

Abstract
Osteoarthritis (OA) is the most common form of arthritis worldwide. In this condition, damage to the extracellular matrix (ECM) of cartilage occurs, resulting in joint destruction. Factors mediating cartilage damage include mechanical injury, cytokine and superoxide release on a background of genetic susceptibility and obesity. Studies of arthritic cartilage show increased production of ECM molecules including type II collagen, cartilage oligomeric matrix protein, fibronectin (FN) and fibromodulin. Recent reports suggest that ECM proteins may become endogenous catabolic factors during joint damage. Activation of pro-inflammatory pathways by ECM proteins has led to their description as damage-associated molecular patterns (DAMPs). The ECM proteins involved include fibromodulin, which activates the complement pathway and may promote the persistence of joint inflammation. Fragmentation of type II collagen, FN and hyaluronan reveals cryptic epitopes that stimulate proteolytic enzymes including matrix metalloproteinases and aggrecanases (ADAMTSs – a disintegrin and metalloproteinase with thrombospondin type 1 motifs). Proteolytic fragments also stimulate the release of nitric oxide, chemokines and cytokines and activation of the MAP kinases. Reports are emerging that the receptors for the fragments described involve interaction with integrins and toll-like receptors. In this review the contribution of endogenous ECM molecules to joint destruction will be discussed. A deeper understanding of the pathways stimulated by endogenous ligands could offer potential avenues for novel therapies in the future.
Keywords: aggrecanases, extracellular matrix, integrins, matrix metalloproteinases, osteoarthritis, toll-like receptors
Introduction
Osteoarthritis (OA) is the most common form of arthritis which causes major disability worldwide (Felson 2009). The hallmark of disease in OA is cartilage destruction resulting in joint space narrowing and loss of function (Mankin & Lippiello 1970). Other features believed to be secondary to primary cartilage damage in OA include periarticular bone sclerosis and cysts (Altman 1991). Synovial thickening and inflammation have also been reported in OA (Belhorn & Hess 1993). The result of ongoing cartilage destruction is irreversible damage to the extracellular matrix (ECM) of cartilage (Figure 1) with ultimate loss of joint function (Hunter & Felson 2006). There are no long-term effective disease modifying treatments for OA and current research is focusing on understanding how the imbalance between specific ECM molecules may influence disease progression. Evidence is emerging to show that endogenous ECM molecules provide signals to damaged cartilage and synovial tissue to promote further cartilage degradation. The aims of this review are to: (i) evaluate the pattern of altered ECM molecules in OA; (ii) discuss the catabolic factors regulating ECM turnover and cartilage destruction; (iii) analyse the effect of endogenous ECM molecules in the progression of cartilage breakdown during OA; and (iv) discuss the impact of ECM breakdown in maintaining ongoing catabolic signals and implications this has for future therapies in OA.
Figure 1.
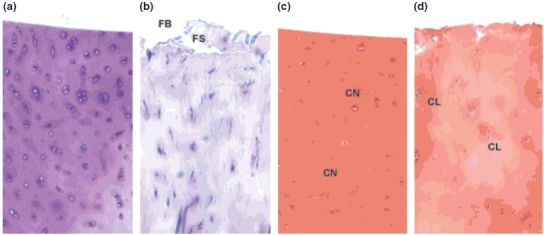
Comparison of the histological appearances of normal and OA cartilage. (a and b) Toluidene blue stain for proteoglycans in cartilage. The normal human cartilage sample (a) shows an even and abundant distribution of aggrecan staining throughout the ECM with a homogeneous distribution of chondrocytes. Cartilage from a patient undergoing joint replacement surgery for OA (panel b) shows a reduction of toluidene blue staining indicating loss of aggrecan and clumping of chondrocytes. The articular surface is damaged and fibrillation (FB) and fissuring (FS) occur. (c and d) Safranin O stain for proteoglycans in cartilage. Normal human cartilage (panel c) is stained homogeneously with safranin O demonstrating chondrocytes (CN) that are evenly distributed within the cartilage. OA cartilage (d) shows proteoglycan loss with paler staining areas and damage to chondrocytes with a clumped (CL) appearance (magnification ×200).
Alteration of cartilage composition in OA
Normal articular cartilage is a hyaline cartilage characterized by its fibrous architecture (Sledge 1975). Aggrecan and type II collagen are the most abundant proteins found within the ECM and are produced by the only cell type within cartilage, the chondrocyte (Hunziker et al. 2002). Collagen fibrils, made predominantly of type II collagen assembled in a triple helix, are linked together by a number of collagen-binding proteins including cartilage oligomeric matrix protein (COMP), chondroadherin and other minor collagens on their surface (Eyre et al. 1991; Vaughan-Thomas et al. 2001). Collagen degradation in cartilage by collagenases is believed to be a critical step in the alteration of cartilage homeostasis in OA (Billinghurst et al. 1997) (Figure 2a). Aggrecan, the other major component of cartilage, is a large aggregating proteoglycan which has an important role in providing a large number of negatively charged groups that attract counterions, thereby creating an osmotic pressure in cartilage that retains water (Sledge 1975). The aggrecan core protein is approximately 230 kDa and contains three globular domains (G1-3) (Figure 2b) (Doege et al. 1991). Aggrecan monomers aggregate by interaction with hyaluronan and link protein to form a large polymeric structure within cartilage (Figure 2b). Destruction of aggrecan and type II collagen are the major features of ECM destruction in OA. Changes in ECM molecules during OA are critical to the progression of the disease and have been investigated at the mRNA and protein level. Cartilage components including type II, III, VI and X collagens, aggrecan, biglycan, decorin, fibronectin (FN) and tenascin-C (TN-C) are all elevated at the mRNA level in OA cartilage (Aigner et al. 2001, 2005, 2006; Kevorkian et al. 2004). More recently, proteomics approaches have allowed a large number of cartilage proteins to be analysed and their expression may be influenced by microRNAs in cartilage (Iliopoulos et al. 2008). A summary of molecules which are regulated in OA cartilage are shown in Table 1. Other studies have attempted to examine the increased synthesis of specific proteins by OA chondrocytes to find that not only are certain ECM components increased in response to injury e.g. type II collagen, COMP, FN and fibromodulin (Stevens et al. 2009), but regulatory molecules are also produced including complement pathway components, growth factors such as IGF-1, TGF-β, activin A, CTGF and chemokines (Hermansson et al. 2004; Stevens et al. 2009). In addition, molecules mediating matrix degradation including matrix metalloproteinase (MMP)-1, MMP-3, MMP-13 and ADAMTSs are also upregulated (Lee et al. 2005; Guo et al. 2008; Jmeian & El Rassi 2008,Vincourt et al. 2008). Changes in the proteins described above are influenced by a number of factors including increased mechanical load on the joint (Loening et al. 2000; Kurz et al. 2001) which is accelerated by obesity (Recnik et al. 2008) and repetitive strain (L’Hermette et al. 2006). Injurious mechanical compression of cartilage explants results in changes at the level of gene transcription that may lead to subsequent degradation of cartilage. For example, Lee et al. (2005) demonstrated that MMP-3 increased approximately 250-fold, ADAMTS-5 increased 40-fold and tissue inhibitor of metalloproteinase 1 (TIMP-1) increased 12-fold above the levels in non-injured cartilage. The up-regulation of the proteinase genes described may eventually lead to matrix degradation and cause a compromise in cartilage structure and function. Increased production of cartilage ECM components may reflect an attempt by damaged cartilage to repair itself. However, as cartilage damage worsens, there is a process of ‘frustrated repair’ whereby chondrocytes are unable to assemble a functional matrix. Genetic susceptibility for OA also compounds the effects of mechanical stress. Disruption of ECM assembly by polymorphisms or mutations in specific proteins may lead to premature OA or cartilage abnormalities in humans. For example, genetic studies have shown the association of a polymorphism in the type II procollagen gene COL2A1 (which encodes the alpha chain of type II collagen) with hereditary forms of premature, polyarticular OA and mild dysplasia (Knowlton et al. 1990), supporting the possible role of a primary abnormality of type II collagen in the cause of rare forms of OA. COMP also has a restricted tissue distribution and is found primarily in cartilage (Riessen et al. 2001). Mutations in human COMP can cause cartilage defects including pseudoachondrodysplasias and multiple epiphyseal dysplasias (Hecht et al. 1995; Jakkula et al. 2003). Matrilin is distributed at the articular surface in human cartilage (Klatt et al. 2002; Strusberg et al. 2002). Its specific function is unknown, but it is believed to facilitate interactions between cells and/or the ECM (Wiberg et al. 2003). Mutations in the vWF A (von Willebrand Factor A) domain of matrilin-3 are associated with human multiple epiphyseal dysplasia (Chapman et al. 2001; Jackson et al. 2004). A missense mutation in the EGF domain of this molecule is also associated with hand OA (Stefansson et al. 2003). Asporin, an ECM protein found in cartilage, is linked to polymorphisms associated with increased OA susceptibility in a Japanese population (Kizawa et al. 2005). However, a European study did not find the same association in subjects with hip and knee OA (Mustafa et al. 2005), suggesting that environmental factors may influence disease susceptibility in diverse ethnic groups or that other gene polymorphisms may be associated with OA in different ethnic groups. This section has outlined how a number of factors including mechanical loading, obesity and genetic changes in ECM genes influence joint damage in OA. As a result of injury, catabolic factors are stimulated within the joint that induce further destruction of cartilage matrix. The agents mediating cartilage breakdown are discussed further in the following section.
Figure 2.
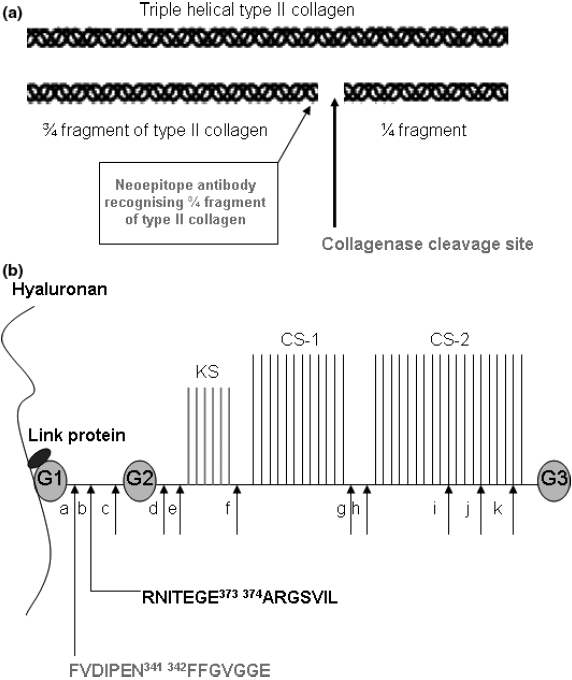
(a) Structure of triple helical collagen and cleavage by collagenases. Schematic representation of fibrillar type II collagen is shown. The 3/4; and ¼. fragments resulting from the cleavage of a triple helical collagen molecule by collagenases and the detection site of neoepitope antibodies is also shown. (b) Structure of aggrecan and cleavage by aggrecanases and MMPs. Schematic drawing showing the structure of aggrecan. G1 – 1st globular domain, G2 – 2nd globular domain, G3 – 3rd globular domain, KS – keratan sulphate glycosaminoglycan (GAG) attachment region, CS-1 – 1st chondroitin sulphate GAG region, CS-2 – 2nd chondroitin sulphate attachment region. Hyaluronan can bind numerous aggrecan molecules to form a large polymeric structure. The interaction of aggrecan with hyaluronan is strengthened by link protein. The reported cleavage sites of aggrecan by MMPs (a, c, d, e, f, g) and aggrecanases (b, h, i, j, k) are shown. The cleavage sites most relevant to OA are in the interglobular domain and the specific amino acid residues involved in cleavage are shown (adapted from Nagase & Kashiwagi 2003).
Table 1.
Genes and proteins reported to be altered in OA are listed according to their function. Although the molecules listed are by no means comprehensive of all proteins described in the literature, those that are considered to be of major significance and that are cited most frequently are described
| Gene | Regulation at mRNA level | Regulation at protein level |
|---|---|---|
| Extracellular matrix | Aggrecan, biglycan, collagen1A2, 2A1, 3A1, 6A1, 6A3,11A2, 6A1, CILP, COMP, decorin, fibronectin, fibromodulin, laminin A4, lumican, matrilin-1, matrilin-3, osteonectin, tenascin-C, SPARC, versican | Aggrecan, Col alpha 1 (1, II, V and XI), Col alpha 2 (I and XI), fibronectin, matrilin-1, matrilin-3, COMP, perlecan, tenascin-C |
| Growth factors | BMP-3B, BMP-2, Fibroblast growth factor receptor 1, insulin-like growth factor binding protein 6, insulin-induced protein 1, oestrogen receptor 1 | Serum amyloid A, chordin like 1, chordin like 2, CTGF, gremlin, HTRA1, IGF-II, IGF-BP3, IGF-BP4, IGF-BP5, IGF-BP6, inhibin beta A, TGFB2 |
| Cytokines and chemokines | CD126, IL-6R, IL-1R, leukaemia inhibitory factor (LIF) precursor, tumour necrosis factor-induced protein | CCL2, CCL20, CCL5, IL-6, IL-17B, CXCL6, M-CSF |
| Proteases and their inhibitors | MMP-1, MMP-2, MMP-3, MMP-8, MMP-9, MMP-10, MMP-11, MMP-13, MMP-14, MMP-17, TIMP-1, TIMP-3, TIMP-4, ADAMTS-1, ADAMTS-2, ADAMTS-4, ADAMTS-5, ADAMTS-9, ADAMTS-12, ADAMTS-14, ADAMTS-5, caspase 10 | MMP-1, MMP-2, MMP-3, MMP-9, MMP-13, MMP-14, TIMP-1, TIMP-2, BMP-1 |
| Complement pathway | Decay accelerating factor, Factor 1 | C1 inhibitor, Factor B, Factor C3, Factor C1r, Factor C1q |
| Other molecules | Superoxide dismutase, S100 calcium-binding protein chitinase 3 like 1, Dual specificity phosphatase 1, FRZB, Wnt-16, Beta catenin, WISP 1 | FRZB, Osteopontin, proenkephalin |
Catabolic factors mediate progression of ECM damage
In OA there is a disturbance in the regulation of synthetic (anabolic) and resorptive (catabolic) activities of cartilage. The end result is a net loss of cartilage with deterioration of its structural and functional properties. The factors influencing cartilage breakdown are cytokines, chemokines and proteinases including the MMPs and aggrecanases. The contribution of these specific molecules to ECM destruction in cartilage is discussed in further detail below.
Cytokines, growth factors and chemokines
Whereas OA is not usually considered to be an inflammatory disease, inflammatory cells, cytokines and growth factors that are not normally present in the cartilage matrix are found (Haynes et al. 2002; reviewed in Dray & Read 2007). Several cytokines may be considered to have catabolic, inhibitory or anabolic/regulatory effects in cartilage (Table 2). In addition, a number of growth factors have anabolic effects, including FGF-2 (Inoue et al. 2006), TGF-β (Blaney Davidson et al. 2006) and IGF-1 (Loeser et al. 2003). Of the cytokines believed to play a role in OA, IL-1 is thought to be major cytokine mediating cartilage destruction. Injection of recombinant IL-1 in rats, mice and rabbits stimulates destruction of articular cartilage (Pettipher et al. 1986; O’Byrne et al. 1990). IL-1 stimulates chondrocytes to produce inflammatory mediators including prostaglandin E2 (PGE2) (Guerne et al. 1999), nitric oxide (NO) (Guerne et al. 1999), superoxides (Tawara et al. 1991) and proteolytic enzymes (Buttle et al. 1993; Powell et al. 2007) that modulate catabolic activities in cartilage.
Table 2.
Classification of cytokines and growth factors in OA. Cytokines and growth factors that are currently believed to have a catabolic, regulatory, anti-catabolic or anabolic role in OA are shown
| Catabolic | Anabolic | Anti-catabolic | Regulatory |
|---|---|---|---|
| IL-1 | Activin | IL-1ra | IL-1 |
| IL-17 IL-18 | BMP-2,-4, -6, -7, -9, -13 | IL-4 | IL-6 |
| OSM | CTGF | IL-10 | LIF |
| TNF | FGF-2 | IL-13 | |
| IGF-1 | |||
| TGF-β |
IL, interleukin; OSM, oncostatin M; LIF, leukaemia inhibitory factor; IL-1ra, IL-1 receptor antagonist; IGF-1, insulin-like growth factor-1; FGF-2, fibroblast growth factor-2; CTGF, connective tissue growth factor.
When IL-1 is combined with TNF-α and injected simultaneously, there is enhanced cartilage damage that exceeds the effects observed with either cytokine alone (Page Thomas et al. 1991). IL-1 and TNF-α can also affect the synthetic activity of chondrocytes by inhibiting the synthesis of proteoglycans and type II collagen (Saklatvala 1986; Goldring et al. 1994). The combination of IL-1 and oncostatin M also upregulates matrix-degrading proteinases in cartilage (Barksby et al. 2006). Other cytokines which are thought to play a catabolic role in cartilage include IL-17 and IL-18 (Van Den Berg 2002). In addition, IL-6 and LIF (leukaemia inhibitory factor) are believed to play a regulatory role. For example, IL-6 is capable of down-regulating type II collagen gene expression in articular chondrocytes (Porée et al. 2008). Furthermore, LIF can upregulate COX-2 and PGE2 synthesis by human OA synovial fibroblasts (Alaaeddine et al. 1999). Recent work has also implicated a role for chemokines in OA. The major function of chemokines is to act as a chemoattractant to guide the migration of cells (Zlotnik et al. 2006). This is of major significance in the arthritic joint, where chondrocytes and synovial cells may be strongly influenced by changes in local concentrations of molecules. Expression of chemokine receptors has been demonstrated in OA chondrocytes (Appleton et al. 2007) and synovial cells (Bruhl et al. 2008). Chemokine signalling pathway molecules e.g. CXCR4 and CCL2 may be upregulated in arthritis by mechanical injury (Appleton et al. 2007). Furthermore, FN fragments in the cell-binding region of FN (discussed in Endogenous ECM proteins acquire biological activity in arthritis) regulate IL-6, IL-8, MCP-1 and growth-related oncogene beta (GRO-beta) via NF-κB mediated pathways (Pulai et al. 2005).
Proteinases
Aggrecan loss from articular cartilage matrix is widely believed to be mediated by aggrecanases and MMPs (reviewed in Nagase & Kashiwagi 2003). The activities of these enzymes may be inhibited by the production of TIMPs (Tissue Inhibitor of MetalloProteinases), but as the disease process ensues, cartilage degradation predominates and impairment of joint function occurs. MMPs can cleave aggrecan at a number of sites with a major site identified at the interglobular domain between residues Asn341 and Phe342 (Fosang et al. 1991) (Figure 2b). At the time that this cleavage was described, Sandy et al. (1991) sequenced aggrecan fragments produced after bovine articular explants were cultured with IL-1. Their results showed that aggrecan fragments are generated by cleavage between Glu373 and Ala374, a site which is not usually cleaved by MMPs (Sandy et al. 1991). The enzyme(s) responsible for this activity were called ‘aggrecanases’. Aggrecan fragments cleaved at the Glu373–Ala374 bond were detected in the synovial fluid of OA subjects (Sandy et al. 1992; Lohmander et al. 1993). More recent work has identified the aggrecanases as members of the disintegrin and metalloproteinase with thrombospondin type 1 motifs (ADAMTS) family of proteinases (Abbaszade et al. 1999; Tortorella et al. 1999). MMPs and aggrecanases cleave aggrecan at sites that can be recognized after cleavage with neoepitope antibodies. Such antibodies detect the newly generated N-terminal or C-terminal sequence of aggrecan fragments (Figure 2b). Aggrecanases cleave the aggrecan core protein in the interglobular domain at the Glu373–Ala374 site to release an aggrecan fragment with an N-terminal sequence 374ARGSV, while MMPs cleave the same region at the Asn341 - Phe342 site to release the 342FFGVG aggrecan fragment (Figure 3). In studies from subjects with OA, immunohistochemistry using neoepitope antibodies showed the presence of MMP-cleaved DIPEN341 and aggrecanase-cleaved NITEGE373 neoepitopes, indicating that both groups of enzymes can act in vivo (Lark et al. 1997). However, until recently it was not clear as to which ADAMTS family member(s) have the highest activity in arthritic tissue. In 2005, two groups showed that the ADAMTS-5 knockout mouse is protected from arthritis, suggesting that this enzyme plays a key role in cartilage breakdown (Glasson et al. 2005; Stanton et al. 2005). In comparison, the ADAMTS-4 knockout mouse did not show any significant suppression of aggrecanase activity in the interglobular domain when challenged in an arthritic model (Glasson et al. 2004). The ADAMTS-4/ADAMTS-5 double knockout mouse was protected from cartilage degradation by IL-1, but not by retinoic acid (Rogerson et al. 2008), suggesting that other aggrecanases apart from ADAMTS-4 and ADAMTS-5 are capable of retinoic acid-induced cartilage breakdown, at least in animal models. It is as yet unclear as to which ADAMTS member(s) is/are critical for human cartilage breakdown in arthritis and further work is required to establish this. The collagenases cleave triple helical collagen at a single site at neutral pH. The degradation of collagen by these proteinases results in typical ¾ and ¼ length collagen fragments (Gross & Nagai 1965). Collagenases have been detected in cartilage from human OA subjects. Using gene expression analysis, Aigner et al. (2001) found that MMP-13 was significantly increased in late-stage specimens. MMP-1 and MMP-8 were not detectable in this study. Expression profiling of MMP genes in other studies are also in keeping with results from Aigner et al. (2001). For example, a study of human cartilage obtained at the time of hip joint replacement surgery for OA showed MMP-13 expression was increased in these late-stage disease specimens (Kevorkian et al. 2004). Expression of MMP-1, MMP-8 and MMP-13 was also detected by immunostaining in the superficial zone of OA cartilage (Tetlow et al. 2001). MMPs must be converted from the pro-form (inactive state) to the active form in order to have proteolytic activity. This process may be initiated by other enzymes in cartilage e.g. MT1-MMP (De Croos et al. 2007). Collagenase expression is detectable in animal models of OA. In the rabbit and canine anterior cruciate ligament incision models of OA, expression of MMP-1 and MMP-13 are increased (Mehraban et al. 1994; Fernandes et al. 1998; Bluteau et al. 2001). In addition, increased mRNA levels of MMP-13 have been found in the middle and deep zones of cartilage of the STR/ort mouse model which spontaneously develops OA, but not in control mice (Flannelly et al. 2002). Studies using synthetic inhibitors of MMPs have been employed to identify the role of collagenases in OA. A compound that selectively inhibits MMP-8 and MMP-13, but inhibits MMP-1 weakly, significantly reduced the spontaneous release of type II collagen fragments in cultured human OA cartilage explants (Dahlberg et al. 2000). Similar results were obtained using a synthetic MMP-13 inhibitor (Billinghurst et al. 1997), suggesting that MMP-13 may play a critical role in collagen destruction in OA. Although pro-inflammatory cytokines such as IL-1 induce strong aggrecanase and collagenase activity in animal models and in vitro, it is unclear as to whether this cytokine is the major mediator of ongoing joint damage during OA. Since OA is a chronic disease, a number of groups have investigated whether endogenous factors exist within the joint that may stimulate protease activity and the activation of pro-inflammatory pathways.
Figure 3.

Detection of aggrecan fragments released from IL-1α-stimulated cartilage by neoepitope antibodies. (a) The cleavage sites of aggrecan in the interglobular domain for MMPs and aggrecanases are shown. A further cleavage site for aggrecanases in the C-terminal region of aggrecan is also illustrated. (b) Porcine cartilage stimulated with control media or IL-1α at 0.1, 1, 10 and 100 ng/ml for 2 days is shown. The conditioned medium was harvested and analysed for GAG release using the DMMB assay (n = 3). (c, d and e) Equal volumes of conditioned media (50 μl) from the experiment shown in (b) were deglycosylated and Western blots performed for aggrecanase-cleaved neoepitopes using the BC-3 antibody (recognizing the N-terminal sequence 374ARGSV) [panel c], and the BC-14 antibody (recognizing the N-terminal sequence 342FFGVG) [panel d]. The Western blot on the far right in panel d shows purified aggrecan cleaved in vitro with recombinant MMP-3, deglycosylated and subjected to Western blotting using the BC-14 antibody.
Endogenous ECM proteins acquire biological activity in arthritis
Among the proteins described in Table 1, several ECM components may be up-regulated and degraded further in OA. The concept that endogenous molecules induce damage-associated molecular patterns (DAMPs) has implicated a number of endogenous factors, including heat shock proteins, uric acid, altered matrix proteins and S100 proteins in mediating joint damage (Foell et al. 2007) and inflammation (Lotze et al. 2007). In OA these molecules include type II collagen, fibromodulin, FN and hyaluronan (Figure 4), which are discussed further below.
Figure 4.

Theoretical model for the degradation of cartilage induced by endogenous ECM fragments. A number of ECM molecules can be cleaved in OA. Cleavage of these proteins by MMPs and/or aggrecanases may lead to the exposure of cryptic epitopes that can engage with ligand receptors e.g. integrins and toll-like receptors on chondrocytes to activate further proteolytic enzyme activity. Initially, this process may be inhibited by TIMPs, but as joint destruction progresses protease activity persists and induces further joint damage.
Type II collagen
Type II collagen is believed to be degraded in cartilage by MMPs (see Catabolic factors mediate progression of ECM damage). Degradation products of collagen type II are generated by cleavage during OA and are secreted into the urine and into the synovial fluid. Collagen-derived peptides, in particular the C-terminal telopeptide, are used as diagnostic tools to follow progressive cartilage breakdown (Hein et al. 1997; Jung et al. 2004). Collagen fragments generated during arthritis may influence matrix turnover. A fragment of type II collagen localizing to the N-terminus has been reported to upregulate mRNA and protein levels of MMP-2, MMP-3, MMP-9 and MMP-13 in bovine chondrocytes and explants (Fichter et al. 2006). Jennings et al. (2001) also demonstrated upregulation of gelatinase activity induced by collagen fragments in human chondrocytes. More recently, cathepsins B, L and K have been shown to be upregulated by an N-terminal 29-mer fragment of type II collagen (Ruettger et al. 2008). Klatt et al. (2009) reported that human chondrocytes cultured with intact monomeric type II collagen produced a dose-dependent induction of MMP-1, MMP-3, MMP-13, MMP-14, as well as cytokines IL-1β, IL-6, and IL-8. Examination of the signalling pathways utilized by monomeric collagen showed involvement of MAPK p38 and NF-κB signalling (Klatt et al. 2009). This study described a sequential activation by type II collagen of initially MMP and pro-inflammatory cytokine production followed by release of collagen II fragments from mature collagen II fibres. These studies suggest that collagen fragments may alter the environment within the ECM in OA to promote a catabolic state with the activation of MMPs and cathepsins.
Fibromodulin
Fibromodulin is a keratan sulphate proteoglycan found in a variety of connective tissues including cartilage, tendon, skin, cornea and sclera (Oldberg et al. 1989). Fibromodulin is a 42-kDa protein which is a member of the leucine rich repeat (LRR) family of extracellular matrix proteoglycans and glycoproteins (Figure 5). The LRR family also includes decorin, biglycan, lumican, chondroadherin, PRELP and epiphycan (Hardingham & Fosang 1992; Roughley 2006). Fibromodulin null mice have abnormally thin type I collagen fibrils in their tendons (Ameye et al. 2002) and increased incidence of arthritis, possibly due to abnormal ligament function. Recent studies have shown that fibromodulin gene expression is upregulated in tissue obtained from arthritic joints (Aigner et al. 2001). Fibromodulin is also degraded when cartilage explants are treated with IL-1 (Heathfield et al. 2004). A similar cleavage product to that generated by IL-1 is obtained when fibromodulin is treated with MMP-13 (Heathfield et al. 2004), and is approximately 10 kDa in size. The fibromodulin fragment cleaved by MMP-13 has been sequenced and used to generate a neoepitope antibody against this peptide (AYGSPPQPEPC). The neoepitope antibody showed similar fragments were generated when cartilage explants were treated with IL-1 (Heathfield et al. 2004). Since fibromodulin exists in cartilage bound to the surface of collagen fibres, the cleavage of fibromodulin may represent a critical early event that disrupts the collagen fibrillar network, leading to the exposure of sites where proteinases can further cleave collagen. Upregulation of fibromodulin in OA may also contribute to ongoing joint damage by stimulating inflammatory pathways. Recent work has shown that fibromodulin can bind C1q and cause direct activation of the complement cascade by this interaction (Sjöberg et al. 2005). The complement system forms a crucial part of the innate immune defence system and complement components have been detected in synovium from subjects with RA and OA (Neumann et al. 2002), where they may contribute to chronic inflammation. Fibromodulin can interact with the globular heads of C1q of complement in a similar manner to other agents that activate C1q, e.g. IgG. Although other ECM molecules e.g. laminin and decorin can also interact with C1q, only fibromodulin is capable of causing complement activation. Fibromodulin can also interact with Factor H, an inhibitory molecule in the complement cascade (Sjöberg et al. 2005). There may therefore be several mechanisms whereby activation of complement pathway components by fibromodulin may contribute to the pathogenesis of arthritis. Since the binding sites for C1q and Factor H on fibromodulin are different, it is possible that proteolytic degradation of fibromodulin leads to differential release of fragments that regulate the complement cascade.
Figure 5.
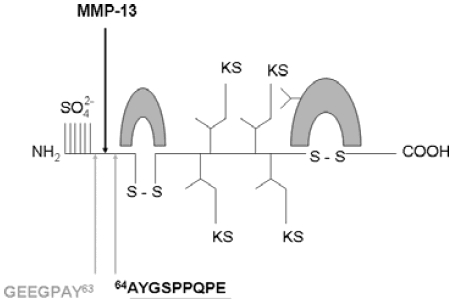
Structure of fibromodulin and cleavage sites. Fibromodulin is a 42 kDa protein with three structural domains. The N-terminal domain has four cysteine residues, two of which are involved in an intrachain disulphide bond. The central domain contains leucine residues in conserved positions. The C-terminal domain contains two cysteine residues which form an intrachain disulphide bond. The cleavage sites reported for bovine fibromodulin which have been used to generate neoepitope antibodies (indicated by the underlined amino acid residues) are shown.
Fibronectin
Fibronectin is a glycoprotein present in tissues and body fluids involved in diverse processes including cellular differentiation, adhesion, migration, wound healing and neoplastic transformation (see Hynes 1990 for review). FN is present in the ECM in an insoluble form, but circulates in plasma as a soluble protein. All FN forms consist of three types of homologous repeating units (types I–III). The protein usually exists as a dimer of 220–250 kDa linked together by a pair of disulfide bonds at the C-terminus (Figure 6). The type III repeats contain the alternatively spliced ED-A and ED-B domains. The ED-A (+) and ED-B (+) isoforms are not expressed in plasma FN, but are often expressed in cellular FN isoforms and in disease (Hynes 1990). The modular domain structure of FN confers a variety of properties to this molecule. FN mRNA and protein are increased up to 10-fold in human OA tissue (Brown & Jones 1990) and FN expression is increased in synovial fluid from OA subjects (Xie et al. 1992). The ED-B (+) FN isoform is also upregulated in OA (Rencic et al. 1995). Furthermore, TGF-β1 increased FN synthesis in canine cartilage explants (Burton-Wurster & Lust 1990). FN can be degraded by a number of proteases. In particular, MMP-1, MMP-3 and MMP-13 can digest FN and all are upregulated in OA cartilage (Okada 2001). Of the aggrecanases, ADAMTS-4 and ADAMTS-5 can also cleave FN albeit more weakly (Gendron et al. 2007). More recently, HtrA1 (high temperature resistant serine proteinase) has also been described in synovial fluid from arthritic joints (Grau et al. 2006). Grau et al. (2006) reported that FNfs generated by HtrA1 could induce the upregulation of MMP-1 and MMP-3 messenger RNA in human synovial fibroblasts. A number of proteolytic FNfs have been used to investigate their role in mediating cartilage degradation in human and animal models (Figure 6). Homandberg et al. (1992) demonstrated that FNfs encompassing the N-terminal 29-kDa fibrin/heparin binding fragment, the 45-kDa collagen-binding and the 120-kDa cell-binding domains stimulate bovine cartilage resorption in culture. These FNfs exerted their activity at micromolar concentrations in cartilage. The 29-kDa N-terminal heparin binding FNf showed the most potent levels of proteoglycan release. This 29-kDa FNf also stimulated the release of IL-1, TNF-α and IL-6 (Homandberg et al. 1997). At nanomolar concentrations the 29-kDa FNf promoted cartilage repair, with the induction of proteoglycan synthesis and release of anabolic factors such as TGF-β and insulin-like growth factor-1 (IGF-1) (Homandberg et al. 1992). It is possible that the accumulation of FNfs in tissue at higher concentrations induced a switch from anabolic to catabolic effects. The catabolic activity induced by the N-terminal 29-kDa FNf was not inhibited by the RGD peptide (Homandberg et al. 1992). The RGD motif represents the known α5β1 integrin binding site in the type III-10 domain of FN (Ruoslahti & Peirschbacher 1987). Subsequently, Homandberg et al. (1993) reported that injection of the N-terminal 29-kDa fragment into rabbit knee joints in vivo caused degradation of cartilage. Other investigators have reported that a recombinant form of the N-terminal 29-kDa FNf induces MAP kinase and IL-1β (Gemba et al. 2002). Stanton et al. (2002) showed the 45-kDa FNf induced MMP-3 and MMP-13 protein expression in chondrocytes in a dose-dependent manner (Stanton et al. 2002). The effects described were independent of IL-1, since they could not be inhibited with the IL-1 receptor antagonist. They also reported that the degradation of aggrecan in porcine cartilage explants induced by this FNf was attributed to the aggrecanases, as evidenced by cleavage at the NITEGE site (detecting aggrecanase cleavage in the interglobular domain), but not the DIPEN site (detecting MMP cleavage in the interglobular domain) using neoepitope antibodies. Werb et al. (1989) reported that a cell-binding FNf induces MMP-1 and MMP-3 expression in rabbit synovial fibroblasts through α5β1 integrins. In addition, they showed that a monoclonal antibody to the FN receptor α5β1 induced gene expression of MMP-1 and MMP-3, suggesting that ligation of the α5β1 integrin receptor enhances MMP gene expression in rabbit synovial fibroblasts. Other groups have also shown a similar cell-binding FNf can induce activation of MAP kinases in human articular chondrocytes (Forsyth et al. 2002). Their study also showed an increased expression of MMP-13 and IL-1β expression after 6 h treatment. The stimulation of MAP kinases was also induced by α5β1 and α2β1 antibodies (Forsyth et al. 2002). Im et al. (2003) reported that the MMP-13 gene upregulation induced by the cell-binding FNf in chondrocytes may be inhibited by exogenous addition of insulin-like growth factor-1 (IGF-1) and osteogenic protein 1 (OP-1) in a dose-dependent manner with corresponding decreases in the protein level of MMP-13. Further work by the same group showed that human articular chondrocytes treated with the cell-binding FN-f caused a >2-fold increase in IL-6, IL-8, and growth-related oncogene beta (GRO-beta) (Pulai et al. 2005). Studies using inhibitors of the NF-κB pathway and IL-1 receptor antagonist revealed that the 110-kDa FNf-induced stimulation of chondrocyte chemokine expression was dependent on NF-κB activity, but independent of IL-1 autocrine signalling. Furthermore, the cell-binding FNf also upregulated RAGE, the receptor for advanced end-glycation products in chondrocytes, to stimulate MMP-13 production via MAP kinase and NF-κB signalling pathways (Loeser et al. 2005). More recently, the same FNf has been shown to stimulate IL-7 production in chondrocytes (Long et al. 2008). The ability of the 110-kDa FNf to stimulate chondrocyte expression of multiple pro-inflammatory cytokines and chemokines suggests that damage to the cartilage matrix is capable of inducing a proinflammatory state responsible for further progressive matrix destruction. Several groups have investigated the role of the 40-kDa C-terminal heparin-binding FNf in cartilage degradation (Figure 6). This domain comprises the type III-12, -13 and -14 repeats which are also known as the Hep II region. Some studies use commercially produced Hep II FNfs which can contain the domain downstream of the III-12, -13 and -14 repeats, also known as the connecting segment (CS) or V region (Yasuda & Poole 2002; Johnson et al. 2004). In one study, degradation of type II collagen was increased in bovine articular cartilage treated with the C-terminal heparin-binding FNf (Yasuda & Poole 2002). MMP-3 and MMP-13 release were also elevated by this FNf in cartilage explant models (Yasuda et al. 2003a) and in synovial cells (Yasuda et al. 2003b). The FNf-induced type II collagen degradation was inhibited by IL-1 receptor antagonist, suggesting the later step of collagen degradation following aggrecan degradation in cartilage was dependent on IL-1 (Yasuda & Poole 2002). The C-terminal 40-kDa heparin-binding FNf increases the release of COMP and chondroadherin in a dose-dependent manner in an equine cartilage explant model (Johnson et al. 2004). The same FNf also induced the degradation of COMP and the production of nitric oxide in a dose-dependent manner. All the effects were observed within 24 h of stimulation by the C-terminal heparin-binding FNf and this FNf had more potent effects than the N-terminal heparin binding FNf (Johnson et al. 2004).
Figure 6.

Structure of fibronectin showing fragments generated by proteolysis. FN consists of a modular structure of type I, II and III repeats. The domains represented in black lines show alternatively spliced variants. The ED-A and ED-B domains are excluded in plasma, whereas most other tissues produce cellular fibronectins that include either one or both of these domains. The ED-B domain is found in up to 30% of cartilage isoforms. The V and C domains are also spliced out of cartilage forms. Fragments reported to have cartilage degrading activity are the 29, 40, 45 and 120-kDa forms and their location is shown by the double headed arrows. The enzymes responsible for generating these fibronectin fragments are depicted in the lower panel (from Hynes 1990; Zhen et al. 2008).
The ED-A isoform of FN is a cellular variant and usually only appears in tissue during specific biological or pathological processes. ED-A isoform expression is also increased in synovial fibroblasts from subjects with rheumatoid arthritis (Hino et al. 1995). In one study a recombinant ED-A domain was expressed and induced increased proteoglycan release in rabbit cartilage explant cultures in a dose-dependent manner (Saito et al. 1999). In addition, recombinant ED-A enhanced MMP-1, MMP-3 and MMP-9 expression by cultured rabbit synovial fibroblasts (Saito et al. 1999). However, recombinant FNfs containing the neighbouring type III-11 and III-12 repeats showed that these domains were unable to induce MMP-1 or MMP-3 mRNA expression (Saito et al. 1999). The MMP-1 release was partially inhibited by 100 ng/ml IL-1 receptor antagonist, suggesting some of the effects of this FNf occurred via an IL-1-mediated pathway. In another study, recombinant ED-A FN, but not full-length FN, induced the upregulation of MMP-9 in fibroblasts (Okamura et al. 2001). The MMP-9 upregulation was dependent on the presence of a toll-like receptor, TLR-4 (Okamura et al. 2001). This study suggested that TLRs may be involved in modulating the response of fibroblasts by the ED-A domain. Although TLRs are usually activated by exogenous ligands including LPS (lipopolysaccharide) to induce innate immune pathways, the study by Okamura et al. (2001) was one of the first to show that endogenous ECM molecules such as FN may utilize receptors such as TLR4. During chronic injury such as occurs in osteoarthritis, endogenous ‘danger signals’ produced by extracellular matrix molecules may activate toll-like receptor pathways (O’Neill 2008). Further evidence for the role of TLRs in OA has been suggested by Zhang et al. (2008) who reported upregulation of TLRs in human chondrocytes and others who have reported the upregulation of TLR-2 by FN fragments (Su et al. 2005). The ED-A FN domain also induced murine mast cells to release pro-inflammatory cytokines via TLR4 (Gondokaryono et al. 2007), suggesting that not only chondrocytes, but other cell types are sensitive to stimulation by FNfs.
Thus, at least five different fragments of FN enhance the expression of matrix-degrading proteinases. The fragmented forms of FN acquire novel functions upon fragmentation that do not exist in the full-length molecule which can further mediate matrix damage during arthritis.
Hyaluronan
Hyaluronan is a large negatively charged molecule that consists of a repeating disaccharide unit β1, 4-glucuronate and β1, 3-N-acteylglucosamine (Toole 1999). Hyaluronan attracts water molecules into the matrix and interacts with aggrecan, thereby retaining this molecule within the collagen structural framework (Quintarelli et al. 1978; Toole 1999) (Figure 3). Hyaluronan is found in synovial fluid, where it potentiates joint lubrication (Toole 1999) and levels are reduced in synovial fluid and cartilage of OA subjects (Rizkalla et al. 1992; Belcher et al. 1997). Several reports implicate the role of hyaluronan fragments in inflammation. One of the first observations was made by Horton et al. (1999), who showed that hyaluronan fragments induce the production of the metalloproteinase murine metalloelastase (MME) in macrophages. Knudson et al. (2000) later showed that hyaluronan fragments induce the degradation of cartilage. It was suggested that uncoupling hyaluronan from its cell–matrix interactions by disruption of CD44-hyaluronan interactions resulted in a catabolic effect mediated by oligosaccharide fragments of hyaluronan (Knudson et al. 2000). In a more recent study by Termeer et al. (2002), hyaluronan fragments induced maturation of dendritic cells via TLR-4. Since this work, other groups have also reported that hyaluronan fragments can induce alloimmunity (Tesar et al. 2006). Evidence of the importance of hyaluronan fragments in other diseases has also been suggested in tumour models e.g melanoma, where hyaluronan fragments have been reported to induce the upregulation of NF-κB, MMP-2 and IL-8 (Voelcker et al. 2008). Hyaluronan is degraded in arthritis (Inerot et al. 1978). There is increasing evidence that uncleaved hyaluronan may be chondroprotective in animal models of arthritis (Takahashi et al. 2001) and intra-articular preparations of hyaluronic acid are being given clinically to reduce pain in subjects with OA (Jüni et al. 2007). These results suggest that fragmented hyaluronan may provide different signals to the chondrocyte in comparison to full-length hyaluronan during inflammation and arthritis.
Conclusions and future directions
From the work described in this review, a model can be proposed whereby the initial injury to cartilage induced by factors such as mechanical injury and superoxide release on a background of genetic susceptibility stimulates chondrocytes and synovial cells within the joint. Activated cells are stimulated to produce cytokines such as IL-1 and matrix-degrading proteinases including MMPs and aggrecanases. The work reviewed here describes how ECM proteins and their fragments can accelerate joint damage. Further inflammation within the joint could be induced by the increased expression of ECM molecules such as fibromodulin that can activate innate immune pathways by complement activation. A number of molecules within cartilage matrix are also susceptible to degradation by proteases including collagen, hyaluronan and FN. When these molecules are degraded, they may reveal cryptic epitopes that acquire new activity hitherto absent in the full-length molecules. Fragments of such proteins may induce endogenous ‘danger signals’ within the joint to stimulate pro-inflammatory pathways via TLR and integrin ligand binding. Inhibition of toll-like receptor and/or integrin pathways may therefore be a potential therapeutic target for halting cartilage destruction. A deeper knowledge of the pathways and receptors used by endogenous proteins in concert with cytokines and chemokines may help lead to the development of newer therapies for arthritis in the future.
Acknowledgments
This work was supported in part by a Kennedy Institute of Rheumatology Trustees’ Fellowship awarded to Dr Nidhi Sofat. Dr Sofat is also the recipient of a Clinical Research Training Fellowship from the Wellcome Trust (Grant number 070848) and a grant from The Hammersmith Hospitals Trustees’ Research Committee. The author would like to thank Mr David Essex for preparing and staining cartilage sections and Dr Robert Visse for assistance with preparing illustrations. Professor Bruce Caterson and Dr Clare Hughes are acknowledged for their gift of BC-3 and BC-14 antibodies. Mr Jonathan Jones, Consultant Orthopaedic Surgeon, is acknowledged for obtaining human tissue samples, under full Ethics Approval, from Heatherwood and Wexham Park Hospitals NHS Trust.
References
- Abbaszade I, Liu RQ, Yang F, et al. Cloning and characterization of ADAMTS11, an aggrecanase from the ADAMTS family. J. Biol. Chem. 1999;274:23443–23450. doi: 10.1074/jbc.274.33.23443. [DOI] [PubMed] [Google Scholar]
- Aigner T, Zien A, Gehrsitz A, Gebhard PM, Mckenna L. Anabolic and catabolic gene expression pattern analysis in normal versus osteoarthritic cartilage using complementary DNA-array technology. Arthritis Rheum. 2001;44:2777–2789. doi: 10.1002/1529-0131(200112)44:12<2777::aid-art465>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Aigner T, McKenna L, Zien A, Fan Z, Gebhard PM, Zimmer R. Gene expression profiling of serum- and interleukin-1 beta-stimulated primary human adult articular chondrocytes – a molecular analysis based on chondrocytes isolated from one donor. Cytokine. 2005;31:227–240. doi: 10.1016/j.cyto.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Aigner T, Fundel K, Saas J, et al. Large scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 2006;54:3533–3544. doi: 10.1002/art.22174. [DOI] [PubMed] [Google Scholar]
- Alaaeddine N, Di Battista JA, Pelltier J-P, Kiansa K, Cloutier J-M, Martel-Pelletier J. Differential effects of IL-8, LIF (Pro-Inflammatory) and IL-11 (anti-inflammtory) on TNFα-induced PGE2 release and on signalling pathways in human OA synovial fibroblasts. Cytokine. 1999;11:1020–1030. doi: 10.1006/cyto.1999.0505. [DOI] [PubMed] [Google Scholar]
- Altman RD. Classification of disease: osteoarthritis. Semin. Arthritis Rheum. 1991;20:40–47. doi: 10.1016/0049-0172(91)90026-v. [DOI] [PubMed] [Google Scholar]
- Ameye L, Aria D, Jepson K, Oldberg A, Xu T, Young MF. Abnormal collagen fibrils in tendons of biglycan/fibromodulin-deficient mice lead to gait impairment, ectopic ossificationm and osteoarthritis. FASEB J. 2002;16:673–680. doi: 10.1096/fj.01-0848com. [DOI] [PubMed] [Google Scholar]
- Appleton CT, Pitelka V, Henry J, Beier F. Global analyses of gene expression in early experimental osteoarthritis. Arthritis Rheum. 2007;56:1854–1868. doi: 10.1002/art.22711. [DOI] [PubMed] [Google Scholar]
- Barksby HE, Hui W, Wappler I, et al. Interleukin-1 in combination with oncostatin M up-regulates multiple genes in chondrocytes: implications for cartilage destruction and repair. Arthritis Rheum. 2006;54:540–550. doi: 10.1002/art.21574. [DOI] [PubMed] [Google Scholar]
- Belcher C, Yaqub R, Fawthrop F, Bayliss M, Doherty M. Synovial fluid chondroitin and keratan sulphate epitopes, glycosaminoglycans, and hyaluronan in arthritic and normal knees. Ann. Rheum. Dis. 1997;56:299–307. doi: 10.1136/ard.56.5.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belhorn LR, Hess EV. Erosive osteoarthritis. Semin. Arthritis Rheum. 1993;22:298–306. doi: 10.1016/s0049-0172(05)80009-5. [DOI] [PubMed] [Google Scholar]
- Billinghurst RC, Dahlberg L, Ionescu M, et al. Enhanced cleavage of type II collagen by collagenases in osteoarthritic articular cartilage. J. Clin. Invest. 1997;99:1534–1545. doi: 10.1172/JCI119316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaney Davidson EN, Vitters EL, van den Berg WB, van der Kraan PM. TGF beta-induced cartilage repair is maintained but fibrosis is blocked in the presence of Smad7. Arthritis Res. Ther. 2006;8:R65. doi: 10.1186/ar1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluteau G, Conrozier T, Mathieu P, Vignon E, Herbage D, Mallein-Gerin F. Matrix metalloproteinase-1, -3, -13 and aggrecanase-1 and -2 are differentially expressed in experimental osteoarthritis. Biochim. Biophys. Acta. 2001;1526:147–158. doi: 10.1016/s0304-4165(01)00122-2. [DOI] [PubMed] [Google Scholar]
- Brown RA, Jones KL. The synthesis and accumulation of fibronectin by human articular cartilage. J. Rheumatol. 1990;17:65–72. [PubMed] [Google Scholar]
- Bruhl H, Mack M, Niedermeier M, Lochbaum D, Scholmerisch J, Straub RH. Functional expression of the chemokine receptor CCR7 on fibroblast-like synoviocytes. Rheumatology. 2008;47:1771–1774. doi: 10.1093/rheumatology/ken383. [DOI] [PubMed] [Google Scholar]
- Burton-Wurster N, Lust G. Fibronectin and proteoglycan synthesis in long term cultures of cartilage explants in Ham’s F12 supplemented with insulin and calcium: effects of the addition of TGF-beta. Arch. Biochem. Biophys. 1990;283:27–33. doi: 10.1016/0003-9861(90)90607-z. [DOI] [PubMed] [Google Scholar]
- Buttle DJ, Handley CJ, Ilic MZ, Saklatvala J, Muata M, Barrett AJ. Inhibition of cartilage proteoglycan release by a specific inactivator of cathepsin B and an inhibitor of matrix metalloproteinases. Evidence for two converging pathways of chindrocyte-mediated proteoglycan degradation. Arthritis Rheum. 1993;36:1709–1717. doi: 10.1002/art.1780361210. [DOI] [PubMed] [Google Scholar]
- Chapman KL, Mortier GR, Chapman K, Loughlin J, Grant ME, Briggs MD. Mutations in the region encoding the von Willebrand factor A domain of matrilin-3 are associated with multiple epiphyseal dysplasia. Nat. Genet. 2001;28:393–396. doi: 10.1038/ng573. [DOI] [PubMed] [Google Scholar]
- Dahlberg L, Billinghurst RC, Manner P, et al. Selective enhancement of collagenase-mediated cleavage of resident type II collagen in cultured osteoarthritic cartilage and arrest with a synthetic inhibitor that spares collagenase 1 (matrix metalloproteinase 1) Arthritis Rheum. 2000;43:673–682. doi: 10.1002/1529-0131(200003)43:3<673::AID-ANR25>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- De Croos JN, Jang B, Dhaliwal SS, Grynpas MD, Pilliar RM, Kandel RA. Membrane type-1 matrix metalloproteinase is induced following cyclic compression of in vitro grown bovine chondrocytes. Osteoarthritis Cartilage. 2007;15:1301–1310. doi: 10.1016/j.joca.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Doege KJ, Sasaki M, Kimura T, Yamada Y. Complete coding sequence and deduced primary structure of the human cartilage large aggregating proteoglycan, aggrecan. Human-specific repeats, and additional alternatively spliced forms. J. Biol. Chem. 1991;266:894–902. [PubMed] [Google Scholar]
- Dray A, Read SJ. Arthritis and pain. Future targets to control osteoarthritis pain. Arthritis Res. Ther. 2007;9:212. doi: 10.1186/ar2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre DR, Woods JJ, Weis MA. The cartilage collagens and joint degeneration. Br. J. Rheumatol. 1991;30(Suppl. 17):10–15. [PubMed] [Google Scholar]
- Felson DT. Developments in the clinical understanding of osteoarthritis. Arthritis Res. Ther. 2009;11:203. doi: 10.1186/ar2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes JC, Martel-Pelletier J, Lascau-Coman V, et al. Collagenase-1 and collagenase-3 synthesis in normal and early experimental osteoarthritic canine cartilage: an immunohistochemical study. J. Rheumatol. 1998;25:1585–1594. [PubMed] [Google Scholar]
- Fichter M, Körner U, Schömburg J, Jennings L, Cole AA, Mollenhauer J. Collagen degradation products modulate matrix metalloproteinase expression in cultured articular chondrocytes. J. Orthop. Res. 2006;24:63–70. doi: 10.1002/jor.20001. [DOI] [PubMed] [Google Scholar]
- Flannelly J, Chambers MG, Dudhia J, et al. Metalloproteinase and tissue inhibitor of metalloproteinase expression in the murine STR/ort model of osteoarthritis. Osteoarthritis Cartilage. 2002;10:722–733. doi: 10.1053/joca.2002.0818. [DOI] [PubMed] [Google Scholar]
- Foell D, Wittkowski H, Roth J. Mechanisms of disease: a ‘DAMP’ view of inflammatory arthritis. Nat. Clin. Pract. Rheumatol. 2007;3:382–390. doi: 10.1038/ncprheum0531. [DOI] [PubMed] [Google Scholar]
- Forsyth CB, Pulai J, Loeser RF. Fibronectin fragments and blocking antibodies to alpha2beta1 and alpha5beta1 integrins stimulate mitogenactivated protein kinase signaling and increase collagenase 3 (matrix metalloproteinase 13) production by human articular chondrocytes. Arthritis Rheum. 2002;46:2368–2376. doi: 10.1002/art.10502. [DOI] [PubMed] [Google Scholar]
- Fosang AJ, Neame PJ, Hardingham TE, Murphy G, Hamilton JA. Cleavage of cartilage proteoglycan between G1 and G2 domains by stromelysins. J. Biol. Chem. 1991;266:15579–15582. [PubMed] [Google Scholar]
- Gemba T, Valbracht J, Alsalameh S, Lotz M. Focal adhesion kinase and mitogen-activated protein kinases are involved in chondrocyte activation by the 29-kDa amino-terminal fibronectin fragment. J. Biol. Chem. 2002;277:907–911. doi: 10.1074/jbc.M109690200. [DOI] [PubMed] [Google Scholar]
- Gendron C, Kashiwagi M, Lim NH, et al. Proteolytic activities of human ADAMTS-5: comparative studies with ADAMTS-4. J. Biol. Chem. 2007;282:18294–18306. doi: 10.1074/jbc.M701523200. [DOI] [PubMed] [Google Scholar]
- Glasson SS, Askew R, Sheppard B, et al. Characterization of and osteoarthritis susceptibility in ADAMTS-4-knockout mice. Arthritis Rheum. 2004;50:2547–2558. doi: 10.1002/art.20558. [DOI] [PubMed] [Google Scholar]
- Glasson SS, Askew R, Sheppard B, et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005;434:644–648. doi: 10.1038/nature03369. [DOI] [PubMed] [Google Scholar]
- Goldring MB, Fukuo K, Birkhead JR, Dudek E, Sandell LJ. Transcriptional suppression by interleukin-1 and interferon-gamma of type II collagen gene expression in human chondrocytes. J. Cell. Biochem. 1994;54:85–99. doi: 10.1002/jcb.240540110. [DOI] [PubMed] [Google Scholar]
- Gondokaryono SP, Ushio H, Niyonsaba F, et al. The extra domain A of fibronectin stimulates murine mast cells via toll-like recptor 4. J. Leukoc. Biol. 2007;82:657–665. doi: 10.1189/jlb.1206730. [DOI] [PubMed] [Google Scholar]
- Grau S, Richards PJ, Kerr B, et al. The role of human HtrA1 in arthritic disease. J. Biol. Chem. 2006;281:6124–6129. doi: 10.1074/jbc.M500361200. [DOI] [PubMed] [Google Scholar]
- Gross J, Nagai Y. Specific degradation of the collagen molecule by tadpole collagenolytic enzyme. Proc. Natl. Acad. Sci. U.S.A. 1965;54:1197–1204. doi: 10.1073/pnas.54.4.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerne PA, Desgeorges A, Jaspar JM, et al. Effects of IL-6 and its soluble receptor on proteoglycan synthesis and NO release by human articular chondrocytes: comparison with IL-1. Modulation by dexamethasone. Matrix Biol. 1999;18:253–260. doi: 10.1016/s0945-053x(99)00021-9. [DOI] [PubMed] [Google Scholar]
- Guo D, Tan W, Wang F, et al. Proteomic analysis of human articular cartilage: identification of differentially expressed proteins in knee osteoarthritis. Joint Bone Spine. 2008;75:439–444. doi: 10.1016/j.jbspin.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Hardingham TE, Fosang AJ. Proteoglycans: many forms and many functions. FASEB J. 1992;6:861–870. [PubMed] [Google Scholar]
- Haynes MK, Hume EL, Smith JB. Phenotypic characterization of inflammatory cells from osteoarthritic synovium and synovial fluids. Clin. Immunol. 2002;105:315–325. doi: 10.1006/clim.2002.5283. [DOI] [PubMed] [Google Scholar]
- Heathfield TF, Onnerfjord P, Dahlberg L, Heinegard D. Cleavage of fibromodulin in cartilage explants involves removal of the N-terminal tyrosine sulfate-rich region by proteolysis at a site that is sensitive to matrix metalloproteinase-13. J. Biol. Chem. 2004;279:6286–6295. doi: 10.1074/jbc.M307765200. [DOI] [PubMed] [Google Scholar]
- Hecht JT, Nelson LD, Crowder E, et al. Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nat. Genet. 1995;10:325–329. doi: 10.1038/ng0795-325. [DOI] [PubMed] [Google Scholar]
- Hein G, Franke S, Müller A, Bräunig E, Eidner T, Stein G. The determination of pyridinium crosslinks in urine and serum as a possible marker of cartilage degradation in rheumatoid arthritis. Clin. Rheumatol. 1997;16:167–172. doi: 10.1007/BF02247846. [DOI] [PubMed] [Google Scholar]
- Hermansson M, Sawaji Y, Bolton M, et al. Proteomic analysis of articular cartilage shows increased type II collagen synthesis in osteoarthritis and expression of inhibin betaA9 (activin A), a regulatory molecule for chondrocytes. J. Biol. Chem. 2004;279:43514–43521. doi: 10.1074/jbc.M407041200. [DOI] [PubMed] [Google Scholar]
- Hino K, Shiozawa S, Kuroki Y, et al. EDA-containing fibronectin is synthesized from rheumatoid synovial fibroblast-like cells. Arthritis Rheum. 1995;38:678–683. doi: 10.1002/art.1780380516. [DOI] [PubMed] [Google Scholar]
- Homandberg GA, Meyers R, Xie DL. Fibronectin fragments cause chondrolysis of bovine articular cartilage slices in culture. J. Biol. Chem. 1992;267:3597–3604. [PubMed] [Google Scholar]
- Homandberg GA, Meyers R, Williams JM. Intraarticular injection of fibronectin fragments causes severe depletion of cartilage proteoglycans in vivo. J. Rheumatol. 1993;20:1378–1382. [PubMed] [Google Scholar]
- Homandberg GA, Hui F, Wen C, et al. Fibronectin-fragment-induced cartilage chondrolysis is associated with release of catabolic cytokines. Biochem. J. 1997;321:751–757. doi: 10.1042/bj3210751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton MR, Shapiro S, Bao C, Lowenstein CJ, Noble PW. Induction and regulation of macrophage metalloelastase by hyaluronan fragments in mouse macrophages. J. Immunol. 1999;162:4171–4176. [PubMed] [Google Scholar]
- Hunter DJ, Felson DT. Osteoarthritis. BMJ. 2006;332:632–642. doi: 10.1136/bmj.332.7542.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker EB, Quinn TM, Hauselmann HJ. Quantitative structural organization of normal adult human articular cartilage. Osteoarthritis Cartilage. 2002;10:564–572. doi: 10.1053/joca.2002.0814. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Fibronectins. New York: Springer; 1990. [Google Scholar]
- Iliopoulos D, Malizos KN, Oikonomou P, Tsezou A. Integrative microRNA and proteomic approaches to identify novel osteoarthritis genes and their collaborative metabolic and inflammatory networks. PLoS ONE. 2008;3:e3740. doi: 10.1371/journal.pone.0003740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im HJ, Pacione C, Chubinskaya S, Van Wijnen AJ, Sun Y, Loeser RF. Inhibitory effects of insulin-like growth factor-1 and osteogenic protein-1 on fibronectin fragment- and interleukin-1beta-stimulated matrix metalloproteinase-13 expression in human chondrocytes. J. Biol. Chem. 2003;278:25386–25394. doi: 10.1074/jbc.M302048200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inerot S, Heinegard D, Audell L, Olsson SE. Articular-cartilage proteoglycans in aging and osteoarthritis. Biochem. J. 1978;169:143–156. doi: 10.1042/bj1690143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Takahashi KA, Arai Y, et al. The therapeutic effects of basic fibroblast growth factor contained in gelatine hydrogel microspheres on experimental osteoarthritis in the rabbit knee. Arthritis Rheum. 2006;54:264–270. doi: 10.1002/art.21561. [DOI] [PubMed] [Google Scholar]
- Jackson GC, Barker FS, Jakkula E, et al. Missense mutations in the beta strands of the single A-domain of matrilin-3 result in multiple epiphyseal dysplasia. J. Med. Genet. 2004;41:52–59. doi: 10.1136/jmg.2003.011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakkula E, Lohiniva J, Capone A, et al. A recurrent R718W mutation in COMP results in multiple epiphyseal dysplasia with mild myopathy: clinical and pathogenetic overlap with collagen IX mutations. J. Med. Genet. 2003;40:942–948. doi: 10.1136/jmg.40.12.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings L, Wu L, King KB, Hämmerle H, Cs-Szabo G, Mollenhauer J. The effects of collagen fragments on the extracellular matrix metablosim of bovine and human chondrocytes. Connect. Tissue Res. 2001;42:71–86. doi: 10.3109/03008200109014250. [DOI] [PubMed] [Google Scholar]
- Jmeian Y, El Rassi Z. Micro-high-performance liquid chromatography platform for the depletion of high-abundance proteins and subsequent on-line concentration/capturing of medium and low-abundance proteins from serum. Application to profiling of protein expression in healthy and osteoarthritis sera by 2-D gel electrophoresis. Electrophoresis. 2008;29:2801–2811. doi: 10.1002/elps.200800039. [DOI] [PubMed] [Google Scholar]
- Johnson A, Smith R, Saxne T, Hickery M, Heinegard D. Fibronectin fragments cause release and degradation of collagen-binding molecules from equine explant cultures. Osteoarthritis Cartilage. 2004;12:149–159. doi: 10.1016/j.joca.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Jung M, Christgau S, Lukoschek M, Henroksen D, Richter W. Increased urinary concentration of collagen type II C-telopeptide fragments in patients with osteoarthritis. Pathobiology. 2004;71:70–76. doi: 10.1159/000074419. [DOI] [PubMed] [Google Scholar]
- Jüni P, Reichenbach S, Trelle S, et al. Efficacy and safety of intraarticular hylan or hyaluronic acids for osteoarthritis of the knee: a randomized controlled trial. Arthritis Rheum. 2007;56:3610–3619. doi: 10.1002/art.23026. [DOI] [PubMed] [Google Scholar]
- Kevorkian L, Young DA, Darrah C, et al. Expression profiling of metalloproteinases and their inhibitors in cartilage. Arthritis Rheum. 2004;50:131–141. doi: 10.1002/art.11433. [DOI] [PubMed] [Google Scholar]
- Kizawa H, Kou I, Iida A, et al. An aspartic acid repeat polymorphism in asporin inhibits chondrogenesis and increases susceptibility to osteoarthritis. Nat. Genet. 2005;37:138–144. doi: 10.1038/ng1496. [DOI] [PubMed] [Google Scholar]
- Klatt AR, Paulsson M, Wagener R. Expression of matrilins during maturation of mouse skeletal tissues. Matrix Biol. 2002;21:289–296. doi: 10.1016/s0945-053x(02)00006-9. [DOI] [PubMed] [Google Scholar]
- Klatt AR, Paul-Klausch B, Klinger G, et al. A critical role for collagen II in cartilage matrix degradation: collagen II induces pro-inflammatory cytokines and MMPs in primary human chondrocytes. J. Orthop. Res. 2009;27:65–70. doi: 10.1002/jor.20716. [DOI] [PubMed] [Google Scholar]
- Knowlton RG, Katzenstein PL, Moskowitz RW, et al. Genetic linkage of a polymorphism in the type II procollagen gene (COL2A1) to primary osteoarthritis associated with mild chondrodysplasia. N. Engl. J. Med. 1990;322:526–530. doi: 10.1056/NEJM199002223220807. [DOI] [PubMed] [Google Scholar]
- Knudson W, Casey B, Nishida Y, Eger W, Kuettner KE, Knudson CB. Hyaluronan oligosaccharides perturb cartilage matrix homeostasis and induce chondrocytic chondrolysis. Arthritis Rheum. 2000;43:1165–1174. doi: 10.1002/1529-0131(200005)43:5<1165::AID-ANR27>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Kurz B, Jin M, Patwari P, Cheng DM, Lark MW, Grodzinsky AJ. Biosynthetic response and mechanical properties of articular cartilage after injurious compression. J. Orthop. Res. 2001;19:1140–1146. doi: 10.1016/S0736-0266(01)00033-X. [DOI] [PubMed] [Google Scholar]
- L’Hermette M, Polle G, Tourny-Chollet C, Dujardin F. Hip passive range of motion and frequency of radiographic hip osteoarthritis in former elite handball players. Br. J. Sports Med. 2006;40:45–49. doi: 10.1136/bjsm.2005.019026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lark MW, Bayne EK, Flanagan J, et al. Aggrecan degradation in human cartilage. Evidence for both matrix metalloproteinase and aggrecanase activity in normal, osteoarthritic, and rheumatoid joints. J. Clin. Invest. 1997;100:93–106. doi: 10.1172/JCI119526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Fitzgerald JB, Dimicco MA, Grodzinsky AJ. Mechanical injury of cartilage explants causes specific time-dependent changes in chondrocyte gene expression. Arthritis Rheum. 2005;52:2386–2395. doi: 10.1002/art.21215. [DOI] [PubMed] [Google Scholar]
- Loening AM, James IE, Levenston ME, et al. Injurious mechanical compression of bovine articular cartilage induces chondrocyte apoptosis. Arch. Biochem. Biophys. 2000;381:205–212. doi: 10.1006/abbi.2000.1988. [DOI] [PubMed] [Google Scholar]
- Loeser RF, Todd MD, Seely BL. Prolonged treatment of human osteoarthritic chondrocytes with insulin-like growth factor-1 stimulates proteoglycan synthesis but not proteoglycan matrix accumulation in alginate cultures. J. Rheumatol. 2003;30:1565–1570. [PubMed] [Google Scholar]
- Loeser RF, Yammani RR, Carlson CS, et al. Articular chondrocytes express the receptor for advanced glycation products: Potential role in osteoarthritis. Arthritis Rheum. 2005;52:2376–2385. doi: 10.1002/art.21199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmander LS, Neame PJ, Sandy JD. The structure of aggrecan fragments in human synovial fluid. Evidence that aggrecanase mediates cartilage degradation in inflammatory joint disease, joint injury, and osteoarthritis. Arthritis Rheum. 1993;36:1214–1222. doi: 10.1002/art.1780360906. [DOI] [PubMed] [Google Scholar]
- Long D, Blake S, Song XY, Lark M, Loeser RF. Human articular chondrocytes produce IL-7 and respond to IL-7 with increased production of matrix metalloproteinase-13. Arthritis Res. Ther. 2008;10:R23. doi: 10.1186/ar2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotze MT, Zeh HJ, Rubartelli A, et al. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol. Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. Review. [DOI] [PubMed] [Google Scholar]
- Mankin HJ, Lippiello L. Biochemical and metabolic abnormalities in articular cartilage from osteo-arthritic human hips. J. Bone Joint Surg. Am. 1970;52:424–434. [PubMed] [Google Scholar]
- Mehraban F, Kuo SY, Riera H, Chang C, Moskowitz RW. Prostromelysin and procollagenase genes are differentially up-regulated in chondrocytes from the knees of rabbits with experimental osteoarthritis. Arthritis Rheum. 1994;37:1189–1197. doi: 10.1002/art.1780370813. [DOI] [PubMed] [Google Scholar]
- Mustafa Z, Dowling B, Chapman K, Sinsheimer JS, Carr A, Loughlin J. Investigating the aspartic acid (D) repeat of asporin as a risk factor for osteoarthritis in a UK Caucasian population. Arthritis Rheum. 2005;52:3502–3506. doi: 10.1002/art.21399. [DOI] [PubMed] [Google Scholar]
- Nagase H, Kashiwagi M. Aggrecanases and cartilage matrix degradation. Arthritis Res. Ther. 2003;5:94–103. doi: 10.1186/ar630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann E, Barnum SR, Tarner IH, et al. Local production of complement proteins in rheumatoid arthritis synovium. Arthritis Rheum. 2002;46:934–945. doi: 10.1002/art.10183. [DOI] [PubMed] [Google Scholar]
- O’Byrne EM, Blancuzzi V, Wilson DE, Wong M, Jeng AY. Elevated substance P and accelerated cartilage degradation in rabbit knees injected with interleukin-1 and tumor necrosis factor. Arthritis Rheum. 1990;33:1023–1028. doi: 10.1002/art.1780330715. [DOI] [PubMed] [Google Scholar]
- O’Neill L. Primer: Toll-like receptor signalling pathways – what do rheumatologists need to know? Nat. Clin. Pract. Rheumatol. 2008;4:319–327. doi: 10.1038/ncprheum0802. [DOI] [PubMed] [Google Scholar]
- Okada Y. Proteinases and matrix degradation. In: Ruddy S, Harris ED, Sledge C, editors. From Kelley’s Textbook of Rheumatology. Philadelphia: Saunders; 2001. pp. 55–73. [Google Scholar]
- Okamura Y, Watari M, Jerud ES, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J. Biol. Chem. 2001;276:10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- Oldberg A, Antonsson P, Lindblom K, Heinegard D. A collagenbinding 59-kd protein (fibromodulin) is structurally related to the small interstitial proteoglycans PG-S1 and PG-S2 (decorin) EMBO J. 1989;8:2601–2604. doi: 10.1002/j.1460-2075.1989.tb08399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page Thomas DP, King B, Stephens T, Dingle JT. In vivo studies of cartilage regeneration after damage induced by catabolin/interleukin-1. Ann. Rheum. Dis. 1991;50:75–80. doi: 10.1136/ard.50.2.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettipher ER, Higgs GA, Henderson B. Interleukin 1 induces leukocyte infiltration and cartilage proteoglycan degradation in the synovial joint. Proc. Natl. Acad. Sci. U.S.A. 1986;83:8749–8753. doi: 10.1073/pnas.83.22.8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porée B, Kypriotou M, Chadjichristos C, et al. Interleukin-6 (IL-6) and/or soluble IL-6 receptor down-regulation of human type II collagen gene expression in articular chondrocytes requires a decrease of Sp1.Sp3 ratio and of the binding activity of both factors to the COL2A1 promoter. J. Biol. Chem. 2008;283:4850–4865. doi: 10.1074/jbc.M706387200. [DOI] [PubMed] [Google Scholar]
- Powell AJ, Little CB, Hughes CE. Low molecular weight isoforms of the aggrecanases are responsible for the cytokine-induced proteolysis of agrecan in a porcine chondrocyte culture system. Arthritis Rheum. 2007;56:3010–3019. doi: 10.1002/art.22818. [DOI] [PubMed] [Google Scholar]
- Pulai JI, Chen H, Im HJ, et al. NF-kappa B mediates the stimulation of cytokine and chemokine expression by human articular chondrocytes in response to fibronectin fragments. J. Immunol. 2005;174:5781–5788. doi: 10.4049/jimmunol.174.9.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintarelli G, Vocaturo A, Roden L, Bellocci M, Vassallo LM. Role of hyaluronic acid in the in vivo aggregation of cartilage proteoglycans. Connect. Tissue Res. 1978;5:237–248. doi: 10.3109/03008207809152278. [DOI] [PubMed] [Google Scholar]
- Recnik G, Kralj-Iglic V, Iglic A, et al. The role of obesity, biomechanical constitution of the pelvis and contact joint stress in progression of hip osteoarthritis. Osteoarthritis Cartilage. 2008;17:865–868. doi: 10.1016/j.joca.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Rencic A, Gehris AL, Lewis SD, Hume EL, Bennett VD. Splicing patterns of fibronectin mRNA from normal and osteoarthritic human articular cartilage. Osteoarthritis Cartilage. 1995;3:187–196. doi: 10.1016/s1063-4584(05)80053-6. [DOI] [PubMed] [Google Scholar]
- Riessen R, Fenchel M, Chen H, Axel DI, Karsch KR, Lawler J. Cartilage oligomeric matrix protein (thrombospondin-5) is expressed by human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2001;21:47–54. doi: 10.1161/01.atv.21.1.47. [DOI] [PubMed] [Google Scholar]
- Rizkalla G, Reiner A, Bogoch E, Poole AR. Studies of the articular cartilage proteoglycan aggrecan in health and osteoarthritis. Evidence for molecular heterogeneity and extensive molecular changes in disease. J. Clin. Invest. 1992;90:2268–2277. doi: 10.1172/JCI116113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogerson FM, Stanton H, East CJ, et al. Evidence of a novel aggrecan-degrading activity in cartilage. Studies of mice deficient in both ADAMST-4 and ADAMTS-5. Arthritis Rheum. 2008;58:1664–1673. doi: 10.1002/art.23458. [DOI] [PubMed] [Google Scholar]
- Roughley PJ. The structure and function of cartilage proteoglycans. Eur. Cell. Mater. 2006;12:92–101. doi: 10.22203/ecm.v012a11. [DOI] [PubMed] [Google Scholar]
- Ruettger A, Schueler S, Mollenhauer JA, Wiederanders B. Cathepsins B, K, and L are regulated by a defined collagen type II peptide via activation of classical protein kinase C and p38 MAP kinase in articular chondrocytes. J. Biol. Chem. 2008;283:1043–1051. doi: 10.1074/jbc.M704915200. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E, Peirschbacher MD. New perspectives in cell adhesion: RGD and intergins. Science. 1987;238:491–497. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- Saito S, Yamaji N, Yasunaga K, et al. The fibronectin extra domain A activates matrix metalloproteinase gene expression by an interleukin-1-dependent mechanism. J. Biol. Chem. 1999;274:30756–30763. doi: 10.1074/jbc.274.43.30756. [DOI] [PubMed] [Google Scholar]
- Saklatvala J. Tumour necrosis factor alpha stimulates resorption and inhibits synthesis of proteoglycan in cartilage. Nature. 1986;322:547–549. doi: 10.1038/322547a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandy JD, Neame PJ, Boynton RE, Flannery CR. Catabolism of aggrecan in cartilage explants. Identification of a major cleavage site within the interglobular domain. J. Biol. Chem. 1991;266:8683–8685. [PubMed] [Google Scholar]
- Sandy JD, Flannery CR, Neame PJ, Lohmander LS. The structure of aggrecan fragments in human synovial fluid. Evidence for the involvement in osteoarthritis of a novel proteinase which cleaves the Glu 373-Ala 374 bond of the interglobular domain. J. Clin. Invest. 1992;89:1512–1516. doi: 10.1172/JCI115742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöberg A, Onnerfjord P, Mörgelin M, Heinegård D, Blom AM. The extracellular matrix and inflammation: fibromodulin activates the classical pathway of complement by directly binding C1q. J. Biol. Chem. 2005;280:32301–32308. doi: 10.1074/jbc.M504828200. [DOI] [PubMed] [Google Scholar]
- Sledge CB. Structure, development, and function of joints. Orthop. Clin. North Am. 1975;6:619–628. [PubMed] [Google Scholar]
- Stanton H, Ung L, Fosang AJ. The 45 kDa collagen-binding fragment of fibronectin induces matrix metalloproteinase-13 synthesis by chondrocytes and aggrecan degradation by aggrecanases. Biochem. J. 2002;364:181–190. doi: 10.1042/bj3640181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton H, Rogerson FM, East CJ, et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature. 2005;434:648–652. doi: 10.1038/nature03417. [DOI] [PubMed] [Google Scholar]
- Stefansson SE, Jonsson H, Ingvarsson T, et al. Genomewide scan for hand osteoarthritis: a novel mutation in matrilin-3. Am. J. Hum. Genet. 2003;72:1448–1459. doi: 10.1086/375556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens AL, Wishnok JS, White FM, Grodzinsky AJ, Tannenbaum SR. Mechanical injury and cytokines cause loss of cartilage integrity and upregulate proteins associated with catabolism, immunity, inflammation and repair. Mol. Cell. Proteomics. 2009 doi: 10.1074/mcp.M800181-MCP200. Feb 4 (epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strusberg I, Sembaj A, Tabares S, et al. Association analysis of genotypic frequencies of matrilin-1 gene in patients with osteoarthritis. Clin. Exp. Rheumatol. 2002;20:543–545. [PubMed] [Google Scholar]
- Su SL, Tsai CD, Lee CH, Salter DM, Lee HS. Expression and regulation of Toll-like receptor 2 by IL-1beta and fibronectin fragments in human articular chondrocytes. Osteoarthritis Cartilage. 2005;13:879–886. doi: 10.1016/j.joca.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Hashimoto S, Kubo T, Hirasawa Y, Lotz M, Amiel D. Hyaluronan suppressed nitric oxide production in the meniscus and synovium of rabbit osteoarthritis model. J. Orthop. Res. 2001;19:500–503. doi: 10.1016/S0736-0266(00)90024-X. [DOI] [PubMed] [Google Scholar]
- Tawara T, Shingu M, Nobunaga M, Naono T. Effects of recombinant human IL-1 beta on production of prostaglandin E2, leukotriene B4, NAG, and superoxide by human synovial cells and chondrocytes. Inflammation. 1991;15:145–157. doi: 10.1007/BF00917509. [DOI] [PubMed] [Google Scholar]
- Termeer C, Benedix F, Sleeman J, et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J. Exp. Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesar BM, Jiang D, Liang J, Palmer SM, Noble PW, Goldstein DR. The role of hyaluronan degradation products as innate alloimmune agonists. Am. J. Transplant. 2006;6:2622–2635. doi: 10.1111/j.1600-6143.2006.01537.x. [DOI] [PubMed] [Google Scholar]
- Tetlow LC, Adlam DJ, Woolley DE. Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum. 2001;44:585–594. doi: 10.1002/1529-0131(200103)44:3<585::AID-ANR107>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Toole B. Hyaluronan, hyaluronan synthesis, and hyaluronan-binding proteins. In: Kreis T, Vale R, editors. Guidebook to the Extracellular Matrix, Anchor and Adhesion Proteins. New York: Oxford University Press; 1999. pp. 430–433. [Google Scholar]
- Tortorella MD, Burn TC, Pratta MA, et al. Purification and cloning of aggrecanase-1: a member of the ADAMTS family of proteins. Science. 1999;284:1664–1666. doi: 10.1126/science.284.5420.1664. [DOI] [PubMed] [Google Scholar]
- Van Den Berg WB. Lessons from animal models of arthritis. Curr. Rheumatol. Rep. 2002;4:232–239. doi: 10.1007/s11926-002-0070-5. [DOI] [PubMed] [Google Scholar]
- Vaughan-Thomas A, Young RD, Phillips AC, Duance VC. Characterization of type XI collagen-glycosaminoglycan interactions. J. Biol. Chem. 2001;276:5303–5309. doi: 10.1074/jbc.M008764200. [DOI] [PubMed] [Google Scholar]
- Vincourt JB, Vignaud JM, Lionneton F, et al. Increased expression of matrilin-3 not only in osteoarthritic articular cartilage but also in cartilage-forming tumors, and down-regulation of SOX9 via epidermal growth factor domain 1-dependent signalling. Arthritis Rheum. 2008;58:2798–2808. doi: 10.1002/art.23761. [DOI] [PubMed] [Google Scholar]
- Voelcker V, Gebhardt C, Averback M, et al. Hyaluronan fragments induce cytokine and metalloprotease upregulation in human melanoma cells in part by signalling via TLR4. Exp. Dermatol. 2008;17:100–107. doi: 10.1111/j.1600-0625.2007.00638.x. [DOI] [PubMed] [Google Scholar]
- Werb Z, Tremble PM, Behrendtsen O, Crowley E, Damsky CH. Signal transduction through the fibronectin receptor induces collagenase and stromelysin gene expression. J. Cell Biol. 1989;109:877–889. doi: 10.1083/jcb.109.2.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiberg C, Klatt AR, Wagener R, et al. Complexes of matrilin-1 and biglycan or decorin connect collagen VI microfibrils to both collagen II and aggrecan. J. Biol. Chem. 2003;278:37698–37704. doi: 10.1074/jbc.M304638200. [DOI] [PubMed] [Google Scholar]
- Xie D-L, Meyers R, Homandberg GA. Fibronectin fragments in osteoarthritic synovial fluid. J. Rheumatol. 1992;19:1448–1452. [PubMed] [Google Scholar]
- Yasuda T, Poole AR. A fibronectin fragment induces type II collagen degradation by collagenase through an interleukin-1-mediated pathway. Arthritis Rheum. 2002;46:138–148. doi: 10.1002/1529-0131(200201)46:1<138::AID-ART10051>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Yasuda T, Poole AR, Shimizu M, et al. Involvement of CD44 in induction of matrix metalloproteinases by a COOH-terminal heparin-binding fragment of fibronectin in human articular cartilage in culture. Arthritis Rheum. 2003a;48:1271–1280. doi: 10.1002/art.10951. [DOI] [PubMed] [Google Scholar]
- Yasuda T, Shimizu M, Nakagawa T, Julovi SM, Nakamura T. Matrix metalloproteinase production by COOH-terminal heparin-binding fibronectin fragment in rheumatoid synovial cells. Lab. Invest. 2003b;83:153–162. doi: 10.1097/01.lab.0000056999.08437.b2. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Hui W, Litherland GJ, et al. Differential Toll-like receptor-dependent collagenase expression in chondrocytes. Ann. Rheum. Dis. 2008;67:1633–1641. doi: 10.1136/ard.2007.079574. [DOI] [PubMed] [Google Scholar]
- Zhen EY, Brittain IJ, Laska DA, et al. Characterization of metalloprotease cleavage products of human articular cartilage. Arthritis Rheum. 2008;58:2420–2431. doi: 10.1002/art.23654. [DOI] [PubMed] [Google Scholar]
- Zlotnik A, Yoshie O, Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 2006;7:243. doi: 10.1186/gb-2006-7-12-243. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]