

Abstract
Oxidative stress is characterized as the imbalance between the cellular production of oxidants and cellular antioxidant defenses and contributes to the development of numerous cardiovascular and metabolic disorders, including hypertension and insulin resistance. The effects of prolonged oxidant stress in vitro on the insulin-dependent glucose transport system in mammalian skeletal muscle are not well understood. The current study examined the in vitro effects of low-level oxidant stress (60-90 μM, H2O2) for 4 hr on insulin-stimulated (5 mU/ml) glucose transport activity (2-deoxyglucose uptake) and on protein expression of critical insulin signaling factors (insulin receptor (IR), IR substrates IRS-1 and IRS-2, phosphatidylinositol-3-kinase (PI3-kinase), Akt, and glycogen synthase kinase-3 (GSK-3)) in isolated soleus muscle of lean Zucker rats. This oxidant stress exposure caused significant (50%, p<0.05) decreases in insulin-stimulated glucose transport activity that was associated with selective loss of IRS-1 (59%) and IRS-2 (33%) proteins, increased (64%) relative IRS-1 Ser307 phosphorylation, and decreased phosphorylation of Akt Ser473 (50%) and GSK-3ß Ser9 (43%). Moreover, enhanced (37%) phosphorylation of p38 mitogen-activated protein kinase (p38 MAPK) was observed. Selective inhibition of p38 MAPK (10 μM A304000) prevented a significant portion (29%) of the oxidant stress-induced loss of IRS-1 (but not IRS-2) protein and allowed partial recovery of the impaired insulin-stimulated glucose transport activity. These results indicate that in vitro oxidative stress in mammalian skeletal muscle leads to substantial insulin resistance of distal insulin signaling and glucose transport activity, associated with a selective loss of IRS-1 protein, in part due to a p38 MAPK-dependent mechanism.
Keywords: Soleus muscle, insulin resistance, hydrogen peroxide, insulin signaling
INTRODUCTION
Defects in the expression and functionality of critical elements of the canonical insulin signaling cascade, including the insulin receptor (IR), IR substrates 1and 2 (IRS-1 and IRS-2), phospatidylinositol-3-kinse (PI-3 kinase), Akt, and glycogen synthase kinase-3 (GSK-3), are known to cause insulin resistance in mammalian skeletal muscle, and these impairments in insulin action are associated with the development of pre-diabetes and type 2 diabetes (reviewed in refs. 1-4). One condition contributing to the multifactorial etiology of insulin resistance is oxidative stress, the imbalance between the cellular production of reactive oxygen species and the antioxidant defenses in cells [5-9]. Previous investigations, using either cultured cell lines or isolated mammalian skeletal muscle, have demonstrated that in vitro exposure to a low-level oxidant stress (H2O2) leads to diminished insulin-stimulated glucose transport activity [10-15], attributed to dysfunctions in the normal engagement of the insulin signaling pathway [11-13, 14-17]. Additional investigations using 3T3-L1 adipocytes and rat hepatoma cells have shown that exposure to this type of oxidant stress leads to a reduction in the protein expression of IRS-1 protein [16, 17], possibly by modulation of IRS-1 serine phosphorylation [16, 17]. However, the impact of this type of in vitro oxidative stress on the expression of critical insulin signaling factors in mammalian skeletal muscle is not currently known.
Several investigations have focused on the potential roles of various stress-activated serine kinases in the development of oxidative stress-associated insulin resistance (see ref. 5). We have shown recently that short-term (2 hr) exposure to a low-level oxidant stress in vitro in mammalian skeletal muscle is associated with decreased insulin suppression of the activity of the serine kinase GSK-3, and that selective inhibition of GSK-3 rescues about 20% of the impaired insulin action on glucose transport activity [15]. Another stress-activated serine kinase, p38 mitogen-activated protein kinase (p38 MAPK), is also thought to play a role in the development of insulin resistance [6, 7, 18-20]. Blair et al. [12] and Maddux et al. [14], in experiments using L6 myocytes, have shown that exposure to a low-level of oxidant stress leads to activation of the serine kinase p38 MAPK. Moreover, we have shown that this same in vitro oxidant stress in isolated rat skeletal muscle leads to engagement of p38 MAPK [15, 21]. Interestingly, the activation of p38 MAPK is inappropriately upregulated in fat cells [18] and skeletal muscle [22] of type 2 diabetic patients, and in cultured fat cells p38 MAPK overactivity is associated with reduced expression of GLUT-4 protein [18]. Using a model of insulin resistance in 3T3-L1 adipocytes, Carlson and colleagues [19] have shown that selective pharmacological inhibition of p38 MAPK improves insulin-stimulated glucose transport activity, an effect associated with the increased expression of GLUT-4, but not IRS-1, protein. However, the specific role of p38 MAPK in the etiology of the oxidant-induced insulin resistance in mammalian skeletal muscle is not well understood.
In the context of the foregoing information, the primary specific aim of the present investigation was to assess the effect of in vitro exposure of skeletal muscle from the lean Zucker rat to a low-grade oxidant stress (60-90 μM H2O2) for up to 4 hr on the protein expression of critical elements of the canonical insulin signaling cascade, including IRß, IRS-1 and IRS-2, PI3-kinase, Akt, and GSK-3, on GLUT-4 protein expression, and on insulin-dependent glucose transport activity. Moreover, an important associated specific aim was to address the specific role of p38 MAPK, which is engaged by oxidants in insulin-sensitive cell lines and tissues [5, 12, 14, 15, 21], in the induction of insulin resistance by this oxidant intervention in this mammalian skeletal muscle preparation.
MATERIALS AND METHODS
Animals
The University of Arizona Animal Use and Care Committee approved all procedures used in this study. Female lean (Fa/-) Zucker rats, aged 7-9 weeks, were obtained from Harlan (Indianapolis, IN) one week in advance of experiments, and they were used when they had body weights of 150-170 g. Animals were housed in a temperature-controlled room (20-22°C) with a 12:12-h light-dark cycle (lights on from 7AM to 7PM) at the University of Arizona Central Animal Facility. The animals had free access to chow (Teklad 7001, Madison, WI) and water. At 5 PM the evening before each experiment, animals were restricted to 4 g of chow, which was consumed immediately. Experiments began at 8 AM the next morning.
Skeletal muscle incubations and oxidant exposure
The animals were deeply anesthetized using an intraperitoneal injection of pentobarbital sodium (50 mg/kg body wt), and both soleus muscles were dissected and prepared for incubation. Each muscle was split into two strips (~25-35 mg each). Muscles were then incubated for 2 or 4 hrs at 37°C in 3 ml of oxygenated (95% O2-5% CO2) in Krebs-Henseleit buffer (KHB) supplemented with 8 mM glucose, 32 mM mannitol, and 0.1% BSA (radioimmunoassay grade, Sigma Chemical), in the absence or presence of a maximally effective concentration of insulin (5 mU/ml; Humulin, Eli Lilly, Indianapolis, IN), and without or with 100 mU/ml glucose oxidase (MP Biomedicals, Solon, OH), which produced the oxidant hydrogen peroxide (H2O2) at a concentration of 60-90 μM [15, 21]. The incubation medium was changed after each hour of treatment. In some experiments, the selective p38 MAPK inhibitor A304000 (kindly provided by Abbott Laboratories, Abbott Park, IL) was added to the incubation medium.
Determination of glucose transport activity
Following the initial 2- or 4-hr period of incubation, muscles were rinsed for 10 min at 37°C in 3 ml of KHB containing 40 mM mannitol, 0.1% BSA, and insulin, glucose oxidase, and inhibitor, if present previously. The muscles were then transferred to 2 ml of KHB containing 1 mM 2-deoxy-[1,2-3H]glucose (2-DG) (300 μCi/mmol; Sigma Chemical), 39 mM [U-14C]mannitol (0.8 mCi/mmol; ICN Radiochemicals, Irvine, CA), 0.1% BSA, and insulin, glucose oxidase, and inhibitor, if present previously. After this final 20-min incubation at 37°C, the muscles were removed, trimmed of excess fat and connective tissue, and quickly frozen between aluminum blocks cooled in liquid nitrogen. The specific intracellular accumulation of 2-DG was determined as described previously [23].
Signaling protein expression and functionality
Some muscles were frozen after the initial incubation period, weighed, and stored at -80°C until analysis. These muscles were homogenized in 8 volumes of ice-cold lysis buffer (50 mM HEPES, 150 mM NaCl, 20 mM Na pyrophosphate, 20 mM β-glycerophosphate, 10 mM NaF, 2 mM Na3VO4, 2 mM EDTA, 1% Triton X-100, 10% glycerol, 1 mM MgCl2, 1 mM CaCl2, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 0.5 μg/ml pepstatin, and 2 mM PMSF). Following homogenization, the homogenate was placed on ice for 20 min. The samples were then centrifuged at 13,000 × g for 20 min at 4°C. The supernatant was removed and stored at -80°C.
Protein concentration was determined using the BCA method (Sigma Chemical). Using 7.5% or 12% polyacrylamide gels (Bio-Rad Laboratories, Hercules, CA), the signaling proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were incubated with the appropriate dilution of commercially available antibodies to determine protein expression of insulin receptor β-subunit (IR-β) (Santa Cruz Biotechnology, Santa Cruz, CA), IRS-1 (Upstate Biotechnology, Lake Placid, NY), the p85 regulatory subunit of PI3-kinase (Upstate Biotechnology), total Akt1/2 and phospho-Akt (Ser473) (Cell Signaling Technology, Beverly, MA), total GSK-3β and phospho-GSK-3 α/ß (Ser21/Ser9) (Upstate Biotechnology), total p38 MAPK (Santa Cruz Biotechnology) and phospho-p38 MAPK (Thr180/Tyr182) (Cell Signaling Technology), and total GLUT-4 (Morphosys, Kingston, NH). For assessment of Ser307 phosphorylation of IRS-1, IRS-1 was immunoprecipitated as described previously [24] and immunoblotting was done with an antibody against IRS-1 phosphorylated on the Ser307 residue (Upstate Biotechnology). The membranes were then incubated in secondary goat anti-rabbit antibody conjugated with HRP (Chemicon, Temecula, CA) following the incubation with the primary antibody. Proteins were visualized on Kodak X-Omat AR film (Kodak, Rochester, NY) using an enhanced chemiluminescence detection system (Amersham Pharmacia, Piscataway, NJ). Densitometry was performed on the autoradiographs using a Bio-Rad imaging densitometer (Model GS-800) with Quantity One software.
Statistical analysis
Data are presented as means ± SE. Paired Student’s t-tests were employed to determine statistically significant differences in group means when soleus splits derived from the same muscle were used to assess the specific effects of the oxidant stress (Figs. 1-6) or the p38 MAPK inhibitor A304000 (Figs. 7-8). A p-value less than 0.05 was considered statistically significant.
Fig. 1.
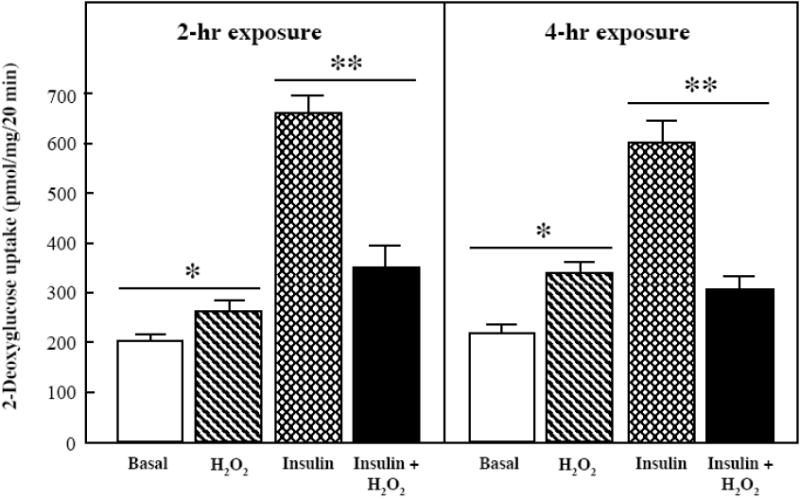
Effects of an oxidant stress on glucose transport activity in mammalian skeletal muscle.
Basal and insulin-stimulated (5 mU/ml) rates of 2-deoxyglucose uptake were determined in soleus muscle preparations from lean Zucker rats following 2-hr and 4-hr exposures to an in vitro oxidant stress (60-90 μM H2O2). Values are means ± SE for 6-9 muscles per group. *p<0.05 vs. basal in the absence of H2O2. **p<0.05 vs. insulin-stimulated in the absence of H2O2.
Fig. 6.
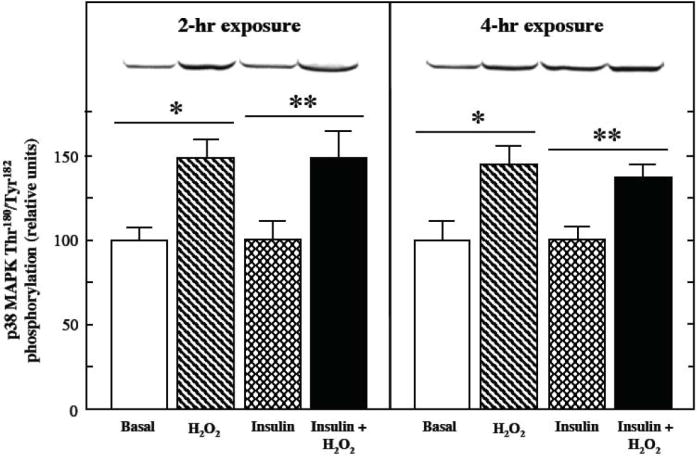
Effects of an oxidant stress on p38 MAPK phosphorylation in mammalian skeletal muscle. The extent of p38 MAPK Thr180/Tyr182 phosphorylation was measured in soleus muscle preparations from lean Zucker rats following 2-hr or 4-hr exposures to an oxidant stress (60-90 μM H2O2) in the presence of insulin (5 mU/ml). Values are means ± SE for 4-5 muscles per group. *p<0.05 vs. basal in the absence of H2O2. **p<0.05 vs. insulin-stimulated in the absence of H2O2.
Fig. 7.
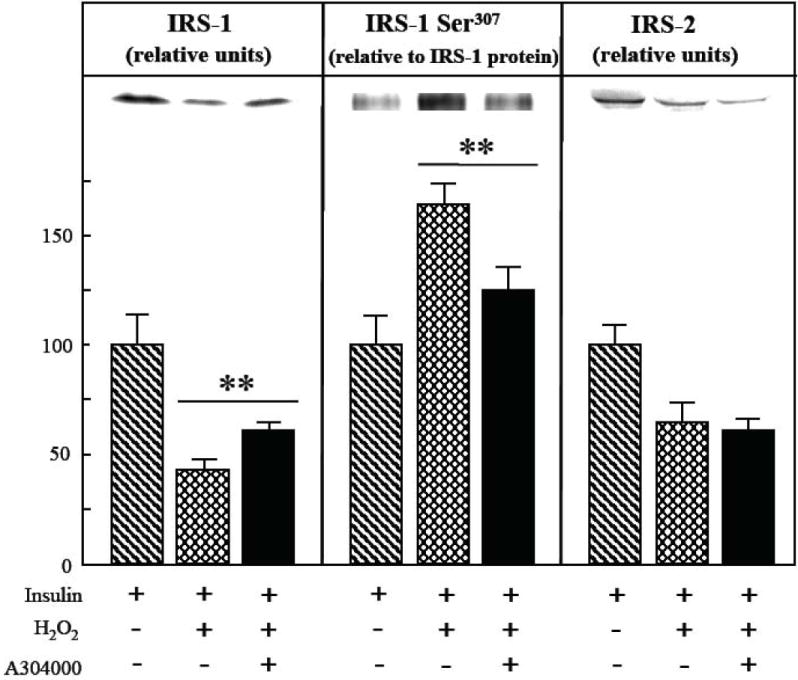
Effects of selective p38 MAPK inhibition on oxidant-associated loss of IRS-1 and IRS-2 protein and increased IRS-1 Ser307 phosphorylation in mammalian skeletal muscle.
The expression of IRS-1 and IRS-2 protein and level of Ser307 phosphorylation on IRS-1 in soleus muscle preparations from lean Zucker rats was determined following a 4-hr exposure to an oxidant stress (60-90 μM H2O2) and insulin (5 mU/ml) in the absence or presence of the selective p38 MAPK inhibitor A304000 (10 μM). IRS-1 Ser307 phosphorylation is expressed relative to total IRS-1 protein levels in the samples. Values are means ± SE for 4-5 muscles per group. **p<0.05 vs. insulin-and H2O2-stimulated in the absence of p38 MAPK inhibitor.
Fig. 8.
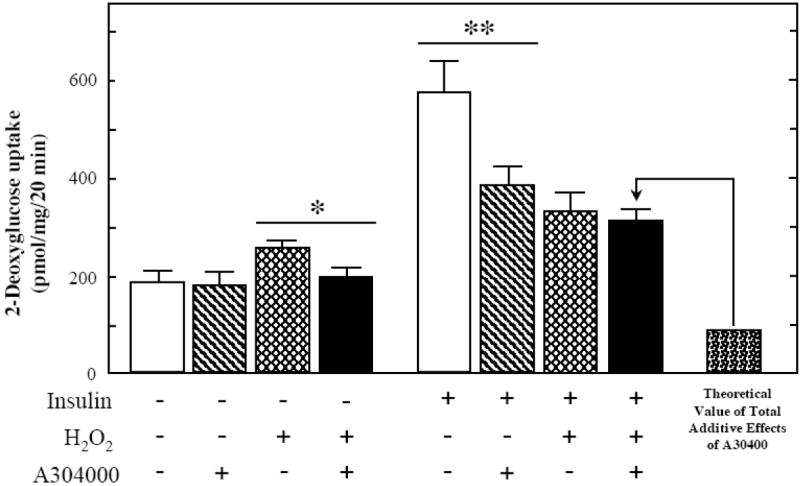
Effects of selective p38 MAPK inhibition in the presence of an oxidant stress on glucose transport activity in skeletal muscle.
Basal and insulin-stimulated rates of 2-deoxyglucose uptake were determined in soleus muscle preparations from lean Zucker rats following a 4-hr exposure to an in vitro oxidant stress (60-90 μM H2O2) in the absence or presence of the selective p38 MAPK inhibitor A304000 (10 μM). Values are means ± SE for 4-5 muscles per group. *p<0.05 vs. H2O2-stimulated in the absence of p38 MAPK inhibitor. **p<0.05 vs. insulin-stimulated in the absence of p38 MAPK inhibitor.
RESULTS
Effects of 4-hr oxidant stress on glucose transport activity
The effect of 2- and 4-hr exposures of the oxidant H2O2 on basal and insulin-stimulated glucose transport activity in mammalian skeletal muscle is shown in Fig. 1. In the absence of insulin, there was a significant increase (23%, p<0.05) in glucose transport activity in the soleus muscle following a 2-hr oxidant exposure, and a 38% increase (p<0.05) following the 4-hr exposure. In contrast, insulin-stimulated glucose transport activity was decreased by 50% (p<0.05) following a 2-hr oxidant exposure, and by 47% (p<0.05) following a 4-hr exposure to the oxidant. At the 4-hr time point, this represented a complete inhibition of insulin action, whereas after 2 hr the suppression of insulin-stimulated glucose transport activity was only ~75%.
Effects of 4-hr oxidant stress on insulin signaling element protein expression and functionality
The impact of the oxidant treatment on the protein expression of the two major IRS isoforms in mammalian skeletal muscle is shown in Fig. 2. In the absence of insulin, there was no significant alteration in IRS-1 protein expression after either a 2-hr or 4-hr exposure to the oxidant (Fig. 2, upper panel). In contrast, under insulin-stimulated conditions, IRS-1 protein expression tended (p<0.10) to be reduced by 22% following a 2-hr H2O2 exposure, and was decreased by 59% (p<0.05) following a 4-hr exposure to the oxidant. Under insulin-stimulated conditions, IRS-1 Ser307 phosphorylation, expressed relative to total IRS-1 protein, was increased by 38% and 64% (both p<0.05) after 2-hr or 4-hr exposure to the oxidant stress, respectively (Fig. 3). There was no significant alteration in IRS-2 protein expression after either the 2-hr or 4-hr incubation with the oxidant in the absence of insulin (Fig. 2, lower panel). Under insulin-stimulated conditions, however, IRS-2 protein expression was decreased by 33% (p<0.05) following the 4-hr oxidant treatment.
Fig. 2.
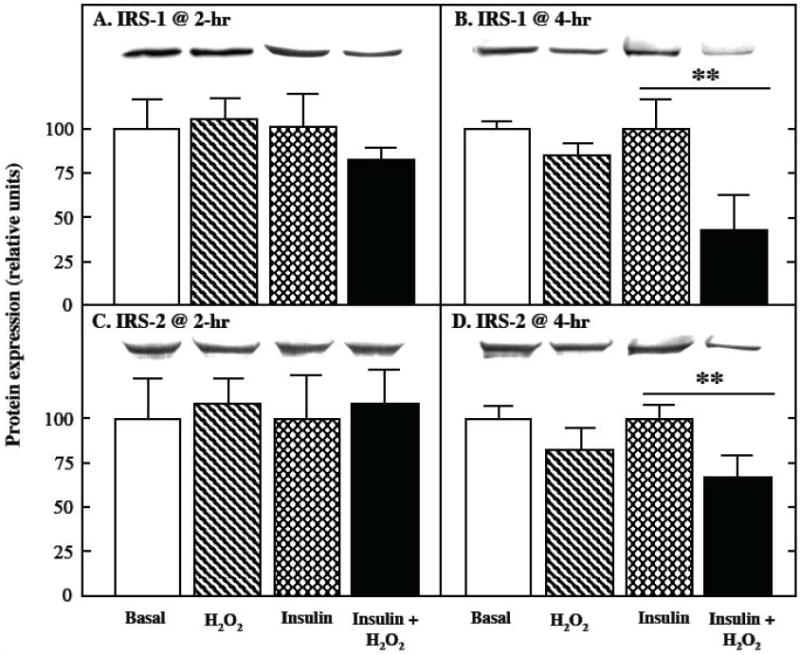
Effects of an oxidant stress on IRS-1 and IRS-2 protein expression in mammalian skeletal muscle. The expression of IRS-1 (upper panels) and IRS-2 (lower panels) protein was assessed in soleus muscle preparations from lean Zucker rats following 2-hr (left panels) or 4-hr (right panels) exposures to an oxidant stress (60-90 μM H2O2) in the absence or presence of insulin (5 mU/ml). Values are means ± SE for 4-6 muscles per group. **p<0.05 vs. insulin-stimulated in the absence of H2O2.
Fig. 3.
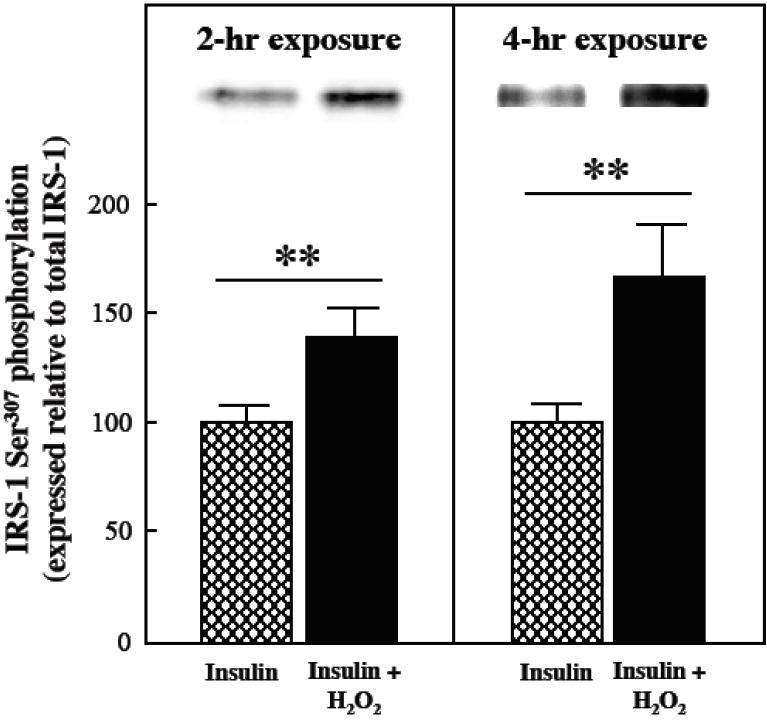
Effects of an oxidant stress on IRS-1 Ser307 phosphorylation in mammalian skeletal muscle. The phosphorylation of IRS-1 on Ser307 was assessed in soleus muscle preparations from lean Zucker rats following 2-hr (left panels) or 4-hr (right panels) exposures to an oxidant stress (60-90 μM H2O2) in the presence of insulin (5 mU/ml). IRS-1 Ser307 phosphorylation is expressed relative to total IRS-1 protein levels in the samples. Values are means ± SE for 4-5 muscles per group. **p<0.05 vs. insulin-stimulated in the absence of H2O2.
The effects of the 4-hr exposure to the oxidant H2O2 on the protein expression of four other critical elements of the canonical insulin signaling cascade (IR-β, the p85 regulatory subunit of PI3-kinase, Akt, and GSK-3) and on GLUT-4 in mammalian skeletal muscle are shown in Fig. 4. In the absence or presence of insulin, there were no significant changes in the protein expression of these insulin signaling factors and this glucose transporter isoform in response to the oxidant stress.
Fig. 4.
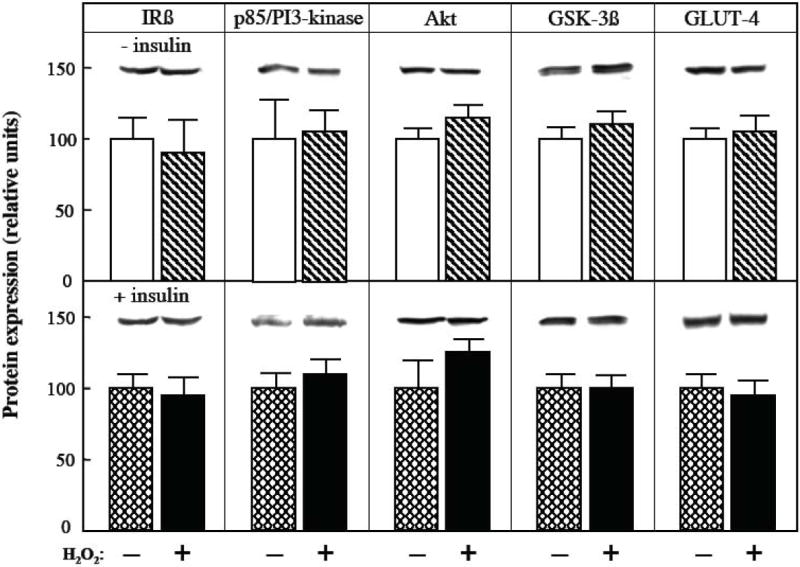
Effects of an oxidant stress on protein expression of insulin signaling factors and GLUT-4 in mammalian skeletal muscle. The protein expression of four critical insulin signaling elements (IRß, p85 regulatory subunit of PI3-kinase, Akt, and GSK-3ß) and GLUT-4 was determined in soleus muscle preparations from lean Zucker rats following a 4-hr exposure to an oxidant stress (60-90 μM H2O2) in the absence (upper panel) or presence (lower panel) of insulin (5 mU/ml). Values are means ± SE for 4-6 muscles per group.
The 4-hr exposure to the oxidant stress had a marked effect on the functionality of distal insulin signaling under insulin-stimulated conditions. As shown in Fig. 5 (left panel), whereas basal Akt phosphorylation was not altered by the 4-hr exposure to the oxidant stress, insulin-stimulated Akt phosphorylation was reduced by 50% (p<0.05). Likewise, the basal phosphorylation of GSK-3ß was not significantly changed by the 4-hr oxidant exposure, but this intervention diminished insulin-stimulated GSK-3ß phosphorylation by 43% (p<0.05).
Fig. 5.
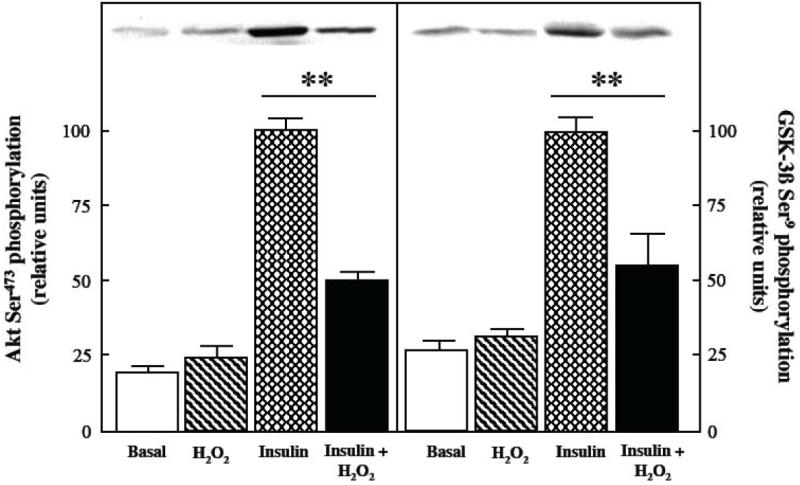
Effects of an oxidant stress on Akt and GSK-3ß phosphorylation in mammalian skeletal muscle.
The phosphorylation of Akt Ser473 and GSK-3ß Ser9 was assessed in soleus muscle preparations from lean Zucker rats following a 4-hr exposure to an oxidant stress (60-90 μM H2O2) in the absence or presence of insulin (5 mU/ml). Values are means ± SE for 4-6 muscles per group. **p<0.05 vs. insulin-stimulated in the absence of H2O2.
Effects of 4-hr oxidant stress on engagement of p38 MAPK
The impact of the oxidant stress on p38 MAPK phosphorylation in the basal and insulin-stimulated states in mammalian skeletal muscle was investigated (Fig. 6). Phosphorylation of this stress-activated serine kinase was increased (p<0.05) in the absence or presence of insulin after both 2 hr (50% and 49%) and 4 hr (45% and 37%) of oxidant exposure. The protein expression of p38 MAPK was not affected by the oxidant stress exposure (data not shown).
Role of p38 MAPK in oxidant-induced insulin resistance
In order to evaluate the specific contribution of this p38 MAPK activation to the etiology of the oxidant-associated insulin resistance, the selective p38 MAPK inhibitor A304000 (10 μM) was used [21, 25-27]. In the presence of insulin and the oxidant, the inhibition of p38 MAPK increased IRS-1 protein expression by 41% (p<0.05) (Fig. 7, left panel), thereby preventing 29% of the oxidant stress-induced loss of IRS-1. Moreover, IRS-1 Ser307 phosphorylation in the presence of insulin and the oxidant stress was reduced by 30% (p<0.05) by treatment with the p38 MAPK inhibtor (Fig. 7, center panel). In contrast, p38 MAPK inhibition had no effect on the oxidant-associated loss of IRS-2 in the presence of insulin (Fig. 7, right panel).
The selective inhibition of p38 MAPK had important and diverse effects on the actions of the oxidant stress and insulin, individually and in combination, on glucose transport activity in isolated mammalian skeletal muscle (Fig. 8). The inhibition of p38 MAPK prevented the increase in basal glucose transport activity due to the oxidant stress and substantially reduced insulin stimulation of this variable, consistent with our previous findings [21, 26, 27]. Importantly, the rate of 2-DG uptake in skeletal muscle treated with insulin and the oxidant stress in combination in the presence of A304000 (307 ± 24 pmol/mg muscle/20 min) was markedly greater than the theoretical additive value (92 pmol/mg muscle/20 min). This theoretical value was calculated by subtracting the individual inhibitory effects of A304000 on oxidant-induced 2-DG uptake (no insulin + H2O2 minus no insulin + H2O2 + A304000, or 250 − 200 = 50 pmol/mg muscle/20 min) and on insulin-stimulated 2-DG uptake (+ insulin minus + insulin + A304000, or 575 − 387 = 188 pmol/mg muscle/20 min) from the value experimentally measured in the presence of H2O2 and insulin (330 ± 36 pmol/mg muscle/20 min) (i.e., 330 − 50 − 188 = 92 pmol/mg muscle/20 min). This latter observation provides evidence that the deleterious effect of the oxidant stress on insulin-stimulated glucose transport activity is associated with a p38 MAPK-dependent mechanism.
DISCUSSION
The present investigation has shown for the first time that a 4-hr exposure of isolated insulin-sensitive skeletal muscle from the lean Zucker rat to a low-grade oxidant stress (60-90 μM H2O2) induces a complete insulin resistance of glucose transport activity (Fig. 1) that is associated with significant and progressive losses of both IRS-1 and IRS-2 proteins (Fig. 2) and with marked impairment of distal insulin signaling (Fig. 5). We have also demonstrated that this oxidant-induced loss of IRS proteins in insulin-stimulated skeletal muscle is selective, as the protein expression of other critical insulin signaling elements (IRß, p85/PI3-kinase, Akt, GSK-3ß) (Fig. 4), the GLUT-4 glucose transporter isoform (Fig. 3), and p38 MAPK was not significantly affected by this intervention. The loss of IRS-1 protein under insulin-stimulated conditions in response to the oxidant stress was associated with enhanced IRS-1 Ser307 phosphorylation. Moreover, the progressive loss of IRS proteins during this period of oxidant exposure appears to have physiological significance, as the insulin resistance of skeletal muscle glucose transport caused by the oxidant exposure was more severe at the 4-hr time point (100%) compared to the 2-hr time point (~75%). However, the loss of IRS proteins can account for only about 25% of this oxidant-induced insulin resistance, and other factors must be contributing to this dysfunction in insulin action.
These results in skeletal muscle are similar to the recent findings of Demozay and colleagues [28], who showed that treatment of 3T3-L1 adipocytes with 4-hydroxynonenal, an end production of lipid peroxidation considered to be an oxidant stress, leads to selective degradation of IRS-1 and IRS-2 protein, but not ERK1/2 MAPK. However, Potashnik et al. [17] and Bloch-Damti et al. [16] demonstrated in 3T3-L1 adipocytes and rat hepatoma cells that exposure to the oxidant H2O2 leads to a reduction in the protein expression of IRS-1, but not IRS-2. The discrepant results between these studies regarding IRS-2 degradation may relate to the differing types of oxidants employed and durations of exposure.
An additional novel finding of the present investigation was the observation that p38 MAPK contributes to the insulin resistance elicited by the oxidative stress in skeletal muscle. We observed that the oxidant stress caused significant increases in p38 MAPK phosphorylation at both the 2-hr and 4-hr time points (Fig. 6), consistent with our previous findings in isolated rat skeletal muscle [15, 21] and with results reported by other research groups using L6 myotubes [12, 14], perfused rat hearts [29], 3T3-L1 adipocytes [30], and cultured human lymphoid cells [31]. Importantly, when p38 MAPK was selectively inhibited using the aza-azulene-based molecule A304000 [21, 25, 26], the loss of IRS-1 protein (but not IRS-2 protein) induced by the oxidant stress and the increase in IRS-1 Ser307 phosphorylation was partially ameliorated (Fig. 7), and insulin action on glucose transport activity was increased relative to the theoretical value in the presence of insulin, the oxidant stress, and the p38 MAPK inhibitor (Fig. 8). In isolated rat skeletal muscle, it appears that a prolonged engagement of p38 MAPK in the presence of insulin is required for its role in the enhanced oxidant-induced loss of IRS-1 protein, as p38 MAPK was already activated by 2 hr of oxidant exposure, yet significant loss of IRS-1 protein was not detected until the 4-hr time point. Interestingly, in 3T3-L1 adipocytes, selective p38 MAPK inhibition with A304000 ameliorated the insulin-dependent loss of GLUT-4 protein, but not the loss of IRS-1 protein, and resulted in increased insulin-stimulated glucose transport activity [18]. Collectively, the results of the present study support the concept that, in mammalian skeletal muscle, engagement of a p38 MAPK-dependent mechanism is needed, at least in part, for the deleterious effects of the oxidant stress on IRS-1 degradation and for exacerbation of the oxidant-induced insulin resistance to be realized. However, it is clear that there are other, p38 MAPK-independent mechanisms for the oxidant-induced insulin resistance in mammalian skeletal muscle. The results of this study also indicate that in vitro oxidative stress impacts regulatory mechanisms differently in freshly isolated mammalian skeletal muscle compared to cultured cells such as the 3T3-L1 adipocyte.
One might suggest that the difference between the theoretical value and the experimental value shown in Fig. 8 is due solely to the decrease in basal transport brought about by A304000. The data do not support this contention. The magnitude of the difference between the theoretical value and the experimental value is much greater (~215 pmol/mg/20 min) than the A304000-mediated reduction in glucose transport activity stimulated by the oxidant alone (~50 pmol/mg/20 min). Moreover, in a separate study from our research group (Durazo, D. E.; Kim, J. S; Henriksen, E. J., unpublished data) investigating the effects of a 6-hr exposure to a lower level of oxidant (30-40 μM H2O2) on insulin action in skeletal muscle, we have demonstrated that this oxidant exposure does not increase basal glucose transport activity at the 6 hr time point, but does reduce insulin-mediated glucose transport activity by ~75% without a decline in IRS-1 protein expression. Importantly, inhibition of p38 MAPK with A304000 again diminishes the magnitude of the oxidant-associated insulin resistance, even under conditions when the oxidant had no effect on basal glucose transport.
The exact cellular mechanism for the preferential loss of IRS-1 and IRS-2 in mammalian skeletal muscle during in vitro oxidative stress is unclear. In skeletal muscle, there are three primary intracellular systems that regulate protein degradation: the ATP-dependent ubiquitin-proteasome system, the lysosomal system, and the calcium-activated protease system [32]. Low-grade oxidant stress has been shown to upregulate activity of the ubiquitin-dependent proteolytic pathway [33], as has exposure of L6 myotubes to reactive nitrogen species [34]. A limited amount of information indicates that the accelerated degradation of IRS-1 in a variety of cultured cell lines exposed to chronic insulin or the inflammatory cytokine TNF-α is mediated by the ubiquitin-proteasome system [33-35], whereas another study has demonstrated in 3T3-L1 adipocytes and rat hepatoma cells that IRS-1 degradation in response to a low-grade oxidant stress occurs by a proteasome-independent mechanism, despite an increase in protein ubiquination [17]. Future investigations should address the cellular mechanisms for the oxidant-induced loss of IRS proteins in mammalian skeletal muscle.
The results from the present study and from our previous investigation [15] provides further support for the concept advanced by Bloch-Damti and Bashan [5] that engagement of stress-activated serine kinases, with subsequent modulation of IRS-1 protein levels and functionality, is important in mediating the effects of oxidative stress on insulin action. Previous investigations have demonstrated that exposure of hepatoma cells [16] and FAO cells [5] to the oxidant H2O2 leads to engagement of various serine kinases, including p38 MAPK, c-Jun N-terminal kinase (JNK), Iκkinase ß (IKKß), and ERK1/2, with subsequent phosphorylation of IRS-1 on Ser307 and selective degradation of IRS-1 [17], leading ultimately to insulin resistance [10, 11, 13]. It should be noted, however, that loss of IRS-1 protein in skeletal muscle is not always associated with decreased insulin action. Indeed, whereas caloric restriction in rats leads to a decrease in muscle IRS-1 protein expression, insulin-stimulated glucose transport activity is actually increased [36, 37]. We have demonstrated in isolated rat skeletal muscle that the loss of IRS-1 protein in response to the oxidant stress under insulin-stimulated conditions is associated with increased IRS-1 Ser307 phosphorylation and activation of p38 MAPK. Other serine kinases were not evaluated in the present study, although in a previous investigation from our research group using isolated rat skeletal muscle [15], engagement of the serine kinase GSK-3ß was induced by shorter-term (2 hr) oxidative stress and selective GSK-3 inhibition resulted in enhanced insulin signaling and improved insulin-dependent glucose transport activity. The effect of selective GSK-3 inhibition on longer periods of oxidant exposure leading to loss of IRS proteins has not been investigated in mammalian skeletal muscle. An additional aspect that should be studied is the potential involvement of p38 MAPK as a regulator of GSK-3 activity under conditions of oxidative stress.
In conclusion, we have demonstrated in the present study using isolated rat skeletal muscle that a 4-hr exposure to a low-grade oxidant stress induces severe insulin resistance of glucose transport activity that is associated with loss of IRS-1 and IRS-2 proteins and impaired engagement of distal elements (Akt, GSK-3ß) of the insulin signaling cascade. We provide evidence that the loss of IRS-1 (but not IRS-2) is due, at least in part, to a p38 MAPK-dependent mechanism. However, it is clear that other mechanisms, both identified (such as GSK-3ß [15]) and yet unidentified, contribute to the etiology of oxidant-associated insulin resistance in mammalian skeletal muscle.
Acknowledgments
The study was supported by NIH grant DK063967 and a grant from Viatris AG, Frankfurt, Germany, to E.J.H.
Abbreviations
- 2-DG
2-deoxyglucose
- GSK-3
glycogen synthase kinase-3
- KHB
Krebs-Henseleit buffer
- IR
insulin receptor
- IRS-1
insulin receptor substrate-1
- IRS-2
insulin receptor substrate-2
- p38 MAPK
p38 mitogen-activated protein kinase
- PI3-kinase
phosphatidylinositol-3-kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zierath JR, Krook A, Wallberg-Henriksson H. Insulin action and insulin resistance in human skeletal muscle. Diabetologia. 2000;43:821–835. doi: 10.1007/s001250051457. [DOI] [PubMed] [Google Scholar]
- 2.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henriksen EJ, Dokken BB. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Current Drug Targets. 2006;7:1435–1442. doi: 10.2174/1389450110607011435. [DOI] [PubMed] [Google Scholar]
- 4.Karlsson HKR, Zierath JR. Insulin signaling and glucose transport in insulin resistant human skeletal muscle. Cell Biochem Biophys. 2007;48:103–113. doi: 10.1007/s12013-007-0030-9. [DOI] [PubMed] [Google Scholar]
- 5.Bloch-Damti A, Bashan N. Proposed mechanisms for the induction of insulin resistance by oxidative stress. Antioxidant Redox Signaling. 2005;7:1553–1567. doi: 10.1089/ars.2005.7.1553. [DOI] [PubMed] [Google Scholar]
- 6.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocrin Rev. 2002;23:599–622. doi: 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- 7.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signaling pathways mediators of insulin resistance and ß-cell dysfunction? Diabetes. 2003;52:1–8. doi: 10.2337/diabetes.52.1.1. [DOI] [PubMed] [Google Scholar]
- 8.Henriksen EJ. Exercise training and the antioxidant alpha-lipoic acid in the treatment of insulin resistance and type 2 diabetes. Free Rad Biol Med. 2006;40:3–12. doi: 10.1016/j.freeradbiomed.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 9.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;13(440):944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 10.Rudich A, Kozlovsky N, Potashnik R, Bashan N. Oxidant stress reduces insulin responsiveness in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab. 1997;272:E935–E940. doi: 10.1152/ajpendo.1997.272.5.E935. [DOI] [PubMed] [Google Scholar]
- 11.Rudich A, Tirosh A, Potashnik R, Hemi R, Kanety H, Bashan N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes. 1998;47:1562–1569. doi: 10.2337/diabetes.47.10.1562. [DOI] [PubMed] [Google Scholar]
- 12.Blair AS, Hajduch E, Litherland GJ, Hundal HS. Regulation of glucose transport and glycogen synthesis in L6 muscle cells during oxidative stress. Evidence for cross-talk between the insulin and SAPK2/p38 mitogen-activated protein kinase signaling pathways. J Biol Chem. 1999;274:36293–36299. doi: 10.1074/jbc.274.51.36293. [DOI] [PubMed] [Google Scholar]
- 13.Rudich A, Tirosh A, Potashnik R, Khamaisi M, Bashan N. Lipoic acid protects against oxidative stress induced impairment in insulin stimulation of protein kinase B and glucose transport in 3T3-L1 adipocytes. Diabetologia. 1999;42:949–957. doi: 10.1007/s001250051253. [DOI] [PubMed] [Google Scholar]
- 14.Maddux BA, See W, Lawrence JC, Jr, Goldfine AL, Goldfine ID, Evans JL. Protection against oxidative stress-induced insulin resistance in rat L6 muscle cells by micromolar concentrations of α-lipoic acid. Diabetes. 2001;50:404–410. doi: 10.2337/diabetes.50.2.404. [DOI] [PubMed] [Google Scholar]
- 15.Dokken BB, Saengsirisuwan V, Kim JS, Teachey MK, Henriksen EJ. Role of glycogen synthase kinase-3 in oxidative stress-induced insulin resistance in skeletal muscle. Am J Physiol Endocrinol Metab. 2008;294:E615–E621. doi: 10.1152/ajpendo.00578.2007. [DOI] [PubMed] [Google Scholar]
- 16.Bloch-Damti A, Potashnik R, Gual P, Le Marchand-Brustel Y, Tanti JF, Rudich A, Bashan N. Differential effects of IRS1 phosphorylated on Ser307 or Ser632 in the induction of insulin resistance by oxidative stress. Diabetologia. 2006;49:2463–2473. doi: 10.1007/s00125-006-0349-6. [DOI] [PubMed] [Google Scholar]
- 17.Potashnik R, Bloch-Damti A, Bashan N. IRS1 degradation and increased serine phosphorylation cannot predict the degree of metabolic insulin resistance induced by oxidative stress. Diabetologia. 2003;46:639–648. doi: 10.1007/s00125-003-1097-5. [DOI] [PubMed] [Google Scholar]
- 18.Carlson CJ, Koterski S, Sciotti RJ, Poccard GB, Rondinone CM. Enhanced basal activation of mitogen-activated protein kinases in adipocytes from type 2 diabetes: potential role of p38 in the downregulation of GLUT4 expression. Diabetes. 2003;52:634–641. doi: 10.2337/diabetes.52.3.634. [DOI] [PubMed] [Google Scholar]
- 19.Carlson CJ, Rondinone CM. Pharmacological inhibition of p38 MAP kinase results in improved glucose uptake in insulin-resistant 3T3-L1 adipocytes. Metabolism. 2005;54:895–901. doi: 10.1016/j.metabol.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 20.O’Keefe MP, Perez FR, Kinnick TR, Tischler ME, Henriksen EJ. Development of whole-body and skeletal muscle insulin resistance after one day of hindlimb suspension. Metabolism. 2004;53:1215–1222. doi: 10.1016/j.metabol.2004.02.025. [DOI] [PubMed] [Google Scholar]
- 21.Kim JS, Saengsirisuwan V, Sloniger JA, Teachey MK, Henriksen EJ. Stimulation of muscle glucose transport by an oxidant stress: roles of insulin signaling and p38 MAP kinase. Free Rad Biol Med. 2006;41:818–824. doi: 10.1016/j.freeradbiomed.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 22.Koistinen HA, Chibalin AV, Zierath JR. Aberrant p38 mitogen-activated protein kinase signaling in skeletal muscle from Type 2 diabetic patients. Diabetologia. 2003;46:1324–1328. doi: 10.1007/s00125-003-1196-3. [DOI] [PubMed] [Google Scholar]
- 23.Henriksen EJ, Halseth AE. Early alteration in soleus GLUT-4, glucose transport, and glycogen in voluntary running rats. J Appl Physiol. 1994;76:1862–1867. doi: 10.1152/jappl.1994.76.5.1862. [DOI] [PubMed] [Google Scholar]
- 24.Henriksen EJ, Teachey MK. Short-term in vitro inhibition of glycogen synthase kinase-3 potentiates insulin signaling in skeletal muscle of Zucker Diabetic Fatty rats. Metabolism. 2007;56:931–938. doi: 10.1016/j.metabol.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Somwar R, Koterski S, Sweeney G, Sciotti R, Djuric S, Berg C, Trevillyan J, Scherer PE, Rondinone CM, Klip A. A dominant-negative p38 MAPK mutant and novel selective inhibitors of p38 MAPK reduce insulin-stimulated glucose uptake in 3T3-L1 adipocytes without affecting GLUT4 translocation. J Biol Chem. 2002;277:50386–50395. doi: 10.1074/jbc.M205277200. [DOI] [PubMed] [Google Scholar]
- 26.Macko AR, Beneze AN, Teachey MK, Henriksen EJ. Roles of insulin signaling and p38 MAPK in the activation by lithium of glucose transport in insulin-resistant rat skeletal muscle. Arch Physiol Biochem. 2008;114:331–339. doi: 10.1080/13813450802536067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harrell NB, Teachey MK, Gifford NJ, Henriksen EJ. Essential role of p38 MAPK for activation of skeletal muscle glucose transport by lithium. Arch Physiol Biochem. 2007;113:221–227. doi: 10.1080/13813450701783158. [DOI] [PubMed] [Google Scholar]
- 28.Demozay D, Mas J-C, Rocchi S, Van Obberghen E. FALDH reverses the deleterious action of oxidant stress induced by lipid peroxidation product 4-hydroxynonenal on insulin signaling in 3T3-L1 adipocytes. Diabetes. 2008;57:1216–1226. doi: 10.2337/db07-0389. [DOI] [PubMed] [Google Scholar]
- 29.Clerk A, Fuller SJ, Michael A, Sugden PH. Stimulation of “stress-regulated” mitogen-activated protein kinases (stress-activated protein kinases/c-Jun N-terminal kinases and p38-mitogen-activated protein kinases) in perfused rat hearts by oxidative and other stresses. J Biol Chem. 1998;273:7228–7234. doi: 10.1074/jbc.273.13.7228. [DOI] [PubMed] [Google Scholar]
- 30.Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem. 1996;271:4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- 31.Kurata S. Selective activation of p38 MAPK cascade and mitotic arrest caused by low level oxidative stress. J Biol Chem. 2000;275:23413–23416. doi: 10.1074/jbc.C000308200. [DOI] [PubMed] [Google Scholar]
- 32.Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- 33.Shang F, Gong X, Taylor A. Activity of ubiquitin-dependent pathway in response to oxidative stress. Ubiquitin-activating enzyme is transiently up-regulated. J Biol Chem. 1997;272:23086–23093. doi: 10.1074/jbc.272.37.23086. [DOI] [PubMed] [Google Scholar]
- 34.Bar-Shai M, Reznick AZ. Reactive nitrogen species induce nuclear factor-kB-mediated protein degradation in skeletal muscle cells. Free Rad Biol Med. 2006;40:2112–2125. doi: 10.1016/j.freeradbiomed.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 35.Sun XJ, Goldberg JL, Qiao LY, Mitchell JJ. Insulin-induced insulin receptor substrate-1 degradation is mediated by the proteasome degradation pathway. Diabetes. 1999;48:1359–1364. doi: 10.2337/diabetes.48.7.1359. [DOI] [PubMed] [Google Scholar]
- 36.Gazdag AC, Dumke CL, Kahn CR, Cartee GD. Calorie restriction increases insulin-stimulated glucose transport in skeletal muscle from IRS-1 knockout mice. Diabetes. 1999;48:1930–1936. doi: 10.2337/diabetes.48.10.1930. [DOI] [PubMed] [Google Scholar]
- 37.McCurdy CE, Davidson RT, Cartee GD. Calorie restriction increases the ratio of phosphatidylinositol 3-kinase catalytic to regulatory subunits in rat skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:E996–E1001. doi: 10.1152/ajpendo.00566.2004. [DOI] [PubMed] [Google Scholar]