

Abstract
The genetic structure of a global sample of 170 clinical and nonclinical Saccharomyces cerevisiae isolates was analyzed using 12 microsatellite markers. High levels of genetic diversity were revealed both among the clinical and among the nonclinical S. cerevisiae isolates without significant differentiation between these two groups of isolates, rendering a single origin of pathogenic isolates unlikely. This suggests that S. cerevisiae is a true opportunistic pathogen, with a diversity of unrelated genetic backgrounds able to cause infections in humans, and that the ability of S. cerevisiae isolates to cause infections is likely due to a combination of their phenotypic plasticity and the immune system status of the exposed individuals. As was previously reported for bread, beer and wine strains and for environmental S. cerevisiae isolates, the microsatellite genotypes indicated ploidy level variation, from possibly haploid up to tetraploid, among clinical S. cerevisiae isolates. However, rather than haploid, sporulation proficiency and spore viability data indicated that most S. cerevisiae isolates that were monoallelic at all examined microsatellite loci were likely homothallic and self-diploidized. Interestingly, the proportion of heterozygous clinical isolates was found to be significantly higher than the proportion of heterozygous nonclinical isolates, suggesting a selective advantage of heterozygous S. cerevisiae yeasts in clinical environments.
Keywords: Saccharomyces cerevisiae, clinical isolates, microsatellites, genetic diversity, ploidy level, heterosis
Introduction
Saccharomyces cerevisiae is a well-studied model organism that has aided our understanding of nearly all eukaryotic cellular processes. It provided the first eukaryotic genome to be completely sequenced (Dujon, 1996) and two-thirds of the approximately 6000 open reading frames have been characterized (Kellis et al., 2003). However, it is only recently that S. cerevisae gained interest as a model for studies in ecological and evolutionary genetics (Landry et al., 2006).
S. cerevisiae is a diplontic, naturally homothallic yeast with a predominantly clonal reproduction, and a recent study showed that, besides natural genetic drift and migration, human technology has been highly influential on its genetic diversity (Legras et al., 2007). Domesticated strains of S. cerevisiae have been used for centuries in baking, brewing, distilling and wine-making, and it is believed that these strains were derived from natural populations not necessarily associated with human activities (Fay and Benavides, 2005; Legras et al., 2007). Although strain isolation from nature is rare and its natural environments remain uncertain, S. cerevisiae has been isolated from a wide variety of substrates, e.g. damaged fruits and trees, soil associated with oak trees, a river and insects (Slavikova and Vadkertiova, 1997; Mortimer and Polsinelli, 1999; Sniegowski et al., 2002; Naumov et al., 2003; Aa et al., 2006).
For years, S. cerevisiae was considered to be a nonpathogenic yeast, with GRAS (“generally recognized as safe”) status in the food industry. However, because of an increase in the number of reports of mucosal and systemic infections, S. cerevisiae is now considered to be an opportunistic pathogen of low virulence (de Hoog, 1996). Most incidences of human infection involve immunocompromised patients or patients who were already severely ill, but fatal S. cerevisiae infections in apparently immunocompetent patients have been described (Smith et al., 2002). Infection is believed to occur by oral administration or other ways of exogenous inoculation, and although the source of emergent S. cerevisiae pathogens is still unclear, there is evidence that at least some of these are food and drink-related (de Llanos et al., 2004; de Llanos et al., 2006). In particular, the probiotic S. cerevisiae var. boulardii strain, which is orally administered to treat antibiotic-associated diarrhoea and Clostridium difficile infections (Guslandi, 2006), has been directly associated with fungaemia in multiple patients (Bassetti et al., 1998; Munoz et al., 2005).
Here, we report the use of 12 microsatellite markers to describe the genetic structure of a global sample of S. cerevisiae isolates obtained from clinical and a variety of nonclinical sources. Our results illustrate the high levels of genetic diversity, more-or-less global panmixia and ploidy level variation in S. cerevisiae, in addition to the absence of strong differentiation between clinical and nonclinical S. cerevisiae isolates and a possible selective advantage for heterozygous isolates in clinical environments.
Material and Methods
Saccharomyces cerevisiae isolates
One hundred and seventy S. cerevisiae isolates of diverse geographical origins, obtained from our own collection and from other laboratories or public collections, were used in this study. Taxonomic identity of the isolates was confirmed using a multispecies based microarray and interspecific hybrids were excluded from this study (Muller and McCusker, 2009). Eighty-seven isolates were of clinical origin, while 83 isolates were obtained from a variety of nonclinical environments (see Table S1, Supplementary Material; please direct strain requests to the appropriate culture collections or laboratories). Sporulation proficiency and spore viability of the yeast isolates were recorded using methods described by Sherman (1991), and percentages of viable spores were calculated based on the analysis of at least 12 tetrads. For sporulation proficient isolates that produced high frequencies of viable spores, the genotype at the HO locus was determined by analysing the sporulation proficiency of the isolates' segregants. Sporulation proficient segregants were considered HO (homothallic) and sporulation deficient segregants were considered ho (heterothallic) if mating type tests (Sherman, 1991) showed them to be mating-competent. Isolates that exhibited 2:2 segregation of HO:ho were considered heterozygous at HO. All isolates were kept in glycerol (15% v/v) at -80°C, or for short-term storage on yeast extract, peptone and 2% dextrose (YPD) agar medium.
Microsatellite analysis
DNA was extracted from overnight cultures grown in 50 ml YPD medium using the QIAGEN Genomic-tip 100/G (QIAGEN) following the manufacturer's instructions. Twelve microsatellite loci dispersed over 7 chromosomes were used to characterize the yeast isolates (see Table 1 for chromosomal locations): C3, C4, C5, C6, C8, C9, C11, SCAAT1, SCAAT5, SCYOR267c, YKL172w and YPL009c, described by Legras et al. (2005). Polymerase chain reactions (PCRs) were set up in total volumes of 20 μl containing 0.5 μM of each primer (see Legras et al. (2005) for primer sequences), 1 U Taq DNA polymerase, 10 mM Tris-HCl pH 9, 15 mM MgCl2, 50 mM KCl, 200 μM of all four dNTPs and approximately 2 ng of genomic DNA template. The following PCR temperature profile was used: initial denaturation at 95 °C for 2 min; 34 cycles of 95 °C for 30 s, 57 °C for 30 s, 72 °C for 1 min; final extension at 72 °C for 30 min. The PCR products were diluted 50 fold and 1 μl of the dilution was added to 18.75 μl of formamide and 0.25 μl of HD400ROX size standard. After denaturation, the PCR products were sized on an ABI 3730 xl DNA Analyzer and allele classes were designated using genemarker v1.71 software (Softgenetics).
Table 1.
Characteristics of microsatellite markers
| Observed heterozygosity (HO) | ||||
|---|---|---|---|---|
| Name of marker | Chromosome no. | No. of alleles found | Clinical isolates | Nonclinical isolates |
| C5 C3 |
VI VII |
33 21 |
0.69 0.43 |
0.51 0.4 |
| C8 | VII | 19 | 0.84 | 0.54 |
| C11 | X | 28 | 0.8 | 0.69 |
| YKL172w | XI | 18 | 0.43 | 0.11 |
| SCAAT1 | XIII | 40 | 0.71 | 0.49 |
| C4 | XV | 40 | 0.51 | 0.29 |
| C9 | XV | 18 | 0.38 | 0.26 |
| SCYOR267c | XV | 42 | 0.44 | 0.17 |
| C6 | XVI | 20 | 0.48 | 0.49 |
| SCAAT5 | XVI | 16 | 0.56 | 0.23 |
| YPL009c | XVI | 32 | 0.64 | 0.66 |
| Average | 27 | 0.57 | 0.4 | |
Characteristics of 12 microsatellite markers (see Legras et al., 2005) used to genotype a global sample of 170 clinical and nonclinical Saccharomyces cerevisiae isolates; name, chromosome number, observed number of alleles and observed heterozygosities for the subsets of diploid clinical and nonclinical isolates are given for each marker.
Statistical analysis
Isolate ploidy levels (di-, tri- and tetraploidy) were estimated based on the maximum number of alleles observed at the microsatellite loci. Estimation of ploidy level was not possible for yeast isolates with monoallelic profiles at all 12 microsatellite loci, although sporulation proficiency data suggested a diploid state for most of these isolates (see Results).
As suggested by Kloda et al. (2008) for the comparison of genetic diversity across ploidy levels, a matrix of composite genotypes was constructed by scoring the microsatellite alleles as either present (1) or absent (0). Correspondence analysis, implemented by the “dudi.coa” routine in r v2.7.2 (R Development Core Team, 2008), was used to explore the binary microsatellite allele presence/absence data. A dendrogram based on pairwise Jaccard coefficients between the isolates was constructed using the UPGMA method implemented in the “neighbor” package of phylip v3.68 (Felsenstein, 1989) and drawn using the “drawgram” package through the mobyle portal (http://mobyle.pasteur.fr/cgibin/MobylePortal/portal.py).
Observed heterozygosities (HO) were calculated for the subsets of diploid clinical and nonclinical S. cerevisiae isolates, and analysis of molecular variance (amova; Excoffier et al., 1992), as implemented in the software arlequin v3.11 (Excoffier et al., 2005), was applied to the nontransformed microsatellite data of the diploid S. cerevisiae isolates in order to partition the genotypic variance into components attributable to different hierarchical levels. Two models were examined in which amova partitioned the total variance into components due to differences between groups of yeast isolates and differences between isolates within groups: a first model which grouped the isolates based on their geographical origin (only American and European isolates were included) and a second model which considered a group of clinical and a group of nonclinical isolates. In a third model, nested analysis of molecular variance was applied to estimate the genetic differentiation between clinical and nonclinical S. cerevisiae isolates when they are grouped according to their geographical origin (only American and European isolates were included). In all cases, pairwise distances were defined as the sum of squared differences in allele size (RST) and the significance of the variance components at the different hierarchical levels was assessed with a permutation procedure (15,000 permutations).
Results
Genotyping 170 S. cerevisiae isolates of clinical and nonclinical origin at 12 microsatellite loci resulted in 161 unique multilocus genotypes, with 15 isolates having genotypes identical to others in the study sample. All isolates with identical genotypes were obtained from similar ecological niches in the same geographical location (see Table S1, Supplementary Material). High levels of allelic diversity were observed at all microsatellite loci, with 16 (SCAAT5) to 42 (SCYOR267c) alleles per locus and an average of 27 alleles per locus (see Table 1). The maximum number of alleles per locus and per isolate varied between 1 and 4, suggesting variability of the ploidy level among the yeast isolates. Based on their multilocus genotypes, 96 isolates (56%) were predicted to be diploid (bi-allelic at ≥ 1 locus), 23 isolates (14%) to be triploid (tri-allelic at ≥ 1 locus) and 18 isolates (11%) to be tetraploid (tetra-allelic at ≥ 1 locus). For the subsets of diploid clinical and nonclinical isolates, the average observed heterozygosities across the microsatellite loci were respectively 0.57 and 0.40 (see Table 1).
The remaining 33 isolates (19%) had monoallelic profiles for all examined microsatellite markers, and there was a significant difference between the number of clinical and the number of nonclinical isolates with exclusively monoallelic profiles (χ2d.f.=1 = 29.03, P-value < 0.01): 3 of the 87 clinical isolates (3%) showed only monoallelic profiles versus 30 of the 83 nonclinical isolates (36%; see Table 2). Because S. cerevisiae isolates can be homothallic or heterothallic, prediction of the ploidy level based on their multilocus genotypes was not possible for the isolates with only monoallelic marker profiles; however, the high proportion of sporulation proficiency among these isolates (82%) and their high average spore viability (90%) indicated that most of these yeasts are likely diploid, homothallic (HO/HO) and self-diploidized.
Table 2.
Microsatellite-predicted ploidy levels of clinical and nonclinical S. cerevisiae isolates
| Origin | ||
|---|---|---|
| Ploidy state | Clinical | Nonclinical |
| Diploid | 59 (68%) | 37 (45%) |
| Triploid | 13 (15%) | 10 (12%) |
| Tetraploid | 12 (14%) | 6 (7%) |
| Unknown (monoallelic)* | 3 (3%) | 30 (36%) |
| Total | 87 (100%) | 83 (100%) |
Predicted ploidy levels of 170 clinical and nonclinical S. cerevisiae isolates, based on the maximum number of alleles observed per individual among 12 microsatellite marker loci;
, unknown ploidy level due to monoallelism of the profiles of all 12 microsatellite markers.
The genotype at the HO locus, which affects the mating system in S. cerevisiae, was determined for a subset of the isolates to test whether sporulation proficient isolates with exclusively monoallelic microsatellite marker profiles were homothallic. For most isolates, sporulation incompetence or low spore viability prohibited unambiguous determination of the genotype at the HO locus. However, precise determination of the genotype at the HO locus was possible for 36 isolates, i.e. 8 clinical and 28 nonclinical isolates. Among the 8 clinical isolates, 3 were homothallic (HO/HO), 4 were heterothallic (ho/ho) and 1 was heterozygous (HO/ho) at HO, while 27 of the 28 nonclinical isolates were homothallic (HO/HO) and 1 was heterothallic (ho/ho; see Table 3).
Table 3.
Genotypes at the HO locus of S. cerevisiae isolates
| Genotype at HO | ||||
|---|---|---|---|---|
| Origin of isolates | Microsatellite profiles | HO/HO | HO/ho | ho/ho |
| Clinical | Polyallelic (diploid) | 2 | 1 | 4 |
| Monoallelic | 1 | 0 | 0 | |
| Nonclinical | Polyallelic (diploid) | 4 | 0 | 1 |
| Monoallelic | 23 | 0 | 0 | |
Genotypes at the HO locus of 36 clinical and nonclinical Saccharomyces cerevisiae isolates with either exclusively monoallelic profiles at 12 examined microsatellite loci or polyallelic profiles suggestive of a diploid state of the isolates; the genotype at the HO locus was determined by analysing the sporulation proficiency of the isolates' segregants; sporulation proficient segregants were considered HO (homothallic) and sporulation deficient segregants were considered ho (heterothallic); isolates that exhibited 2:2 segregation of HO:ho were considered heterozygous at HO, and sporulation deficient segregants were tested for heterothallism and haploidy by mating type tests.
Consistent with their being homozygous throughout their genomes (except for the MAT locus) due to self-diploidization, 24 of the 36 HO-genotyped isolates had exclusively monoallelic microsatellite marker profiles and were all homothallic (HO/HO; see Table 3). Of the remaining 12 isolates, which were predicted to be diploid based on their microsatellite marker profiles, 6 were homothallic (HO/HO), while 5 were heterothallic (ho/ho) and 1 was heterozygous (HO/ho) at the HO locus (see Table 3). For 2 of the 6 homothallic (HO/HO) isolates with heterozygous microsatellite loci, heterozygosity was restricted to 1 of the 12 microsatellite marker loci and involved alleles that differed in length by only one repeat unit. In contrast, heterozygosity was more extensive (at least half of the microsatellite loci were heterozygous) and involved alleles that differed in length by multiple repeat units for the remaining 4 homothallic (HO/HO) isolates with heterozygous microsatellite loci.
One hundred and fourty-seven of the 170 yeast isolates (86%) were sporulation proficient and 120 of the sporulation proficient isolates (82%) produced viable spores. No significant difference in sporulation proficiency was found across the three ploidy levels (χ2d.f.=2 = 0.0577, P-value = 0.97), but, as expected, significantly fewer sporulation proficient triploid isolates (26%) produced viable spores compared to diploid (73%) and tetraploid isolates (78%; χ2d.f.=2 = 19.40, P-value < 0.01). In accordance, the average percentages of viable spores produced by sporulation proficient diploid, triploid and tetraploid isolates were respectively 41%, 3% and 42%. No significant difference was found between the percentages of viable spores produced by sporulation proficient clinical and nonclinical diploid isolates (data not shown).
Figure 1 illustrates the results of the correspondence analysis as a plane projection of the two most informative axes accounting for the genetic structure among the 170 S. cerevisiae isolates of clinical and nonclinical origin. In total, the first two factors accounted for approximately 10% of the total variation in the sample. No separate clustering of clinical and nonclinical isolates of S. cerevisiae was obvious, an observation which was also apparent in the UPGMA tree (see Figure S1, Supplementary Material), and the genetic diversity of clinical isolates appeared at least as high as the diversity of nonclinical isolates.
Figure 1.
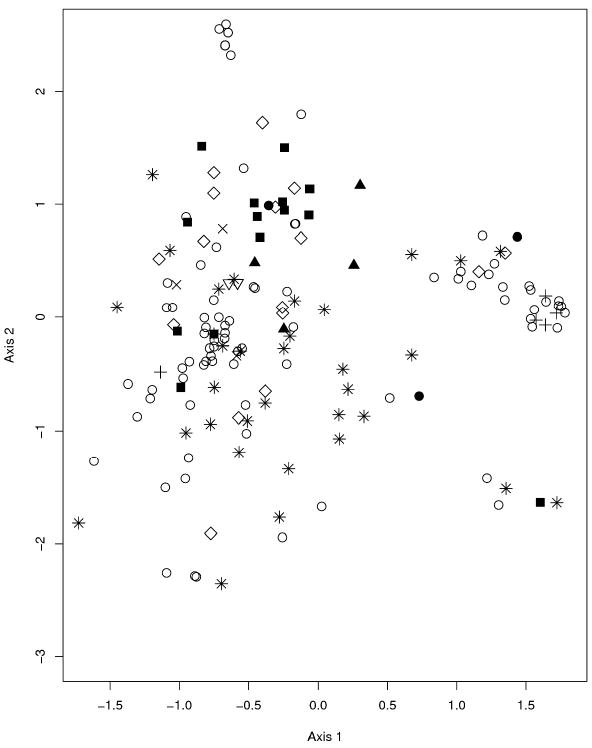
Correspondence analysis of microsatellite allele presence/absence data in clinical and nonclinical S. cerevisiae isolates
Results of a correspondence analysis performed with the “dudi.coa” routine in R (R Development Core Team, 2007) using microsatellite allele presence/absence data of 12 microsatellite loci in 170 clinical and nonclinical Saccharomyces cerevisiae isolates of diverse geographical origins; “■”, wine strain; “▲”, distillery strain; “○”, clinical strain; “◊”, fruit strain; “△”, S. boulardii; “●”, brewery strain; “+”, commercial strain; “×”, lab strain; “*”, other.
In all three models examined, amova indicated that most of the genetic variation in the subset of diploid S. cerevisiae isolates was due to differences within groups or subgroups of geographically or environmentally (clinical versus nonclinical) related isolates (between 97% and 99%; see Table 4). Differentiation between European and American isolates was found to be low and nonsignificant (3%, P-value = 0.15; see Table 4a), as was the case for the differentiation between clinical and nonclinical isolates in the global sample (1%, P-value = 0.21; see Table 4b). Nested amova showed that differentiation between groups and subgroups was also nonsignificant when clinical and nonclinical S. cerevisiae isolates were grouped separately according to their geographical origins (see Table 4c).
Table 4.
Analysis of molecular variance
| Source of variation | d.f. | Sum of squares | Variance component | % of total | P-value |
|---|---|---|---|---|---|
| (a) Grouping according to geographical origin | |||||
| Among groups | 1 | 9468.13 | 96.44 | 2.65 | 0.15 |
| Within groups | 122 | 432480.92 | 3544.93 | 97.35 | |
| Total | 123 | 441949.05 | 3641.36 | ||
| (b) Clinical versus nonclinical isolates | |||||
| Among groups | 1 | 8499.57 | 54.13 | 1.47 | 0.21 |
| Within groups | 190 | 689280.75 | 3627.79 | 98.53 | |
| Total | 191 | 697780.32 | 3681.92 | ||
| (c) Clinical versus nonclinical isolates grouped according to geographical origin | |||||
| Among groups | 1 | 9467.66 | 61.83 | 1.7 | 0.33 |
| Among subgroups | 2 | 9684.49 | 56.53 | 1.55 | 0.51 |
| Within subgroups | 120 | 422796.43 | 3523.3 | 96.75 | |
| Total | 123 | 441948.58 | 3641.66 | ||
Analysis of molecular variance (amova; Excoffier et al., 1992) with data from 12 microsatellite loci in diploid Saccharomyces cerevisiae isolates of clinical and nonclinical origin. (a) amova with data of European (28) and American (34) isolates grouped separately; (b) amova with data of clinical (60) and nonclinical (36) isolates grouped separately; (c) nested analysis with data of clinical and nonclinical isolates (subgroups) grouped separately according to geographical origin. Data show the degrees of freedom (d.f.), the sum of squared deviations, the variance component estimates, the percentage of total variance contributed by each component and the significance of the variance components (P-value) estimated computing 15000 permutations.
Discussion
In this study, we used data from 12 polymorphic microsatellite marker loci to analyze the genetic diversity among clinical and nonclinical S. cerevisiae isolates of diverse geographical origins. A high level of intraspecific genetic diversity was revealed, at least as high as described in previous studies (Malgoire et al., 2005; Ezov et al., 2006; Legras et al., 2007), with 161 unique multilocus genotypes detected among the global sample of 170 isolates.
The maximum number of alleles observed per microsatellite locus and per isolate varied between one and four, and it has been demonstrated that this number is a good indicator of the ploidy level of S. cerevisiae isolates which is known to vary from haploid up to tetraploid (Ezov et al., 2006). Based on their multilocus genotypes, approximately 55% of the S. cerevisiae isolates included in this study were predicted to be diploids and approximately 25% were predicted to be either triploid or tetraploid, with very similar distributions across the different ploidy levels for heterozygous clinical and nonclinical isolates. Earlier studies reported significant proportions of bread, beer and wine strains to be polyploid, as well as significant proportions of natural S. cerevisiae populations, and this was suggested to reflect the higher adaptive potential of heterozygous polyploids in combination with a higher flexibility of populations consisted of individuals with different ploidy levels (Ezov et al., 2006; Legras et al., 2007). Predicted triploid isolates produced a significantly lower proportion of viable spores when compared with either predicted diploid or tetraploid isolates, and it has been shown in earlier studies that this low spore viability is due to the random 2:1 segregation of chromosomes in triploid isolates that leads to the generation of genetically imbalanced, aneuploid spores (Loidl, 1995).
Approximately 20% of the isolates had only monoallelic marker profiles at the examined microsatellite loci, which rendered estimation of their ploidy levels based on the multilocus genotypes impossible. However, based on their high proportions of sporulation proficiency and spore viability, most of these isolates are expected to be diploid. The fact that the proportion of viable spores produced by these isolates is markedly higher than the proportion of viable spores produced by the heterozygous isolates illustrates the inverse relation between the heterozygosity and the spore viability of S. cerevisiae isolates as reported by Mortimer et al. (1994). Interestingly, the frequency of clinical isolates with exclusively monoallelic marker profiles was significantly lower than the frequency of nonclinical isolates with only monoallelic profiles, which may indicate a selective advantage of heterozygous S. cerevisiae isolates in clinical environments. This is also supported by the higher average observed heterozygosity of the microsatellite loci in the heterozygous clinical isolates compared to the heterozygous nonclinical isolates.
S. cerevisiae is known to be a mainly homothallic yeast species, producing haploid spores that are normally able to switch mating-type and self-diploidize thus resulting in genome-wide homozygosity (except at the mating-type locus). Although most S. cerevisiae isolates are homothallic, several have been shown to be heterozygous at one or more loci, and this heterozygosity in homothallic isolates has been attributed to both mutations that occur during mitotic growth of homozygous diploid isolates (Mortimer et al., 1994; Johnston et al., 2000) and to outcrossing of homothallic isolates (Clemons et al., 1997; McCullough et al., 1998; McCusker, 2006). In this study, we found that the majority of the tested isolates with exclusively monoallelic microsatellite marker profiles, were homothallic (HO/HO) and thus likely formed by self-diploidization. However, several homothallic (HO/HO) isolates were found to contain heterozygous microsatellites, and this heterozygosity was either restricted to a single microsatellite locus with alleles differing in length by only one repeat unit or was more extensive with multiple microsatellite loci being heterozygous and alleles differing in length by multiple repeat units. This suggests that both of the aforementioned scenarios of the formation of heterozygous homothallic isolates can occur. Although the number of clinical isolates tested for homothallism was relatively low, the frequency of heterothallism and outcrossing among these isolates appears to be higher than among nonclinical isolates. Heterozygosity due to outcrossing can increase the adaptive potential of natural populations and heterozygous progeny may exhibit heterosis or hybrid vigor. It has been demonstrated that crosses between unrelated S. cerevisiae strains exhibit increased fitness in terms of their ability to grow at high temperatures, a putative virulence trait, and their in vivo survival (Clemons et al., 1994; McCusker et al., 1994a,b), and the increased heterozygosity of clinical S. cerevisiae isolates, possibly due to an increased rate of outcrossing, revealed in this study is consistent with such a selective advantage.
In addition to an increased fitness of outcrossed heterozygous isolates and a possible co-occuring indirect selection of heterothallism over homothallism, the increased heterozygosity of clinical S. cerevisiae isolates may also be (partly) due to increased outbreeding in response to passage through the human gastrointestinal tract. As shown for the probiotic S. cerevisiae var. boulardii, ingestion of live yeasts may lead to fungemia (Bassetti et al., 1998; Munoz et al., 2005). Studies in the fruit fly Drosophila melanogaster, which uses yeasts as a food source, have shown that the ascal wall is digested in the insect gut and that viable, single spores can be recovered from fly faeces (Reuter et al., 2007; Coluccio et al., 2008). Moreover, it was demonstrated that the release of individual spores from tetrads in the digestive tract of D. melanogaster promotes outbreeding by increasing the chance of mating between spores with different genetic backgrounds (Reuter et al., 2007). Although it remains to be demonstrated whether spores actually germinate in vivo and whether the spore load in the human digestive tract is high enough for mating to occur, passage through the human gastrointestinal tract of ingested asci may have a similar effect on the outbreeding rate of clinical S. cerevisiae isolates.
Although previous studies based on microsatellite markers and chromosome length polymorphisms reported significant genetic differentiation between clinical and nonclinical S. cerevisiae isolates (Malgoire et al., 2005; Klingberg et al., 2008), this study revealed a more-or-less single global population under panmixia without this differentiation. No separate clustering of clinical and nonclinical isolates was observed in either the results of the correspondence analysis or the UPGMA clustering, and amova indicated that most of the genetic variation in the sample was due to differences between the yeast isolates and not between groups of isolates based on their geographical origin or their clinical versus nonclinical background, suggesting high levels of gene flow across geographical locations and source substrates. The high level of genetic diversity among pathogenic S. cerevisiae isolates suggests that a single origin of these isolates is unlikely and that many isolates can be pathogens when given the opportunity. This is in agreement with a study by Klingberg et al. (2008), in which it was concluded that the immunological status of an exposed individual may be a more significant factor in the development of S. cerevisiae infections than the yeast strain characteristics, and reflects a more general trend of high phenotypic plasticity among opportunistic fungal pathogens (Rypien et al., 2008).
Supplementary Material
Acknowledgments
The authors wish to thank the many colleagues and culture collection curators who provided yeast isolates. The work was supported by the National Institutes of Health grants GM070541 and GM081690.
References
- Aa E, Townsend JP, Adams RI, Nielsen KM, Taylor JW. Population structure and gene evolution in Saccharomyces cerevisiae. FEMS Yeast Research. 2006;6:702–715. doi: 10.1111/j.1567-1364.2006.00059.x. [DOI] [PubMed] [Google Scholar]
- Bassetti S, Frei R, Zimmerli W. Fungemia with Saccharomyces cerevisiae after treatment with Saccharomyces boulardii. American Journal of Medicine. 1998;105:71–72. doi: 10.1016/s0002-9343(98)00133-8. [DOI] [PubMed] [Google Scholar]
- Clemons KV, McCusker JH, Davis RW, Stevens DA. Comparative pathogenesis of clinical and nonclinical isolates of Saccharomyces cerevisiae. Journal of Infectious Diseases. 1994;169:859–867. doi: 10.1093/infdis/169.4.859. [DOI] [PubMed] [Google Scholar]
- Clemons KV, Park P, McCusker JH, McCullough MJ, Davis RW, Stevens DA. Application of DNA typing methods and genetic analysis to epidemiology and taxonomy of Saccharomyces isolates. Journal of Clinical Microbiology. 1997;35:1822–1828. doi: 10.1128/jcm.35.7.1822-1828.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coluccio AE, Rodriguez RK, Kernan MJ, Neiman AM. The yeast spore wall enables spores to survive passage through the digestive tract of Drosophila. PLoS ONE. 2008;3:e2873. doi: 10.1371/journal.pone.0002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoog GS. Risk assessment of fungi reported from humans and animals. Mycoses. 1996;39:407–417. doi: 10.1111/j.1439-0507.1996.tb00089.x. [DOI] [PubMed] [Google Scholar]
- de Llanos R, Querol A, Peman J, Gobernado M, Fernandez-Espinar MT. Food and probiotic strains from the Saccharomyces cerevisiae species as a possible origin of human systemic infections. International Journal of Food Microbiology. 2006;110:286–290. doi: 10.1016/j.ijfoodmicro.2006.04.023. [DOI] [PubMed] [Google Scholar]
- de Llanos R, Querol A, Planes AM, Fernandez-Espinar MT. Molecular characterization of clinical Saccharomyces cerevisiae isolates and their association with non-clinical strains. Systematic and Applied Microbiology. 2004;27:427–435. doi: 10.1078/0723202041438473. [DOI] [PubMed] [Google Scholar]
- Dujon B. The yeast genome project: what did we learn? Trends in Genetics. 1996;12:263–270. doi: 10.1016/0168-9525(96)10027-5. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Laval G, Schneider S. Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezov TK, Boger-Nadjar E, Frenkel Z, Katsperovski I, Kemeny S, Nevo E, Korol A, Kashi Y. Molecular-genetic biodiversity in a natural population of the yeast Saccharomyces cerevisiae from “Evolution Canyon”: microsatellite polymorphism, ploidy and controversial sexual status. Genetics. 2006;174:1455–1468. doi: 10.1534/genetics.106.062745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Benavides JA. Evidence for domesticated and wild populations of Saccharomyces cerevisiae. PLoS Genetics. 2005;1:66–71. doi: 10.1371/journal.pgen.0010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- Guslandi M. Are probiotics effective for treating Clostridium difficile disease and antibiotic-associated diarrhea? Nature Clinical Practice Gastroenterology & Hepatology. 2006;3:606–607. doi: 10.1038/ncpgasthep0627. [DOI] [PubMed] [Google Scholar]
- Johnston JR, Baccari C, Mortimer RK. Genotypic characterization of strains of commercial wine yeasts by tetrad analysis. Research in Microbiology. 2000;151:583–590. doi: 10.1016/s0923-2508(00)00228-x. [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Klingberg TD, Lesnik U, Arneborg N, Raspor P, Jespersen L. Comparison of Saccharomyces cerevisiae strains of clinical and nonclinical origin by molecular typing and determination of putative virulence traits. FEMS Yeast Research. 2008;8:631–640. doi: 10.1111/j.1567-1364.2008.00365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloda JM, Dean PD, Maddren C, MacDonald DW, Mayes S. Using principle component analysis to compare genetic diversity across polyploidy levels within plant complexes: an example from British Restharrows (Ononis spinosa and Ononis repens) Heredity. 2008;100:253–260. doi: 10.1038/sj.hdy.6801044. [DOI] [PubMed] [Google Scholar]
- Landry CR, Townsend JP, Hartl DL, Cavalieri D. Ecological and evolutionary genomics of Saccharomyces cerevisiae. Molecular Ecology. 2006;15:575–591. doi: 10.1111/j.1365-294X.2006.02778.x. [DOI] [PubMed] [Google Scholar]
- Legras JL, Merdinoglu D, Cornuet JM, Karst F. Bread, beer and wine: Saccharomyces cerevisiae diversity reflects human history. Molecular Ecology. 2007;16:2091–2102. doi: 10.1111/j.1365-294X.2007.03266.x. [DOI] [PubMed] [Google Scholar]
- Legras JL, Ruh O, Merdinoglu D, Karst F. Selection of hypervariable microsatellite loci for the characterization of Saccharomyces cerevisiae strains. International Journal of Food Microbiology. 2005;102:73–83. doi: 10.1016/j.ijfoodmicro.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Loidl J. Meiotic chromosome pairing in triploid and tetraploid Saccharomyces cerevisiae. Genetics. 1995;139:1511–1520. doi: 10.1093/genetics/139.4.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malgoire JY, Bertout S, Renaud F, Bastide JM, Mallie M. Typing of Saccharomyces cerevisiae clinical strains by using microsatellite sequence polymorphism. Journal of Clinical Microbiology. 2005;43:1133–1137. doi: 10.1128/JCM.43.3.1133-1137.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough MJ, Clemons KV, Farina C, McCusker JH, Stevens DA. Epidemiological investigation of vaginal Saccharomyces cerevisiae isolates by a genotypic method. Journal of Clinical Microbiology. 1998;36:557–562. doi: 10.1128/jcm.36.2.557-562.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker JH. Saccharomyces cerevisiae: an emerging and model pathogenic fungus. In: Heitman J, Filler SG, Mitchell AP, editors. Molecular principles of fungal pathogenesis. ASM Press; Washington, DC: 2006. pp. 245–259. [Google Scholar]
- McCusker JH, Clemons KV, Stevens DA, Davis RW. Genetic characterization of pathogenic Saccharomyces cerevisiae isolates. Genetics. 1994a;136:1261–1269. doi: 10.1093/genetics/136.4.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker JH, Clemons KV, Stevens DA, Davis RW. Saccharomyces cerevisiae virulence phenotype as determined with CD-1 mice is associated with the ability to grow at 42 degrees C and form pseudohyphae. Infection and Immunity. 1994b;62:5447–5455. doi: 10.1128/iai.62.12.5447-5455.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer R, Polsinelli M. On the origins of wine yeast. Research in Microbiology. 1999;150:199–204. doi: 10.1016/s0923-2508(99)80036-9. [DOI] [PubMed] [Google Scholar]
- Mortimer RK, Romano P, Suzzi G, Polsinelli M. Genome renewal: a new phenomenon revealed from a genetic study of 43 strains of Saccharomyces cerevisiae derived from natural fermentation of grape musts. Yeast. 1994;10:1543–1552. doi: 10.1002/yea.320101203. [DOI] [PubMed] [Google Scholar]
- Muller LAH, McCusker JH. A multispecies-based taxonomic microarray reveals interspecies hybridization and introgression in Saccharomyces cerevisiae. FEMS Yeast Research. 2009;9:143–152. doi: 10.1111/j.1567-1364.2008.00464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz P, Bouza E, Cuenca-Estrella M, Eiros JM, Perez MJ, Sanchez-Somolinos M, Rincon C, Hortal J, Pelaez T. Saccharomyces cerevisiae fungemia: an emerging infectious disease. Clinical Infectious Diseases. 2005;40:1625–1634. doi: 10.1086/429916. [DOI] [PubMed] [Google Scholar]
- Naumov GI, Gazdiev DO, Naumova ES. Identification of the yeast species Saccharomyces bayanus in Far East Asia. Mikrobiologiia. 2003;72:834–839. [PubMed] [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna: 2008. [Google Scholar]
- Reuter M, Bell G, Greig D. Increased outbreeding in yeast in response to dispersal by an insect vector. Current Biology. 2007;17:R81–R83. doi: 10.1016/j.cub.2006.11.059. [DOI] [PubMed] [Google Scholar]
- Rypien KL, Andras JP, Harvell CD. Globally panmictic population structure in the opportunistic fungal pathogen Aspergillus sydowii. Molecular Ecology. 2008;17:4068–4078. doi: 10.1111/j.1365-294X.2008.03894.x. [DOI] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. In: Guthrie C, Fink GR, editors. Methods in enzymology. Academic Press; New York: 1991. pp. 3–21. [DOI] [PubMed] [Google Scholar]
- Slavikova E, Vadkertiova R. Seasonal occurrence of yeasts and yeast-like organisms in the river Danube. Antonie Van Leeuwenhoek. 1997;72:77–80. doi: 10.1023/a:1000287005253. [DOI] [PubMed] [Google Scholar]
- Smith D, Metzgar D, Wills C, Fierer J. Fatal Saccharomyces cerevisiae aortic graft infection. Journal of Clinical Microbiology. 2002;40:2691–2692. doi: 10.1128/JCM.40.7.2691-2692.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski PD, Dombrowski PG, Fingerman E. Saccharomyces cerevisiae and Saccharomyces paradoxus coexist in a natural woodland site in North America and display different levels of reproductive isolation from European conspecifics. FEMS Yeast Research. 2002;1:299–306. doi: 10.1111/j.1567-1364.2002.tb00048.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.