

Abstract
We recently reported a heretofore unknown role for the aryl hydrocarbon receptor in host resistance to listeriosis in mice. Hepatocytes are an important site for Listeria monocytogenes multiplication in vivo. In this study, we investigated whether activation of AhR in TIB73 murine embryonic hepatocytes affects the ingestion and intracellular multiplication of L. monocytogenes. Treatment of TIB73 cells with the AhR agonist β-naphthoflavone (BNF) significantly inhibited the ingestion and intracellular growth of L. monocytogenes. The inhibitory effects of BNF were dose-dependent and correlated with up-regulation of CYP1A1. Surprisingly, pretreatment with AhR antagonists (3′-MNF or α-naphthoflavone) or knocking-down of AhR with siRNA did not abolish the inhibitory effects of BNF. Moreover, the inhibitory effects of BNF on invasion and intracellular growth of L. monocytogenes by BNF were observed in AhR-deficient (CRL-2710), or ARNT-dysfunctional (CRL-2717) Hepa cells. We also observed similar inhibitory effects of BNF treatment using primary hepatocytes recovered from AhR+/- or AhR-/- mice. Moreover, the prototypic AhR agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) did not inhibit the invasion and intracellular growth of L. monocytogenes in TIB73 cells. Mechanistic studies demonstrated that ROS, but not TNF-α or iNOS, plays an important role in mediating BNF-induced inhibition. In conclusion, BNF caused an AhR-independent inhibition of ingestion and intracellular multiplication of L. monocytogenes in murine hepatocytes, mediated in part by production of ROS.
Keywords: aryl hydrocarbon receptor, Listeria monocytogenes, β-naphthoflavone, hepatocytes
1. Introduction
The aryl hydrocarbon receptor (AhR), together with its nuclear partner (ARNT), provides a powerful signaling system that mediates many of the biological and toxic effects of halogenated aromatic hydrocarbons (HAHs), polycyclic aromatic hydrocarbons (PAHs), and other structurally diverse ligands[1,2]. Once exogenous ligands such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), or endogenous ligands like 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) [3] or 6-formylindole[3,2-b]carbazole (FICZ) [4] bind to AhR, the cytoplasmic AhR complex alters its conformation and translocates into the nucleus. There it forms a heterodimer with ARNT that recognizes the upstream xenobiotic response elements (XRE, whose core motif is 5′-GCGTG-3′) of target genes [5]. Many cytokines (e.g., TNF-α, IL-1, IFN-γ, IL-8, IL-6, CCL1) [6,7,8,9,10] and immune cell surface receptors (e.g., Fas) [11] can be modulated by the AhR signaling pathway, indicating a regulatory role for the AhR in inflammation and immunity.
Several studies demonstrated that the AhR is involved in host resistance to infectious diseases. L. monocytogenes is a gram-positive, facultative intracellular bacterium that causes an important food-borne disease (listeriosis), which results in considerable morbidity and a relatively high mortality rate [12]. Early studies using in vivo treatment with TCDD or PAHs yielded inconsistent results on susceptibility to infection with L. monocytogenes [13,14,15,16,17], which were dependent on the AhR agonists used or their routes of administration. Activation of AhR by TCDD decreased the survival of mice infected with influenza virus, which correlated with increased levels of IFN-γ [18]. In contrast, TCDD treatment substantially improved the survival of mice infected with Streptococcus pneumoniae, albeit without evidence of a more vigorous inflammatory response to the bacterial cells [19]. More recently, we demonstrated that AhR null mice exhibit decreased resistance to Listeria monocytogenes infection, as evidenced by the greater numbers of L. monocytogenes in the livers and spleens of infected AhR null mice [20]. Further in vivo and ex vivo analyses did not reveal significant differences between AhR null and wild-type (WT) mice with respect to their ability to produce proinflammatory cytokines or the antimicrobial activity of their macrophages [20].
The liver is one of the principal target organs of L. monocytogenes infection. Liver parenchymal cells (hepatocytes), which constitute ∼77% of the total hepatic cells, can support intracellular multiplication of L. monocytogenes in vivo and in vitro [21]. Hepatocytes can be activated to restrict the intracellular multiplication of L. monocytogenes following stimulation with IFN-γ [22]. Therefore, in a broad sense, hepatocytes can be considered part of the “innate” immune system, especially in the local milieu at the liver. Because AhR is richly expressed in hepatocytes, and hepatocytes are highly responsive to AhR agonists, we hypothesized that AhR activation might influence the ability of hepatocytes to restrict L. monocytogenes infection.
Beta-naphthoflavone (BNF) has been widely used as an AhR agonist [23,24,25]. In this study, we investigated whether AhR activation by BNF would inhibit the ingestion and intracellular growth of L. monocytogenes in murine hepatocytes. Indeed, BNF significantly decreased the uptake and multiplication of L. monocytogenes in TIB73 cells, a murine embryonic hepatocyte cell line. As expected, BNF-dependent inhibition corresponded with the upregulation of CYP1A1, a biomarker for AhR activation. However, further mechanistic studies using pharmacological inhibitors, AhR-deficient Hepa cells, knock-down of AhR by siRNA, or primary hepatocytes isolated from AhR null mice, all strongly support the inference that BNF-mediated inhibition of L. monocytogenes is AhR-independent. Moreover, inhibition was not seen when TIB73 cells were treated with the potent AhR agonist TCDD. Pretreatment of TIB73 cells with the ROS scavenger N-acetyl-L-cysteine (NAC), a ROS scavenger completely blocked the effects of BNF, indicating that reactive oxygen species (ROS) are an important player in mediating BNF-induced inhibition of L. monocytogenes.
2. Results
2.1 BNF inhibits the ingestion and intracellular multiplication of L. monocytogenes in TIB73 cells
TIB73 cell monolayers were pretreated with an AhR agonist BNF (10 μM), or with a selective AhR antagonist 3′-MNF (1 μM) for 5 h, prior to infection with 105 CFU L. monocytogenes strain EGD (log-phase). Control monolayers were incubated with vehicle (DMSO) for 5 h before infection. BNF treated and control monolayers were then assessed for ingestion and intracellular multiplication of L. monocytogenes. Parallel sets of TIB73 cell cultures were used to isolate total RNA for real-time RT-PCR analyses of CYP1A1, as an indicator of AhR activation. As shown in Fig. 1A, BNF treatment (10 μM) significantly inhibited the ingestion and dramatically reduced the survival of L. monocytogenes in TIB73 cells. In contrast, the selective AhR antagonist 3′-MNF (1 μM) did not show any inhibitory effects. Nor was any inhibitory effect observed when 3′-MNF was used at a higher concentration (5 μM) (data not shown). Inhibition of L. monocytogenes by BNF corresponded with an upregulation of CYP1A1 (Fig. 1B). This finding is consistent with the inference that activation of AhR by BNF resulted in a significant inhibition of L. monocytogenes infection in TIB73 cells. Further experiments demonstrated that BNF-induced inhibition was dose-dependent. While inhibition of ingestion was observed only at 10 μM BNF, both 5 and 10 μM BNF significantly reduced intracellular multiplication of L. monocytogenes in TIB73 cells (Fig. 2A). Similar to the time-course studies (Fig. 1), dose-dependent inhibition of L. monocytogenes by BNF was associated with upregulation of CYP1A1 (Fig. 2B).
Figure 1.

Time-course studies of the invasion and intracellular multiplication of L. monocytogenes in TIB73 cells after BNF treatment. TIB73 cells were pretreated with BNF (10 μM), 3′-MNF (1 μM), or DMSO for 5 h before being infected with 105 log-phase L. monocytogenes for 2 h (Ingestion). Cells were then washed extensively to remove extracellular Listeriae and incubated in media with 5 μg/mL gentamycin for 2 to 24 h. A. CFU of L. monocytogenes recovered from infected TIB73 cells: Infected cells were lysed in sterile H2O to determine the CFU. Symbols indicate: BNF (10 μM) ( ), 3′-MNF (1 μM) (
), 3′-MNF (1 μM) ( ), or DMSO (
), or DMSO ( ). Data represent the mean±SEM Log10CFU/mL of six independent experiments. *: p<0.05, as compared to the control group. B. Fold change in CYP1A1 mRNA after being normalized to the control (GAPDH). Identically-treated TIB73 cells were used to isolate RNA for real-time RT-PCR analyses of CYP1A1. Data represent mean±SEM fold change of six independent experiments that were performed. Symbols indicate BNF (10 μM) (
). Data represent the mean±SEM Log10CFU/mL of six independent experiments. *: p<0.05, as compared to the control group. B. Fold change in CYP1A1 mRNA after being normalized to the control (GAPDH). Identically-treated TIB73 cells were used to isolate RNA for real-time RT-PCR analyses of CYP1A1. Data represent mean±SEM fold change of six independent experiments that were performed. Symbols indicate BNF (10 μM) ( ), 3′-MNF (1 μM) (
), 3′-MNF (1 μM) ( ), or DMSO (
), or DMSO ( ).
).
Figure 2.

BNF dose-dependently inhibits the invasion and intracellular growth of L. monocytogenes in TIB73 cells. TIB73 cells were treated with 1 ( ), 5 (), or 10 μM (
), 5 (), or 10 μM ( ) of BNF for 5 h prior to infection with 105 log-phase L. monocytogenes for 2 h (Ingestion). Cells were then washed extensively to remove extracellular Listeriae and incubated in media with 5 μg/mL gentamycin for an additional 8 h. DMSO was included as a vehicle control (). A. Infected cells were lysed in sterile water to quantify the number of viable L. monocytogenes. Data represent the mean±SEM Log10CFU/mL of four independent experiments that were performed. *: p<0.05 in comparison to the vehicle control. B. Parallel sets of TIB73 cells were used to isolate total RNA for real-time RT-PCR analyses of CYP1A1. CYP1A1 mRNA expression was presented as the mean±SEM fold change, relative to control samples, after normalization to that of GAPDH in the same samples.
) of BNF for 5 h prior to infection with 105 log-phase L. monocytogenes for 2 h (Ingestion). Cells were then washed extensively to remove extracellular Listeriae and incubated in media with 5 μg/mL gentamycin for an additional 8 h. DMSO was included as a vehicle control (). A. Infected cells were lysed in sterile water to quantify the number of viable L. monocytogenes. Data represent the mean±SEM Log10CFU/mL of four independent experiments that were performed. *: p<0.05 in comparison to the vehicle control. B. Parallel sets of TIB73 cells were used to isolate total RNA for real-time RT-PCR analyses of CYP1A1. CYP1A1 mRNA expression was presented as the mean±SEM fold change, relative to control samples, after normalization to that of GAPDH in the same samples.
2.2 BNF-mediated inhibition of L. monocytogenes is AhR-independent
Because BNF has been widely used as an AhR agonist, we first assumed the inhibitory effects of BNF on the ingestion and intracellular growth of L. monocytogenes in TIB73 cells were AhR-mediated. This was consistent with the correlation between BNF inhibition and CYP1A1 upregulation. To better establish a causal relationship between AhR activation and BNF-mediated inhibition of L. monocytogenes, we treated TIB73 cells with BNF (10 μM) in combination with the AhR antagonists 3′-MNF (5 μM) or α-naphthoflavone (ANF) (20 μM). To our surprise, neither 3′-MNF nor ANF abolished the inhibitory effects of BNF (Fig. 3). We tested various combinations of 3′-MNF or ANF with BNF, and found none of them blocked the inhibition associated with BNF. On the contrary, we observed an even stronger inhibition when 3′-MNF was combined with BNF, suggesting interactive effects between BNF and 3′-MNF. Because 3′-MNF [26] and ANF [27] can function as partial AhR agonist, we next used siRNA to knock down AhR expression in TIB73 cells. As shown by immunoblot (Fig. 4A), transfection of TIB73 cells with siRNAs against AhR for 72 h significantly reduced AhR expression by more than 90%. However, as shown by Fig. 4B, knocking down AhR with siRNAs did not block the inhibition of L. monocytogenes invasion and intracellular growth by BNF, which further supported that the inhibitory effects of BNF occurred independently of the AhR signaling pathway.
Figure 3.

AhR antagonists 3′-MNF and ANF do not block the inhibitory effects of BNF. TIB73 cells were treated with DMSO (), BNF (10 μM) alone (), or BNF (10 μM) in combination with 3′-MNF (1 μM) () or ANF (20 μM) ( ) for 5 h, followed by a 2-h inoculation with 105 log-phase L. monocytogenes. Ingestion and intracellular multiplication of L. monocytogenes were determined as described in Figure 1. Data represent the mean±SEM Log10 CFU/mL of four separate experiments that were performed. *: p<0.05, as compared to the vehicle control.
) for 5 h, followed by a 2-h inoculation with 105 log-phase L. monocytogenes. Ingestion and intracellular multiplication of L. monocytogenes were determined as described in Figure 1. Data represent the mean±SEM Log10 CFU/mL of four separate experiments that were performed. *: p<0.05, as compared to the vehicle control.
Figure 4.
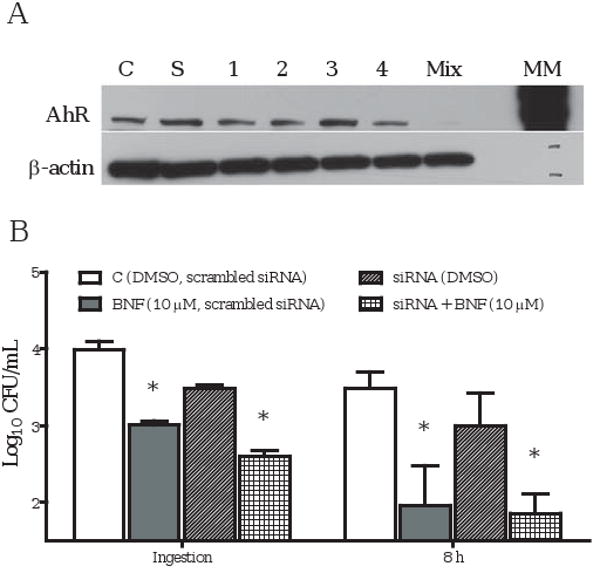
Knocking down AhR with siRNAs did not abolish BNF inhibition. Low-confluence TIB73 cells (∼20%) were transfected with a set of four ON-TARGETplus siRNAs against mouse AhR, or non-specific scrambled siRNAs, using Lipofectamine 2000. After 72 h, siRNA-treated TIB73 cells were used to isolate total protein (A), or were subjected to BNF treatment (B). A. AhR protein expression in TIB73 cells was decreased greater than 90% following the treatment with a mixture of 4 ON-TARGETplus siRNA duplexes (numbered 1-4) (Western Blot). B. Scrambled siRNA-transfected TIB73 cells treated with DMSO () or BNF (10 μM) (), or ON-TARGET siRNA-transfected TIB73 cells treated with DMSO () or BNF (10 μM) ( ), were infected with L. monocytogenes, as described in the methods. The data depict the mean±SEM Log10CFU/mL of three independent experiments that were performed. *: p<0.05, as compared to the corresponding DMSO controls.
), were infected with L. monocytogenes, as described in the methods. The data depict the mean±SEM Log10CFU/mL of three independent experiments that were performed. *: p<0.05, as compared to the corresponding DMSO controls.
We then investigated further using AhR-deficient and ARNT-dysfunctional Hepa cell lines. We observed a significant reduction in the numbers of L. monocytogenes recovered from BNF-treated Hepa-1 cells (wild type) (Fig. 5A), similar to what was observed for TIB73 cells. BNF treatment also inhibited L. monocytogenes invasion and multiplication in AhR-deficient and ARNT dysfunctional Hepa cells (Fig. 5B), further demonstrating that BNF-induced inhibition is not due to AhR activation.
Figure 5.

BNF inhibits the ingestion and intracellular multiplication of L. monocytogenes in AhR-deficient and ARNT-dysfunctional Hepa cells. Hepa-1 (wild type) and AhR-deficient and ARNT-dysfunctional Hepa cells were treated with DMSO or BNF (10 μM) for 5 h before being infected with 105 log-phase L. monocytogenes for 2 h (Ingestion). Cells were then washed extensively to remove extracellular Listeriae and incubated in media with 5 μg/mL gentamycin for 8 h. Cells were then lysed in sterile H2O to determine the CFU of L. monocytogenes. Data illustrate the mean±SEM Log10CFU/mL of three separate experiments that were performed. *: p<0.05, as compared to the corresponding controls. A. BNF () inhibited the ingestion and intracellular growth of L. monocytogenes in the WT Hepa-1 cells at all the times tested as compared to the DMSO control (). B. The inhibitory effects of BNF on L. monocytogenes persist as seen in AhR-deficient and ARNT-dysfunctional Hepa cells. Used symbols indicate: : Ingestion, DMSO control; : Ingestion, BNF (10 μM); : 8 h, DMSO control; : 8 h, BNF (10 μM).
One might argue that embryonic or transformed hepatocytes (TIB73 or Hepa cells, respectively) might behave differently from primary adult hepatocytes in terms of their ability to ingest and restrict the intracellular growth of L. monocytogenes, and the effects of BNF on these processes. We, therefore, isolated primary hepatocytes from AhR+/- and AhR-/- mice and tested these for the ability of BNF to inhibit ingestion and multiplication of L. monocytogenes. The inhibitory effects of BNF on the intracellular growth of L. monocytogenes were seen in both AhR+/- and AhR-/- hepatocytes (Fig. 6B). These data provide further evidence that BNF inhibition of L. monocytogenes is not mediated by AhR activation. However, unlike the TIB73 and Hepa cell lines, BNF treatment did not inhibit the ingestion of L. monocytogenes by primary hepatocytes. Indeed, we found several distinct differences between primary and transformed hepatocytes: 1. Most primary hepatocytes have more than one nucleus (Fig. 6A); 2. Primary hepatocytes killed rather than inhibited intracellular L. monocytogenes; and 3. BNF treatment rendered these cells even more efficient at killing intracellular L. monocytogenes (Fig. 6B). This observation is consistent with a previous report for IFN-γ activated primary hepatocytes [28]. In comparison, L. monocytogenes multiplied about 100-fold at 8 h post-inoculation in the hepatocyte cell lines (refer to Figs. 1A, 2A, and 5A&B).
Figure 6.
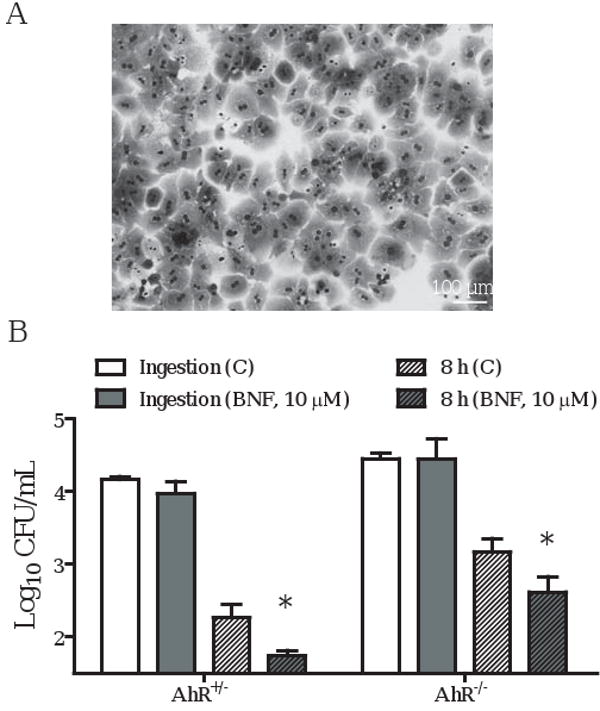
BNF inhibits the intracellular growth of L. monocytogenes in AhR-/- primary hepatocytes. Primary hepatocytes from AhR+/- or AhR-/- mice were prepared as described in the Material and Methods. A. Morphology of primary hepatocytes: most of the primary hepatocytes are bi-nucleated. B. Primary hepatocyte monolayers were pretreated with DMSO (Ctrl) or BNF (10 μM). The treated cells were then infected with 105 log-phase L. monocytogenes for 2 h (Ingestion). Cells were then washed extensively to remove extracellular Listeriae and incubated in media with 5 μg/mL gentamycin for 8 h. Cells were then lysed in sterile H2O to determine the CFU of L. monocytogenes. Symbols indicate: : Ingestion, DMSO control; : Ingestion, BNF (10 μM); : 8 h, DMSO control; : 8 h, BNF (10 μM). Data illustrate the mean±SEM Log10 CFU/mL from three separate experiments. *: p<0.05, as compared to the corresponding controls.
2.3 TCDD does not inhibit the ingestion and intracellular growth of L. monocytogenes
TCDD is the prototypical AhR agonist, whose biological effects and toxicity are thought to be mediated through the AhR signaling pathway. Here, we investigated whether activation of AhR with TCDD would alter the ingestion and intracellular growth of L. monocytogenes in TIB73 cells. TCDD treatment did not inhibit the invasion and intracellular growth of L. monocytogenes (Fig. 7A), despite significant up-regulation of CYP1A1 expression (Fig. 7B). These data provide yet another line of evidence that AhR activation does not necessarily inhibit ingestion and intracellular growth of L. monocytogenes in TIB73 cells.
Figure 7.

TCDD does not inhibit the ingestion and intracellular multiplication of L. monocytogenes. A. TIB73 cells were pretreated with DMSO (), BNF (10 μM) (), or TCDD (10 nM) () for 5 h prior to infection with 105 log-phase L. monocytogenes for 2 h (Ingestion). Cells were then washed extensively to remove extracellular Listeriae and incubated in media with 5 μg/mL gentamycin for 8 h. Cells were then lysed in sterile H2O to determine the CFU of L. monocytogenes. B. Identically-treated and infected TIB73 cells were used to isolate total RNA for real-time RT-PCR analyses of CYP1A1. Data in A represent the mean±SEM Log10CFU/mL of six independent experiments. *: p<0.05, as compared to the control. Data in B represent the fold change in CYP1A1 mRNA after TCDD or BNF treatment, after being normalized to the endogenous control (GAPDH).
2.4 Reactive oxygen species (ROS) are important mediators in BNF-induced inhibition
Because our data indicated BNF inhibition of ingestion and intracellular multiplication of L. monocytogenes is AhR-independent, we considered other possible mechanisms. First we applied various inhibitors in our system, including neutralizing anti-TNF-α antibody, NF-κB inhibitor (SN50), iNOS inhibitor (L-NIL: L-N6-(1-Iminoethyl)lysine hydrochloride), and an ROS scavenger (NAC: N-Acetyl-L-cysteine) to assess their effects on BNF-induced inhibition of L. monocytogenes. Pretreatment with the ROS scavenger NAC (1 mM) substantially blocked the inhibition of L. monocytogenes by BNF (Fig. 8A). At 20 μM, L-NIL did not abolish the inhibitory effects of BNF on Listeria monocytogenes although a marginal blocking effect on BNF-induced inhibition of intracellular multiplication was observed (Fig. 9). The other treatments did not produce noticeable significant effects on BNF inhibition of Listeria monocytogenes (data not shown). Utilizing a sensitive fluorescent dye (DFF) to quantify ROS production, we observed a significant increase in ROS levels in TIB73 cells after BNF treatment (Fig. 8B). When the thio-based antioxidant NAC (1 mM) was added together with BNF, ROS production returned to the baseline level (Fig. 8B). These findings suggest that ROS play an important role in mediating the inhibitory effects of BNF on the invasion and intracellular multiplication of L. monocytogenes in TIB73 cells.
Figure 8.

NAC treatment blocks the inhibition of L. monocytogenes induced by BNF. TIB73 cells were pretreated with DMSO, BNF alone (10 μM), NAC alone (1 mM), or BNF (10 μM) and NAC (1 mM) in combination for 5 h. A. Pretreated cells were infected with 105 log-phase L. monocytogenes for 2 h (Ingestion). Cells were washed extensively to remove extracellular L. monocytogenes. Infected cells were lysed in sterile H2O to estimate the ingested L. monocytogenes or incubated in media with 5 μg/mL gentamycin for 8 h to determine the intracellular multiplication of L. monocytogenes (8 h). Used symbols indicate: DMSO (), BNF alone (10 μM) (), NAC alone (1 mM) (), or BNF (10 μM) and NAC (1 mM) in combination (). Data represent the mean±SEM Log10CFU/mL of three representative experiments. *: p<0.05 as compared to the control group. B. Pretreated cells were loaded with 200 μM of DFF in growth medium for 1 h. The DFF loading medium was replaced with pre-warmed growth medium supplemented with BNF (10 μM) (), 3′-MNF (1 μM) (), or DMSO () after 1× wash with pre-warmed HBSS. Cells treated with PMA (0.5 μM) served as a positive control ( ). The production of ROS was monitored using a fluorescent microplate reader after addition of 105 log-phase L. monocytogenes. Data represent the mean±SEM fold change of 3 representative experiments, relative to the control by arbitrarily setting the control level at 1. *: p<0.05, as compared to the control.
). The production of ROS was monitored using a fluorescent microplate reader after addition of 105 log-phase L. monocytogenes. Data represent the mean±SEM fold change of 3 representative experiments, relative to the control by arbitrarily setting the control level at 1. *: p<0.05, as compared to the control.
Figure 9.

L-NIL treatment does not diminish the inhibition of Listeria monocytogenes induced by BNF. TIB73 cells were pretreated with DMSO, BNF alone (10 μM), L-NIL alone (20 μM), or BNF (10 μM) and NAC (20 μM) in combination for 5 h. A. Pretreated cells were infected with 105 log-phase L. monocytogenes for 2 h (Ingestion). Cells were washed extensively to remove extracellular L. monocytogenes. Infected cells were lysed in sterile H2O to estimate the ingested L. monocytogenes or incubated in media with 5 μg/mL gentamycin for 8 h to determine the intracellular multiplication of L. monocytogenes (8 h). Used symbols indicate: C (DMSO) (), BNF alone (10 μM) (), L-NIL alone (20 μM) (), or BNF (10 μM) and L-NIL (20 μM) in combination (). Data represent the mean±SEM Log10CFU/mL of three representative experiments. *: p<0.05 as compared to the control group; #: p<0.05 as compare to the BNF alone group at the same time point.
3. Discussion
The results of this study demonstrate that the well-recognized AhR agonist, beta-naphthoflavone inhibits the ingestion and intracellular multiplication of L. monocytogenes in hepatocytes. Surprisingly, we found that the inhibitory effects of BNF are AhR independent. This conclusion is based on five separate lines of evidence. 1) the selective AhR antagonists (3′-MNF or ANF) were without effect; 2) knocking-down of AhR with siRNAs did not prevent BNF mediated inhibition; 3) BNF induced inhibition of L. monocytogenes was observed in AhR-deficient or ARNT-dysfunctional Hepa cells; 4) the prototypical AhR agonist TCDD did not inhibit the ingestion and intracellular growth of L. monocytogenes in TIB73 cells; 5) inhibitory effects of BNF were observed in primary hepatocytes derived from AhR null mice.
BNF has been widely used as an AhR agonist [23,24,25]. It is believed that the majority of its biological effects are AhR-dependent. For example, BNF represses Dystrophin Dp71 expression in Hepa-1 cells in an AhR-dependent manner [23]. BNF treatment attenuated stressor-induced elevations in plasma cortisol and glucose levels in rainbow trout, which were blocked by the AhR inhibitor resveratrol [28]. However, we report here that BNF inhibited L. monocytogenes infection in various types of murine hepatocytes through an AhR-independent mechanism. To the best of our knowledge, this is the first study demonstrating BNF-induced effects that are not AhR-mediated. Surprisingly, increased inhibition of L. monocytogenes rather than an antagonistic effect was noted when TIB73 cells were treated with both BNF and the presumed AhR antagonists (3′-MNF or ANF). This observation is similar in principle to a previous study performed in zebrafish [29]. A possible explanation for the increased inhibition of L. monocytogenes upon combined treatment with 3′-MNF or ANF and BNF is that 3′-MNF or ANF inhibited CYP1A1 activity, which resulted in a longer biological half-life of BNF and thus in turn enhanced the inhibitory activity of BNF on L. monocytogenes infection, similar to what was reported previously [30].
Mechanistic studies indicated that BNF-induced inhibition is neither due to a direct inhibitory effect of BNF for L. monocytogenes, nor its ability to prevent the internalization of L. monocytogenes by hepatocytes (data not shown). One mediator that is essential for resistance to L. monocytogenes infection [31] is TNF-alpha, which can dramatically interrupt upregulation of CYP1A1 by BNF [32]. We, therefore, tested the role of TNF-α in BNF mediated inhibition of L. monocytogenes by adding neutralizing anti-TNF-α antibody to TIB73 cells prior to BNF treatment and infection with L. monocytogenes. Neutralization of TNF-α did not prevent the inhibition of L. monocytogenes associated with BNF (data not shown). Transgenic mice possessing a mutant inactive NF-κB signaling pathway (restricted to hepatocytes) were unable to clear L. monocytogenes from the liver and succumbed to sepsis, despite having ample numbers of immune and inflammatory cells in the liver [33]. We asked whether normal activation of NF-κB signaling by L. monocytogenes infection is differentially regulated by BNF, and thus contributes to the BNF-induced inhibition of L. monocytogenes in murine hepatocytes. After applying a selective NF-κB inhibitor (SN50), we did not observe any diminution in BNF inhibition of L. monocytogenes. Another important mediator involved in anti-Listeria resistance is nitric oxide (NO), which is produced by inducible nitric oxide synthase (iNOS) [34]. To clarify the role of NO in BNF-induced inhibition, we utilized a specific iNOS inhibitor L-NIL (L-N6-(1-Iminoethyl)lysine hydrochloride) and found that pretreatment of TIB73 cells with L-NIL only slightly reduced the inhibitory effects of BNF (Fig. 9). We also investigated whether another group of antimicrobial mediators, reactive oxygen species (ROS), could play an important role as effector molecules. The rationales were that ROS production is increased in microsomes isolated from the livers of BNF-treated rats (in vivo) [35], and Hepa-1 cells in vitro [36]. Although ROS often are thought of in the context of phagocyte antibacterial activity [37], there is evidence that they are produced by other non-phagocytic cells. For example, oxidative stress is of importance in damage to endothelial cells and has been reported for hepatocytes as well [38]. L. monocyogenes is also reported to increase its virulence by augmenting its capacity to counteract ROS [39]. Our data (Fig. 8) demonstrate that ROS plays a pivotal role in mediating BNF-induced inhibition, as NAC pretreatment blocked the inhibitory effects of BNF treatment. It is not clear how BNF affects ROS production, although our data convincingly suggest it is not due to AhR activation. Because oxidative phosphorylation by the mitochondrial respiratory chain is the main cellular source of ROS, future studies should focus on how BNF influences the mitochondrial respiratory chain.
Although hepatocyte cell lines have been widely used as an in vitro experimental system, our data argue that distinct differences exist between the antimicrobial activity of transformed cell lines and primary hepatocytes. Primary hepatocytes usually possessed more than one nucleus and killed rather than inhibited the ingested L. monocytogenes. It is notable that BNF inhibited the ingestion of Listeria in transformed hepatocyte cell lines, but not in primary hepatocytes. Our data also suggest that to some extent BNF can enhance the listericidal ability of primary hepatocytes, similar to what was reported for IFN-γ [28]. Moreover, we found that AhR+/- hepatocytes kill the ingested L. monocytogenes more efficiently than AhR-/- hepatocytes, which could at least in part explain the increased susceptibility of AhR-/- mice to listeriosis, as we reported previously [20]. This observation indicates the importance of constitutive AhR in controlling L. monocytogenes infection in hepatocytes. However, inhibitory effects of BNF on ingestion and intracellular growth of Listeria in hepatocytes are not dependent on the AhR signaling pathway.
4. Conclusions
In this study we demonstrated that a widely used AhR agonist, BNF could inhibit L. monocytogenes infection in hepatocytes in AhR-independent manner. This inhibition is mediated at least in part by ROS production. Using primary hepatocytes, we were able to provide further in vitro evidence that AhR is important for the optimal anti-listeria ability of hepatocytes, in support of our published in vivo data [20]. Further efforts should be directed to examine how BNF affects ROS production, as our data indicate it is not because of AhR activation. Since oxidative phosphorylation by the mitochondrial respiratory chain is the main cellular source of ROS, it would be interesting to see how BNF influence the mitochondrial respiratory chain. In addition, based on these findings, a potential anti-listeria therapy using flavonoids such as BNF could be developed.
5. Materials and Methods
5.1 Materials
AhR-/- mice were received from Dr. Christopher Bradfield at the McArdle lab, University of Wisconsin-Madison. 3′-methoxy-4′-nitroflavone (3-MNF) was a generous gift from Dr. Thomas Gasiewicz at the University of Rochester. BEAR-1 rabbit polyclonal antibody against mouse AhR (1:300) was generously provided by Dr. Richard Pollenz at the University of South Florida. The TIB73 (ATCC TIB73) and Hepa cell lines (ATCC CRL-2026, CRL-2710, and CRL-2717) were obtained from ATCC (Manassas, VA). Chemically synthesized siRNAs were purchased from Darmacon (Chicago, IL). Dulbecco modified Eagle tissue culture medium (DMEM), α-minimum essential medium without ribonucleosides and deoxyribonucleosides, L-glutamine, fetal bovine serum, and gentamicin were purchased from Mediatech (Manassas, VA). BNF, TCDD, and α-naphthoflavone (ANF) were originally purchased from Sigma (St. Louis, MO). These were dissolved in DMSO as stock solution and stored in -80 °C. Lipofectamine 2000 were purchased from Invitrogen (Carlsbad, CA), N-Acetyl-L-cysteine (NAC) was from Calbiochem (San Diego, CA), and difluorodihyrdofluorescein (DFF) was originally purchased from Molecular Probes (Eugene, OR).
5.2 Animal care and breeding
C57Bl/6J-AhR-deficient (AhR-/-) mice, created by deletion of exon 2 of the AhR gene, were obtained from Dr. C. Bradfield (Madison, WI) and maintained as a breeding colony in the animal care facility at the University of Wisconsin School of Veterinary Medicine. The AhR-/- mice could not be bred efficiently by homozygous mating. Instead, AhR+/- and AhR-/- littermates were generated by breeding AhR+/- females with AhR-/- males. The offspring were genotyped by PCR. All mice were provided either regular chow or breeder chow (Harlan Teklad 8604 rodent diet, Madison, WI) and water ad libitum. All animal handling and experimental processes were performed in accordance with an IACUC approved protocol.
5.3 Isolation of primary hepatocytes from AhR+/- or AhR-/- mice
Hepatocytes were isolated following perfusion of livers with collagenase using a two-step technique described previously with minor modifications [41]. In brief, the livers of euthanized 4-8 months old AhR+/- or AhR-/- mice were perfused with warm (37°C) Ca2+- and Mg2+-free HBSS, followed by warm HBSS containing 0.6 mg/mL collagenase (type IV) and 0.25 mg/mL trypsin inhibitor (Sigma, St. Louis, MO). The perfused livers were dissected, the gall bladders and extraneous tissue were removed, and the livers were immediately teased apart. The resulting cell suspensions (total hepatic cells) were centrifuged at 30 × g for 5 min at 4°C. The cell pellet was washed once and re-suspended in DMEM supplemented with 10% FBS, 1% P/S, and L-glutamine (2 mM). About 106 cells in 1 mL of culture medium were added into each well of 24-well culture plate and incubated overnight in a humidified incubator with 95% air-5% CO2 at 37°C. Cell monolayers (∼70% confluence) were subject to the treatments and infection with L. monocytogenes described below. The majority of cells (>95%) were identified to be hepatocytes as assessed by morphology. Primary hepatocytes were used within 2 days following seeding to retain their physiological characteristics.
5.4 Preparation of log-phase L. monocytogenes
Stock cultures of L. monocytogenes were maintained at -70 °C. To prepare the log-phase L. monocytogenes, the frozen cultures were diluted 1:500 in brain heart infusion broth (BHI) and grown to mid-logarithmic phase (4.5-7 h at 37 °C) on a rotating platform. The bacterial cells were pelleted by centrifugation (1500 × g for 10 min), washed twice with sterile PBS, and diluted in PBS to the appropriate concentration for inoculation. The number of viable L. monocytogenes in the inoculum was confirmed by plating serial dilutions on blood agar.
5.5 Infection of hepatocyte cells
TIB73 murine embryonic hepatocyte cells were cultured as described previously [42]. In brief, TIB73 cells were cultured in Dulbecco modified Eagle tissue culture medium (DMEM) (Sigma) supplemented with 2 mM glutamine and 10% heat-inactivated fetal bovine serum (FBS). Hepa cells, including wild type (ATCC CRL-2026), AhR-deficient (ATCC CRL-2710), and ARNT-dysfunctional (ATCC CRL-2717) cells, were cultured in α-minimum essential medium without ribonucleosides and deoxyribonucleosides, supplemented with 2 mM L-glutamine and 10% heat-inactivated fetal bovine serum according to data sheets from ATCC. One mL of hepatocyte suspensions were added to each well of 24-well tissue culture plates and incubated overnight in a humidified incubator with 95% air-5% CO2 at 37°C. After an overnight incubation, monolayers with a ∼ 70% confluence were pretreated with BNF, 3′-MNF, ANF, TCDD, or combined treatments for 5 h as described in the individual experiments. Monolayers in each well were then infected with 105 CFU log-phase L. monocytogenes for 2 h. Following infection, extracellular listeriae were removed by washing at least 4 times with 1 mL of pre-warmed DMEM. The infected hepatocyte monolayers were then either lysed in sterile H2O or incubated in DMEM supplemented with 5 μg/ml of gentamicin for additional time points (2 to 24 h) as designated in the individual experiments. At the indicated time, hepatocytes were washed 4 × with PBS and then lysed in 1ml of sterile water. The lysates were serially diluted in PBS and plated in duplicate on blood agar (Becton, Dickinson and Company, Sparks, MD). The blood agar plates were incubated for 24 to 48 h at 37°C and colony forming units (CFU) were counted. Results were expressed as the mean±SEM log10 CFU/mL of L. monocytogenes. Three wells (triplicates) were used for each treatment group in an experiment. Each experiment was repeated at least three times.
5.6 Quantitative real-time RT-PCR analyses of CYP1A1 and GAPDH
The transcription of genes encoding CYP1A1 and GAPDH was quantified using real-time reverse transcriptase-polymerase chain reaction (RT-PCR). Briefly, total RNA was isolated from TIB73 cells treated with BNF (10 μM), TCDD (10 nM), or DMSO using RNeasy columns (Qiagen, Palo Alto, CA). RNA (1 μg) was reverse transcribed into cDNA using the reverse transcription system from Promega (Madison, WI). The primers for target genes were designed using Primer Express 3.0 software (Applied Biosystems, Foster City, CA): for CYP1A1, 5′-TGAGTCAGCAGTATGGGGACG (forward), 5′-AGGGCCTGCTTGATGGTGTT (reverse); for GAPDH, 5′-CTCCACTCACGGCAAATTCA (forward), 5′-CGCTCCTGGAAGATGGTGAT (reverse). The ABsolute QPCR SYBR green Mix kit (ABgene, Rochester, New York) was used for real-time RT-PCR analyses. The amplification was carried out in the 7300 real time PCR system (Applied biosystems). Amplification conditions were 2 min at 50 °C, 15 min at 95 °C, followed by 40 cycles of 15 sec at 95 °C and 1 min at 60 °C. All the primers for real-time RT-PCR analyses were synthesized by IDT (Coralville, IA).
5.7 Knocking-down of AhR in TIB73 cells with siRNAs
Chemically synthesized, double-stranded ON-TARGETplus siRNA duplexes targeting mouse AhR (NM_013464), as well as control siRNA were purchased from Dharmacon (Chicago, IL). Low-confluence TIB73 cell cultures (∼20%) in 24-well plate were transfected with a set of 4 ON-TARGETplus duplexes (100 nM) with sense sequences: 5′-CGACAUAACGGACGAAAUCUU, 5′-GUGCAGAGUUGAGGUGUUUUU, 5′-UCAGAGCUCUUUCCGGAUAUU, and 5′-CAAGGGAGGUUAAAGUAUCUU, or control siRNAs (5′-GAAACGCCGAAGCUAGAACUU and 5′-GACGUCAAUCCGCAGUGACUU) after complex formation with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) for 20 min. Twenty-four hours later, siRNAs were removed by replacing old medium with fresh medium and the transfected cells were incubated further for 2 days. Parallel sets of siRNA-treated TIB73 cells were then subjected to BNF (10 μM) or DMSO (vehicle control) treatment for 5 h, followed by a 2-h inoculation with 105 log-phase L. monocytogenes. Ingestion and intracellular multiplication were determined as described above. Another set of wells in each experiment were used to isolate total protein for Western Blot analyses of AhR and β-actin as described below.
5.8 Western Blot Analyses of AhR
Total proteins were isolated by re-suspending siRNA-treated TIB73 cells, as described above, in 1 mL homogenization buffer containing 5 μM pepstatin A, 10 μM leupeptin, 7.5 μM trans-epoxysuccinyl-L-leucylamido(4-guanidino) butane (E-64), 25 μM bestatin, 0.4 μM aprotinin, 500 μM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF), 1 mM EGTA, 1 mM EDTA, 1 mM sodium vanadate, 1 mM phenylmethylsulfonyl fluoride (PMSF), 15 mM 2-mercaptoethanol (ME), 0.1% Triton X-100, 150 mM NaCl, and 0.05% SDS in 50 mM Tris-HCl buffer, pH 7.5. After 3 cycles of freezing-thawing, cell homogenates were centrifuged at 10,000 × g for 10 min at 4 °C, and the supernatant was collected and stored at -80°C until analyzed further. Protein concentration was determined by BCA protein assay (Pierce, Rockford, IL). Equal amounts of proteins were loaded onto 4-20% Tris-HCl SDS-PAGE gel (Bio-Rad) for AhR (∼95 kD) and β-actin (∼42 kD) detection. After electrophoresis, gels were immunoblotted with the BEAR-1 rabbit polyclonal antibody against mouse AhR (1:300) (a gift received from Dr. Pollenz of University of South Florida). Membranes were further treated with a secondary antibody (goat anti-mouse IgG) and visualized using an ECL detection kit (Amersham Biosciences). As an internal standard, the same membranes were stripped with Western blot stripping buffer (Pierce) and immunoblotted with the monoclonal anti-β-actin antibody (1:4000) (Sigma). The density of bands was analyzed using ImageJ. The values of AhR proteins were normalized by those of β-actin in the same gel. The decrease of AhR protein in the AhR-targeting siRNA-treated samples was assessed by comparing to the level in the scrambled siRNA-treated controls.
5.9 Detection of ROS
TIB73 cells were pretreated with BNF (10 μM), or BNF (10 μM) combined with 1 mM of NAC (N-acelyl-L-cysteine), a thio-based antioxidant. Same cells were incubated with DMSO (vehicle) as the control. After 5 h, cells were loaded for 1 h with 200 μM of difluorodihyrdofluorescein (DFF) in complete culture medium. DFF passively diffuses across the cell membrane. Non-specific esterases then cleave lipophilic blocking groups and produce a charged form of the dye that does not readily diffuse out of the cell. After 1 h incubation at 37°C, hepatocytes were washed 1× with pre-warmed HBSS and then cultured in pre-warmed fresh growth medium supplemented with BNF, BNF and NAC, or DMSO. Treated cells were inoculated with 105 log-phase L. monocytogenes, and bacterial ingestion and intracellular growth determined as described above. Some monolayers were incubated with PMA (0.5 μM) as a positive control for ROS. ROS production was immediately detected using a fluorescent plate reader (emission wavelength: 485 nm; absorption wavelength: 525 nm).
5.10 Statistical Analysis
Statistical comparisons were performed using one-way ANOVA (Prism 5.0, GraphPad software Inc., San Diego, CA) or Student t-test, whichever was appropriate. A probability of p<0.05 was considered statistically significant. Data are reported as mean ± SEM for the indicated number of experiments.
Acknowledgments
This work was supported by funds from the National Institute of Health [grant number R21AI059656 to C.J.C.], the USDA National Research Institute [grant number 2005-35201-15313 to C.J.C.], the National Alliance for Food Safety and Security [grant number 58-1935-4450 to C.J.C.], and the Walter and Martha Renk Endowed Laboratory for Food Safety [to C.J.C.]. We would like to thank Dr. Christopher Bradfield for providing us the AhR-/- mouse breeders. We thank Dr. Thomas Gasiewicz for offering us the selective AhR antagonist, 3′-MNF. We thank Dr. Richard Pollenz for providing the BEAR-1 rabbit polyclonal antibody against mouse AhR. We are grateful to Dr. Colin Jefcoate for constructive discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Lewis Zhichang Shi, Email: lewis.shi@stjude.org.
Charles J. Czuprynski, Email: czuprync@svm.vetmed.wisc.edu.
References
- 1.Safe S. Polychlorinated biphenyls (PCBs), dibenzo-p-dioxins (PCDDs) dibenzofurans (PCDFs), and related compounds: environmental and mechanistic considerations which support the development of toxic equivalency factors (TEFs) Crit Rev Toxicol. 1990;21:51–88. doi: 10.3109/10408449009089873. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci USA. 1996;93:6731–6. doi: 10.1073/pnas.93.13.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song J, Clagett-Dame M, Peterson RE, Hahn ME, Westler WM, Sicinski RR, DeLuca HF. A ligand for the aryl hydrocarbon receptor isolated from lung. Proc Natl Acad Sci USA. 2002;99:14694–9. doi: 10.1073/pnas.232562899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fritsche E, Schafer C, Calles C, Bernsmann T, Bernhausen T, Wurm M, et al. Lightening up the UV response by identification of the aryl hydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc Natl Acad Sci USA. 2007;104:8851–6. doi: 10.1073/pnas.0701764104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol. 1995;35:307–40. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- 6.Fan F, Yan B, Wood G, Viluksela M, Rozman KK. Cytokines (IL-1beta and TNF alpha) in relation to biochemical and immunological effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in rats. Toxicology. 1997;116:9–16. doi: 10.1016/s0300-483x(96)03514-7. [DOI] [PubMed] [Google Scholar]
- 7.Jensen BA, Leeman RJ, Schlezinger JJ, Sherr DH. Aryl hydrocarbon receptor (AhR) agonists suppress interleukin-6 expression by bone marrow stromal cells: an immunotoxicology study. Environ Health. 2003;2:16. doi: 10.1186/1476-069X-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.N'Diaye M, Le Ferrec E, Lagadic-Gossmann D, Corre S, Gilot D, Lecureur V, et al. Aryl hydrocarbon receptor- and calcium-dependent induction of the chemokine CCL1 by the environmental contaminant benzo[a]pyrene. J Biol Chem. 2006;281:19906–15. doi: 10.1074/jbc.M601192200. [DOI] [PubMed] [Google Scholar]
- 9.Vogel CF, Sciullo E, Wong P, Kuzmicky P, Kado N, Matsumura F. Induction of proinflammatory cytokines and C-reactive protein in human macrophage cell line U937 exposed to air pollution particulates. Environ Health Perspect. 2005;113:1536–41. doi: 10.1289/ehp.8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warren TK, Mitchell KA, Lawrence BP. Exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) suppresses the humoral and cell-mediated immune responses to influenza A virus without affecting cytolytic activity in the lung. Toxicol Sci. 2000;56:114–23. doi: 10.1093/toxsci/56.1.114. [DOI] [PubMed] [Google Scholar]
- 11.Singh NP, Nagarkatti M, Nagarkatti PS. Role of dioxin response element and nuclear factor-kappaB motifs in 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated regulation of Fas and Fas ligand expression. Mol Pharmacol. 2007;71:145–57. doi: 10.1124/mol.106.028365. [DOI] [PubMed] [Google Scholar]
- 12.Kathariou S. Listeria monocytogenes virulence and pathogenicity, a food safety perspective. J Food Prot. 2002;65:1811–29. doi: 10.4315/0362-028x-65.11.1811. [DOI] [PubMed] [Google Scholar]
- 13.Hinsdill RD, Couch DL, Speirs RS. Immunosuppression in mice induced by dioxin (TCDD) in feed. J Environ Pathol Toxicol. 1980;4:401–25. [PubMed] [Google Scholar]
- 14.Luster MI, Boorman GA, Dean JH, Harris MW, Luebke RW, Padarathsingh ML, Moore JA. Examination of bone marrow, immunologic parameters and host susceptibility following pre- and postnatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Int J Immunopharmacol. 1980;2:301–10. doi: 10.1016/0192-0561(80)90030-2. [DOI] [PubMed] [Google Scholar]
- 15.Sugita-Konishi Y, Kobayashi K, Naito H, Miura K, Suzuki Y. Effect of lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin on the susceptibility to Listeria infection. Biosci Biotechnol Biochem. 2003;67:89–93. doi: 10.1271/bbb.67.89. [DOI] [PubMed] [Google Scholar]
- 16.Vos JG, Kreeftenberg JG, Engel HW, Minderhoud A, Van Noorle Jansen LM. Studies on 2,3,7,8-tetrachlorodibenzo-p-dioxin induced immune suppression and decreased resistance to infection: endotoxin hypersensitivity, serum zinc concentrations and effect of thymosin treatment. Toxicology. 1978;9:75–86. doi: 10.1016/0300-483x(78)90033-1. [DOI] [PubMed] [Google Scholar]
- 17.Ward EC, Murray MJ, Lauer LD, House RV, Irons R, Dean JH. Immunosuppression following 7,12-dimethylbenz[a]anthracene exposure in B6C3F1 mice. I. Effects on humoral immunity and host resistance. Toxicol Appl Pharmacol. 1984;75:299–308. doi: 10.1016/0041-008x(84)90212-6. [DOI] [PubMed] [Google Scholar]
- 18.Neff-LaFord H, Teske S, Bushnell TP, Lawrence BP. Aryl hydrocarbon receptor activation during influenza virus infection unveils a novel pathway of IFN-gamma production by phagocytic cells. J Immunol. 2007;179:247–55. doi: 10.4049/jimmunol.179.1.247. [DOI] [PubMed] [Google Scholar]
- 19.Vorderstrasse BA, Lawrence BP. Protection against lethal challenge with Streptococcus pneumoniae is conferred by aryl hydrocarbon receptor activation but is not associated with enhanced inflammatory response. Infect Immun. 2006;74:5679–86. doi: 10.1128/IAI.00837-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi LZ, Faith NG, Kakayama Y, Suresh M, Steinberg H, Czuprynski CJ. The aryl hydrocarbon receptor is required for optimal resistance to Listeria monocytogenes infection in mice. J Immunol. 2007;179:6952–62. doi: 10.4049/jimmunol.179.10.6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gregory SH, Barczynski LK, Wing EJ. Effector function of hepatocytes and Kupffer cells in the resolution of systemic bacterial infections. J Leukoc Biol. 1992;51:421–4. doi: 10.1002/jlb.51.4.421. [DOI] [PubMed] [Google Scholar]
- 22.Gregory SH, Wing EJ. IFN-gamma inhibits the replication of Listeria monocytogenes in hepatocytes. J Immunol. 1993;151:1401–9. [PubMed] [Google Scholar]
- 23.Bermudez de Leon M, Gomez P, Elizondo G, Zatarain-Palacios R, Garcia-Sierra F, Cisneros B. Beta-naphthoflavone represses dystrophin Dp71 expression in hepatic cells. Biochim Biophys Acta. 2006;1759:152–8. doi: 10.1016/j.bbaexp.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 24.Hollingshead BD, Patel RD, Perdew GH. Endogenous hepatic expression of the hepatitis B virus X-associated protein 2 is adequate for maximal association with aryl hydrocarbon receptor-90-kDa heat shock protein complexes. Mol Pharmacol. 2006;70:2096–107. doi: 10.1124/mol.106.029215. [DOI] [PubMed] [Google Scholar]
- 25.Patel RD, Hollingshead BD, Omiecinski CJ, Perdew GH. Aryl hydrocarbon receptor activation regulates constitutive androstane receptor levels in murine and human liver. Hepatology. 2007;46:209–18. doi: 10.1002/hep.21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zatloukalova J, Svihalkova-Sindlerova L, Kozubik A, Krcmar P, Machala M, Vondracek J. beta-Naphthoflavone and 3′-methoxy-4′-nitroflavone exert ambiguous effects on Ah receptor-dependent cell proliferation and gene expression in rat liver ‘stem-like’ cells. Biochem Pharmacol. 2007;73:1622–34. doi: 10.1016/j.bcp.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 27.Santostefano M, Merchant M, Arellano L, Morrison V, Denison MS, Safe S. alpha-Naphthoflavone-induced CYP1A1 gene expression and cytosolic aryl hydrocarbon receptor transformation. Mol Pharmacol. 1993;43:200–6. [PubMed] [Google Scholar]
- 28.Aluru N, Vijayan MM. Aryl hydrocarbon receptor activation impairs cortisol response to stress in rainbow trout by disrupting the rate-limiting steps in steroidogenesis. Endocrinology. 2006;147:1895–903. doi: 10.1210/en.2005-1143. [DOI] [PubMed] [Google Scholar]
- 29.Billiard SM, Timme-Laragy AR, Wassenberg DM, Cockman C, Di Giulio RT. The role of the aryl hydrocarbon receptor pathway in mediating synergistic developmental toxicity of polycyclic aromatic hydrocarbons to zebrafish. Toxicol Sci. 2006;92:526–36. doi: 10.1093/toxsci/kfl011. [DOI] [PubMed] [Google Scholar]
- 30.Timme-Laragy AR, Cockman CJ, Matson CW, Di Giulio RT. Synergistic induction of AHR regulated genes in developmental toxicity from co-exposure to two model PAHs in zebrafish. Aquat Toxicol. 2007;85:241–50. doi: 10.1016/j.aquatox.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakane A, Minagawa T, Kato K. Endogenous tumor necrosis factor (cachectin) is essential to host resistance against Listeria monocytogenes infection. Infect Immun. 1988;56:2563–9. doi: 10.1128/iai.56.10.2563-2569.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muntane-Relat J, Ourlin JC, Domergue J, Maurel P. Differential effects of cytokines on the inducible expression of CYP1A1, CYP1A2, and CYP3A4 in human hepatocytes in primary culture. Hepatology. 1995;22:1143–53. [PubMed] [Google Scholar]
- 33.Lavon I, Goldberg I, Amit S, Landsman L, Jung S, Tsuberi BZ, et al. High susceptibility to bacterial infection, but no liver dysfunction, in mice compromised for hepatocyte NF-kappaB activation. Nat Med. 2000;6:573–7. doi: 10.1038/75057. [DOI] [PubMed] [Google Scholar]
- 34.Remer KA, Juni TW, Fatzer R, Tauber MG, Leib SL. Nitric oxide is protective in listeric meningoencephalitis of rats. Infect Immun. 2001;69:4086–93. doi: 10.1128/IAI.69.6.4086-4093.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dewa Y, Nishimura J, Muguruma M, Jin M, Saegusa Y, Okamura T, et al. beta-Naphthoflavone enhances oxidative stress responses and the induction of preneoplastic lesions in a diethylnitrosamine-initiated hepatocarcinogenesis model in partially hepatectomized rats. Toxicology. 2008;244:179–89. doi: 10.1016/j.tox.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 36.Elbakai RH, Korashy HM, Wills K, Gharavi N, El-Kadi AO. Benzo[a]pyrene, 3-methylcholanthrene and beta-naphthoflavone induce oxidative stress in hepatoma heap 1c1c7 Cells by an AHR-dependent pathway. Free Radic Res. 2004;38:1191–200. doi: 10.1080/10715760400017319. [DOI] [PubMed] [Google Scholar]
- 37.Heyworth PG, Cross AR, Curnutte JT. Chronic granulomatous disease. Curr Opin Immunol. 2003;15:578–84. doi: 10.1016/s0952-7915(03)00109-2. [DOI] [PubMed] [Google Scholar]
- 38.Lajarin F, Rubio G, Lorenzo N, Gámiz P, Hernandez-Caselles T, Garcia-Peñarrubia P. Implication of reactive oxygen species in the antibacterial activity against Salmonella typhimurium of hepatocyte cell lines. Free Radic Biol Med. 1999;27(910):1008–18. doi: 10.1016/s0891-5849(99)00148-3. [DOI] [PubMed] [Google Scholar]
- 39.Archambaud C, Nahori MA, Pizarro-Cerda J, Cossart P, Dussurget O. Control of Listeria superoxide dismutase by phosphorylation. J Biol Chem. 2006;281:31812–22. doi: 10.1074/jbc.M606249200. [DOI] [PubMed] [Google Scholar]
- 40.Klaunig JE, Goldblatt PJ, Hinton DE, Lipsky MM, Chacko J, Trump BF. Mouse liver cell culture. I. Hepatocyte isolation. In vitro. 1981;17:913–25. doi: 10.1007/BF02618288. [DOI] [PubMed] [Google Scholar]
- 41.Haponsaph R, Czuprynski CJ. Inhibition of the multiplication of Listeria monocytogenes in a murine hepatocyte cell line (ATCC TIB73) by IFN-gamma and TNF-alpha. Microb Pathog. 1996;20:287–95. doi: 10.1006/mpat.1996.0027. [DOI] [PubMed] [Google Scholar]