagar A. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocr Rev. 2005;26:380–392. doi: 10.1210/er.2004-0028. [DOI] [PubMed] [Google Scholar]
- [19].Walley KR, Lukacs NW, Standiford TJ, Strieter RM, Kunkel SL. Balance of inflammatory cytokines related to severity and mortality of murine sepsis. Infect Immun. 1996;64:4733–4738. doi: 10.1128/iai.64.11.4733-4738.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circulation Research. 2002;91:988–998. doi: 10.1161/01.res.0000043825.01705.1b. [DOI] [PubMed] [Google Scholar]
- [21].Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- [22].Levi M, van der Poll T, ten Cate H, Van Deventer SJH. The cytokine-mediated imbalance between coagulant and anticoagulant mechanisms in sepsis and endotoxemia. Eur J Clin Invest. 1997;27:3–9. doi: 10.1046/j.1365-2362.1997.570614.x. [DOI] [PubMed] [Google Scholar]
- [23].Benjamin CF, Hogaboam CM, Kunkel SL. The chronic consequences of severe sepsis. Journal of Leukocyte Biology. 2004;75:408–412. doi: 10.1189/jlb.0503214. [DOI] [PubMed] [Google Scholar]
- [24].Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and duidelines for the use of innovative therapies in sepsis. Chest. 1992;101:1664–1655. doi: 10.1378/chest.101.6.1644. The ACCP/SCCM consensus conference committee. American college of chest physicians/society of critical care medicine. [DOI] [PubMed] [Google Scholar]
- [25].Maier S, Traeger T, Entleutner M, Westerholt A, Kleist B, Huser N, Holzmann B, Stier A, Pfeffer K, Heidecke C. Cecal ligation and puncture versus colon ascendens stent peritonitis: Two distinct animal models for polymicrobial sepsis. Shock. 2004;21:505–511. doi: 10.1097/01.shk.0000126906.52367.dd. [DOI] [PubMed] [Google Scholar]
- [26].Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, RueIII LW, Bland KI, Chaudry IH. Cecal ligation and puncture. Shock. 2005;24:52–57. doi: 10.1097/01.shk.0000191414.94461.7e. [DOI] [PubMed] [Google Scholar]
- [27].Wang P, Chaudry IH. A single hit model of polymicrobial sepsis: cecal ligation and puncture. Sepsis. 1998;2:227–233. [Google Scholar]
- [28].Damas P, Ledoux D, Nys M, Vrindts Y, De Groote D, Franchimont P, Lamy M. Cytokine serum level during sepsis in human IL-6 as a marker of severity. Ann Surg. 1992;215:356–362. doi: 10.1097/00000658-199204000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ho E, Bray TM. Antioxidants, NF-κB Activation, and Diabetogenesis. Proceedings of the Society for Experimental Biology and Medicine. 1999;222:205–213. doi: 10.1046/j.1525-1373.1999.d01-137.x. [DOI] [PubMed] [Google Scholar]
- [30].Ramnath RD, Ng SW, Guglielmotti A, Bhatia M. Role of MCP-1 in endotoxemia and sepsis. Int Immunopharmacol. 2008;8:810–818. doi: 10.1016/j.intimp.2008.01.033. [DOI] [PubMed] [Google Scholar]
- [31].Yang S, Hu S, Hsieh Y, Choudhry MA, RueIII LW, Balnd KI, Chaudry IH. Mechanism of IL-6-mediated cardiac dysfunction following trauma-hemorrhage. Journal of Molecular and cellular cardiology. 2006;40:570–579. doi: 10.1016/j.yjmcc.2006.01.008. [DOI] [PubMed] [Google Scholar]
- [32].Ritter C, Andrades ME, Reinke A, Menna-Barreto S, Moreira JF, Dal-Pizzol F. Treatment with N-acetylcysteine plus deferoxamine protects rats against oxidative stress and improves survival in sepsis. Crit Care Med. 2004;32:342–349. doi: 10.1097/01.CCM.0000109454.13145.CA. [DOI] [PubMed] [Google Scholar]
- [33].Galley HF, Howdle PD, Walker BE, Webster NR. The effects of intravenous antioxidants in patients with septic shock. Free Radic Biol Med. 1997;23:768–774. doi: 10.1016/s0891-5849(97)00059-2. [DOI] [PubMed] [Google Scholar]
- [34].Spapen H, Zhang H, Demanet C, Vleminckx W, Vincent JL, Huyghens L. Does N-acetyl-l-cysteine influence cytokine response during early human septic shock? Chest. 1998;113:1616–1624. doi: 10.1378/chest.113.6.1616. [DOI] [PubMed] [Google Scholar]
- [35].Konukoglu D, Iynem H, Ziylan E. Antioxidant status in experimental peritonitis: Effects of alpha tocopherol and taurolin. Pharmacological Research. 1999;39:247–251. doi: 10.1006/phrs.1998.0439. [DOI] [PubMed] [Google Scholar]