

Abstract
Using iTRAQ labeling and mass spectrometry on an LTQ-Orbitrap with HCD capability, we assessed relative changes in protein phosphorylation in the mitochondria upon physiological perturbation. As a reference reaction, we monitored the well-characterized regulation of pyruvate dehydrogenase (PDH) activity via phosphorylation/dephosphorylation by pyruvate dehydrogenase kinase/pyruvate dehydrogenase phosphatase in response to dichloroacetate, de-energization and Ca2+. Relative quantification of phosphopeptides of PDH-E1α subunit from porcine heart revealed dephosphorylation at three serine sites (Ser231, Ser292 and Ser299). Dephosphorylation at Ser292 (i.e., the inhibitory site) with DCA correlated with an activation of PDH activity as previously reported, consistent with our de-energization data. Calcium also dephosphorylated (i.e., activated) PDH thus confirming calcium activation of PDP. With this approach, we successfully monitored other phosphorylation sites of mitochondrial proteins including adenine nucleotide translocase, malate dehydrogenase and mitochondrial creatine kinase, etc. Among them four proteins exhibited phosphorylation changes with these physiological stimuli: (1) BCKDH-E1α subunit increased phosphorylation at Ser337 with DCA and de-energization; (2) apoptosis-inducing factor phosphorylation was elevated at Ser345 with calcium; (3) ATP synthase F1 complex α subunit and (4) mitofilin dephosphorylated at Ser65 and Ser264 upon de-energization. This screening validated the iTRAQ/HCD technology as a method for functional quantitation of mitochondrial protein phosphorylation as well as providing insights into the regulation of mitochondria via phosphorylation.
Keywords: Quantitative Phosphoproteomics, iTRAQ, HCD, LTQ-Orbitrap, Mitochondria
INTRODUCTION
Phosphorylation events regulated by networks of kinases and phosphatases are currently believed to be among the one of the most prevalent acute regulatory modifications within the cell (1–3). Mass spectrometric screening tools have begun to show a large population of protein phosphorylation in both cytosol and mitochondrial matrix (4–6). However, it is unclear which modified sites actually play a significant role in protein function rather than basal level phosphorylation by non-specific actions of some kinases present in the system. One approach to address this problem is to challenge biological systems and then quantitatively monitor the alterations in specific protein phosphorylation sites. This screening approach could be one way to focus on phosphorylation sites that may potentially modulate specific signaling pathways. Herein, we utilized iTRAQ labeling techniques combined with 2D-LC-MS/MS-HCD analysis on a high-resolution LTQ-Orbitrap XL hybrid instrument to quantitatively measure protein phosphorylation during experimental perturbations in intact biological systems.
The model system we have selected is the isolated porcine heart mitochondria. In addition to being a critical element in the function of the cell such as energy metabolism and apoptosis, mitochondria can be isolated in vitro for well-controlled experimental perturbations that can be screened for post-translational modifications (PTMs). It has previously been demonstrated that the heart mitochondrial proteins are extensively phosphorylated (7, 8, 9) using phosphor-specific dyes, 32P labeling and mass spectroscopy. One of the best characterized protein phosphorylation systems in the mitochondria is the pyruvate dehydrogenase kinase (PDK) and phosphatase (PDP) system that regulate pyruvate dehydrogenase activity via phosphorylation at Ser292 of E1α subunit of PDC (10–15). This phosphorylation site can be challenged with the addition of Ca2+ to activate PDP or DCA to inhibit PDK. Thus, the mitochondrial PDH system provides an excellent opportunity to validate this quantitative approach using the modulation of PDH phosphorylation and activity in this relatively simple system.
In the iTRAQ methodology, peptides are labeled with isobaric chemical tags, which avoid mass heterogeneity for the parent ions and rather rely on quantitation of reporter groups released from the chemical tag during collision-induced dissociation (CID) (16). Among the advantages of this method is the ability to use up to four to eight isotope tags in a single four-plex or eight-plex experiment. Quadrupole time-of-flight (Q-TOF) mass spectrometers such as Q-TOF Ultima Global (Waters) and Q-STAR (Applied Biosystems) used to be the choice of instruments for iTRAQ quantitation because of the poor recovery of low mass fragments observed in tandem mass spectra acquired on ion trap (IT) mass spectrometers. The “one-third rule” on an ion trap instrument precluded the use of these reagents on this widely available instrument platform. It was only until recently that the Pulsed Q Dissociation (PQD) technique on an ion trap allowed negotiating this limitation but it still suffers from poor fragmentation efficiency, which has raised doubts in the community as to its practical utility. The LTQ-Orbitrap XL-ETD hybrid instrument equipped with a High Energy Collision Dissociation (HCD) cell can produce rich “triple-quadrupole” like fragmentation patterns including fragments in the low m/z range in a similar fashion to a Q-TOF instrument (17). Exploiting this capability, we performed quantitative phosphoproteomic analysis of porcine heart mitochondria using iTRAQ in response to DCA, de-energization without carbon substrates as well as calcium. We successfully monitored relative changes in phosphopeptides from mitochondrial proteins upon physiological perturbation which directly corresponded to changes in biological activities.
EXPERIMENTAL PROCEDURES
Materials
All HPLC solvents were of the highest grade purchased from JT Baker (New Jersey). Porcine trypsin gold (mass spectrometry grade) was obtained from Promega (Madison, WI). DTT was from Thermo-Fisher Scientific and IAM was from Sigma (St. Louis, MO). The iTRAQ four-plex reagent kits were purchased from Applied Biosystems, Inc. (Framingham, MA). Oasis HLB desalting cartridges were purchased from Waters (Milford, MA). Titanium dioxide pre-packed toptips were obtained from Glygen (Columbia, MD). Bovine serum albumin (BSA) tryptic digest was purchased from Michrom Bioresources Inc. (Auburn, CA).
Isolation of Porcine Heart Mitochondria
All procedures were approved by the NHLBI ACUC and performed in accordance with the guidelines described in the Animal Care and Welfare Act (7 U.S.C. 2142 § 13). Porcine heart mitochondria were isolated from perfused tissue as previously described (9, 18). Mitochondrial preparations were checked for viability using respiratory control ratio (RCR) measurements by taking the ratio of the oxygen consumption rate with and without 1 mM ADP at 37°C in buffer B* (137 mM KCl, 10 mM HEPES and 2.5 mM MgCl2, pH 7.1) containing 5 mM potassium-glutamate and 5 mM potassium-malate (G/M) and 1 mM inorganic phosphate (Pi). Mitochondrial preparation with RCR > 8 was rendered as an acceptable preparation. Protein concentration was determined using a Bradford assay (USB Quant Kit, Cleveland, OH).
In order to test for differences in protein phosphorylation, mitochondria were incubated under several different conditions: (1) in the presence and absence of DCA, which is known to suppress pyruvate dehydrogenase (PDH) phosphorylation (19); (2) under energized versus de-energized states; and (3) with and without calcium, another player shown to alter mitochondrial protein phosphorylation via calcium-sensitive kinases and phosphatases (20, 21). For DCA treatments, mitochondria (1 mg/ml) were incubated in an oxygen-saturated buffer C (125 mM KCl, 15 mM NaCl, 20 mM HEPES, 1 mM EGTA, 1 mM EDTA, 5 mM MgCl2, pH 7.1) with the addition of 5 mM G/M, 5 mM Pi and 1 mM DCA for 20 min at 37°C with 100% oxygen blowing over the top of the reaction chamber whereas paired control experiments were conducted in the absence of DCA. For calcium experiments, mitochondria (1 mg/ml) were incubated in the same oxygen-saturated buffer C at 37°C for 6 min to deplete calcium prior to the addition of 5 mM G/M (glutamate/malate), 5 mM Pi, 10 mM ATP and 0.65 µM free calcium. The reaction mixture was allowed to incubate for 5 min with oxygen as previously described (7). Similarly, the paired control experiments were conducted without calcium. Free calcium levels were determined using a calcium electrode under these experimental conditions. For the energized versus de-energized experiments, mitochondria (1 mg/ml) were energized with 5 mM G/M, 5 mM Pi and 10 mM ATP for 20 min as described above while de-energization of mitochondria was conducted in pairs by incubating mitochondria in oxygen-saturated buffer C without the addition of G/M, Pi or ATP.
2D-DIGE Upon DCA Treatment
Mitochondrial samples (1 mg) were precipitated using a standard TCA/acetone procedure and re-suspended in 100 µl of lysis buffer (15 mM Tris-HCl, 7 M urea, 2 M thiourea and 4% CHAPS (w/v), pH 8.5) as previously described (9). Briefly, control and DCA-treated samples were labeled with CyDyes and prior to isoelectric focusing (IEF) with a 24-cm Immobiline DryStrip gels (pH 3–10 NL, GE Healthcare, Piscataway, NJ) and electrophoresis with 10–15% SDS gels (Nextgen Sciences, Ann Arbor, MI) in the second dimension as previously detailed (9, 22). Gels were then scanned on a Typhoon 9400 variable mode imager (GE Healthcare) at a resolution of 100 µm using the appropriate parameters for each CyDye. Proteins of interest were picked and identified from the 2D gels as previously described (7).
iTRAQ Labeling of Pig Heart Mitochondria
Approximately 1 mg of each intact mitochondrial pellet was lysed in 100 µl of lysis buffer containing 8 M urea, 2 M thiourea, 4% CHAPS (w/v), 2% maltoside (w/v), 2 mM PMSF, 2 mM KF, 5 mM sodium orthovanadate, 0.01% (v/v) protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) with sonication on ice. Both lysates were spun down at 17,000g for 1 h at 4°C. Bradford assay was performed to obtain the protein concentration in the range of 5–8 mg/ml (USB Corporation, Cleveland, OH). Approximately 500 µg heart mitochondrial lysates was reduced and alkylated as previously described (23). Trypsin digestion (protein: enzyme ratio of 25:1) was performed at 35°C overnight. Tryptic digests were desalted on Waters Oasis HLB 1cc cartridges (Milford, MA) following manufacturer’s instructions. Eluents were dried completely followed by re-dissolution in 150 µl of iTRAQ dissolution buffer (30 µl per 100 µg proteins). Five vials of iTRAQ reagent 115 was used to label pig heart control sample while 5 vials of iTRAQ reagent 117 labeled pig heart sample with treatment at RT for 2 h (iTRAQ pairs were always chosen between 114/116 or 115/117 to avoid isotopic correction during data analysis). Subsequently, both samples were combined and taken up to a total of 21 ml with 0.1% TFA prior to cleanup on Waters Oasis HLB cartridges 6cc. The eluted peptides were dried by SpeedVac.
Strong Cation Exchange Chromatography (SCX) of iTRAQ-labeled Peptides
The dried peptides were taken up in 300 µl SCX solvent A (5 mM KPi, pH 3.0, 25% ACN) and injected onto a PolySulfoethyl A SCX column (4.6 mm id × 20 cm length, 5 µm particle size, 300 Å pore size). SCX chromatography was carried out on an Agilent HP1100 system at 1 ml/min flow rate using the following gradient: 0% B for 2 min; 0–20% B for 40 min; 20–100% B for 5 min; 100% B held for 5 min (solvent A: 5 mM KPi, pH 3.0, 25% ACN; solvent B: 5 mM KPi, pH 3.0, 25% ACN, 350 mM KCl). UV absorbance at 214 nm was monitored while fractions were collected at 1.5-ml intervals and dried down by SpeedVac. All 15 fractions were desalted on Vivapure C18 micro spin columns according to manufacturer’s instructions (Sartorius-Stedim Biotech, Edgewood, NY) prior to phosphopeptide enrichment.
Hydrophilic Interaction Chromatography (HILIC) of iTRAQ-labeled Peptides
The dried peptides were taken up in 300 µl of 88% ACN, 0.1% TFA solvent and injected onto a TSKgel Amide-80 HILIC column (4.6 mm id × 25 cm length, 5 µm particle size, 80 Å pore size, Tosoh Biosciences LLC, Montgomeryville, PA). HILIC gradient was performed on an Agilent HP1100 system at 1 ml/min flow rate as previously described (24). Briefly, the following gradient was used: 250 µl of samples were loaded in 80% B; 80% B held for 5 min; 80–60% B for 40 min; 60–0% B for 5 min; 0% B held for 5 min (solvent A: 98% water, 0.1% TFA; solvent B: 98% ACN, 0.1% TFA). UV absorbance at 214 nm was monitored while fraction collector collected a total of 15 fractions. All dried fractions were taken up in 20 µl of solvent A (2% ACN, 0.1% HCOOH) for LC-MS/MS analysis.
Phosphopeptide Enrichment by TiO2
iTRAQ-labeled samples were cleaned up by Oasis HLB cartridges (6 cc, Waters) according to manufacturer’s instruction. Dried samples were taken up in 200 µl of loading buffer (65% ACN, 2% TFA) prior to loading onto TiO2 toptips (Glygen, Columbia, MD) pre-equilibrated with 200 µl of the same loading buffer. The samples were loaded slowly using a microcentrifuge (Thermo Sorvall Legend Micro 17R, Thermo Fisher Scientific LLC, Ashville, NC) at 100 g for 20 min. Tips were washed once with 200 µl loading buffer, once with 200 µl of wash buffer 1 (65% ACN, 0.5% TFA) followed by another wash with wash buffer 2 (65% ACN, 0.1% TFA). Elution of phosphopeptides was carried out using 150 µl of 0.3 M NH4OH each for three times. Combined eluents were dried down by SpeedVac and re-acidified with 10% formic acid (25).
LC-MS/MS Analysis on LTQ-Orbitrap
All dried fractions were analyzed on a LTQ-Orbitrap XL (Thermo-Fisher Scientific, LLC) interfaced with an Eksigent nano-LC 1D plus system (Eksigent Technologies, LLC, Dublin, CA) using high energy collision dissociation (HCD). Briefly, samples were loaded onto an Agilent Zorbax 300SB-C18 trap column (0.3 mm id × 5 mm length, 5 µm particle size) at a flow rate of 5 µl/min for 10 min. Reversed-phase C18 chromatographic separation of peptides was carried out on a pre-packed BetaBasic C18 PicoFrit column (75 µm id × 10 cm length, New Objective, Woburn, MA) at 300 nl/min using the following gradient: 2–5% B for 5 min; 5–40% B for 40 min; 40–50% B for 10 min; 50–95% B for 5 min (solvent A: 0.1% formic acid in 98% water, 2% ACN; solvent B: 0.1% formic acid in 100% ACN). LTQ-Orbitrap settings were as follows: spray voltage 1.2 kV, 1 microscan for MS1 scans at 30,000 resolution (FWHM at m/z 400), 2 microscans for MS2 at 7500 resolution (FWHM at m/z 400); Full MS mass range: m/z 300–2000; MS/MS mass range: m/z 100–2000. The LTQ-Orbitrap was operated in a data-dependent mode, i.e., one MS1 FTMS scan for precursor ions followed by three data-dependent HCD-MS2 scans for precursor ions above a threshold ion count of 5000 with collision energy of 45%.
Database Search and iTRAQ Quantification
Three search algorithms were used: Mascot, Sequest and InsPecT. For Mascot, Mascot generic format files (.mzData files) from off-line SCX and HILIC fraction analyses were generated using Bioworks version 3.3.1 SP1 (Thermo Fisher Scientific, LLC, Ashville, NC) and combined using mzData Combinator software (version 1.0.6) prior to submitting search to our six-processor Mascot cluster at NIH (http://biospec.nih.gov, version 2.2) using the following criteria: database: NCBInr; taxonomy: mammalia (mammals); enzyme: trypsin; miscleavages: 2; fixed modifications: carbamidomethylation (+57 Da), N-terminal iTRAQ4plex (+144 Da), lysyl iTRAQ4plex (+144 Da); variable modifications: methionine oxidation (+16 Da); STY phosphorylation (+80 Da); MS peptide tolerance as 15 ppm; MS/MS tolerance as 0.05 Da. Mammalian protein database was used in this case because porcine protein sequences is not fully available at the present time. The same raw data files were also searched against a compiled pig plus human protein decoy database (45559 sequence entries) using InsPecT (http://bix.ucsd.edu, University of California, San Diego) via http://biowulf.nih.gov and Sequest (Bioworks version 3.3.1 SP1, Thermo Fisher Scientific LLC, Ashville, NC) with similar search parameters to Mascot search. Peptides reported via all search engines were accepted only if they met the false discovery rate of p<0.05 (target decoy database). Manual inspection of all phosphopeptide mass spectra was conducted. For iTRAQ quantification, the ratios of iTRAQ reporter ion intensities in MS/MS spectra (m/z 114.11–117.11) from raw datasets were used to calculate fold changes in phosphorylation between control and treatment upon correcting discrepancies resulting from Bradford assay. Standard deviation (SD) was only calculated if a phosphopeptide has been observed in all three biological replicates. With Sequest results, PepQuan in Bioworks 3.3.1 SP1 was used for iTRAQ quantitation after filtering data with the following parameters: XCorr vs. charge state (XCorr of 1.5, 2.0, 2.5 and 3.0 for +1, +2, +3, ≥+3 charges respectively); RSn of 10; delta CN of 0.1).
Luciferin-Luciferase Assay for [ATP]
Mitochondrial samples were prepared by placing them immediately into 6% Perchloric acid (at 4°C) followed by neutralization to pH 7.0 with KOH. Samples were frozen at −80°C and assayed within 24 h. ATP was determined using an ATP determination kit by following manufacturer’s instructions (Invitrogen, Carlsbad, CA) as previously described (9). Luciferin fluorescence was determined using a Glomax Luminometer (Promega, Madison, WI).
Pyruvate Dehydrogenase Assay
PDH activity was determined by following the rate of NADH production in the presence of pyruvate, coenzyme A, and NAD as previously described (7, 26). Briefly, PDH activity was assayed in a reaction mixture (pH 8.0) containing 50 mM Tris, 10 mM pyruvate, 0.2 mM coenzyme A, 2 mM NAD, 2 mM cocarboxylase, 1 mM MgCl2, and pulverized mitochondria at a concentration of 0.2–0.4 nmol of cyt a/mL. The reaction was carried out at 37°C and was initiated with coenzyme A. Production of NADH was assessed spectrophotometrically by monitoring A350. PDH activity was expressed as nmol NADH produced/min/mg mitochondrial protein. Three biological replicates were conducted to calculate an averaged activity ± SEM. Statistical analysis was performed using two-tailed unpaired Student t-test by GraphPad Prizm (version 4.0, San Diego, CA; **p<0.01; *p<0.05 when treatments were compared with their pairing controls).
RESULTS
DIGE Comparison between Control and DCA-treated Mitochondria
In mammalian tissue, PDH activity has been shown to be regulated by its own kinase/phosphatase system. This protein is active in its dephosphorylated state and becomes inactive upon phosphorylation. Treatment of mitochondria with DCA has been shown to suppress PDH phosphorylation by inhibition of its kinase (15). Comparing a control and a sample treated with 1 mM DCA using 2D-DIGE revealed a specific alteration to the phosphorylation status of PDH E1α subunit (middle section of the gel, Figure 1). Specifically in the control sample, the isoelectric variants (IEVs) of PDH E1α subunit (labeled red) were shifted toward the acidic region of the gel which is consistent with PDH E1α subunit phosphorylation. Conversely, the IEVs of PDH E1α subunit in our DCA-treated sample (labeled green) were shifted toward the basic region of the gel which agrees with the notion of its extensive de-phosphorylation.
Figure 1.

2D-DIGE image comparing control porcine heart mitochondria (labeled red, Cy3) and DC-Atreated mitochondria (labeled green, Cy5). Proteins were separated in the horizontal direction by isoelectric focusing point (pI) from pH ~3 to 10, and vertically by molecular weight from ~150 to 10 kDa.
Optimization of iTRAQ Signals Using LTQ-Orbitrap HCD
Since the LTQ-Orbitrap XL is equipped with a HCD cell behind the C-trap that allows selected ions for fragmentation without the loss of low mass ion information (16), we chose HCD as the method of fragmentation due to the need for low mass tag detection for iTRAQ quantitation in MS2 mode with better accuracy and precision than commonly used Pulse-Q dissociation (PQD) fragmentation in the ion trap (IT). Bovine serum albumin tryptic digest (1 pmol) derivatized with iTRAQ reagent 114 was used to optimize HCD parameters for iTRAQ reporter ions and overall peptide fragmentation pattern such as number of microscans, Automatic Gain Control (AGC) settings and collision energy (CE). Nano-LC analyses of derivatized BSA tryptic digests (Michrom Bioresources, Inc., Auburn, CA) were also performed using different parameter settings including minimum ion threshold counts for MS2, dynamic exclusion and isolation window in XCalibur software (version 2.0.7) in order to optimize iTRAQ and peptide fragmentation as well as reporter ion detection. We determined the optimal conditions for high quality iTRAQ HCD-MS/MS spectra at 2 microscans (MS/MS), 45% CE, minimal ion counts of 5000 and an isolation window of 3 Da. Technical variability of HCD experiments was assessed by running triplicate injections (500 fmol per injection) of a combined BSA tryptic digest spanning a dynamic range of 10-fold that were labeled with two different iTRAQ reagents (A: 1 µM BSA digest labeled with iTRAQ 114; B: 0.1 µM BSA digest labeled with iTRAQ 116). Our results showed an average ratio of 9.65 ± 0.18 indicating that this quantitative analysis is accurate, precise and reproducible (data not shown).
SCX/HILIC Fractionation of iTRAQ-labeled Porcine Mitochondrial Phosphoproteome
A total of 1 mg of pig heart mitochondrial proteins trypsinized, iTRAQ labeled and enriched for phosphopeptides using pre-packed TiO2 toptips yielded 31 phosphorylation sites with iTRAQ mass tags from 18 proteins in DCA samples, 27 phosphosites from 19 proteins in energized/de-energized mitochondria, and 31 phosphosites in 21 proteins in calcium samples (see Table I & Supplemental Tables I–III). The workflow for iTRAQ labeling and peptide fractionation is illustrated in Figure 2A. Figure 2B is a representative SCX chromatograph (UV absorbance at 214 nm) of iTRAQ-labeled pig heart mitochondrial control combined with calcium-treated samples using a shallow salt gradient from 0–70 mM KCl in 40 min. The diversity and complexity of proteins and peptides in biological systems requires powerful liquid chromatography-based separations to optimize resolution and detection of components. Proteomics strategies often combine two orthogonal separation modes to meet this challenge. SCX takes advantage of the fact that tryptic peptides carrying different net charges (e.g., phosphopeptides vs. non-phosphopeptides) elute at different salt concentration (i.e., ionic strength). Villén et. al. has shown the powerful utility of SCX in combination with phosphopeptide enrichment with IMAC in phosphoproteomic screening of complex mixtures (27). Another mode of separation for complex mixtures is HILIC columns using a shallow inverse organic gradient. This is based on the overall hydrophilicity of tryptic peptides, a separation truly orthogonal to reverse phase (RP). It has been demonstrated that HILIC columns retain hydrophilic phosphopeptides (20). In our case, the use of SCX and HILIC pre-fractionation prior to TiO2 enrichment expanded our coverage of mitochondrial phosphoproteome by approximately 3-fold with a combination of three search algorithms (a total of 56 phosphorylation sites in 38 proteins).
Table I.
Porcine Heart Mitochondrial Proteins with Phosphorylation Changes Upon Physiological Perturbation
| Proteins | Accession# | Phosphopeptides | m/z/z | aiTRAQ Ratios |
dIon Score/ Expect |
||
|---|---|---|---|---|---|---|---|
| CTL/DCA | CTL/Ca2+ | D/E | |||||
| PDH E1α subunit |
gi|266686 | 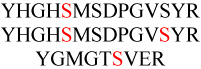 |
908.88/2 | 3.0±0.28 | 3.3±0.26 | 0.23±0.03 | 42/0.0041 |
| 632.91/3 | 15.2±1.27 | 4.4±0.11 | 0.20±0.06 | 36/0.013 | |||
| 612.26/2 | 5.2±0.49 | 2.7±0.31 | 0.15±0.05 | 36/0.0014 | |||
| BCKDH E1α subunit |
gi|129032 | 586.25/3 | 0.30 | cND | 1.50±0.71 | 42/0.021 | |
| ATP synthase, F1 α subunit |
gi|187370717 | 808.39/2 | 1.26 | 0.94±0.27 | 0.33 | 66/0.0006 | |
| Mitofilin | gi|154354964 | 490.63/3 | cND | cND | 0.15 | 31/0.29 | |
| Apoptosis- inducing factor |
gi|115607158 | 947.03/2 | cND | 0.53 | cND | 24/0.4 | |
Nomenclature used here applies to all tables unless otherwise stated. Fold changes in phosphorylation were expressed as the iTRAQ reporter ion ratios of controls in reference to their respective treatments (Mean ± SD). Serine (S), Threonine (T) and Tyrosine (Y) labeled in red are phosphorylated. All residue numbers are based on the mature protein sequences from NCBInr database.
CTL: Control; D: de-energization; E: energization; m/z precursor ion mass; z charge state; SD (standard deviation) was calculated only if a phosphopeptide was observed in all three pig heart mitochondrial preparations.
iTRAQ ratios
Ac-: N-terminal acetylation of proteins; M*: methionine oxidation; C*: cysteine carbamidomethylation
ND: not determined due to lack of iTRAQ reporter ions
Ion score/Expect (Mascot); p-value (InsPecT); X-Corr (Sequest).
Figure 2.

(A). The workflow of an iTRAQ-based phosphoproteomic profiling experiment in response to DCA treatment; (B). A typical SCX chromatograph of a combined iTRAQ-labeled mitochondrial sample using KCl salt elution gradient (0–350 mM). The dashed line denotes the salt gradient across the chromatograph.
Mitochondrial Phosphopeptide Analysis Upon DCA Treatment and De-energization
As expected, the α subunit of E1 component of pyruvate dehydrogenase complex (PDC) was clearly phosphorylated as shown by the doubly and triply charged phosphopeptide YHGHS*MSDPGVSYR at m/z 606.26 and 908.08 respectively (Figure 3A, top inset). The LTQ-Orbitrap HCD cell used for fragmentation revealed the presence of b1–2, b5-98, b6-98, b8-98, y10-98, [b5-98]2+ and [b8-98]2+ ions clearly indicating the site of phosphorylation at Ser292 (Figure 3A). In addition, the neutral loss of phosphoric acid (−98 Da) from its precursor ion at m/z 606.26, a signature ion for this phosphopeptide, was observed at m/z 573.56. The relative abundances of iTRAQ reagent tags at m/z 115.11 and 117.11 calculated by peak height showed a 2.8-fold dephosphorylation at this phosphoserine site upon DCA treatment (Figure 3A, bottom inset). Biological replicates from three different pig heart mitochondrial preparations showed an average fold of dephosphorylation of 3.0 ± 0.28 at Ser292 (Table I). Interestingly, our PDH assay showed a corresponding activation of PDH activity of 3.8-fold (black solid bar) in reference to its control (blue solid bar) presumably due to this dephosphorylation event (Figure 4, left panel). This observation is in an excellent agreement with previously published results using 32P. An additional phosphoserine site encompassing the same tryptic peptide was observed as a doubly and triply charged parent ion at m/z 948.87 and m/z 632.91 (YHGHS*MSDPGVS*YR, Figure 3B inset, Table I). The MS/MS spectrum clearly showed a phosphosite at Ser299 by the presence of y4–y9 as well as doubly charged b10 and b12 ions having lost phosphoric acid (−98 Da), in addition to the initial pSer292 site as shown by the doubly charged b5–b8 as well as y10 and y12 ions minus 98 Da (Figure 3B). Dephosphorylation at both serine sites amounted to a total of 15.2-fold in the presence of 1 mM DCA (Table I; Figure 3B, bottom inset). However, the second site of dephosphorylation was presumed to be irresponsible for PDH activation according to previous studies (13). The third phosphosite in PDH E1α subunit was observed in the peptide YGMGTS*VER as observed by a +2 charge precursor ion at m/z 612.26 (Figure 3C, top inset). The analysis of dephosphorylation by iTRAQ tags clearly showed an average of 5.2-fold change upon 1 mM DCA addition (Table I). Again, dephosphorylation at these sites presumably did not contribute to activation of PDH activity (13). All these data supported earlier literature on PDH phosphorylation resulting in an inactivation of PDH activity. Interestingly, when mitochondria were de-energized without carbon substrates, we observed a similar dephosphorylation phenomenon at Ser292, Ser292/Ser299 and Ser231 by 4.3-, 5.0- and 6.7-fold as with DCA (Table I). This is in accordance with our previous finding of matrix ATP depletion by DCA possibly resulting in de-energized mitochondria similarly to the absence of carbon substrates (Figure 4C). To our knowledge, our data is the first report that coupled quantitative mass spectrometric analysis to the regulation of enzyme activity of PDH via phosphorylation/dephosphorylation. This method thus has the potential as a functional proteomic tool to discover phosphoprotein targets with regulatory roles in metabolic and signaling pathways.
Figure 3.



HCD-MS/MS spectra of iTRAQ-labeled phosphopeptides identified from pig heart mitochondrial proteins. Insets showed the precursor ion masses of phosphopeptides and iTRAQ ratios representing the change between control mitochondria and mitochondria treated with 1 mM DCA. (A). YHGHS*MSDPGVSYR (PDH E1α subunit) showed 2.8-fold dephosphorylation at Ser292; (B).YHGHS*MSDPGVS*YR (PDH E1α subunit) demonstrated a total of 15.2-fold dephosphorylation at both Ser292 and Ser299; (C). YGMGTS*VER (PDH E1α subunit) showed 5.2-fold dephosphorylation at Ser231; (D). IGHHS*TSDDSSAYR (BCKDH E1α subunit) showed increase in phosphorylation at Ser293 by 3.4-fold; (E). AAYFGVY*DTAK (solute carrier family 25) showed no change in phosphorylation at Tyr195 site; (F). TGTAEVSS*ILEER (ATP synthase F1 complex α subunit) exhibited 3.0-fold dephosphorylation upon de-energization.
Figure 4.


(A). PDH Activity of Porcine Heart Mitochondria. Enzyme activity was monitored by following the rate of NADH production at A340nm (expressed as nmol/min/mg mitochondrial protein). Each dataset represents a replicate of three biological samples (**p<0.01; *p<0.05 when comparing treatments with their paired controls using unpaired two-tailed Student’s t-test (GraphPad Prism 4.0, San Diego, CA). Error bars were calculated as SEM. The left panel is PDH activity comparison between paired control (blue solid bar) and 1 mM DCA (black solid bar) while right panel represents activity differences between control (blue solid bar) and 0.65 µM Ca2+ (black solid bar); (B). Summary of identified phosphorylation sites in PDH E1α subunit showing averaged fold changes upon various treatments: left panel, 1 mM DCA; middle panel, 0.65 µM calcium; right panel, de-energization. When relative ratio of control in reference to its pairing treatment is > 1, dephosphorylation occurred upon perturbation whereas phosphorylation took place with a ratio < 1. Error bars represent SD from 3 biological replicates; (C). The effect of DCA on Matrix [ATP] was measured by a luciferin-luciferase ATP assay (Mean ± SD).
Further analysis of iTRAQ data revealed no additional DCA-sensitive phosphoproteins at our detection level except for E1α subunit of BCKDH (Table I). Surprisingly, DCA treatment increased the phosphorylation of this protein at Ser337 in the peptide IGHHS*TSDDSSAYR by 3.4-fold as indicated by the ratio of m/z 115.11/117.11 (Figure 3D, right inset). This observation is in agreement with our results where de-energized mitochondria showed a 1.5-fold increase in phosphorylation. Both results were contrary to the belief that BCKDH activity is regulated by BDK/BDP in a similar way as PDH. Other mitochondrial proteins with identified phosphosites included solute carrier family 25, MDH, etc. (Supplemental Table I & III). Figure 3E illustrated the MS/MS spectrum of a doubly charged precursor ion of the phosphopeptide AAYFGVY*DTAK at m/z 787.38 identified (top inset) from adenine nucleotide translocator (ANT). The MS/MS spectrum indicated that phosphorylation occurred at Tyr195 rather than Tyr191 by the presence of y5-98, y7-98, y8-98 as well as b3–b5 ions (Figure 3E). The immonium ion of phosphotyrosine at m/z 216.14 as a signature ion for tyrosine phosphorylation was also present. From the iTRAQ ratios of 115.11 and 117.11 (Figure 3E, bottom inset), it was clear that phosphorylation between control and DCA samples showed virtually no change. Our result confirms the belief that DCA only targets PDH by inhibiting PDK. However, the unexpected increase in BCKDH E1α subunit phosphorylation suggests the possibility of dual regulation of these matrix dehydrogenases by physiological PDK inhibitors. Phosphorylation sites in PDH showing significant changes with DCA and de-energization were summarized in a bar graph in Figure 4B, left panel (DCA) and right panel (de-energization).
Interestingly, de-energization of mitochondria did alter phosphorylation in two more proteins: ATP synthase F1 complex α subunit (TGTAEVSS*ILEER, Figure 3F & Table I) and mitofilin (KS*IQSGPLK) (Supplemental Figure 2A & Table I). The MS/MS spectrum of m/z 808.402+ clearly showed dephosphorylation at Ser65 (y6–y12 ions minus 98 Da) in ATP synthase F1 complex α subunit by 3.0-fold. Previously we have demonstrated that ATP synthase α subunit was autophosphorylated in the presence of radioactive ATP (9). This observation suggests that Ser65 may be a matrix phosphate sensor because dephosphorylation at this site could be a consequence of matrix [ATP] decrease in de-energized mitochondria. Mitofilin is a transmembrane protein of the inner mitochondrial membrane with critical functions in mitochondrial morphology, mitochondrial fusion and fission, specifically in the formation of tubular cristae and cristae junctions. Significant dephosphorylation at Ser264 by 6.7-fold in de-energized mitochondria can possibly change the morphology of mitochondrial membrane structure.
Mitochondrial Phosphopeptide Analysis in Response to Calcium
A total of 1 mg of pig heart mitochondrial proteins enriched for phosphopeptides showed 32 phosphorylation sites with iTRAQ mass tags from 21 proteins in calcium samples treated with 0 and 0.65 µM free calcium (Supplemental Table II). Again, α subunit of E1 component of pyruvate dehydrogenase complex (PDC) was clearly phosphorylated as shown by the doubly and triply charged precursor ions of the phosphopeptide YHGHS*MSDPGVSYR at m/z 606.26 and 908.08 respectively (Supplemental Figure 1A, inset). Dephosphorylation of this phosphoserine site occurred in a dose-dependent manner (data not shown). The HCD fragmentation pattern resembled that in Figure 3A indicating a phosphosite at Ser292. In addition, the neutral loss of phosphoric acid (−98 Da) from its precursor ion at m/z 606.26 was observed at m/z 573.56. The relative abundances of iTRAQ reagent tags at m/z 114.11 and 116.11 representing control and 0.65 µM free Ca2+ showed a 3.3-fold dephosphorylation at this site in response to 0.65 µM of free calcium (Supplemental Figure 1A, top inset). Accordingly, PDH assay showed an activation of PDH activity of 3.3-fold (black solid bar) compared to its paired control (blue solid bar) as a result of dephosphorylation (Figure 4, right panel). The additional serine site on the same phosphopeptide was observed as a doubly charged precursor ion at m/z 948.87 (YHGHS*MSDPGV*SYR, Supplemental Figure 1B, top inset; Table I). The total change of phosphorylation at both sites was approximately 4.4-fold indicating no significant change in dephosphorylation at Ser299 downstream from the regulatory site. This implies that Ser299 was not responsible for PDH activation in response to physiological concentration of calcium. The third site of phosphorylation in PDH as observed in the case of DCA was again in the peptide YGMGTS*VER at m/z 612.26 (+2 charge, Supplemental Figure 1C, top inset). Dephosphorylation as analyzed by iTRAQ tags demonstrated about 2.7-fold change (Supplemental Figure 1C, bottom inset), however, we did not observe a correlation between dephosphorylation at this site and PDH activity. Further analysis of other phosphopeptides detected in this screening revealed no calcium sensitive sites of other mitochondrial proteins including ATP synthase F1 complex α (Supplemental Figure 1D) and γ subunits (Table I & Supplemental Table II) to a significant extent (≥1.5-fold). Figure 4B bar graphs in the middle panel (calcium) illustrated phosphorylation sites in PDH with significant fold changes. The lack of detectable changes in mitochondrial protein phosphorylation in response to calcium except for PDH and AIF (Table I) may be attributed to residual calcium in the tissue and/or the observation of a non-exchanging pool of protein phosphorylation.
DISCUSSION
The current study aimed to extend our knowledge of mitochondrial phosphoproteome in the porcine heart by measuring the dynamic response of specific phosphorylation sites to well-characterized perturbations using iTRAQ and LC-MS/MS-HCD on an LTQ-Orbitrap. This approach is superior to 2D electrophoresis-based Pro-Q Diamond staining or even 32P labeling methods in that the dynamics of particular sites within a protein can be monitored rather than the integrated signal from all of its phosphorylation sites. This is particularly important in proteins with multiple phosphorylation sites that may not share the same regulatory network. Thus, this quantitative mass spectroscopic technique will provide not only a determination of specific phosphorylation sites but also insights into which sites are labile to significant physiological perturbations because these labile sites may be directly linked to the function of proteins and warrant further investigation.
The mitochondrial PDH catalyzes the oxidative decarboxylation of pyruvate, linking glycolysis to the tricarboxylic acid cycle (TCA) and fatty acid (FA) synthesis. Mammalian pyruvate dehydrogenase complexes contain three enzymes that catalyze the conversion of pyruvate, CoA and NAD+ into acetyl-CoA, NADH and CO2. The mammalian complex also contains an intrinsic PDK, which catalyses an MgATP2−-dependent phosphorylation of the α subunit of the pyruvate decarboxylase (E1), thereby inactivating the overall reaction (10–13). Two further phosphorylation events can then occur without any effect on PDC activity. The non-inactivating phosphorylations are thought to be related to the regulation of the rate of dephosphorylation and re-activation of the phosphorylated complex by PDP (10–15). Furthermore, BCKDH, another mitochondrial matrix enzyme complex that catalyzes the degradation of branched-chain amino acids (leucine, valine, isoleucine) to generate NADH and CO2, is believed to be regulated in an analogous manner to the structurally similar PDH (28, 29). Both of them serve as excellent reference points for validating our quantitative approach.
We challenged the porcine heart mitochondria with DCA, de-energization and calcium while quantitatively monitoring the change in peptide phosphorylation using iTRAQ. DCA is an investigational drug for the treatment of genetic mitochondrial diseases by targeting PDH complex, which it stimulates by altering its phosphorylation state and stability (30). Ca2+ is a well-recognized second messenger in the control of mitochondrial function. Ca2+ action has often been linked to protein phosphorylation events via Ca2+-sensitive kinases and phosphatases including PDK and PDP (20, 21), as well as directly affects α-ketoglutarate dehydrogenase (KDH) and NAD+-isocitrate dehydrogenase (31). Here we demonstrated the ability of these isobaric chemical tags to detect and quantify mitochondrial phosphopeptides with an excellent dynamic range, accuracy and reproducibility which directly linked to protein function. Briefly, DCA dephosphorylated the primary phosphorylation site on Ser292 of PDH E1α subunit which in turn activated PDC. Two additional phosphorylation sites on Ser231 and Ser299 in PDH were both dephosphorylated with DCA. However, they were not responsible for the activation of PDH activity but rather concerned with the regulation of the rate of dephosphorylation and reactivation of the phosphorylated PDC complex by pyruvate dehydrogenase phosphatase (13). Similarly, PDH dephosphorylation at all three serine sites occurred in de-energized mitochondria. Previously, we observed a decrease in matrix [ATP] in the presence of DCA (32). It is plausible that DCA effect on PDH can be two-fold, i.e., inhibiting PDK and/or depleting ATP resulting in dephosphorylation of E1α subunit. BCKDH E1α subunit, however, demonstrated an increase in phosphorylation at Ser337 with both DCA and de-energization. This is contrary to previous reports showing similar regulation of BCKDH to PDH via phosphorylation/dephosphorylation by BDK/BDP (28, 29). Five phosphorylation sites in E1α subunit of BCKDH have been identified in human, rat and mouse (www.phosphosite.org), four of which locate on the same tryptic peptide IGHHS*T*S*DDSSAY*R. Ser337 and Ser339 were thought to be two important regulatory sites responsible for activity according to 32P studies (33). Olson et. al. reported that pyruvate caused both inactivation and inhibition of the flux through BDC indicating a reciprocal regulatory mechanism of PDC and BDC exists via physiologically important inhibitors such as pyruvate and DCA of their respective kinases (34, 35). Although the exact mechanism of this dual regulation remains unknown, one speculation is that DCA and pyruvate may act as activators of BDK at low concentrations but as inhibitors at high concentrations.
Calcium studies revealed that PDH dephosphorylation at Ser292 resulted in an activation of PDC by inhibiting Ca2+-sensitive PDP. Other phosphopeptides (a total of 31 additional phosphosites) revealed no calcium sensitive sites of detected mitochondrial phosphoproteins under our experimental condition except for AIF. Notably, phosphorylation sites identified from the α and γ subunits of ATP synthase, an important player in energy production, did not change with Ca2+, thereby suggesting an unlikely regulatory role of these phosphorylation sites on complex V activity. Although we previously demonstrated that calcium induced dephosphorylation of the γ subunit of ATP synthase by Pro-Q Diamond staining (7) which may contribute to the activation of Complex V activity, it does not appear that Ser146 is likely the phosphate turnover site (Supplemental Table II). Previously, Thr144 in the tryptic peptide T*HSDQFLVTFK has been found to be phosphorylated by Villén et. al. (5). Future experiments are currently underway to examine the possible role of phosphate turnover site(s) of γ subunit in regulating Complex V activity.
Previous mitochondrial phosphoproteomic studies have revealed up to 80 phosphorylation sites in yeast (36) and 84 sites in mouse liver (4, 37) using LC-MS/MS. In our study, a total of 57 phosphorylation sites from 38 proteins with iTRAQ tags were detected, some of which were protein contaminants from the mitochondrial isolation process. We speculate that this discrepancy was possibly due to several factors: (1) concentration; (2) labeling efficiency; (3) incomplete protein sequence information on porcine; (4) loss of sensitivity using iTRAQ/HCD for more accurate quantification compared to PQD or CID approaches; (5) differences in the phosphoproteomes between heart (38) and other tissues such as liver and brain (37, 39). Although not all of the previously reported phosphopeptides of the mitochondrial matrix were detected in our study, it is of interest that the effects of DCA, de-energization and Ca2+ in causing significant fold changes were limited to only a few of proteins. This is consistent with our previous observation that not all proteins labeled with Pro-Q Diamond identified phosphorylation exchange with [γ32P]-ATP (7, 9) suggesting that many of these sites may not undergo dynamic modifications. These stable sites may very well represent events associated with early folding processes of protein complexes that are no longer accessible to phosphatases, those by non-specific protein kinase activity or simply active sites that the perturbations used did not result in significant quantitative changes. We speculate that the non-specific protein kinase activity could contribute significantly to this non-exchanging pool of protein phosphorylation since a protein phosphorylation that has no effect on protein function would have little negative effect on the protein as well as almost no energetic consequences.
Although the iTRAQ labeling technique is powerful in conjunction with LTQ-Orbitrap-HCD in the application of quantitative phosphoproteomic analysis as demonstrated here, there are complications that may interfere with iTRAQ labeling efficiency that will ultimately affect this quantitative analysis. One such complication is the possibility of negatively charged phosphates on phosphoamino acids at the N-termini of peptides interfering with iTRAQ labeling at the α-amino group of a peptide. Whether or not iTRAQ labeling at this N-terminal site is efficient may also depend on the chemical environment of other amino acids in the peptide. The same scenario can occur on phosphoamino acids near the C-terminal lysine resulting from trypsin digestion, which can be labeled with iTRAQ at the ε-amino group of the lysyl side chain. Although it did not interfere with iTRAQ labeling at some N-terminal phosphoamino acids in our samples, one instance where we did fail to label was a phosphopeptide S*HGS*HETDEEFDAR from cytochrome c oxidase (COX) subunit Va in both DCA and calcium samples. This subunit of Complex IV of the electron transport chain (ETC) has been positively stained by Pro-Q Diamond (7). Here we observed a phosphoserine site at position 1 in addition to the previously published Ser4 site (40), but there were no iTRAQ tags derivatized to the α-amino group of the N-terminus for quantitative purposes. Moreover, it is conceivably difficult to quantify each phosphosite within the same peptide where multiple sites are phosphorylated upon biological perturbation especially if these multi-phosphosites change in opposite directions unless each singly phosphorylated species is detected. Another potential problem with iTRAQ-HCD approach is poor signal intensities for low mass iTRAQ reporter ions and peptide fragmentation pattern of large peptides as one would encounter from any multipole collision cell. This may be circumvented by the application of newly developed Electron Transfer Dissociation (ETD) on the LTQ-Orbitrap XL-ETD.
ETD has been widely used for post-translational modifications (PTMs) of peptides such as phosphorylation. ETD is advantageous for PTMs because of the high energy impact of electron dissociation preserving labile phosphate groups on the peptides without causing “neutral losses”. However, it was unclear whether ETD would be compatible with iTRAQ since the fragmentation mechanisms and pathways of ETD differ significantly from CID. Most recently, Phanstiel et. al. (41) demonstrated that ETD of iTRAQ labeled peptides produces c- and z-type fragment ions as well as reporter ions that were unique from those produced by CID. The downside of this approach is (1) ETD works poorly with lower precursor charge states (≤+2); (2) Since the 116 and 117 tags produce the same reporter ions after fragmentation by ETD, the user is limited to only three channels of relative quantification rather than four.
CONCLUSION
iTRAQ combined with HCD on a LTQ-Orbitrap XL-ETD mass spectrometer provides researchers with a useful quantitative tool to analyze proteomes of their interest with good dynamic range, accuracy and precision. It is potentially very useful for screening labile phosphorylation sites that may be functionally significant and have sufficiently detectable dynamic range. With ETD capability in analyzing PTMs such as phosphorylation and quantifying with iTRAQ, this hybrid instrument is very powerful in phosphoproteomic profiling, and more importantly in integrating mass spectrometric analysis with biological function.
Supplementary Material
Acknowledgments
Funding: This work was supported by the Intramural Funding of the Division of Intramural Research, NHLBI, NIH, DHHS.
ABBREVIATIONS
- PDH
pyruvate dehydrogenase
- PDK
pyruvate dehydrogenase kinase
- PDC
pyruvate dehydrogenase complex
- PDP
pyruvate dehydrogenase phosphatase
- BCKDH
branched chain alpha keto-acid dehydrogenase
- BDC
branched chain alpha keto-acid dehydrogenase complex
- BDK
branched chain alpha keto-acid dehydrogenase kinase
- BDP
branched chain alpha keto-acid dehydrogenase phosphatase
- PTM
post-translational modification
- DCA
dichloroacetate
- G/M
glutamate/malate
- MS
mass spectrometry
- HCD
high energy collision dissociation
- ETD
electron transfer dissociation
- PQD
Pulsed-Q Dissociation
- CID
Collisionally Induced Dissociation
- CE
collision energy
- DIGE
difference in gel electrophoresis
- ETC
electron transfer chain
- IEV
isoelectric variants
- IMAC
Immobilized Metal Affinity Chromatography
- SCX
Strong Cation Exchange Chromatography
- HILIC
Hydrophilic Interaction Chromatography
- RP
Reverse Phase
- DTT
dithiothreitol
- IAM
iodoacetamide
- ROS
Reactive Oxygen Species
- 2D-GE
two-dimensional gel electrophoresis
- IEF
isoelectric focusing
- IEVs
isoelectric variants
- ANT
adenosine nucleotide translocase
- BSA
bovine serum albumin
- COX
cytochrome c oxidase
- KDH
alpha keto-acid dehydrogenase
- IDH
isocitrate dehydrogenase
- MDH
malate dehydrogenase
- CK
creatine kinase
- AIF
apoptosis-inducing factor
- SD
standard deviation
- SEM
standard error of the mean
Footnotes
FIGURES: Symbols * & ** in MS/MS spectra (Figure 3A–3F; Supplemental Figure 1A–1D & 2A–2B) denote precursor and/or fragment ions with one or two phosphoric acid (−98 Da) losses unless otherwise stated. Superscripts (+2, +3) indicate the charge states of these ions. S*/T*/Y* in figure legends represent serine, threonine and tyrosine residues in their phosphorylated form.
REFERENCES
- 1.Hunter T. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 2.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 3.Cohen P. Nat. Cell Biol. 2002;4:E127–E130. doi: 10.1038/ncb0502-e127. [DOI] [PubMed] [Google Scholar]
- 4.Lee J, Xu Y, Chen Y, Sprung R, Kim SC, Xie S, Zhao Y. Mol Cell Proteomics. 2007;6(4):669–676. doi: 10.1074/mcp.M600218-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Villén J, Beausoleil SA, Gerber SA, Gygi SP. Proc Natl Acad Sci U S A. 2007;104(5):1488–1493. doi: 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins MO, Yu L, Campuzano I, Grant SG, Choudhary JS. Mol Cell Proteomics. 2008;7(7):1331–1348. doi: 10.1074/mcp.M700564-MCP200. [DOI] [PubMed] [Google Scholar]
- 7.Hopper RK, Carroll S, Aponte AM, Johnson DT, French S, Shen RF, Witzmann FA, Harris RA, Balaban RS. Biochemistry. 2006;45(8):2524–2536. doi: 10.1021/bi052475e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schulenberg B, Goodman TN, Aggeler R, Capaldi RA, Patton WF. Electrophoresis. 2004;25(15):2526–2532. doi: 10.1002/elps.200406007. [DOI] [PubMed] [Google Scholar]
- 9.Aponte AM, Phillips D, Hopper RK, Johnson DT, Harris RA, Blinova K, Boja ES, French SA, Balaban RS. J. Proteome Res. 2009 doi: 10.1021/pr800913j. Manuscript in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris RA, Popov KM, Zhao Y, Kedishvili NY, Shimomura Y, Crabb DW. Adv. Enzyme Regul. 1995;35:147–162. doi: 10.1016/0065-2571(94)00020-4. [DOI] [PubMed] [Google Scholar]
- 11.Yeaman SJ, Hutcheson ET, Roche TE, Pettit FH, Brown JR, Reed LJ, Watson DC, Dixon GH. Biochemistry. 1978;17(12):2364–2370. doi: 10.1021/bi00605a017. [DOI] [PubMed] [Google Scholar]
- 12.Sugden PH, Hutson NJ, Kerbey AL, Randle PJ. Biochem J. 1978;169(2):433–435. doi: 10.1042/bj1690433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sugden PH, Randle PJ. Biochem J. 1978;173(2):659–668. doi: 10.1042/bj1730659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel MS, Korotchkina LG. Biochem Soc Trans. 2006;34(Pt 2):217–222. doi: 10.1042/BST20060217. [DOI] [PubMed] [Google Scholar]
- 15.Roche TE, Reed LJ. Biochem Biophys Res Commun. 1974;59(4):1341–1348. doi: 10.1016/0006-291x(74)90461-6. [DOI] [PubMed] [Google Scholar]
- 16.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Mol Cell Proteomics. 2004;3(12):1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Bantscheff M, Boesche M, Eberhard D, Matthieson T, Sweetman G, Kuster B. Mol Cell Proteomics. 2008;7(9):1702–1713. doi: 10.1074/mcp.M800029-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.French SA, Territo PR, Balaban RS. Am. J. Physiol. 1998;275(3 Pt 1):C900–C909. doi: 10.1152/ajpcell.1998.275.3.C900. [DOI] [PubMed] [Google Scholar]
- 19.Whitehouse S, Cooper RH, Randle PJ. Biochem J. 1974;141(3):761–774. doi: 10.1042/bj1410761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balaban RS. J. Mol. Cell Cardiol. 2002;34:1259–1271. doi: 10.1006/jmcc.2002.2082. [DOI] [PubMed] [Google Scholar]
- 21.Tang H, Zhao ZJ, Landon EJ, Inagami T. J. Biol. Chem. 2000;275(12):8389–8396. doi: 10.1074/jbc.275.12.8389. [DOI] [PubMed] [Google Scholar]
- 22.Schieke SM, Phillips D, McCoy JP, Jr., Aponte AM, Shen RF, Balaban RS, Finkel T. J Biol Chem. 2006;281(37):27643–27652. doi: 10.1074/jbc.M603536200. [DOI] [PubMed] [Google Scholar]
- 23.Boja ES, Hoodbhoy T, Fales HM, Dean J. J Biol Chem. 2003;278(36):34189–34202. doi: 10.1074/jbc.M304026200. [DOI] [PubMed] [Google Scholar]
- 24.McNulty DE, Annan RS. Mol Cell Proteomics. 2008;7(5):971–980. doi: 10.1074/mcp.M700543-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Wu J, Shakey Q, Liu W, Schuller A, Follettie MT. J Proteome Res. 2007;6(12):4684–4689. doi: 10.1021/pr070481m. [DOI] [PubMed] [Google Scholar]
- 26.Popov KM, Shimomura Y, Harris RA. Protein Expr Purif. 1991;2(4):278–286. doi: 10.1016/1046-5928(91)90084-v. [DOI] [PubMed] [Google Scholar]
- 27.Villén J, Gygi SP. Nat. Protoc. 2008;3(10):1630–1638. doi: 10.1038/nprot.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel TB, Olson MS. Biochemistry. 1982;21:4259–4265. doi: 10.1021/bi00261a012. [DOI] [PubMed] [Google Scholar]
- 29.Lau KS, Cooper AJ, Chuang DT. Biochim Biophys Acta. 1990;1038(3):360–366. doi: 10.1016/0167-4838(90)90249-f. [DOI] [PubMed] [Google Scholar]
- 30.Stacpoole PW, Kurtz TL, Han Z, Langaee T. Adv Drug Deliv Rev. 2008;60(13–14):1478–1487. doi: 10.1016/j.addr.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nichols BJ, Denton RM. Mol Cell Biochem. 1995;149–150:203–212. doi: 10.1007/BF01076578. [DOI] [PubMed] [Google Scholar]
- 32.Aponte AM, Phillips D, Harris RA, Blinova K, French S, Johnson DT, Balaban RS. Methods in Enzymology. 2009;457(Chapter 4) doi: 10.1016/S0076-6879(09)05004-6. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paxton R, Kuntz M, Harris RA. Arch Biochem Biophys. 1986;244(1):187–201. doi: 10.1016/0003-9861(86)90108-6. [DOI] [PubMed] [Google Scholar]
- 34.Buxton DB, Barron LL, Taylor MK, Olson MS. Biochem. J. 1984;221:593–599. doi: 10.1042/bj2210593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olson MS. Annals of the New York Academy of Sciences. 1989;573:218–229. doi: 10.1111/j.1749-6632.1989.tb14999.x. [DOI] [PubMed] [Google Scholar]
- 36.Reinders J, Wagner K, Zahedi RP, Stojanovski D, Eyrich B, Van der Laan M, Rehling P, Sickmann A, Pfanner N, Meisinger C. Mol Cell Proteomics. 2007;6(11):1896–1906. doi: 10.1074/mcp.M700098-MCP200. [DOI] [PubMed] [Google Scholar]
- 37.Villén J, Beausoleil SA, Gerber SA, Gygi SP. Proc Natl Acad Sci U S A. 2007;104(5):1488–1493. doi: 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng J, Zhu M, Schaub MC, Gehrig P, Roschitzki B, Lucchinetti E, Zaugg M. Cardiovasc Res. 2008;80(1):20–29. doi: 10.1093/cvr/cvn161. [DOI] [PubMed] [Google Scholar]
- 39.Lewandrowski U, Sickmann A, Cesaro L, Brunati AM, Toninello A, Salvi M. FEBS Lett. 2008;582(7):1104–1110. doi: 10.1016/j.febslet.2008.02.077. [DOI] [PubMed] [Google Scholar]
- 40.Helling S, Vogt S, Rhiel A, Ramzan R, Wen L, Marcus K, Kadenbach B. Mol Cell Proteomics. 2008;7(9):1714–1724. doi: 10.1074/mcp.M800137-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phanstiel D, Zhang Y, Marto JA, Coon JJ. J Am Soc Mass Spectrom. 2008;19(9):1255–1262. doi: 10.1016/j.jasms.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.