

Abstract
Although many nervous system disorders are associated with N-methyl-d-aspartate (NMDA) receptor overactivation, pharmacological inhibition of NMDA receptors has typically demonstrated limited clinical value due to debilitating psychotomimetic side-effects. Memantine, however, induces far fewer behavioural side-effects than other NMDA receptor channel blockers such as ketamine, and slows the progressive cognitive decline associated with Alzheimer's disease. Memantine and ketamine inhibit NMDA receptors with similar affinity and kinetics. A prominent mechanistic difference between memantine and ketamine is the degree to which they are ‘trapped’ within the closed channel of NMDA receptors following removal of agonist: ketamine becomes trapped in nearly all NMDA receptors to which it was bound before agonist removal, whereas some bound memantine molecules dissociate after agonist removal, a phenomenon called partial trapping. Here we investigated the mechanism underlying partial trapping of memantine by recombinant NR1/2A NMDA receptors. We found that memantine dissociation from NR1/2A receptors after agonist removal (the process that results in partial trapping) followed an exponential time course with τ= 0.79 ± 0.32 s. Neither membrane voltage depolarization nor maintained presence of memantine after agonist removal affected partial trapping, suggesting that partial trapping does not result from memantine escape through open channels. We tested the hypothesis that partial trapping results from binding of memantine to two sites, a superficial ‘non-trapping’ site and a deep ‘trapping’ site, which cannot be occupied simultaneously. This hypothesis was supported by the lack of ketamine binding to the superficial site, the voltage dependence of partial trapping, and the effect on partial trapping of a mutation near the deep site. The superficial binding site for memantine may, by causing partial trapping, contribute to memantine's unique therapeutic utility.
Most excitatory neurotransmission occurring within the vertebrate central nervous system (CNS) is mediated by the neurotransmitter glutamate. The NMDA receptor subtype of the glutamate receptor family possesses unique properties that contribute to many physiological (e.g. development, learning and memory) and pathological (e.g. ischaemic cell death, neurodegenerative disorders) processes (Bliss & Collingridge, 1993; Dingledine et al. 1999; Hardingham & Bading, 2003; Olney, 2003). Activated NMDA receptors are highly calcium (Ca2+)-permeable. NMDA receptor-mediated Ca2+ influx is necessary for many types of synaptic plasticity, which may underlie some forms of learning and memory; however, excessive rises in intracellular Ca2+ concentration ([Ca2+]i) may lead to cell death. Inhibition of NMDA receptors by endogenous extracellular magnesium 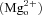 is important for preventing excessive Ca2+ influx and accumulation of
is important for preventing excessive Ca2+ influx and accumulation of 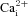 to toxic levels. Drugs that inhibit NMDA receptors, such as the channel blockers ketamine and phencyclidine (PCP), also can prevent excessive Ca2+ influx; however, their therapeutic usefulness has been plagued by their tendency to induce symptoms similar to schizophrenia in healthy adults (Krystal et al. 2003). Ketamine and PCP nevertheless have been used to expand our understanding of the mechanisms underlying schizophrenia. The NMDA receptor channel blocker memantine, which is used to treat Alzheimer's disease, generally lacks the undesirable side-effects of other inhibitors, and has provided new hope for NMDA receptor modulation as a viable treatment strategy.
to toxic levels. Drugs that inhibit NMDA receptors, such as the channel blockers ketamine and phencyclidine (PCP), also can prevent excessive Ca2+ influx; however, their therapeutic usefulness has been plagued by their tendency to induce symptoms similar to schizophrenia in healthy adults (Krystal et al. 2003). Ketamine and PCP nevertheless have been used to expand our understanding of the mechanisms underlying schizophrenia. The NMDA receptor channel blocker memantine, which is used to treat Alzheimer's disease, generally lacks the undesirable side-effects of other inhibitors, and has provided new hope for NMDA receptor modulation as a viable treatment strategy.
Despite differences in tolerability, memantine and ketamine interact with NMDA receptors with comparable IC50 values and kinetics. Both drugs require agonist binding and channel opening for access to their channel blocking site (Chen et al. 1992; Parsons et al. 1995; Blanpied et al. 1997; Sobolevsky & Koshelev, 1998; Mealing et al. 1999; Bolshakov et al. 2003; Johnson & Kotermanski, 2006; but see Sobolevsky et al. 1998). Channel closure and agonist dissociation can occur while either drug is bound, trapping the drug within the receptor's channel until subsequent agonist binding and channel opening permit blocker dissociation. The tendency of channel blockers to become trapped varies among NMDA receptor channel blockers. Ketamine has been classified as a ‘full trapping’ channel blocker because all (or nearly all) molecules bound to NMDA receptors become trapped following rapid removal of agonist and blocker (Mealing et al. 1999). Memantine has been classified as a ‘partial trapping’ channel blocker because a fraction of memantine molecules bound to NMDA receptors unbind following rapid removal of agonist and blocker, and thus are not trapped (Blanpied et al. 1997; Chen & Lipton, 1997; Sobolevsky & Koshelev, 1998; Mealing et al. 1999).
Several hypotheses have been proposed to explain how the non-trapped memantine molecules dissociate from NMDA receptors. Memantine, which has faster unbinding kinetics than many full trapping blockers, has been hypothesized to dissociate via ‘open-channel escape’ from NMDA receptors with channels that remain open temporarily after agonist removal (Blanpied et al. 1997; Bolshakov et al. 2003). Some blocker may escape from open channels immediately after rapid removal of agonist and blocker, an idea supported by the reported correlation between blocker unbinding kinetics and partial trapping (Mealing et al. 2001; Bolshakov et al. 2003). However, because memantine unbinds much more slowly than agonists (especially NMDA) unbind and channels close, open-channel escape hypotheses generally involve a mechanism that temporarily holds channels open after agonist removal. Mechanisms proposed to hold channels open include (1) memantine stabilizing the channel open state when blocking NMDA receptors at a deep site (Blanpied et al. 1997) and (2) memantine binding simultaneously to a deep site and to a superficial site, occupancy of which prevents channel closure (Bolshakov et al. 2003). Although capable of explaining how partial trapping can occur, these hypotheses are not entirely consistent with experimental observations. If open-channel escape occurs, then receptors that release memantine after removal of agonist should mediate a surge in current before subsequent channel closure. This ‘tail current’, which is observed for sequential (foot-in-the-door) blockers that do not allow channel closure while bound such as 9-aminoacridine (Benveniste & Mayer, 1995), generally is not observed with memantine (Blanpied et al. 1997).
A modified open-channel escape mechanism has been proposed based on the hypothesis that NMDA receptors have two gates (Mealing et al. 2001). After removal of agonist, a deep channel gate has been proposed to terminate current flow quickly. A superficial ‘trapping gate’ then would close slowly compared to the memantine unbinding rate, permitting some memantine to escape. Here we tested the open-channel escape hypotheses.
Memantine passage through a lipophilic pathway is another possible mechanism of partial trapping. However, previous studies have shown no correlation between the lipophilicity of a compound and its degree of trapping (Mealing et al. 1999, 2001). The observation that memantine does not unbind during extended wash periods (Blanpied et al. 1997; but see Sobolevsky et al. 1998) also argues against a lipophilic pathway for blocker escape.
Here we further explore the possibility that the ability of memantine to inhibit NMDA receptors at two separate sites (Blanpied et al. 1997; Sobolevsky & Koshelev, 1998; Sobolevsky et al. 1998; Chen & Lipton, 2005) may contribute to partial trapping. The principal binding site for memantine (as well as ketamine) is located deep in the voltage field of NMDA receptor channels, is only accessible after channel opening, and overlaps the 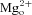 binding site (Johnson & Kotermanski, 2006). The second memantine binding site is at a shallower location in the voltage field of NMDA receptors than the deep site (Blanpied et al. 1997; Sobolevsky & Koshelev, 1998; Chen & Lipton, 2005). Memantine can bind to and unbind from this superficial site in the absence of agonists (Blanpied et al. 1997). Whether ketamine can bind to the superficial site has not previously been investigated. Our results support the idea that interaction of memantine with the superficial site contributes to partial trapping.
binding site (Johnson & Kotermanski, 2006). The second memantine binding site is at a shallower location in the voltage field of NMDA receptors than the deep site (Blanpied et al. 1997; Sobolevsky & Koshelev, 1998; Chen & Lipton, 2005). Memantine can bind to and unbind from this superficial site in the absence of agonists (Blanpied et al. 1997). Whether ketamine can bind to the superficial site has not previously been investigated. Our results support the idea that interaction of memantine with the superficial site contributes to partial trapping.
Methods
Cell culture and transfection
Experiments were performed on the human embryonic kidney (HEK) 293T cell line. Cells were enzymatically dissociated, plated onto glass coverslips coated with poly-d-lysine and collagen (Sigma, St Louis, MO, USA), and incubated at 37°C for 24 h prior to transfection. Transfection of the NR1-1a (GenBank accession number (ACCN) X63255, in pcDM8) and NR2A (ACCN M91561, in pcDM8) NMDA receptor subunit cDNAs, along with eGFP cDNA for identification of successfully transfected cells, was performed with Lipofectamine (Invitrogen Corp., Carlsbad, CA, USA) at a ratio of 1 eGFP: 3 NR1: 6 NR2A. dl-2-Amino-5-phosphonopentanoic acid (dl-APV, Sigma, 200 μm) was added to culture medium at the end of transfection to limit excitotoxic activation of expressed NMDA receptors. cDNA for the NR1(N616Q) mutant (residue numbering starting from initiating methionine) subunit (in pRCCMVαα) was kindly provided by Pierre Paoletti (Ecole Normale Supérieure) and was transfected at the same ratio as wild-type NR1.
Solutions
Solutions were prepared from concentrated stock solutions. The external bath solution (normal Ringer solution) contained (in mm): 140 NaCl, 2.8 KCl, 1 CaCl2 and 10 Hepes, with pH adjusted to 7.20 ± 0.05 using 6.0 n NaOH and osmolality adjusted to 290 ± 10 mosmol kg−1 with sucrose. Internal solution contained (in mm): 125 CsCl, 10 BAPTA and 10 Hepes, with pH adjusted to 7.20 ± 0.05 with CsOH and an osmolality of 275 ± 10 mosmol kg−1. A frozen aliquot of internal solution was thawed for each day of experiments. Correction for the junction potential (measured as −6 mV) was applied to all data.
Electrophysiology
Whole-cell patch-clamp recordings were performed on transiently transfected HEK293T cells expressing either NR1/2A or NR1(N616Q)/2A receptors. Recording electrodes were pulled from standard-walled borosilicate glass capillary tubes with filaments (Warner Instruments, Hamden, CT, USA). Electrode tips were heat polished and had a resistance of 2–6 MΩ. Series resistance was compensated 80–95% in all experiments. Whole-cell current responses were recorded with an Axopatch-1D amplifier (Molecular Devices, Sunnyvale, CA, USA), printed on a thermal arraycorder (Graphtech Corp., Irvine, CA, USA) for monitoring of data quality during experiments, and stored on VHS tape for additional analysis.
A seven-barrelled gravity-fed fast perfusion system (Blanpied et al. 1997) was used for solution application to patch-clamped cells. Currents were activated by application of 10 μm NMDA and 10 μm glycine (referred to as ‘agonist’). Recordings were made at −66 mV unless otherwise stated. For experiments testing the effect of a depolarizing voltage step on recovery from memantine inhibition, a voltage step to +54 mV for 5 s was applied using pCLAMP 9.2 (Molecular Devices).
Valid interpretation of several experimental protocols required adequately rapid solution exchange. For valid interpretation of the experiments shown in Fig. 3, the wash with normal Ringer solution following application of blocker (50 μm memantine or 15 μm ketamine) needed to effectively eliminate blocker to avoid binding during agonist reapplication. To determine the minimum adequate wash duration we estimated the time constant (τ) of solution exchange as follows. External solution was switched from normal Ringer solution containing agonist to a solution identical except that 140 mm NMDG replaced NaCl. The resulting decay τ of the NMDA receptor-mediated response (∼16 ms) was used as an estimate of the τ of solution exchange. Based on this estimate, a minimum wash duration of 0.1 s (>5 times solution exchange τ) was used in Fig. 3. The same minimum wash duration was used in Fig. 4 to ensure that agonist had been removed before the depolarizing step was initiated. The experiments shown in Fig. 6 had a more stringent requirement for solution exchange because up to 500 μm memantine or ketamine had to be eliminated before reapplication of agonist. To estimate the wash duration needed to eliminate these high blocker concentrations, we measured the decay of current following removal of 10 mm NMDA (hundreds of times its EC50) in the continuous presence of 10 μm glycine. We found that the NMDA response decayed to less than 10% of its steady-state value in 0.20 s (average of 3; range 0.14–0.30 s). Based on these data, we used a minimum wash duration of 0.4 s for the experiments shown in Figs 6 and 8B.
Figure 3. Time course of partial trapping development.

A and B, double-pulse protocol current traces used to measure time course of the recovery from inhibition that results in partial trapping. NR1/2A receptor-mediated currents at −66 mV were inhibited by 50 μm memantine (A) or 15 μm ketamine (B). The break in the traces represents the wash duration, which was varied (a wash duration of 15 s is shown in the sample traces). C, plot of the time dependence of the fractional recovery from inhibition (eqn (2); mean ±s.e.m.) by memantine (filled circles; n= 6) or ketamine (open circles; n= 4). Continuous and dashed lines are single exponential fits to memantine and ketamine data, respectively. Fractional recovery differed significantly between drugs at each wash duration tested (Student's t-test). #Values for memantine significantly different from value for briefest (0.1 s) wash (one-way ANOVA with Bonferroni's post hoc test). Although there also was a significant effect of wash duration on ketamine fractional recovery (one-way ANOVA), no other values were significantly different from value at the briefest (0.1 s) wash (Bonferroni's post hoc test).
Figure 4. Open-channel escape does not explain partial trapping.

A, voltage (upper) and current (lower) traces during protocol used to examine effect on partial trapping of depolarization after removal of agonist and memantine. Voltage was held at −66 mV, then depolarized to +54 mV for 5 s beginning 0.1–10 s after removal of agonist and blocker. Break in the traces represents the variable delay from removal of agonist and memantine until the depolarization (sample trace shows a 10 s wait before voltage step application). B, depolarizations applied after removal of agonist and memantine had no significant effect (one-way ANOVA) on fractional recovery (n= 5). Control data represent measurements made without a voltage step. C, current trace during the modified trapping protocol, which resembled the double-pulse protocol (Fig. 3) except that blocker application was extended 10 s beyond the end of the agonist application. Thus, blocker was present throughout the time when partial trapping developed (Fig. 3C). D, effects of the modified trapping protocol on fractional recovery from memantine inhibition. The continued presence of 50 μm memantine during the development of partial trapping (grey bar) had no significant effect (Student's t-test) on memantine trapping (n= 4).
Figure 6. NMDA receptor inhibition by memantine but not ketamine at the superficial site.

A and B, NR1/2A receptor current traces demonstrating the protocol used to estimate memantine and ketamine inhibition at the superficial site at −66 mV. The wash duration following a 60 s application of 500 μm memantine (A) or 500 μm ketamine (B) and before the subsequent application of agonist was varied to measure the time constant of blocker unbinding from the superficial site. The traces presented are examples of a 0.4 s wash, the minimum duration wash used to remove the very high blocker concentrations (see Methods). C, plot of the time dependence of the fractional response after superficial site inhibition (eqn (3); mean ±s.e.m.) by either 500 μm memantine (filled circles; n= 4) or 500 μm ketamine (open circles; n= 4). Continuous and dashed lines are single exponential fits to memantine and ketamine data, respectively. *Significant difference between drugs (Student's t-test). #Values for memantine significantly different from value for briefest (0.4 s) wash (one-way ANOVA with Bonferroni's post hoc test). Fractional response after superficial site inhibition by ketamine did not depend significantly on wash duration (one-way ANOVA). D, memantine concentration–inhibition curve for the superficial site. Measurements were gathered with the protocol demonstrated in A (wash duration of 0.4 s between memantine application and subsequent agonist application). Continuous line is the fit of eqn (4) to the data (IC50= 79.1 ± 20.2 μm; nH= 2.2 ± 1.3; C1= 0.48 ± 0.07). E, the effect of the duration of memantine application in the absence of agonist on inhibition of NR1/2A receptors by 50 μm memantine (n= 3). The protocol was similar to that shown in A, with the duration of memantine application varied; wash between memantine application and subsequent agonist application was 0.4 s. There were no significant differences (one-way ANOVA) between the data points.
Figure 8. Effect of NR1(N616Q) mutation on memantine trapping.

A, memantine concentration–inhibition curves from HEK293T cells expressing mutant NR1(N616Q)/2A or wild-type NR1/2A receptors. Wild-type NR1/2A data (filled circles, continuous line) are replotted from Fig. 2C. The memantine IC50 for NR1(N616Q)/2A receptors (open squares, dashed lines), 2.88 ± 0.45 μm, was 2.3-fold larger than the memantine IC50 for wild-type receptors (1.25 ± 0.04 μm). B, the effect of the NR1(N616Q) mutation on superficial site inhibition by memantine. No significant difference (Student's t-test) between superficial site inhibition of wild-type NR1/2A and mutant NR1(N616Q)/2A receptors was observed with 100 μm or 500 μm memantine. C, current trace recorded from HEK293T cells expressing NR1(N616Q)/2A receptors during double-pulse protocol used to measure fractional recovery from inhibition at the deep site. D, the effect of the NR1(N616Q) mutation on memantine trapping. NR1/2A normal trapping data replotted from Fig. 4D. Fractional recovery from memantine inhibition was significantly greater for NR1(N616Q)/2A receptors (black hatched bar) than for wild-type NR1/2A receptors (black bar). Use of the modified trapping protocol (grey hatched bar; see Fig. 4C) with NR1(N616Q)/2A receptors did not have a significant effect on fractional recovery from memantine inhibition. *Significantly different (Student's t-test).
Analysis
Data were played back from tape, low-pass filtered (Bessel) at 2.5 kHz, digitized using pCLAMP 9.2 with a Digidata 1200 or 1440A (Molecular Devices) at 5 kHz or 400 Hz. For most analysis and display, data subsequently were refiltered at 100 Hz and decimated using Clampfit 9.2 (Molecular Devices).
Current measurements used here are defined below and in Fig. 1. All currents were measured relative to the baseline current (500 ms current average preceding first agonist application). Peak current response to the initial agonist application (Ip) was the average current during a 50 ms window centred on the time of peak current. The difference between the time of perfusion barrel movement and the time of peak current was termed tP and will be used below. Three steady-state currents (ISS, steady-state response to initial agonist application; IBSS, steady-state response in agonist and blocker; and ISS2, steady-state response in agonist after recovery from inhibition) were 500 ms current averages. Data were rejected if the ISS2 did not recover to 60% of the previous ISS. IB is the initial current jump activated by reapplication of agonist. The time after barrel movement when IB was measured equalled tP (50–250 ms after barrel movement). To determine IB, a single or double exponential was fitted to the initial 1 s (beginning tp after barrel movement) of the current activated by the second agonist application. IB was taken as the initial value of the exponential fit.
Figure 1. Double-pulse protocol.

Sample trace shows the double-pulse protocol used for measuring the fractional recovery of NR1/2A and NR1(N616Q)/2A receptors from inhibition by memantine or ketamine (NR1/2A receptors and 50 μm memantine were used in this example). The lines above the current trace in this and subsequent figures show time of agonist and blocker applications. IP, ISS, IBSS, IB and ISS2 identify the time when each type of current measurement used in eqns (1)–(4) (Methods) was made.
For concentration–inhibition curve measurements, ISS and IBSS were measured using the protocol shown in Fig. 2A and B. Concentration–inhibition curves for the deep site were fitted using the equation:
| (1) |
where [B] is the blocker concentration, and nH is the Hill coefficient. Free variables during fitting were nH and IC50.
Figure 2. NMDA receptor inhibition by memantine and ketamine.

A and B, representative current traces recorded at −66 mV from transfected HEK293T cells used to measure concentration–inhibition curves for memantine (A) and ketamine (B). C, memantine (filled circles) and ketamine (open circles) concentration–inhibition curves. Continuous and dashed lines are fits (eqn (1)) to the memantine (IC50= 1.25 ± 0.04; nH= 0.99 ± 0.03; n= 4) and ketamine (IC50= 0.35 ± 0.01; nH= 0.82 ± 0.02; n= 4) data, respectively.
The fractional current recovery that results from partial trapping of blocker as measured in the double-pulse protocol (Fig. 1) was calculated as:
| (2) |
IB is multiplied by ISS/IP to account for recovery from NMDA receptor desensitization between agonist applications.
To measure inhibition at the superficial site, blocker had to be applied in the absence of agonist to prevent binding of blocker to the deep site. Inhibition was then quantified (Figs 6 and 8B) based on the response to a subsequent application of agonist using the equation:
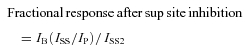 |
(3) |
where IB, ISS2, IP and ISS are defined in Fig. 1 but measured using the protocol shown in Fig. 6A and B. Concentration–inhibition curves for superficial site inhibition were fitted using the equation:
| (4) |
where C1 is the value of IB(ISS/IP)/ISS2 at high [B]. Free variables during fitting were C1, nH and IC50.
Where appropriate, Student's two-tailed t-test or one- or two-way ANOVA with Bonferroni's post-hoc test was used. Significance level was set at P < 0.05.
Results
We performed whole-cell voltage-clamp recordings on HEK293T cells expressing recombinant NR1/2A receptors to examine the mechanism of partial trapping. We first compared concentration–inhibition curves of memantine, a partially trapped blocker, and ketamine, a fully trapped blocker. Fitting of concentration–inhibition curves gave IC50 values of 1.25 ± 0.04 μm for memantine and 0.35 ± 0.01 μm for ketamine (Fig. 2), which are similar to previously reported values (Parsons et al. 1995; Blanpied et al. 1997; Sobolevsky & Koshelev, 1998; Sobolevsky et al. 1998; Mealing et al. 1999; Kashiwagi et al. 2002; Kotermanski & Johnson, 2009). In subsequent experiments we studied partial trapping of memantine at 50 μm, a concentration used in previous work (Blanpied et al. 1997). We studied partial trapping of ketamine at 15 μm so that ketamine and memantine were used at similar concentrations relative to their IC50 values.
To further characterize the response to memantine, we performed double exponential fitting of the NMDA receptor current decay during the onset of block induced by several memantine concentrations. The results are presented as online supplemental material (Table S1).
Time course of development of partial trapping
To characterize partial trapping, we measured the time course of the recovery from memantine inhibition that results in partial trapping. A double-pulse protocol was used (Figs 1 and 3A and B). The initial current activated by the second application of agonist (IB) was larger than the previous steady-state current in the presence of agonist + memantine (IBSS) as a result of partial trapping. Our goal was to examine the time course of the increase of the response from IBSS to IB, which we refer to as the time course of development of partial trapping. The interval between the removal of agonist + memantine and the subsequent application of agonist was varied, with a minimum interval of 0.1 s (see Methods). Our data show that recovery from memantine inhibition developed with a τ of 0.79 ± 0.32 s (Fig. 3A and C). When the same protocol was used to examine recovery from ketamine inhibition, a small but significant effect of wash duration on recovery from inhibition was observed (τ= 0.53 ± 0.31 s; Fig. 3B and C). The fractional recovery from inhibition was significantly lower with ketamine than memantine at all wash durations tested (Fig. 3C). These results suggest that ketamine does exhibit some partial trapping, although much less than memantine. The magnitude of trapping may depend on the protocol used (see Mealing et al. 1999). We speculate that, although it is a relatively small fraction of the NMDA response, the difference in fractional recovery from inhibition by memantine and ketamine may contribute to differences in their clinical effects (see Discussion).
Partial trapping is not the result of open-channel escape
We next tested the hypothesis that partial trapping occurs by open-channel escape (see Introduction). The unbinding rate of memantine from open NMDA receptor channels increases as voltage depolarizes (Parsons et al. 1993, 1995, 1996; Blanpied et al. 1997). Thus, if open-channel escape contributes to partial trapping, then depolarization shortly after removal of agonist and blocker from our cells (when recovery from inhibition occurs) should increase fractional recovery from inhibition. We applied a 5 s voltage jump to +54 mV (Fig. 4A) during and after the time our data suggested partial trapping develops (Fig. 3C). There was no significant effect of the depolarizing voltage steps on fractional recovery from memantine inhibition (Fig. 4B).
To further examine the contribution of open-channel escape to partial trapping, we modified the double-pulse protocol by continuing memantine application beyond the end of the agonist application (Fig. 4C), similar to the approach used by Mealing et al. (2001). Thus, in the modified trapping protocol a constant concentration of memantine was present throughout the time when memantine dissociates in the absence of agonists (Fig. 3C). If partial trapping results from memantine unbinding from the deep (trapping) site before channels close after agonist removal, then the continued presence of memantine should maintain occupancy of the deep site, decreasing fractional recovery. We found that extending the duration of memantine application beyond the time of agonist removal had no significant effect on fractional recovery (Fig. 4D). The data presented in Fig. 4 provide strong evidence that, despite the relatively fast unbinding rate of memantine, open-channel escape is not principally responsible for partial trapping.
NR1/2A receptor inhibition in the absence of agonist
Our finding that open-channel escape cannot fully account for the partial trapping of memantine led us to consider hypotheses in which memantine unbinds from closed channels. We explored the possibility that non-trapped memantine dissociates from a second inhibitory site from which unbinding can occur when the channel is closed. We hypothesized that, when the channel is open, memantine can bind to either a non-trapping (superficial) site or the trapping (deep) site (Fig. 5, top). For this hypothesis to explain partial trapping, binding of memantine to the two sites must be competitive (in contrast to the hypothesis of Sobolevsky & Koshelev (1998)), so that binding at the superficial site prevents memantine from binding to the deep site (Fig. 5, bottom). The ability of high concentrations of memantine to inhibit NMDA responses in the absence of agonist has been previously demonstrated (Blanpied et al. 1997; Sobolevsky & Koshelev, 1998; Sobolevsky et al. 1998; Chen & Lipton, 2005). We next tested the hypothesis that memantine binding to this superficial site contributes to partial trapping.
Figure 5. Competitive binding hypothesis of partial trapping.

We hypothesized that partial trapping of memantine by NMDA receptors is the result of competitive binding while the channel is open at two inhibitory binding sites that cannot be occupied simultaneously (upper panels). Blocker (represented by an open circle with +) can only be trapped by channel closure (bottom left) when occupying the deep trapping site. Blocker binding and unbinding at the deep site requires that the channel be open, and is strongly voltage dependent. Memantine or ketamine can bind at the deep site (left panels). Binding and unbinding of blocker at the superficial non-trapping site (right panels) may occur whether or not channels are open. Memantine, but not ketamine, can bind at the superficial site (right panels), which is located external to the trapping gate (horizontal line at channel entrance in lower panels). Memantine unbinding from the superficial site after channel closure (bottom right) is hypothesized to result in partial trapping.
If partial trapping results from unbinding of memantine from a superficial non-trapping site, then the more fully trapped blocker ketamine should exhibit much lower or no affinity for the superficial site. To test this prediction we compared memantine and ketamine inhibition of NR1/2A receptor currents resulting from binding to the superficial site. Superficial site inhibition was quantified (eqn (3)) as fractional response after blocker application in the absence of agonist using the experimental protocol shown in Fig. 6A and B. To optimize detection of binding at the superficial site we used a high blocker concentration (500 μm) and the briefest wash between blocker application and agonist reapplication that could fully remove blocker (0.4 s; see Methods). Under these conditions the value of fractional response after superficial site inhibition by memantine was 0.52 ± 0.03 (Fig. 6A and C). Thus, we found that memantine can bind to the superficial site, in agreement with previous studies (Blanpied et al. 1997; Sobolevsky & Koshelev, 1998; Bolshakov et al. 2003; Chen & Lipton, 2005). When similar experiments were performed with 500 μm ketamine, fractional response after superficial site inhibition was 1.05 ± 0.03 (Fig. 6B and C), providing no evidence for superficial site inhibition by ketamine.
We then investigated the time course of blocker unbinding from the superficial site. If binding to the superficial site contributes to partial trapping, then the unbinding time course should be similar to the time course for partial trapping development (Fig. 3C). To estimate the time constant for blocker unbinding from the superficial site we varied (0.4—15 s) the duration of the wash between blocker application and agonist reapplication. Memantine was found to unbind from the superficial site with τ= 1.94 ± 0.19 s (Fig. 6C), a value slower than the τ for development of partial trapping (see Discussion).
To estimate the IC50 of memantine inhibition at the superficial site, we used the protocol shown in Fig. 6A (with a 0.4 s wash between application of memantine and reapplication of agonist) and varied the concentration of memantine applied in the absence of agonist. Based on fitting of the data with eqn (4), the memantine IC50 was found to be 79.1 ± 20.2 μm (Fig. 6D).
To determine if memantine applications in the Fig. 6A protocol were long enough to permit superficial site binding to reach steady state, we varied the duration of applications of 50 μm memantine (the lowest concentration tested, which would be expected to have the slowest binding kinetics). The protocol shown in Fig. 6A was used in these experiments, with the duration of memantine application in the absence of agonist varied from 30 to 240 s. We found no significant effect of application duration on inhibition at the superficial site by 50 μm memantine (Fig. 6E), suggesting that the measurements presented in Fig. 6D do represent steady-state inhibition.
Voltage dependence of trapping
Blanpied et al. (1997) showed that memantine binding to the superficial site is less voltage dependent than binding to the deep site. Thus, as voltage is depolarized, memantine binding to the superficial site relative to binding at the deep site should increase. If partial trapping depends on competition between binding at the superficial and deep sites, then depolarization should lead to less complete trapping (greater fractional recovery). Trapping of several NMDA receptor channel blockers has been found to be voltage dependent (Bolshakov et al. 2003). We measured the fractional recovery from inhibition by memantine and by ketamine at voltages of −26, −66 and −106 mV (Fig. 7). Memantine and ketamine concentrations were adjusted in these experiments to achieve similar steady-state inhibition (IBSS/ISS) at each voltage (Table 1). Our measurements demonstrate that, consistent with the competitive binding hypothesis, fractional recovery from inhibition by memantine but not ketamine is voltage dependent (Fig. 7E).
Figure 7. Effect of membrane voltage on fractional recovery from memantine and ketamine inhibition.

A and B, current traces recorded with the double-pulse protocol at −26 mV (A) and −106 mV (B) used to examine voltage dependence of memantine trapping. C and D, current traces recorded at −26 mV (C) and −106 mV (D) used to examine voltage dependence of ketamine trapping. E, the effect of membrane voltage on fractional recovery from inhibition by memantine and ketamine. Inhibitor concentrations used at each voltage are listed in Table 1. Data for memantine at −66 mV are replotted from Fig. 4D. There was a significant drug × voltage interaction; fractional recovery from inhibition by memantine but not ketamine depended significantly on voltage (two-way ANOVA). Fractional recovery differed significantly between drugs at each voltage; #significantly different from other two values for same drug (Bonferroni's post hoc tests).
Table 1.
Steady-state inhibition and unbinding time constant of memantine and ketamine at concentrations used to measure voltage dependence of trapping
| Voltage (mV) | [Mem] (μm) | Memantine IBSS/ISS | Unbinding τ (s), memantine | [Ket] (μm) | Ketamine IBSS/ISS | Unbinding τ (s), ketamine |
|---|---|---|---|---|---|---|
| −26 | 180 | 0.020 ± 0.006 | 4.24 ± .42#* | 55 | 0.035 ± 0.002 | 11.2 ± 1.4* |
| −66 | 50 | 0.020 ± 0.002 | 9.73 ± .49* | 15 | 0.044 ± 0.007 | 15.1 ± 1.4* |
| −106 | 15 | 0.017 ± 0.007 | 8.76 ± 1.23* | 5 | 0.056 ± 0.003 | 25.8 ± 1.5#* |
Values of IBSS/ISS and unbinding time constants are presented as means ±s.e.m.
Significantly different from respective value at −66 mV (one-way ANOVA with Bonferroni's post hoc test).
Significantly different between drugs (Student's t-test).
Depolarization also significantly affected the unbinding time constant of both memantine and ketamine. As noted above, in principle faster unbinding kinetics could lead to decreased trapping by permitting more blocker unbinding at the time of removal of agonist and antagonist. However, Fig. 4B and D provides strong evidence that unbinding rate does not contribute substantially to partial trapping of memantine.
Effect of mutation at deep site on memantine trapping
Inhibition of NMDA receptors by memantine, 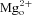 , and other channel blockers is strongly affected by mutation of the ‘N-site’ asparagines in the M2 reentrant loops of NMDA receptors (Burnashev et al. 1992; Kuner & Schoepfer, 1996; Wollmuth et al. 1996, 1998; Dingledine et al. 1999; Kashiwagi et al. 2002). The N-site asparagines of NR1 and NR2 subunits therefore are thought to be critical components of the high-affinity, strongly voltage-dependent (deep) binding site for channel blockers. To further test our hypothesis that partial trapping results from the existence of a deep site and a superficial site at which memantine cannot bind simultaneously (resulting in competitive binding at the two sites), experiments were performed on NR1(N616Q)/NR2A receptors. The N-site mutation in NR1(N616Q) subunits was previously demonstrated to lower memantine IC50∼28-fold when coexpressed with NR2B subunits (Kashiwagi et al. 2002). We predicted that, if the N-site mutation in NR1(N616Q)/2A receptors selectively decreases memantine binding to the deep site, then competitive binding should favour superficial site occupancy, increasing fractional recovery from memantine inhibition.
, and other channel blockers is strongly affected by mutation of the ‘N-site’ asparagines in the M2 reentrant loops of NMDA receptors (Burnashev et al. 1992; Kuner & Schoepfer, 1996; Wollmuth et al. 1996, 1998; Dingledine et al. 1999; Kashiwagi et al. 2002). The N-site asparagines of NR1 and NR2 subunits therefore are thought to be critical components of the high-affinity, strongly voltage-dependent (deep) binding site for channel blockers. To further test our hypothesis that partial trapping results from the existence of a deep site and a superficial site at which memantine cannot bind simultaneously (resulting in competitive binding at the two sites), experiments were performed on NR1(N616Q)/NR2A receptors. The N-site mutation in NR1(N616Q) subunits was previously demonstrated to lower memantine IC50∼28-fold when coexpressed with NR2B subunits (Kashiwagi et al. 2002). We predicted that, if the N-site mutation in NR1(N616Q)/2A receptors selectively decreases memantine binding to the deep site, then competitive binding should favour superficial site occupancy, increasing fractional recovery from memantine inhibition.
The memantine IC50 of NR1(N616Q)/2A mutant receptors was increased 2.3-fold to 2.88 μm from the 1.25 μm IC50 of wild-type NR1/2A receptors (Fig. 8A). We detected no significant difference in superficial site inhibition by memantine between wild-type and mutant NMDA receptors using either 100 μm or 500 μm memantine (Fig. 8B). These results indicate that the N-site mutation in NR1(N616Q)/2A receptors selectively decreased memantine binding at the deep site.
We next determined whether the N-site mutation affected fractional recovery from inhibition by memantine. To ensure that similar levels of inhibition were induced in NR1(N616Q)/2A and wild-type NR1/2A receptors, the memantine concentration used in NR1(N616Q)/2A trapping protocols was increased 2.3-fold to 115 μm. The values of IBSS/ISS for wild-type receptors (0.020 ± 0.002 with 50 μm memantine) and NR1(N616Q)/2A receptors (0.020 ± 0.007 with 115 μm memantine) were not significantly different, confirming the accuracy of memantine IC50 measurements for wild-type and mutant receptors. We found that fractional recovery from memantine inhibition was significantly greater for NR1(N616Q)/2A receptors than for wild-type receptors (Fig. 8D). These data provide strong evidence that memantine binding at the superficial site contributes to partial trapping.
The increase in memantine IC50 for NR1(N616Q)/2A receptors was accompanied by a decrease in memantine unbinding τ to 1.93 s, indicating that memantine unbound from NR1(N616Q)/2A receptors faster than from wild-type NR1/2A receptors. The rate of memantine unbinding from wild-type receptors is not fast enough to account for partial trapping as a result of open-channel escape (Fig. 4). However, it is possible that the faster memantine unbinding from NR1(N616Q)/2A receptors could contribute to their increased fractional recovery from inhibition. We therefore measured the fractional recovery from memantine inhibition from NR1(N616Q)/2A using the modified trapping protocol (see Fig. 4C). As we observed with wild-type receptors, use of the modified protocol had no significant effect on fractional recovery from memantine inhibition (Fig. 8D).
Discussion
A lack of consensus on the mechanism of partial trapping (Blanpied et al. 1997; Chen & Lipton, 1997; Sobolevsky & Koshelev, 1998; Sobolevsky et al. 1998; Mealing et al. 1999, 2001; Bolshakov et al. 2003) and inconsistencies between proposed hypotheses and experimental data led us to explore further partial trapping of memantine by NR1/2A NMDA receptors. We tested the hypothesis that partial trapping of memantine is the result of competitive binding at two sites on NR1/2A receptors that cannot be occupied simultaneously: a deep trapping site accessible only when the channel is open, and a superficial non-trapping site accessible when the channel is closed or open (Fig. 5). The existence of a superficial non-trapping site for memantine, in addition to the deep site, has been demonstrated previously (Blanpied et al. 1997; Sobolevsky & Koshelev, 1998; Chen & Lipton, 2005).
Development of partial trapping
Using a double-pulse protocol (Fig. 1), we determined that non-trapped memantine dissociates from NR1/2A receptors with τ= 0.79 ± 0.32 s (Fig. 3C). The fractional recovery from memantine inhibition (the difference between the response at the end of the agonist + memantine application and the initial response on subsequent reapplication of agonist) was measured to be 0.171, consistent with measurements by others (Blanpied et al. 1997). Fractional recovery from ketamine inhibition is substantially and significantly smaller (7%), and develops with τ= 0.53 ± 0.31 s (Fig. 3C). We found that neither depolarization (Fig. 4A and B) nor maintained application of memantine (Fig. 4C and D) during the time when partial trapping develops affected fractional recovery from memantine inhibition. Fig. 4 suggests that partial trapping does not result from escape of memantine from open channels.
Inaccuracy in partial trapping quantification may have resulted from multiple sources. One source was the correction for recovery from desensitization that occurred between measurements of ISS and of IB (Fig. 1). Recovery from desensitization should have occurred during the wash between application of agonist + blocker and reapplication of agonist, and some recovery (e.g. from Ca2+-dependent desensitization; Dingledine et al. 1999) may also have occurred during application of agonist + blocker. When calculating fractional recovery from inhibition we assumed that responses had fully recovered from desensitization when IB was measured (see eqn (2)). Although this assumption may not always have been correct, any resulting inaccuracy should have been small because corrections for desensitization were not large. Despite this potential inaccuracy, the observation that partial trapping of memantine was substantially greater than that of ketamine using identical procedures provides confidence that memantine partial trapping is significant. Inaccuracy in partial trapping quantification also may have resulted from possible blocker unbinding during the delay (tp; see Methods) between perfusion barrel movement and measurement of IB (Fig. 1). The delay (0.15 ± 0.1 s) was necessary to permit solution exchange and NMDA receptor activation. Slow memantine unbinding kinetics at −66 mV prevents measureable unbinding during this brief delay (Blanpied et al. 1997). However, significant unbinding might occur when memantine's kinetics are accelerated, as observed with NR1(N616Q)/2A receptors (Fig. 8) and, to a lesser extent, at −26 mV (Fig. 7 and Table 1). Using the fastest memantine unbinding τ observed here (τ= 1.93 s for unbinding from NR1(N616Q)/2A receptors) to calculate the maximum artifactual increase in fractional recovery if blocker unbinding were to occur throughout a delay of tp= 0.15 s yields a value of 0.075. This should be an overestimate, since NMDA receptors are not activated at the start of the delay, and is much less than the difference between fractional recovery of wild-type (0.17 ± 0.01) and NR1(N616Q)/2A (0.47 ± 0.05) receptors. Memantine unbinding during the tp delay nevertheless may have led to modest overestimation of fractional recovery in some experiments. It also is possible that the correlation between unbinding kinetics and partial trapping (Mealing et al. 2001; Bolshakov et al. 2003) may result in part from initial blocker unbinding during the agonist application used to measure partial trapping.
Inhibition of NR1/2A receptors at the superficial site
The memantine IC50 we measured for the superficial site (79.1 ± 20.2 μm, Fig. 6D) is lower than the previously reported (Blanpied et al. 1997) IC50 (179 μm). The difference in IC50 could be in part due to preparation differences: superficial site measurements in Blanpied et al. (1997) were made using cultured cortical neurons. However, we feel that the most likely explanation for the difference in IC50 estimates is based on the following differences in how the estimates were made: (1) the highest memantine concentration used in Blanpied et al. (1997) (1 mm) was higher than the highest concentration used here (500 μm); (2) the fitted concentration–inhibition curve assumed full inhibition at high memantine concentrations in Blanpied et al. (1997), but permitted partial inhibition here (eqn (4) and Fig. 6D). Use of a concentration–inhibition curve assuming full inhibition at high memantine concentration was supported by the strong inhibition (∼80%) observed at 1 mm memantine. However, strong inhibition by 1 mm memantine may have resulted in part from binding to the deep site due to difficulty in washing off such a high memantine concentration from primary neuronal cultures. Furthermore, the delay between memantine washout and reapplication of agonist (needed to minimize binding to the deep site) should result in some unbinding of memantine, implying that maximal inhibition must be under 100%. Thus, we believe that our estimate of memantine IC50 is likely to be the more accurate of the two. However, the need to eliminate memantine before measurement of inhibition at the superficial site in both studies made measurements challenging, and both estimates are subject to error.
If the competitive binding hypothesis (Fig. 5) is correct, unbinding from the superficial site and development of partial trapping should follow similar time courses. The unbinding time constant of memantine we measured at −66 mV for unbinding from the superficial site (τ= 1.94 ± 0.19 s, Fig. 6C) is over twofold slower than that measured for the development of partial trapping (τ= 0.79 ± 0.32 s. Fig. 3C). Although the difference is large, each of the time constant measurements were indirect and involved substantial potential for error.
Another prediction of the competitive binding hypothesis is that the more fully trapped ketamine should exhibit weaker binding to the superficial site than memantine. Consistent with this prediction, we measured little or no binding of ketamine to the superficial site (Fig. 6C).
Voltage dependence of trapping
To further test the competitive binding hypothesis, we measured partial trapping over a range of voltages. We predicted that depolarizing membrane voltage would cause a greater increase in the IC50 of memantine for the more strongly voltage-dependent deep site than for the superficial site. As a result, depolarization should favour memantine binding to the superficial site and result in less trapping. Similarly, hyperpolarizing membrane voltage should favour binding to the deep site and result in greater memantine trapping. Consistent with these predictions, fractional recovery from memantine inhibition increased (trapping decreased) significantly with depolarization (Fig. 7E). Fractional recovery from ketamine, in contrast, did not depend significantly on voltage, consistent with evidence that ketamine cannot bind to the superficial site (Fig. 6B and C). Fractional recovery from inhibition also was lower for ketamine than memantine at all voltages tested.
Altering memantine affinity for the deep binding site affects partial trapping
If the competitive binding hypothesis is correct, then any mutation that changes the memantine IC50 only at the deep site or only at the shallow site should affect memantine trapping. There is only limited information on the location of the superficial site, and mutations that modify the superficial site have not been described. However, mutations at the N-site (N616) of the NR1 subunit have been shown to increase the IC50 for memantine (as well as 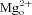 ) for the deep site in NMDA receptors (Kashiwagi et al. 2002). We used an NR1 subunit with an N-site mutation (NR1(N616Q)) that moderately increased memantine IC50 to test the prediction that an increase in memantine IC50 specifically at the deep site leads to less trapping.
) for the deep site in NMDA receptors (Kashiwagi et al. 2002). We used an NR1 subunit with an N-site mutation (NR1(N616Q)) that moderately increased memantine IC50 to test the prediction that an increase in memantine IC50 specifically at the deep site leads to less trapping.
We found that the memantine IC50 was higher for NR1(N616Q)/2A receptors than for wild-type NR1/2A receptors (Fig. 8A). The mutation had no effect on memantine binding at the superficial site (Fig. 8B), supporting the idea that the superficial and deep memantine binding sites are distinct. NR1(N616Q)/2A receptors exhibited significantly greater fractional recovery from memantine inhibition than wild-type receptors (Fig. 8C and D), consistent with the prediction of the competitive binding hypothesis.
The NR1(N616Q) mutation also decreased the τ of memantine unbinding from the deep site. We found, as for wild-type NR1/2A receptors, that fractional recovery from memantine inhibition of NR1(N616Q)/2A receptors was not affected by use of the modified trapping protocol (Fig. 8D). These data suggest that open-channel escape of memantine from NR1(N616Q)/2A receptors does not significantly contribute to partial trapping despite memantine's relatively fast unbinding kinetics. These results also further strengthen the conclusion that open-channel escape of memantine does not contribute to partial trapping by wild-type receptors, since memantine unbinding from wild-type receptors is slower.
The 2.3-fold difference between the memantine IC50 of NR1/2A and NR1(N616Q)/2A receptors that we observed was much smaller than the ∼28-fold difference between NR1/2B and NR1(N616Q)/2B receptors previously observed (Kashiwagi et al. 2002). Two possible causes of the discrepancy in the effect of the NR1(N616Q) mutation on memantine IC50 are coexpression with a different NR2 subunit, or the use of a different expression system (HEK293T cells versus Xenopus oocytes).
Evaluation of the competitive binding hypothesis
The data presented here indicate that partial trapping of memantine is not a result of memantine unbinding from open channels (Fig. 4). An alternative explanation, the competitive binding hypothesis (Fig. 5), is supported by several results: (1) the correlation, using memantine and ketamine, between superficial site binding (Fig. 6A–C) and partial trapping (Fig. 3); (2) the observation that partial trapping of memantine, but not ketamine, is voltage dependent (Fig. 7); (3) the observation that an N-site mutation that decreases memantine binding at the deep site but not at the superficial site powerfully influences partial trapping (Fig. 8).
Another consideration, however, suggests that competitive binding of memantine to the superficial site (as characterized in Fig. 6) and the deep site cannot simply explain partial trapping. The difference between the estimated memantine IC50 at the deep site (1.25 μm) and superficial site (79.1 μm) appears too great to permit adequate binding competition, a point made in Blanpied et al. (1997). To further examine this objection, we used a simplified NMDA receptor kinetic model (data not shown) to estimate the superficial site IC50 that could result in partial trapping as observed with memantine. With a deep site IC50 of 1.25 μm (Fig. 2), model simulations reproduced partial trapping similar to that observed experimentally (∼17%), but only with a superficial site IC50 of ∼30 μm. It is possible that this discrepancy resulted from inaccuracy in our measured superficial site IC50, which we needed to estimate indirectly (Fig. 6). An alternative possible explanation for the discrepancy in measured and estimated IC50 values is that memantine binding to the superficial site may be state dependent. The superficial site is likely to be in the external vestibule of NMDA receptors (Chen & Lipton, 2005), a region that undergoes extensive conformational change during gating (Sobolevsky et al. 2002). We could only estimate memantine interactions with the superficial site with the channel closed to avoid memantine binding to the deep site. Thus, the discrepancy in IC50 values might be eliminated if memantine affinity for the superficial site increases (IC50 decreases) when the channel opens.
The memantine concentrations used here to study partial trapping are well above the extracellular memantine concentration range found in humans during treatment with memantine (Parsons et al. 2007). However, a prediction of the competitive binding hypothesis is that, at equilibrium, the relative occupancy of the two competititive binding sites should be memantine concentration independent. Thus, the partial trapping observed at high memantine concentrations should also decrease trapping of memantine at therapeutic concentrations, and may contribute to memantine's clinical utility.
The difference between fractional recovery from inhibition by memantine and ketamine represents a modest portion of the NMDA receptor response amplitude before inhibition (e.g. Fig. 3). How might this relatively small difference in partial trapping contribute to differences in the clinical effects of memantine and ketamine? It is possible that even a small difference in the fraction of receptors that release blocker (and therefore are available for future activation) can have a significant influence on the function of neuronal circuits. It also is possible that the striking difference between memantine and ketamine inhibition at the superficial site (Fig. 6A–C) can affect blocker action more powerfully than revealed here. For example, during synaptic transmission, NMDA receptors are activated for far shorter periods than in our experiments. If binding occurs more quickly to the superficial than the deep site, then occupation of the superficial site (and fractional recovery from inhibition) may be exaggerated during synaptic transmission. Further research is needed to elucidate the physiological implications of partial trapping and its contribution to the therapeutic properties of memantine.
Acknowledgments
The authors would like to thank Karen Bouch for excellent technical assistance and Jon Berkepile for help with computer simulations. This work was supported by US National Institute of Mental Health grants R01MH045817.
Glossary
Abbreviations
- dl-APV
dl-2-amino-5-phosphonopentanoic acid
- B
blocker
- eGFP
enhanced green fluorescent protein
- Gly
glycine
- HEK 293T
human embryonic kidney cell line that expresses the SV40 large T antigen
- IB
initial current jump activated by agonist reapplication
- IBSS
steady-state current response in agonist and blocker
- Ip
peak current response to the initial agonist application
- ISS
steady-state current response to initial agonist application
- ISS2
steady-state current response in agonist after recovery from inhibition
- Ket
ketamine
- M2
reentrant loop of NMDA receptors
- Mem
memantine
- nH
Hill coefficient
- NMDG
N-methyl-d-glutamine
- NR1/2A
NMDA receptors composed of NR1 and NR2A subunits
- NR1/2B
NMDA receptors composed of NR1 and NR2B subunits
- NR1(N616Q)
NMDA receptor NR1 subunit with glutamine mutation at asparagine 616
- PCP
phencyclidine
- Sup Site Inhib
superficial site inhibition
- tP
difference between the time of perfusion barrel movement and the time of peak current
Author contributions
J.W.J. and S.E.K. contributed to the conception, design and interpretation of all experiments. S.E.K. performed and analysed all experiments except those on NR1(N616Q)/2A receptors. J.T.W. performed, analysed and contributed to the design and interpretation of experiments on NR1(N616Q)/2A receptors. All authors contributed to the drafting and revising, and approved the final version, of the manuscript. Experiments were performed in the Department of Neuroscience, University of Pittsburgh.
Supplemental material
References
- Benveniste M, Mayer ML. Trapping of glutamate and glycine during open channel block of rat hippocampal neuron NMDA receptors by 9-aminoacridine. J Physiol. 1995;483:367–384. doi: 10.1113/jphysiol.1995.sp020591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpied TA, Boeckman FA, Aizenman E, Johnson JW. Trapping channel block of NMDA-activated responses by amantadine and memantine. J Neurophysiol. 1997;77:309–323. doi: 10.1152/jn.1997.77.1.309. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bolshakov KV, Gmiro VE, Tikhonov DB, Magazanik LG. Determinants of trapping block of N-methyl-D-aspartate receptor channels. J Neurochem. 2003;87:56–65. doi: 10.1046/j.1471-4159.2003.01956.x. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Schoepfer R, Monyer H, Ruppersberg JP, Gunther W, Seeburg PH, Sakmann B. Control by asparagine residues of calcium permeability and magnesium blockade in the NMDA receptor. Science. 1992;257:1415–1419. doi: 10.1126/science.1382314. [DOI] [PubMed] [Google Scholar]
- Chen HS, Lipton SA. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. J Physiol. 1997;499:27–46. doi: 10.1113/jphysiol.1997.sp021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HS, Pellegrini JW, Aggarwal SK, Lei SZ, Warach S, Jensen FE, Lipton SA. Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J Neurosci. 1992;12:4427–4436. doi: 10.1523/JNEUROSCI.12-11-04427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HSV, Lipton SA. Pharmacological implications of two distinct mechanisms of interaction of memantine with N-methyl-D-aspartate-gated channels. J Pharmacol Exp Ther. 2005;314:961–971. doi: 10.1124/jpet.105.085142. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003;26:81–89. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- Johnson JW, Kotermanski SE. Mechanism of action of memantine. Curr Opin Pharmacol. 2006;6:61–67. doi: 10.1016/j.coph.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Kashiwagi K, Masuko T, Nguyen CD, Kuno T, Tanaka I, Igarashi K, Williams K. Channel blockers acting at N-methyl-D-aspartate receptors: differential effects of mutations in the vestibule and ion channel pore. Mol Pharmacol. 2002;61:533–545. doi: 10.1124/mol.61.3.533. [DOI] [PubMed] [Google Scholar]
- Kotermanski SE, Johnson JW. Mg2+ imparts NMDA receptor subtype selectivity to the Alzheimer's drug memantine. J Neurosci. 2009;29:2774–2779. doi: 10.1523/JNEUROSCI.3703-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, D'Souza DC, Mathalon D, Perry E, Belger A, Hoffman R. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl) 2003;169:215–233. doi: 10.1007/s00213-003-1582-z. [DOI] [PubMed] [Google Scholar]
- Kuner T, Schoepfer R. Multiple structural elements determine subunit specificity of Mg2+ block in NMDA receptor channels. J Neurosci. 1996;16:3549–3558. doi: 10.1523/JNEUROSCI.16-11-03549.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mealing GA, Lanthorn TH, Murray CL, Small DL, Morley P. Differences in degree of trapping of low-affinity uncompetitive N-methyl-D-aspartic acid receptor antagonists with similar kinetics of block. J Pharmacol Exp Ther. 1999;288:204–210. [PubMed] [Google Scholar]
- Mealing GA, Lanthorn TH, Small DL, Murray RJ, Mattes KC, Comas TM, Morley P. Structural modifications to an N-methyl-D-aspartate receptor antagonist result in large differences in trapping block. J Pharmacol Exp Ther. 2001;297:906–914. [PubMed] [Google Scholar]
- Olney JW. Excitotoxicity, apoptosis and neuropsychiatric disorders. Curr Opin Pharmacol. 2003;3:101–109. [PubMed] [Google Scholar]
- Parsons CG, Gruner R, Rozental J, Millar J, Lodge D. Patch clamp studies on the kinetics and selectivity of N-methyl-D-aspartate receptor antagonism by memantine (1-amino-3,5-dimethyladamantan) Neuropharmacology. 1993;32:1337–1350. doi: 10.1016/0028-3908(93)90029-3. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Panchenko VA, Pinchenko VO, Tsyndrenko AY, Krishtal OA. Comparative patch-clamp studies with freshly dissociated rat hippocampal and striatal neurons on the NMDA receptor antagonistic effects of amantadine and memantine. Eur J Neurosci. 1996;8:446–454. doi: 10.1111/j.1460-9568.1996.tb01228.x. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Quack G, Bresink I, Baran L, Przegalinski E, Kostowski W, Krzascik P, Hartmann S, Danysz W. Comparison of the potency, kinetics and voltage-dependency of a series of uncompetitive NMDA receptor antagonists in vitro with anticonvulsive and motor impairment activity in vivo. Neuropharmacology. 1995;34:1239–1258. doi: 10.1016/0028-3908(95)00092-k. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Stoffler A, Danysz W. Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system – too little activation is bad, too much is even worse. Neuropharmacology. 2007;53:699–723. doi: 10.1016/j.neuropharm.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Sobolevsky A, Koshelev S. Two blocking sites of amino-adamantane derivatives in open N-methyl-D- aspartate channels. Biophys J. 1998;74:1305–1319. doi: 10.1016/S0006-3495(98)77844-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Beck C, Wollmuth LP. Molecular rearrangements of the extracellular vestibule in NMDAR channels during gating. Neuron. 2002;33:75–85. doi: 10.1016/s0896-6273(01)00560-8. [DOI] [PubMed] [Google Scholar]
- Sobolevsky AI, Koshelev SG, Khodorov BI. Interaction of memantine and amantadine with agonist-unbound NMDA- receptor channels in acutely isolated rat hippocampal neurons. J Physiol. 1998;512:47–60. doi: 10.1111/j.1469-7793.1998.047bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmuth LP, Kuner T, Sakmann B. Adjacent asparagines in the NR2-subunit of the NMDA receptor channel control the voltage-dependent block by extracellular Mg2+ J Physiol. 1998;506:13–32. doi: 10.1111/j.1469-7793.1998.013bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmuth LP, Kuner T, Seeburg PH, Sakmann B. Differential contribution of the NR1- and NR2A-subunits to the selectivity filter of recombinant NMDA receptor channels. J Physiol. 1996;491:779–797. doi: 10.1113/jphysiol.1996.sp021257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.