

Abstract
Vitamin D is associated with decreased risks of various cancers, including colon cancer. The vitamin D receptor (VDR) is a transcription factor, which plays an important role in cellular differentiation and inhibition of proliferation. A link between VDR and the RAS-MAPK or phosphatidylinositol 3-kinase (PI3K)-AKT pathway has been suggested. However, the prognostic role of VDR expression or its relationship with PIK3CA or KRAS mutation remains uncertain. Among 619 colorectal cancers in two prospective cohort studies, 233 (38%) tumors showed VDR overexpression by immunohistochemistry. We analyzed for PIK3CA and KRAS mutations and LINE-1 methylation by Pyrosequencing, microsatellite instability (MSI), and DNA methylation (epigenetic changes) in 8 CpG island methylator phenotype (CIMP)-specific promoters [CACNA1G, CDKN2A (p16), CRABP1, IGF2, MLH1, NEUROG1, RUNX3 and SOCS1] by MethyLight (real-time PCR). VDR overexpression was significantly associated with KRAS mutation [odds ratio (OR), 1.55; 95% confidence interval (CI), 1.11-2.16] and PIK3CA mutation (OR, 2.17; 95% CI, 1.36-3.47), both of which persisted in multivariate logistic regression analysis. VDR was not independently associated with body mass index, family history of colorectal cancer, tumor location (colon vs. rectum), stage, tumor grade, signet ring cells, CIMP, MSI, LINE-1 hypomethylation, BRAF, p53, p21, β-catenin or cyclooxygenase-2. VDR expression was not significantly related with patient survival, prognosis or clinical outcome. In conclusion, VDR overexpression in colorectal cancer is independently associated with PIK3CA and KRAS mutations. Our data support potential interactions between the VDR, RAS-MAPK and PI3K-AKT pathways, and possible influence by KRAS or PIK3CA mutation on therapy or chemoprevention targeting VDR.
Keywords: colon cancer, VDR, methylation, PI3K, RAS
Introduction
The vitamin D receptor (VDR) is a member of the steroid hormone receptor superfamily, and regulates gene expression in a ligand-dependent manner (1). Plasma vitamin D level and estimated whole-body vitamin D level have been associated with decreased incidence and mortality in various cancers (1), including colorectal cancer (2, 3). Vitamin D and VDR appear to play important roles in preventing tumor development and progression through induction of cellular differentiation and inhibition of proliferation (1, 4-9). VDR is overexpressed or repressed in various human cancers (1, 10-15). In colorectal cancer, VDR is expressed in early-stage neoplasias, but repressed in high-grade and metastatic cancers (12).
An activation of epidermal growth factor receptor (EGFR) triggers a chain of downstream signaling events that include the phosphatidylinositol 3-kinase (PI3K)/AKT and RAS/MAPK pathways. Accumulating evidence suggests that VDR binds to PI3K and inhibits cell proliferation by inducing differentiation (1, 16), and that the PI3K/AKT pathway is activated by 1,25(OH)2D, the active form of vitamin D (17). In addition, VDR expression has been associated with RAS/MAPK pathway activation in leukemia cells (18). These observations suggest that VDR-expressing cells may need activation of the PI3K/AKT or RAS/MAPK pathway in order to acquire malignant characteristics. Activating mutations in PIK3CA and KRAS play important roles in colorectal carcinogenesis. Thus, we hypothesized that VDR expression is associated with PIK3CA and KRAS mutations in colorectal cancer. If this hypothesis is true, therapy or chemoprevention targeting VDR and downstream pathways may be influenced by PIK3CA or KRAS mutations. In addition, VDR-mediated action of 1,25(OH)2D can limit colon cancer cell growth particularly when induced by activation of EGFR (19). Thus, response to EGFR-targeted therapy may be modified by VDR status in tumor cells.
We therefore utilized 619 colorectal cancers identified in two prospective cohort studies, and examined the relation of VDR expression with patient survival, PIK3CA and KRAS mutations, and other related molecular features including BRAF mutation, microsatellite instability (MSI) and the CpG island methylator phenotype (CIMP), which were potential confounders.
Materials and Methods
Study group
We utilized the databases of two independent prospective cohort studies; the Nurses' Health Study (N = 121,700 women followed since 1976) (20), and the Health Professional Follow-up Study (N = 51,500 men followed since 1986) (20). Data on height and weight were obtained by biennial questionnaire. A subset of the cohort participants developed colorectal cancers during prospective follow-up. Data on tumor location and TNM stage were obtained through medical record review by study physicians. We collected paraffin-embedded tissue blocks from hospitals where cohort participants with colorectal cancers had undergone resections of primary tumors. Up to 2002, there were 1834 incident colorectal cancer cases with adequate clinical information. We successfully retrieved formalin-fixed paraffin-embedded tumor tissue in 998 cases (54%). We have previously shown that there are no differences in demographic, nutritional or exposure features between patients with and without available tumor tissue (20). Among 998 cases with available tumor tissue, we were able to construct tissue microarrays (TMA) using 625 cases. Among them, we obtained valid VDR expression data in 619 cases, which were eligible for the current study (Table 1). In any of our previous studies, we have not examined VDR expression in colorectal cancers. This current analysis represents a new analysis of VDR on the existing colorectal cancer database that have been previously characterized for MSI, KRAS, PIK3CA and BRAF (21, 22), which is analogous to novel analysis using the well-characterized cell lines or animal models. Patients were observed until death or June 30, 2006, whichever came first. Ascertainment of deaths included reporting by the family or postal authorities. In addition, the names of persistent nonresponders were searched in the National Death Index. More than 98% of deaths in the cohorts were identified by these methods. The cause of death was assigned by physicians blinded to information on lifestyle exposures and molecular features in colorectal cancer. Written informed consent was obtained from all study subjects. Tissue collection and analyses were approved by the Harvard School of Public Health and Brigham and Women's Hospital Institutional Review Boards.
Table 1.
Frequency of vitamin D receptor (VDR) expression in colorectal cancer
| Clinical, pathologic or molecular feature | Total N | VDR+ | Univariate OR (95% CI) | P value |
|---|---|---|---|---|
| All cases | 619 | 233 (38%) | ||
| Gender | ||||
| Men | 244 | 83 (34%) | 1 | Reference |
| Women | 375 | 150 (40%) | 1.29 (0.92-1.81) | 0.13 |
| Age at diagnosis (years) | ||||
| ≤59 | 141 | 60 (43%) | 1 | Reference |
| 60-69 | 268 | 104 (39%) | 0.86 (0.57-1.30) | 0.46 |
| ≥70 | 210 | 69 (33%) | 0.66 (0.43-1.03) | 0.065 |
| Body mass index (BMI, kg/m2) | ||||
| <30 | 483 | 178 (37%) | 1 | Reference |
| ≥30 | 100 | 38 (38%) | 1.05 (0.67-1.64) | 0.83 |
| Family history of colorectal cancer | ||||
| (-) | 472 | 180 (38%) | 1 | Reference |
| (+) | 147 | 53 (36%) | 0.91 (0.62-1.34) | 0.65 |
| Tumor location | ||||
| Distal (splenic flexure to rectum) | 325 | 131 (56%) | 1 | Reference |
| Proximal (cecum to transverse) | 287 | 98 (34%) | 0.77 (0.55-1.07) | 0.12 |
| Stage | ||||
| I | 135 | 47 (35%) | 1 | Reference |
| II | 189 | 69 (37%) | 1.08 (0.68-1.71) | 0.75 |
| III | 174 | 79 (45%) | 1.56 (0.98-2.47) | 0.060 |
| IV | 84 | 27 (32%) | 0.89 (0.50-1.58) | 0.68 |
| Tumor grade | ||||
| Low | 546 | 214 (39%) | 1 | Reference |
| High | 53 | 11 (21%) | 0.41 (0.20-0.81) | 0.008 |
| Mucinous component | ||||
| 0% | 317 | 124 (39%) | 1 | Reference |
| ≥1% | 221 | 74 (33%) | 0.78 (0.55-1.12) | 0.18 |
| Signet ring cell component | ||||
| 0% | 461 | 179 (39%) | 1 | Reference |
| ≥1% | 41 | 10 (24%) | 0.51 (0.24-1.06) | 0.068 |
| CIMP status (No. of methylated CIMP markers) | ||||
| CIMP-0 (0) | 266 | 100 (38%) | 1 | Reference |
| CIMP-low (1-5) | 246 | 100 (41%) | 1.14 (0.80-1.62) | 0.48 |
| CIMP-high (6-8) | 91 | 27 (30%) | 0.70 (0.42-1.17) | 0.17 |
| MSI status | ||||
| MSS | 461 | 168 (36%) | 1 | Reference |
| MSI-low | 59 | 30 (51%) | 1.80 (1.05-3.11) | 0.032 |
| MSI-high | 96 | 32 (33%) | 0.87 (0.55-1.39) | 0.56 |
| MSI/CIMP status | ||||
| CIMP-low/0 MSI-low/MSS | 478 | 186 (39%) | 1 | Reference |
| CIMP-high MSI-low/MSS | 30 | 8 (27%) | 0.57 (0.25-1.31) | 0.18 |
| CIMP-low/0 MSI-high | 32 | 12 (38%) | 0.94 (0.45-1.97) | 0.87 |
| CIMP-high MSI-high | 61 | 19 (31%) | 0.71 (0.40-1.26) | 0.24 |
| BRAF mutation | ||||
| (-) | 515 | 194 (38%) | 1 | Reference |
| (+) | 86 | 28 (33%) | 0.80 (0.49-1.30) | 0.36 |
| KRAS mutation | ||||
| (-) | 388 | 131 (34%) | 1 | Reference |
| (+) | 229 | 101 (44%) | 1.55 (1.11-2.16) | 0.010 |
| PIK3CA mutation | ||||
| (-) | 470 | 158 (34%) | 1 | Reference |
| (+) | 84 | 44 (52%) | 2.17 (1.36-3.47) | 0.001 |
| KRAS/PIK3CA mutation | ||||
| KRAS(-)/PIK3CA(-) | 312 | 99 (32%) | 1 | Reference |
| KRAS(-)/PIK3CA(+) | 34 | 15 (44%) | 1.70 (0.83-3.48) | 0.14 |
| KRAS(+)/PIK3CA(-) | 157 | 59 (38%) | 1.30 (0.87-1.94) | 0.21 |
| KRAS(+)/PIK3CA(+) | 50 | 29 (58%) | 2.97 (1.61-5.47) | 0.0003 |
| LINE-1 methylation | ||||
| ≥70% | 95 | 33 (35%) | 1 | Reference |
| 50-69% | 424 | 160 (38%) | 1.14 (0.71-1.81) | 0.58 |
| ≤49% | 77 | 30 (39%) | 1.20 (0.64-2.24) | 0.57 |
| p53 expression | ||||
| (-) | 355 | 133 (37%) | 1 | Reference |
| (+) | 259 | 97 (37%) | 1.00 (0.72-1.39) | 0.99 |
| p21 | ||||
| Expressed | 115 | 36 (31%) | 1 | Reference |
| Lost | 490 | 191 (39%) | 1.40 (0.91-2.16) | 0.13 |
| Nuclear β-catenin expression | ||||
| (-) | 345 | 136 (39%) | 1 | Reference |
| (+) | 207 | 78 (38%) | 0.93 (0.65-1.32) | 0.68 |
| COX-2 expression | ||||
| (-) | 104 | 31 (30%) | 1 | Reference |
| (+) | 511 | 199 (39%) | 1.50 (0.95-2.37) | 0.079 |
CI, confidence interval; OR, odds ratio; CIMP, CpG island methylator phenotype; COX-2, cyclooxygenase-2; MSI, microsatellite instability; MSS, microsatellite stable
Histopathologic evaluations
Hematoxylin and eosin (H&E) stained tissue sections were examined by a pathologist (S.O.) unaware of other data. The tumor grade was categorized as low (≥50% gland formation) vs. high (<50% gland formation). The presence and extent of extracellular mucin and signet ring cells were categorized as 0% (no mucin or signet ring cells), or ≥1% of the tumor volume.
Sequencing of KRAS, BRAF and PIK3CA, and analyses for MSI
DNA was extracted from paraffin-embedded tumor tissue sections, and PCR and Pyrosequencing targeted for KRAS (codons 12 and 13) (23), BRAF (codon 600) (24) and PIK3CA (exons 9 and 20) were performed as previously described (25). Microsatellite instability (MSI) analysis was performed, using 10 microsatellite markers (D2S123, D5S346, D17S250, BAT25, BAT26, BAT40, D18S55, D18S56, D18S67 and D18S487) (26). MSI-high was defined as the presence of instability in ≥30% of the markers. MSI-low was defined as instability in 10-29% of the markers, and “microsatellite stable (MSS)” tumors were defined as tumors with no unstable marker.
Real-time PCR for CpG island methylation and Pyrosequencing to measure LINE-1 methylation
Sodium bisulfite treatment on genomic DNA and subsequent real-time PCR (MethyLight) were validated and performed as previously described (27). We quantified DNA methylation in 8 CIMP-specific promoters [CACNA1G, CDKN2A (p16), CRABP1, IGF2, MLH1, NEUROG1, RUNX3 and SOCS1] (28-30). CIMP-high was defined as the presence of ≥6 of 8 methylated promoters, CIMP-low as the presence of 1/8-5/8 methylated promoters, and CIMP-0 as the absence (0/8) of methylated promoters, according to the previously established criteria (30). In order to accurately quantify relatively high methylation levels in LINE-1 repetitive elements, we utilized Pyrosequencing (31).
Immunohistochemistry for p53, p21, β-catenin, COX-2 and VDR
Tissue microarrays (TMAs) were constructed as previously described (20). Methods of immunohistochemical procedures and interpretations were previously described for p53, p21 (32, 33), β-catenin (34) and COX-2 (20, 26). Appropriate positive and negative controls were included in each run of immunohistochemistry for all markers. For VDR immunohistochemistry, antigen retrieval was performed, and deparaffinized tissue sections in Antigen Retrieval Citra Solution (Biogenex Laboratories, San Ramon, CA) were treated with microwave for 15 min. Tissue sections were incubated with 3% H2O2 (10 min) to block endogenous peroxidase (Dako Cytomation, Carpinteria, CA), with 10% normal goat serum (Vector Laboratories, Burlingame, CA) in phosphate-buffered saline (10 min). Primary antibody against VDR [polyclonal rabbit anti-VDR (C-20), 1:200 dilution; Santa Cruz Biotechnology, San Diego, CA] was applied, and the slides were maintained for 1 hours at room temperature. Next, we applied Envision System HRP labeled polymer anti rabbit (Dako) for 30 min, followed by visualizing signal with diaminobenzidine (5 min) and methyl-green counterstain. Appropriate positive and negative controls were included in each run of VDR immunohistochemistry. VDR overexpression (Figure 1) was evaluated by one of the investigators (K.No.) unaware of other data. Cytoplasmic VDR expression was recorded as no, weak, or moderate/strong expression, and the number of cells with expression was recorded. Among the 619 tumors, 82 showed strong cytoplasmic expression, 139 showed moderate cytoplasmic expression, 12 showed ≥50% of tumor cells with weak cytoplasmic expression, 53 showed 30-49% of tumor cells with weak cytoplasmic expression, 112 showed 1-29% of tumor cells with weak cytoplasmic expression and 221 tumors showed no staining in tumor cells. VDR positivity (i.e., overexpression) was defined as moderate/strong cytoplasmic expression in any fraction of tumor cells or at least 50% of tumor cells with weak cytoplasmic expression. Either the absence of staining or the presence of weak cytoplasmic expression in <50% of tumor cells was interpreted as negative. This cut point was based on the frequency of PIK3CA mutation in colorectal cancer groups categorized by VDR status. The frequency of PIK3CA mutation was; 21% (40/191) in tumors with moderate or strong cytoplasmic expression; 36% (4/11) in tumors with weak cytoplasmic expression in ≥50% of tumor cells, 11% (5/47) in tumors with weak cytoplasmic expression in 30-49% of tumor cells, 12% (12/103) in tumors with weak cytoplasmic expression in 1-29% of tumor cells and 11% (23/202) in tumors with no staining.
Figure 1.
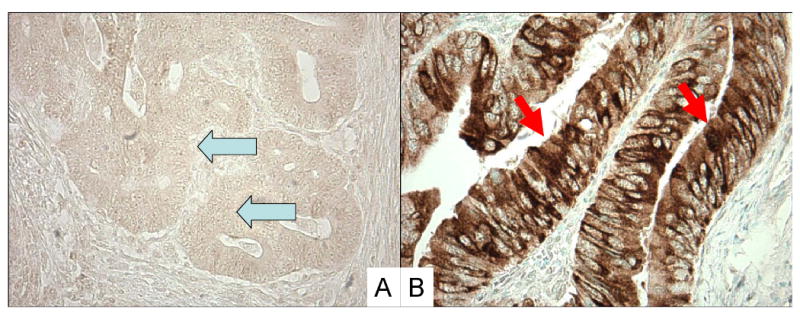
Vitamin D receptor (VDR) expression in colon cancer. A. No overexpression of VDR in cancer cells (arrows). B. Overexpression of VDR in cytoplasm of cancer cells (arrows).
A random selection of 139 cases was examined for VDR by a second observer (Y.B.) unaware of other data, and concordance between the two observers was 0.81 (κ=0.62, p<0.0001), indicating substantial agreement. Each of the other immunohistochemical markers was interpreted by a pathologist unaware of other data (p53, p21 and COX-2 by S.O.; β-catenin by K.No.). For each of the other immunohistochemical markers, a second observer (S.O. for β-catenin; K.S. for p21; K.No. for p53; R. Dehari, Kanagawa Cancer Center, for COX-2) examined a random sample of 108-402 tumors, unaware of other data. The κ coefficient between the two observers was 0.65 for β-catenin (p<0.0001; N=402), 0.62 for p21 (p<0.0001; N=179), 0.75 for p53 (p<0.0001; N=118), and 0.62 for COX-2 (p<0.0001; N=108), indicating substantial agreement.
Statistical analysis
For all statistical analyses, we used SAS program (Version 9.1, SAS Institute, Cary, NC). All p values were two-sided, and statistical significance was set at p ≤ 0.05; however, whenever appropriate, p values were conservatively interpreted considering multiple hypotheses testing. For categorical data, the chi-square test (or Fisher's exact test when any expected cell count was less than 5) was performed. To assess independent relations of VDR with a number of variables, a multivariate logistic regression analysis was performed. Odds ratio (OR) was adjusted for age at diagnosis (<65 vs. ≥65-year-old), sex, tumor location (proximal vs. distal), body mass index (BMI, ≥30 vs. <30 kg/m2), tumor stage (I-II vs. III-IV), grade (low vs. high), family history of colorectal cancer in any first degree relative (present vs. absent), mucinous component (present vs. absent), CIMP status (high vs. low/0), MSI status (high vs. low/MSS), LINE-1 methylation (continuous), β-catenin, COX-2, p53, p21, BRAF, KRAS and PIK3CA. For cases with missing data on KRAS (0.3% missing) and PIK3CA (11% missing), we assigned separate (“missing”) indicator variables, and included those cases in the multivariate model. For cases with missing information in other variables [BMI (5.8% missing), tumor location (1.1%), tumor stage (11%), tumor grade (3.2%), mucinous component (13%), CIMP (2.6%), MSI (0.5%), LINE-1 (3.7%), BRAF (2.9%), β-catenin (11%), p53 (0.8%), p21 (2.3%) and COX-2 (0.6%)], we included those cases in a majority category, in order to minimize the number of indicator variables. We confirmed that excluding cases with missing information in any of the covariates did not substantially alter results (data not shown).
For survival analysis, Kaplan-Meier method was used and log-rank test was used to test significance of a deviation from the null hypothesis. For analyses of colorectal cancer-specific mortality, death as a result of colorectal cancer was the primary end point and deaths as a result of other causes were censored. To assess independent effect of VDR status on mortality, we constructed a multivariate, stage-matched (stratified) Cox proportional hazards model to compute a hazard ratio (HR) according to VDR status, adjusted for age at diagnosis, sex, BMI, year of diagnosis, tumor grade, tumor location, mucinous component, family history, CIMP, MSI, KRAS, BRAF, PIK3CA, p53, β-catenin, p21, COX-2 and LINE-1 methylation. Tumor stage (I, IIA, IIB, IIIA, IIIB, IIIC, IV, unknown) was used as a matching variable using the “strata” option in the SAS “proc phreg” command to minimize residual confounding and overfitting. We also used stage-matched Cox model including only stage as a stratifying variable without any other covariate in the model. The proportionality of hazards assumption was satisfied by evaluating time-dependent variables, which were the cross-product of the VDR variable and survival time (p=0.62 for colorectal cancer-specific mortality; p=0.85 for overall mortality). For cases with missing data in any of the covariates were dealt as in the multivariate logistic regression analysis described above, except for stage, KRAS and PIK3CA. For cases missing data on KRAS (or PIK3CA), we included those cases in the wild-type category. An interaction was assessed by including the cross product of the VDR variable and another variable of interest (excluding data-missing cases) in a multivariate Cox model, and the Wald test was performed.
Results
Vitamin D receptor (VDR) expression in colorectal cancer
We examined VDR overexpression by immunohistochemistry in 619 colorectal cancers identified in two independent prospective cohort studies, and detected VDR overexpression in 233 (38%) tumors. Table 1 shows the frequencies of VDR expression according to various clinical and pathologic features. VDR overexpression was significantly less common among high grade tumors [odds ratio (OR), 0.41; 95% confidence interval (CI), 0.20-0.81; p=0.008].
Relationship of VDR expression with KRAS and PIK3CA mutations
Table 1 shows the frequencies of VDR overexpression according to various molecular features in colorectal cancer. VDR overexpression was significantly more common among KRAS-mutated tumors (OR, 1.55; 95% CI, 1.11-2.16; p=0.010) and PIK3CA-mutated tumors (OR, 2.17; 95% CI, 1.36-3.47; p=0.001). In order to examine combined effect of KRAS and PIK3CA mutations on VDR overexpression, we classified tumors into 4 subtypes according to KRAS and PIK3CA status (Table 1). VDR overexpression was significantly more common in KRAS-mutated/PIK3CA-mutated tumors (58%=29/50; OR, 2.97; 95% CI, 1.61-5.47; p=0.0003) than in KRAS-wild-type/PIK3CA-wild-type tumors (32%=99/312). We also examined the frequency of PIK3CA or KRAS mutation according to VDR staining intensity in tumor cells (Table 2). The frequencies of PIK3CA and KRAS mutations increased as intensity of VDR staining increased, and reached plateau at moderate intensity of staining.
Table 2.
Intensity of vitamin D receptor (VDR) expression in colorectal cancer and mutations in PIK3CA and KRAS
| Total N | VDR expression | ||||
|---|---|---|---|---|---|
| No | Weak | Moderate | Strong | ||
| PIK3CA mutation | |||||
| (-) | 470 (85%) | 179 (89%) | 140 (87%) | 98 (80%) | 53 (78%) |
| (+) | 84 (15%) | 23 (11%) | 21 (13%) | 25 (20%) | 15 (22%) |
| KRAS mutation | |||||
| (-) | 388 (63%) | 154 (70%) | 110 (62%) | 79 (57%) | 45 (56%) |
| (+) | 229 (37%) | 66 (30%) | 67 (38%) | 60 (43%) | 36 (44%) |
A percentage number indicates the frequency of PIK3CA or KRAS mutation among colorectal cancers in a specific category of VDR expression intensity.
Relationship of VDR expression with other molecular variables
We determined CpG island methylator phenotype (CIMP) status using MethyLight assays on a panel of 8 CIMP-specific promoters (CACNA1G, CDKN2A, CRABP1, IGF2, MLH1, NEUROG1, RUNX3 and SOCS1) (29, 30). VDR overexpression was not significantly associated with CIMP (Table 1). On the other hand, VDR overexpression was more common in microsatellite instability (MSI)-low tumors (51%=30/59; OR, 1.80; 95% CI, 1.05-3.11; p=0.032) than in microsatellite stable (MSS) tumors (36%=168/461); nonetheless, this could be a chance association given multiple hypothesis testing on multiple tumor markers (excluding PIK3CA and KRAS).
VDR expression is independently associated with PIK3CA and KRAS mutations
We performed multivariate logistic regression analysis, to confirm independent relations between VDR and mutations of KRAS and PIK3CA after adjusting for clinical, pathologic and other molecular variables (Table 3). VDR overexpression was significantly associated with PIK3CA mutation (adjusted OR, 2.36; 95% CI, 1.43-3.91; p=0.0008) and KRAS mutation (adjusted OR, 1.53; 95% CI, 1.04-2.23; p=0.029).
Table 3.
Multivariate analysis to examine the relations of PIK3CA and KRAS mutations with vitamin D receptor (VDR) expression in colorectal cancer
| Variable independently associated with VDR | Multivariate OR (95% CI) | P value |
|---|---|---|
| PIK3CA mutation | 2.36 (1.43-3.91) | 0.0008 |
| KRAS mutation | 1.53 (1.04-2.23) | 0.029 |
| Other variable | ||
| Mucinous component | 0.64 (0.43-0.96) | 0.029 |
Multivariate logistic regression analysis assessing the relations of PIK3CA and KRAS mutations with VDR included age at diagnosis, sex, BMI, tumor location, tumor stage, grade, mucinous component, family history of colorectal cancer, microsatellite instability, CpG island methylator phenotype, LINE-1, β-catenin, COX-2, p53, p21 and BRAF. Only variables with p<0.05 are listed.
CI, confidence interval; OR, odds ratio.
VDR expression and patient survival
Utilizing our cohort database, we previously demonstrated that molecular features in colon cancer such as BRAF mutation, PIK3CA mutation and LINE-1 hypomethylation were significantly associated with inferior prognosis (21, 22, 35). We assessed the influence of VDR overexpression on survival of patients with stage I-IV colorectal cancers. During follow-up of 599 patients who were eligible for survival analysis, there were 260 deaths, including 158 colorectal cancer-specific deaths. In Kaplan-Meier analysis, VDR status was not significantly associated with colorectal cancer-specific survival (log rank p=0.57) or overall survival (log rank p=0.31) (Figure 2).
Figure 2.

Kaplan-Meier curves for colorectal cancer-specific survival (left panel) and overall survival (right panel) according to tumoral vitamin D receptor (VDR) status.
We performed Cox regression analysis to assess mortality according to VDR status (Table 4). VDR status was not significantly related with patient survival in univariate analysis, stage-matched analysis, or multivariate analysis in colorectal cancer. We also examined whether any of the clinical, pathologic and molecular variables significantly modified the effect of VDR overexpression on patient survival. There was no evidence for a significant interaction between VDR overexpression and any of the variables examined, including KRAS and PIK3CA (all Pinteraction >0.05).
Table 4.
Vitamin D receptor (VDR) expression and patient mortality in colorectal cancer
| Total N | Colorectal cancer-specific mortality | Overall mortality | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Deaths / person- years |
Univariate HR (95% CI) |
Stage- matched HR* (95% CI) |
Multivariate HR^ (95% CI) |
Deaths / person- years |
Univariate HR (95% CI) |
Stage- matched HR* (95% CI) |
Multivariate HR^ (95% CI) |
||
| VDR (-) | 374 (62%) | 101/2284 | 1 (reference) | 1 (reference) | 1 (reference) | 164/2284 | 1 (reference) | 1 (reference) | 1 (reference) |
| VDR (+) | 225 (38%) | 57/1540 | 0.91 (0.66-1.26) | 0.88 (0.63-1.23) | 0.88 (0.61-1.26) | 96/1540 | 0.88 (0.68-1.13) | 0.85 (0.65-1.10) | 0.87 (0.66-1.14) |
The stage-matched (stratified) Cox model included stage categories (I, IIA, IIB, IIIA, IIIB, IIIC, IV, unknown) as a stratifying variable without any other covariate in the model.
The multivariate, stage-matched (stratified) Cox model included the VDR variable stratified by age at diagnosis, year of diagnosis, sex, body mass index, family history of colorectal cancer, tumor location, tumor grade, mucinous component, KRAS, PIK3CA, BRAF, β-catenin, COX-2, p53, p21, LINE-1 methylation, microsatellite instability, and the CpG island methylator phenotype.
CI, confidence interval; HR, hazard ratio.
Discussion
We conducted this study to examine the relationship between vitamin D receptor (VDR) expression and mutations in PIK3CA and KRAS in colorectal cancer. Accumulating evidence indicate a substantial role of vitamin D in prevention of various forms of human cancer (1, 2, 17, 36-38). A potential link between VDR and the PI3K/AKT or RAS/MAPK pathway has been suggested (17, 18). Thus, examining VDR in colorectal cancer may shed lights on biological mechanisms of vitamin D action and its failure. We have found that VDR overexpression in colorectal cancer is significantly associated with both PIK3CA and KRAS mutations, independent of other clinical and molecular features. Our data support the hypothesis that the VDR pathway interact with the PI3K/AKT and RAS/MAPK pathways in colonic neoplastic cells.
Our resource of a large number of colorectal cancers derived from the two prospective cohort studies has enabled us to precisely estimate the frequency of colorectal cancers with a specific molecular feature (such as VDR expression, KRAS mutation, PIK3CA mutation, etc.). The large number of cases has also provided a sufficient power in our multivariate logistic regression analysis and survival analysis.
Studying risk modifying factors or molecular changes is important in cancer research (39-44). Previous studies have consistently reported the preventive effect of vitamin D on colorectal cancers (1, 2, 36-38), and that higher plasma levels of vitamin D confer a greater reduction in the risk of colorectal cancer (2). Studies have reported that 1,25(OH)2D potentiates the effects of many cytotoxic and anti-proliferative drugs (1, 17), and that higher plasma levels of 25(OH)D is associated with a significant reduction in colon cancer mortality (2). Thus, accumulating evidence indicates important roles of vitamin D in preventing the development and progression of colorectal cancer. Interestingly, VDR and 1-α-hydroxylase (encoded by CYP27B1), which converts 25(OH)D into 1,25(OH)2D, are frequently overexpressed in colon cancer cells (1, 4-6, 8, 9). The anti-proliferative action of 1,25(OH)2D appears to depend on VDR expression level and differentiation status of tumor cells (45, 46). Therefore, it is possible that the effect of serum vitamin D level, which is protective against cancer incidence and mortality, may differ according to VDR expression status in colorectal cancer. Additional studies are necessary to address this intriguing hypothesis.
A potential link between VDR and the PI3K/AKT or RAS/MAPK pathway has been suggested (16-18, 47-51). In these pathways, PIK3CA or KRAS mutation plays an important role in the progression of colorectal cancer. Mutant PIK3CA stimulates the downstream AKT pathway, and promotes cell growth in various cancers, including colorectal cancer. The PI3K/AKT pathway has been known to mediate signals from growth factors, which is influenced by the state of energy balance. In addition, PI3K/AKT signaling is influenced by KRAS (52). In fact, PIK3CA mutation is positively associated with KRAS mutation in colorectal cancer (25, 53). Our data support a potential link between PIK3CA and VDR and suggest that VDR expression may affect the regulation of PI3K/AKT pathway in colorectal cancer. In myeloid leukemia cells, VDR activated by 1,25(OH)2D can inhibit tumor cell proliferation by inducing differentiation, which depends on the formation of activated VDR and PI3K complexes (1, 16). A combination of 1,25(OH)2D with AKT pathway inhibitors is strongly anti-proliferative and should be considered for differentiation therapy of myeloid leukemia (17). These previous observations (16, 17) and our data suggest that VDR-expressing cells may need the activation of PI3K/AKT pathway in order to acquire malignant characteristics, and that therapy or chemoprevention targeting VDR and downstream pathways may be influenced by PIK3CA mutation in colon cancer cells.
The vitamin D pathway may also interact with RAS signaling. A recent case-control study has reported that the VDR poly A, rs10735810 (so-called “FokI SNP”) and rs11568820 (so-called “CDX2 SNP”) polymorphisms are associated with KRAS mutation in colon cancer (54). VDR protein expression has been shown to be down-regulated in KRAS-mutated colon cancer cells (47). In contrast, VDR expression has been associated with the activation of the RAS/MAPK pathway in leukemia cells (18), which is in agreement with our current data. In addition, RAS-transformed human keratinocytes are shown to be resistant to the growth-inhibitory effects of 1,25(OH)2D (48-50). Further analysis is needed to clarify how vitamin D and its downstream pathway influence colorectal cancer development in relation to KRAS mutation.
We did not observe a significant relation between VDR expression and microsatellite instability (MSI)-high or the CpG island methylator phenotype (CIMP) in colorectal cancer. A molecular classification of colorectal cancer based on MSI and CIMP is increasingly important (55), because MSI and CIMP status represent genomic and epigenomic alterations, respectively, in tumor cells and largely determine clinical, pathologic and molecular characteristics (55). Nonetheless, our data do not support a link between VDR and CIMP or MSI in colorectal cancer. A recent study has reported that the VDR rs10735810 (so-called “FokI SNP”) polymorphism is significantly associated with CIMP-high and inversely with MSI-high (54); however, either of these could be a chance association given multiple hypothesis testing.
In the current study, we have shown that VDR expression is inversely associated with high grade tumors in univariate analysis, but not in multivariate analysis. Thus, our data are not incompatible with the inverse association between VDR expression and high tumor grade in the previous study (12), but do not support a mechanistic link between VDR loss and high tumor grade. With regard to disease stage, the previous study (12) has shown that VDR expression in colon cancer cells is commonly lost in a metastatic focus, but not in a primary lesion. Therefore, the absence of the relation between loss of VDR expression in primary tumor and advanced disease stage in the current study is not incompatible with the previous data (12).
As one limitation in our cohort studies, data on cancer treatment were limited. Nonetheless, it is unlikely that chemotherapy use differed according to tumoral VDR status, since such data were not available to patients or treating physicians. In addition, beyond cause of mortality, data on cancer recurrences were not available in these cohorts. Nonetheless, given the median survival for metastatic colorectal cancer was approximately 10 to 12 months during much of the time period of this study, colorectal cancer-specific survival should be a reasonable surrogate for colorectal cancer-specific outcomes.
As another limitation of this study, there are currently no standardized methods to assess VDR overexpression in colorectal cancer. Thus, our current study is exploratory by nature, and our data and method to determine a cutpoint for VDR overexpression need to be validated and confirmed by independent datasets.
In summary, this large cohort of colorectal cancers has shown that VDR expression is significantly associated with PIK3CA and KRAS mutations, independent of clinical, pathological and molecular features. On the other hand, VDR expression is not significantly related with patient survival. Our data support the hypothesis that PIK3CA and/or KRAS mutations may influence biological effect of VDR and its downstream pathway. Thus, targeting VDR for chemoprevention or cancer therapy likely needs to consider the effect of KRAS or PIK3CA mutation. Likewise, therapy targeting EGFR or the downstream RAS or PI3K pathway may be influenced by VDR status. Considering that VDR regulates the transcription of various genes involved in cellular differentiation and inhibition of proliferation, our findings may have considerable clinical implications. Further studies are necessary to elucidate exact roles of vitamin D and VDR in prevention of colorectal neoplasias, as well as to examine a potential mechanistic link between VDR and the RAS/MAPK and PI3K/AKT pathways.
Acknowledgments
This work was supported by The U.S. National Institute of Health (NIH) grants P01 CA87969 (to S. Hankinson), P01 CA55075 (to W. Willett), P50 CA127003 (to C.S.F.) and K07 CA122826 (to S.O.), and in part by grants from the Bennett Family Fund and the Entertainment Industry Foundation National Colorectal Cancer Research Alliance. K.No. was supported by a fellowship grant from the Japan Society for Promotion of Science. K.Ng. was supported by the American Society of Clinical Oncology Cancer Foundation Young Investigator Award and the Charles A. King Trust Fellowship Award, Bank of America, Co-Trustee. The funding sponsors had no role or involvement in the study design, the collection, analysis and interpretation of data, or writing and submission of the manuscript. We deeply thank the Nurses' Health Study and Health Professionals Follow-up Study cohort participants who have generously agreed to provide us with biological specimens and information through responses to questionnaires. We thank Frank Speizer, Walter Willett, Susan Hankinson, Meir Stampfer, and many other staff members who implemented and have maintained the cohort studies.
Funding: The U.S. National Institute of Health (P01 CA87969 to S. Hankinson, P01 CA55075 to W. Willett, P50 CA127003 to C.S.F., K07 CA122826 to S.O.); the Bennett Family Fund; and the Entertainment Industry Foundation National Colorectal Cancer Research Alliance. K.No. was supported by a fellowship grant from the Japan Society for Promotion of Science. K.Ng. was supported by the American Society of Clinical Oncology Cancer Foundation Young Investigator Award and the Charles A. King Trust Fellowship Award, Bank of America, Co-Trustee. The content is solely the responsibility of the authors and does not necessarily represent the official views of NCI, NIH or any other funders. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations and the HUGO Gene Nomenclature Committee-approved official gene symbols
- BMI
body mass index
- CI
confidence interval
- CIMP
CpG island methylator phenotype
- COX-2
cyclooxygenase-2 (PTGS2)
- EGFR
epidermal growth factor receptor
- HR
hazard ratio
- MSI
microsatellite instability
- MSS
microsatellite stable
- OR
odds ratio
- VDR
vitamin D receptor
Footnotes
No conflicts of interest exist.
References
- 1.Deeb KK, Trump DL, Johnson CS. Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer. 2007;7:684–700. doi: 10.1038/nrc2196. [DOI] [PubMed] [Google Scholar]
- 2.Ng K, Meyerhardt JA, Wu K, et al. Circulating 25-hydroxyvitamin d levels and survival in patients with colorectal cancer. J Clin Oncol. 2008;26:2984–91. doi: 10.1200/JCO.2007.15.1027. [DOI] [PubMed] [Google Scholar]
- 3.Wu K, Feskanich D, Fuchs CS, Willett WC, Hollis BW, Giovannucci EL. A nested case control study of plasma 25-hydroxyvitamin D concentrations and risk of colorectal cancer. J Natl Cancer Inst. 2007;99:1120–9. doi: 10.1093/jnci/djm038. [DOI] [PubMed] [Google Scholar]
- 4.Spina CS, Ton L, Yao M, et al. Selective vitamin D receptor modulators and their effects on colorectal tumor growth. J Steroid Biochem Mol Biol. 2007;103:757–62. doi: 10.1016/j.jsbmb.2006.12.040. [DOI] [PubMed] [Google Scholar]
- 5.Iseki K, Tatsuta M, Uehara H, et al. Inhibition of angiogenesis as a mechanism for inhibition by 1alpha-hydroxyvitamin D3 and 1,25-dihydroxyvitamin D3 of colon carcinogenesis induced by azoxymethane in Wistar rats. Int J Cancer. 1999;81:730–3. doi: 10.1002/(sici)1097-0215(19990531)81:5<730::aid-ijc11>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 6.Shabahang M, Buras RR, Davoodi F, et al. Growth inhibition of HT-29 human colon cancer cells by analogues of 1,25-dihydroxyvitamin D3. Cancer Res. 1994;54:4057–64. [PubMed] [Google Scholar]
- 7.Lamprecht SA, Lipkin M. Cellular mechanisms of calcium and vitamin D in the inhibition of colorectal carcinogenesis. Ann N Y Acad Sci. 2001;952:73–87. doi: 10.1111/j.1749-6632.2001.tb02729.x. [DOI] [PubMed] [Google Scholar]
- 8.Eisman JA, Barkla DH, Tutton PJ. Suppression of in vivo growth of human cancer solid tumor xenografts by 1,25-dihydroxyvitamin D3. Cancer Res. 1987;47:21–5. [PubMed] [Google Scholar]
- 9.Beaty MM, Lee EY, Glauert HP. Influence of dietary calcium and vitamin D on colon epithelial cell proliferation and 1,2-dimethylhydrazine-induced colon carcinogenesis in rats fed high fat diets. J Nutr. 1993;123:144–52. doi: 10.1093/jn/123.1.144. [DOI] [PubMed] [Google Scholar]
- 10.Cross HS, Bises G, Lechner D, Manhardt T, Kallay E. The Vitamin D endocrine system of the gut--its possible role in colorectal cancer prevention. J Steroid Biochem Mol Biol. 2005;97:121–8. doi: 10.1016/j.jsbmb.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Cross HS, Bareis P, Hofer H, et al. 25-Hydroxyvitamin D(3)-1alpha-hydroxylase and vitamin D receptor gene expression in human colonic mucosa is elevated during early cancerogenesis. Steroids. 2001;66:287–92. doi: 10.1016/s0039-128x(00)00153-7. [DOI] [PubMed] [Google Scholar]
- 12.Matusiak D, Murillo G, Carroll RE, Mehta RG, Benya RV. Expression of vitamin D receptor and 25-hydroxyvitamin D3-1{alpha}-hydroxylase in normal and malignant human colon. Cancer Epidemiol Biomarkers Prev. 2005;14:2370–6. doi: 10.1158/1055-9965.EPI-05-0257. [DOI] [PubMed] [Google Scholar]
- 13.Friedrich M, Rafi L, Mitschele T, Tilgen W, Schmidt W, Reichrath J. Analysis of the vitamin D system in cervical carcinomas, breast cancer and ovarian cancer. Recent Results Cancer Res. 2003;164:239–46. doi: 10.1007/978-3-642-55580-0_17. [DOI] [PubMed] [Google Scholar]
- 14.Mitschele T, Diesel B, Friedrich M, et al. Analysis of the vitamin D system in basal cell carcinomas (BCCs) Lab Invest. 2004;84:693–702. doi: 10.1038/labinvest.3700096. [DOI] [PubMed] [Google Scholar]
- 15.Reichrath J, Rafi L, Rech M, et al. Analysis of the vitamin D system in cutaneous squamous cell carcinomas. J Cutan Pathol. 2004;31:224–31. doi: 10.1111/j.0303-6987.2003.00183.x. [DOI] [PubMed] [Google Scholar]
- 16.Hmama Z, Nandan D, Sly L, Knutson KL, Herrera-Velit P, Reiner NE. 1alpha,25-dihydroxyvitamin D(3)-induced myeloid cell differentiation is regulated by a vitamin D receptor-phosphatidylinositol 3-kinase signaling complex. J Exp Med. 1999;190:1583–94. doi: 10.1084/jem.190.11.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Zhang J, Studzinski GP. AKT pathway is activated by 1, 25-dihydroxyvitamin D3 and participates in its anti-apoptotic effect and cell cycle control in differentiating HL60 cells. Cell Cycle. 2006;5:447–51. doi: 10.4161/cc.5.4.2467. [DOI] [PubMed] [Google Scholar]
- 18.Hughes PJ, Brown G. 1Alpha,25-dihydroxyvitamin D3-mediated stimulation of steroid sulphatase activity in myeloid leukaemic cell lines requires VDRnuc-mediated activation of the RAS/RAF/ERK-MAP kinase signalling pathway. J Cell Biochem. 2006;98:590–617. doi: 10.1002/jcb.20787. [DOI] [PubMed] [Google Scholar]
- 19.Sheinin Y, Kaserer K, Wrba F, et al. In situ mRNA hybridization analysis and immunolocalization of the vitamin D receptor in normal and carcinomatous human colonic mucosa: relation to epidermal growth factor receptor expression. Virchows Arch. 2000;437:501–7. doi: 10.1007/s004280000275. [DOI] [PubMed] [Google Scholar]
- 20.Chan AT, Ogino S, Fuchs CS. Aspirin and the Risk of Colorectal Cancer in Relation to the Expression of COX-2. New Engl J Med. 2007;356:2131–42. doi: 10.1056/NEJMoa067208. [DOI] [PubMed] [Google Scholar]
- 21.Ogino S, Nosho K, Kirkner GJ, et al. PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. J Clin Oncol. 2009;27:1477–84. doi: 10.1200/JCO.2008.18.6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogino S, Nosho K, Kirkner GJ, et al. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut. 2009;58:90–6. doi: 10.1136/gut.2008.155473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ogino S, Kawasaki T, Brahmandam M, et al. Sensitive sequencing method for KRAS mutation detection by Pyrosequencing. J Mol Diagn. 2005;7:413–21. doi: 10.1016/S1525-1578(10)60571-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogino S, Kawasaki T, Kirkner GJ, Loda M, Fuchs CS. CpG island methylator phenotype-low (CIMP-low) in colorectal cancer: possible associations with male sex and KRAS mutations. J Mol Diagn. 2006;8:582–8. doi: 10.2353/jmoldx.2006.060082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nosho K, Kawasaki T, Ohnishi M, et al. PIK3CA mutation in colorectal cancer: relationship with genetic and epigenetic alterations. Neoplasia. 2008;10:534–41. doi: 10.1593/neo.08336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogino S, Brahmandam M, Cantor M, et al. Distinct molecular features of colorectal carcinoma with signet ring cell component and colorectal carcinoma with mucinous component. Mod Pathol. 2006;19:59–68. doi: 10.1038/modpathol.3800482. [DOI] [PubMed] [Google Scholar]
- 27.Ogino S, Kawasaki T, Brahmandam M, et al. Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. J Mol Diagn. 2006;8:209–17. doi: 10.2353/jmoldx.2006.050135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogino S, Cantor M, Kawasaki T, et al. CpG island methylator phenotype (CIMP) of colorectal cancer is best characterised by quantitative DNA methylation analysis and prospective cohort studies. Gut. 2006;55:1000–6. doi: 10.1136/gut.2005.082933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–93. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 30.Ogino S, Kawasaki T, Kirkner GJ, Kraft P, Loda M, Fuchs CS. Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. J Mol Diagn. 2007;9:305–14. doi: 10.2353/jmoldx.2007.060170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogino S, Kawasaki T, Nosho K, et al. LINE-1 hypomethylation is inversely associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Int J Cancer. 2008;122:2767–73. doi: 10.1002/ijc.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ogino S, Kawasaki T, Kirkner GJ, et al. Down-regulation of p21 (CDKN1A/CIP1) is inversely associated with microsatellite instability and CpG island methylator phenotype (CIMP) in colorectal cancer. J Pathol. 2006;210:147–54. doi: 10.1002/path.2030. [DOI] [PubMed] [Google Scholar]
- 33.Ogino S, Meyerhardt JA, Cantor M, et al. Molecular alterations in tumors and response to combination chemotherapy with gefitinib for advanced colorectal cancer. Clin Cancer Res. 2005;11:6650–6. doi: 10.1158/1078-0432.CCR-05-0738. [DOI] [PubMed] [Google Scholar]
- 34.Kawasaki T, Nosho K, Ohnishi M, et al. Correlation of beta-catenin localization with cyclooxygenase-2 expression and CpG island methylator phenotype (CIMP) in colorectal cancer. Neoplasia. 2007;9:569–77. doi: 10.1593/neo.07334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ogino S, Nosho K, Kirkner GJ, et al. A cohort study of tumoral LINE-1 hypomethylation and prognosis in colon cancer. J Natl Cancer Inst. 2008;100:1734–8. doi: 10.1093/jnci/djn359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garland C, Shekelle RB, Barrett-Connor E, Criqui MH, Rossof AH, Paul O. Dietary vitamin D and calcium and risk of colorectal cancer: a 19-year prospective study in men. Lancet. 1985;1:307–9. doi: 10.1016/s0140-6736(85)91082-7. [DOI] [PubMed] [Google Scholar]
- 37.Giovannucci E, Liu Y, Rimm EB, et al. Prospective Study of Predictors of Vitamin D Status and Cancer Incidence and Mortality in Men. J Natl Cancer Inst. 2006;98:451–9. doi: 10.1093/jnci/djj101. [DOI] [PubMed] [Google Scholar]
- 38.Garland CF, Comstock GW, Garland FC, Helsing KJ, Shaw EK, Gorham ED. Serum 25-hydroxyvitamin D and colon cancer: eight-year prospective study. Lancet. 1989;2:1176–8. doi: 10.1016/s0140-6736(89)91789-3. [DOI] [PubMed] [Google Scholar]
- 39.Ally MS, Al-Ghnaniem R, Pufulete M. The relationship between gene-specific DNA methylation in leukocytes and normal colorectal mucosa in subjects with and without colorectal tumors. Cancer Epidemiol Biomarkers Prev. 2009;18:922–8. doi: 10.1158/1055-9965.EPI-08-0703. [DOI] [PubMed] [Google Scholar]
- 40.Bapat B, Lindor NM, Baron J, et al. The association of tumor microsatellite instability phenotype with family history of colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2009;18:967–75. doi: 10.1158/1055-9965.EPI-08-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.English DR, Young JP, Simpson JA, et al. Ethnicity and risk for colorectal cancers showing somatic BRAF V600E mutation or CpG island methylator phenotype. Cancer Epidemiol Biomarkers Prev. 2008;17:1774–80. doi: 10.1158/1055-9965.EPI-08-0091. [DOI] [PubMed] [Google Scholar]
- 42.Figueiredo JC, Grau MV, Wallace K, et al. Global DNA hypomethylation (LINE-1) in the normal colon and lifestyle characteristics and dietary and genetic factors. Cancer Epidemiol Biomarkers Prev. 2009;18:1041–9. doi: 10.1158/1055-9965.EPI-08-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poynter JN, Siegmund KD, Weisenberger DJ, et al. Molecular characterization of MSI-H colorectal cancer by MLHI promoter methylation, immunohistochemistry, and mismatch repair germline mutation screening. Cancer Epidemiol Biomarkers Prev. 2008;17:3208–15. doi: 10.1158/1055-9965.EPI-08-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weinstein SJ, Albanes D, Selhub J, et al. One-carbon metabolism biomarkers and risk of colon and rectal cancers. Cancer Epidemiol Biomarkers Prev. 2008;17:3233–40. doi: 10.1158/1055-9965.EPI-08-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larriba MJ, Martin-Villar E, Garcia JM, et al. Snail2 cooperates with Snail1 in the repression of vitamin D receptor in colon cancer. Carcinogenesis. 2009 doi: 10.1093/carcin/bgp140. [DOI] [PubMed] [Google Scholar]
- 46.Gonzalez-Sancho JM, Larriba MJ, Ordonez-Moran P, Palmer HG, Munoz A. Effects of 1alpha,25-dihydroxyvitamin D3 in human colon cancer cells. Anticancer Res. 2006;26:2669–81. [PubMed] [Google Scholar]
- 47.Qi X, Tang J, Pramanik R, et al. p38 MAPK activation selectively induces cell death in K-ras-mutated human colon cancer cells through regulation of vitamin D receptor. J Biol Chem. 2004;279:22138–44. doi: 10.1074/jbc.M313964200. [DOI] [PubMed] [Google Scholar]
- 48.Dunn SE, Ehrlich M, Sharp NJ, et al. A dominant negative mutant of the insulin-like growth factor-I receptor inhibits the adhesion, invasion, and metastasis of breast cancer. Cancer Res. 1998;58:3353–61. [PubMed] [Google Scholar]
- 49.Solomon C, White JH, Kremer R. Mitogen-activated protein kinase inhibits 1,25-dihydroxyvitamin D3-dependent signal transduction by phosphorylating human retinoid X receptor alpha. J Clin Invest. 1999;103:1729–35. doi: 10.1172/JCI6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Solomon C, Kremer R, White JH, Rhim JS. Vitamin D resistance in RAS-transformed keratinocytes: mechanism and reversal strategies. Radiat Res. 2001;155:156–62. doi: 10.1667/0033-7587(2001)155[0156:vdrirt]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 51.Stedman L, Nickel KP, Castillo SS, Andrade J, Burgess JR, Teegarden D. 1,25-dihydroxyvitamin D inhibits vitamin E succinate-induced apoptosis in C3H10T1/2 cells but not Harvey ras-transfected cells. Nutr Cancer. 2003;45:93–100. doi: 10.1207/S15327914NC4501_11. [DOI] [PubMed] [Google Scholar]
- 52.Barault L, Veyrie N, Jooste V, et al. Mutations in the RAS-MAPK, PI(3)K (phosphatidylinositol-3-OH kinase) signaling network correlate with poor survival in a population-based series of colon cancers. Int J Cancer. 2008;122:2255–9. doi: 10.1002/ijc.23388. [DOI] [PubMed] [Google Scholar]
- 53.Velho S, Oliveira C, Ferreira A, et al. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur J Cancer. 2005;41:1649–54. doi: 10.1016/j.ejca.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 54.Slattery ML, Wolff RK, Curtin K, et al. Colon tumor mutations and epigenetic changes associated with genetic polymorphism: Insight into disease pathways. Mutat Res. 2009;660:12–21. doi: 10.1016/j.mrfmmm.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ogino S, Goel A. Molecular classification and correlates in colorectal cancer. J Mol Diagn. 2008;10:13–27. doi: 10.2353/jmoldx.2008.070082. [DOI] [PMC free article] [PubMed] [Google Scholar]