

Abstract
Unlike protein folding, the process by which a large RNA molecule adopts a functionally active conformation remains poorly understood. Chemical mapping techniques, such as Hydroxyl Radical (·OH) footprinting report on local structural changes in an RNA as it folds with single nucleotide resolution. The analysis and interpretation of this kinetic data requires the identification and subsequent optimization of a kinetic model and its parameters. We detail our approach to this problem, specifically focusing on a novel strategy to overcome a factorial explosion in the number of possible models that need to be tested to identify the best fitting model. Previously, smaller systems (less than three intermediates) were computationally tractable using a distributed computing approach. However, for larger systems with three or more intermediates, the problem became computationally intractable. With our new enumeration strategy, we are able to significantly reduce the number of models that need to be tested using non-linear least squares optimization, allowing us to study systems with up to five intermediates. Furthermore, two intermediate systems can now be analyzed on a desktop computer, which eliminates the need for a distributed computing solution for most medium-sized data sets. Our new approach also allows us to study potential degeneracy in kinetic model selection, elucidating the limits of the method when working with large systems. This work establishes clear criteria for determining if experimental ·OH data is sufficient to determine the underlying kinetic model, or if other experimental modalities are required to resolve any degeneracy.
Keywords: RNA folding, kinetic modeling, Tetrahymena thermophila group I intron, distributed computing, ·OH radical footprinting
1. Introduction
Understanding and predicting the process by which a large RNA molecule like the L-21 Tetrahymena thermophila group I intron adopts its catalytically active conformation remains a contemporary challenge in the life sciences [1–5]. Of particular interest are the effects of temperature, the electrostatic environment, and exogenous molecule binding (such as RNA chaperones) on the kinetics of the folding reaction [6–8]. We have shown that changes in the folding conditions (such as variation of the counter-ion concentration and mutation) have a profound effect on the observed rate constants, suggesting an intricate relationship between the structure, environment, and folding dynamics of an RNA molecule [8, 9]. It is now well established that changes in RNA conformation are key regulatory processes in the cell [10]. As a result, quantitative and predictive models of RNA folding kinetics are essential to understanding regulatory processes in the cell [8, 9].
Chemical and enzymatic mapping techniques are particularly well suited for the study of RNA structure and kinetics because modern electrophoretic approaches can achieve single nucleotide resolution for RNAs well over 400 residues in length [11–13]. Coupled with novel bench-top approaches to collect kinetic data with millisecond resolution [14], these experimental approaches are producing large data sets that require significant computational analysis. This manuscript outlines algorithmic developments for determining the underlying kinetic model that best describes the folding of an RNA molecule based on the analysis of time-resolved hydroxyl radical (· OH) footprinting data [8, 9]. Specifically, we focus on a new strategy that simplifies an exhaustive enumeration of possible kinetic models that limited the size and number of RNA molecules that could be analyzed using our original Kinfold algorithm [9].
1.1. Kinetic Models Describe the Folding Reaction
We consider the process by which an RNA molecule adopts a single, native conformation as illustrated in Figure 1A. RNA secondary structure is highly stable and is formed in several microseconds [1, 15 –17]. The folding process we describe here involves the conformational rearrangement of the secondary structure elements (helices and junctions) that is induced by the addition of a counter-ion (in general Magnesium salts [17]) which leads to a native, and therefore active RNA molecule. This process is the rate-limiting step in RNA folding, due to the appearance of multiple, long-lived intermediates along the folding pathways [7]. We describe the folding process by a kinetic model, such as that illustrated in Figure 1A. The RNA begins in the unfolded state (U) and folds either directly or through intermediates (I) to the final, active state (F). Mathematically, the kinetic model can be written as a series of ordinary differential equations, with rate constants (k and r) describing the rate of transition between states in inverse seconds. The rate of change of the unfolded state, U(t) is therefore described by
Figure 1.

Basic premise for describing an RNA folding reaction with a kinetic model. A.) Unfolded RNA (U state) has only secondary structure elements formed (secondary structure diagram shown in the left hand circle) while the folded state (F) has the full complement of tertiary interactions which allow it to adopt a unique three-dimensional structure (shown in the right hand circle). The folding reaction often takes several hours to go to completion due to multiple intermediates I that populate the pathways to the F state. Each transition between state curves is indicated with an arrow and has a rate constant associated with it. For clarity, we only show the forward transitions in this diagram as the forward rates are generally much larger than the reverse rates. B.) Resulting state curves x⃗(t) for U (orange), I1 (magenta), I2 (purple) and F (black) that describe the relative fraction of each species as a function of time for the folding of the L-21 T. thermophila group I intron in the presence 10mM MgCl2 [9]. These curves are obtained from Equation 5. C.) ·OH footprinting curves (C⃗P (t)) for folding of the T. thermophila group I intron in the presence of 10 mM MgCl2. The green curve corresponds to the P4P6 subdomain, red to the periphery, and blue to the catalytic core of the molecule; a corresponding color scheme is used to illustrate these subdomains for the structures shown in A. We assume that the progress curves are a result of differrent fractions of the unfolded state and the fully folded state.
| (1) |
In this case, k ≫ r such that a majority of the molecules ultimately reach the final folded state (F) and the unfolded state (U) is almost completely unpopulated when steady state (t = ∞) conditions are achieved. If we now consider a vector of the individual state curves, x⃗(t), defined as
| (2) |
and further define matrices K and D, as shown
| (3) |
| (4) |
then the kinetic model illustrated in Figure 1A is written as
| (5) |
Numerical integration of Equation 5 for a given K matrix (set of rate constants) yields the state curves (x⃗(t)) illustrated in Figure 1B.
Our computational goal is to determine the underlying kinetic model by fitting time-resolved ·OH data. This data, however, does not directly measure the state curves x⃗(t). Instead, it measures the change in solvent accessibility of specific nucleotides during the folding reaction [14, 18, 19]. As the molecule folds, a subset of the nucleotides become progressively more buried (or more exposed) and therefore less reactive (more reactive) to the ·OH probe. We monitor the change in accessibility of these nucleotides as a function of time, which yields experimental time-progress curves (Figure 1C), which we call C⃗E(t).
As an example, we use data collected for the folding of the L-21 T. thermophila group I intron [8] in the presence of 10 mM MgCl2. The ·OH footprinting curves shown in Figure 1C correspond to individual subdomains of the molecule; in this case the green curve (Figure 1C) is the average change in accessibility of nucleotides in the P4P6 subdomain (Figure 1A, secondary structure), while the red curves correspond to the peripheral helices, and the blue curves correspond to nucleotides in the catalytic core [9]. These assignments are based on a k-means clustering of individual time-progress curves using the Gap statistic described previously [9].
The state curves, x⃗(t) (Figure 1B), and predicted ·OH fooptrinting, C⃗P (t) (Figure 1C), curves are related by
| (6) |
where the matrix 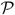 represents the linear combinations of the state curves (x⃗(t)) to generate the progress curves (C⃗P (t)). This matrix has the dimensions of the number of states (U (t), I1(t), …, F (t)) by the number of number of intermediates plus one, which is also the length of C⃗P (t). The matrix for the data set shown in Figure 1 is given by
represents the linear combinations of the state curves (x⃗(t)) to generate the progress curves (C⃗P (t)). This matrix has the dimensions of the number of states (U (t), I1(t), …, F (t)) by the number of number of intermediates plus one, which is also the length of C⃗P (t). The matrix for the data set shown in Figure 1 is given by
| (7) |
The first column in the matrix will always be comprised of zeros, since it corresponds to the unfolded (U(t)) progress curve. As a result, a square and invertible subsection of can be extracted which we denote as P. The sub-matrix of Equation 7 is
| (8) |
The general form of P is a square invertible matrix comprised of ones and zeros, where the columns are linear independent. A priori P is not known for a given RNA folding reaction since only the experimental progress curves, C⃗E(t), (from a ·OH footprinting experiment) are known. We thus aim to identify P and K computationally from the ·OH footprinting data.
We previously implemented a brute force approach for solving this problem that is described in [9]. In this initial implementation we determined an optimal K for all the different possibilities of P. For the L-21 T. thermophila group I intron with two intermediates, 28 individual K matrices were optimized on separate CPUs corresponding to the 28 different P matrices previously enumerated by hand [9]. We used a non-linear least squares optimization that minimizes the difference between the experimental progress curves, C⃗E(t), and the predicted progress curves, C⃗P (t), by adjusting K for a given P matrix. This process is computationally intensive because we perform non-linear optimization to identify an optimal K, requiring multiple solutions of Equation 5 before achieving the convergence criteria. Finally, multiple optimizations are run using different starting values of K to correctly identify a global minimum [9]. Together, identifying the winning combination of P and K can take upwards of 75 CPU days for the T. thermophila group I intron. Given the embarrassingly parallel nature of the problem, however, this calculation is easily accomplished on a distributed computing system. More importantly, this calculation reveals that a single P matrix (of the 28 tested) systematically produces C⃗P (t) curves that are statistically a better fit to the C⃗E(t) when K is optimized, as determined by a three-fold difference in root mean square error (RMSE) [9].
This result suggests that for RNA folding reactions measured by ·OH footprinting, it is possible to identify a single model (combination of K and P matrices) that best describes the experimentally measured data C⃗E(t), and that this result is unique. Although computationally tractable for a system with two intermediates as shown above, our brute force approach becomes limited for larger systems. Furthermore, many of the experimental groups that carry out ·OH footprinting experiments do not have ready access to a computational cluster. There is therefore significant interest in simplifying our approach both to facilitate broader adoption of kinetic modeling, and to allow the study of larger systems such as the Ribosome [20, 21]. Furthermore, a computationally simplified approach will allow us to test the generality of the observation that a single combination of P and K always fits the experimental data significantly better.
2. Implementation
2.1. Factorial Explosion of P
Our new approach is based on the fact that the C⃗P (t) curves are in fact linear combinations of the state curves x⃗ (t) as given by Equation 6. Since the rows in P are linearly independent, we can solve for all but U(t) in x⃗ (t) by inverting P,
| (9) |
In other words, given the experimental data C⃗E(t) we are able to generate time-progress curves for all the species in solution except U(t). The mass-balance equation allows us to generate U(t) by subtracting the other state curves from unity,
| (10) |
and allows the complete recreation of x⃗(t).
We show in the supplementary material that for I intermediates, there are n different P matrices, as given by
| (11) |
In Figure 2, we illustrate the combinatorial explosion of possible P matrices as a function of the number of intermediates with the blue curve. In our original implementation of the Kinfold algorithm [9] we made some simplifications to the combinatorics of P with several assumptions that somewhat offset the explosion (Figure 2, red curve). Nonetheless, we still needed to test a prohibitively large numbers of models for any system with more than two intermediates. We therefore focus our current approach on more efficiently testing all combinations of P.
Figure 2.

Illustration of the combinatorial explosion in the enumeration of all possible P matrices as a function of the number of intermediates I. In blue we plot n as given by equation Equation 11 which we derive in the supplementary material. The red curve represents the number of models (P matrices) that are tested when using the previous implementation of Kinfold by non-linear least squares optimization [9]. The black curve is the average number of models that now need to test based on a sampling of 100 random data sets using our new approach. Error bars represent three standard deviations and the light gray shadow the maximum and minimum values of n for each I.
2.2. Testing P without Fitting K
Applying Equation 9 and Equation 10 to C⃗E(t) for all P and inspecting the resulting x⃗ (t) curves reveals the key to greatly simplifying our approach. Indeed, certain putative state curves drop below zero, which is physically non-sensical. This inspection allows us to eliminate the vast majority of models (P matrices) that result in at least one of the state curves having a negative component. Since all possible P models are tested, individual combinations of state curves are only eliminated when all the models that contain them are eliminated. This is illustrated in Figure 3 for our two intermediate test case, where only two P matrices result in all positive state curves.
Figure 3.

Illustration of the application of Equation 9 to C⃗E(t) (center panel) to generate possible x⃗(t) state curves (external panels); colors are identical to those used in Figure 1. Both and matrices generate negative state curves allowing us to eliminate them without the need to optimize K with non-linear least squares. and yield identical curve shapes, but I1 and I2 are inverted. In this case, these two matrices yield degenerate models that are in fact equivalent, such that a single kinetic model describes the RNA folding reaction.
Since we now fit x⃗ (t) directly, only two non-linear least-squares minimizations are required to determine K. Previously, 28 different models would have been tested [9]. For this simple case, we already have a significant improvement in the computational requirements of the method. Furthermore, we have eliminated the need for a distributed computing solution to analyze this data. We have so far, however, only presented the concepts behind our fitting strategy using idealized data (i.e. C⃗E(t) = C⃗P(t)). Real ·OH data is noisier than the curves we have presented; we thus begin our validation of the algorithm by fitting experimentally obtained data.
3. Validation and Results
3.1. Experimentally Acquired ·OH Data
Time-resolved ·OH radical footprinting data measures the nucleotide solvent accessibility of the RNA molecule [22]. This technique can be done at multiple times during the folding process resulting in time-progress curves such as those illustrated in Figure 1 where the fraction folded is zero at t = 0, and one when t = ∞[18, 19]. The noise in the data is mostly a result of the subsequent analysis of the fragmentation reaction using electrophoretic separation [13, 23]. The progress curves we analyze are averaged over multiple nucleotides, which has a smoothing effect on the data, but nonetheless still results in some jaggedness, as illustrated in Figure 4A.
Figure 4.

Illustration of current algorithm when applied to actual experimentally obtained ·OH footprinting data; curve colors are consistent with Figure 1. A) Time-progress curves with experimental noise. B) Resulting state curves x⃗(t) determined by applying Equation 9 to the raw data and only selecting the P matrices that satisfy the AUC criteria. C) Optimized fit using non-linear least squares minimization of K of state curves. D) K matrices obtained using the old and new approaches to determining P and K, which yield identical results within error. Error on the rate values vary between 5 and 20%.
To determine if we can still identify a single set of state curves x⃗(t) from this data, we performed the same analysis as that described above. When we apply Equation 9 for all P to the data, again a majority of the resulting x⃗ (t) curves dip below zero. However, because of the noise in the data, even the correct state curves have a small negative component. If we define the criteria that up to 10% of the area under the curve (AUC) can be negative before we eliminate the corresponding P matrix as a potential model, then we identify a single set of unique state curves (Figure 4B). If we now optimize K using non-linear least square regression on the resulting x⃗ (t), we are able to fit these data accurately (Figure 4C). Furthermore, the values we obtain for K (Figure 4D) are equivalent within error to those obtained using the original Kinfold [9] algorithm.
Adjusting the AUC criteria allows us to fine tune the sensitivity of our approach. In this particular example, we chose 10% as this correctly identified the set of state curves corresponding to the folding model. Had we chosen a less stringent criteria, such as 50% negative AUC, we would have identified additional models (P matrices) that require testing using non-linear optimization of the K matrix. In essence, raising the AUC criteria results in having to test more models with least-squares optimization, and thus makes the problem more computationally intensive. When using our approach with a novel data set, an AUC criteria can be picked such that a minimal number of models need to be tested. In practice however, it will generally be more desirable to test several models to evaluate the significance in the difference in root mean square error (RMSE) of the fit. The AUC criteria allows users of the algorithm to balance computational cost and desire to comprehensively test all models with the number of models that pass the AUC criteria is highly dependent on the experimental data.
As we move beyond two intermediate systems to larger and more complex molecules, one question that remains is the true information content of a series of C⃗E(t), and whether we can always resolve a single kinetic model from them. Independent of the noise in the data, we now aim to better understand if a unique model can always be found from experimental curves(C⃗E(t)), or if there are cases where there are degenerate models that fit the data with equivalent RMSE. In this case, we do not mean degenerate models like those illustrated in Figure 3 for P2 and P3 (where the curve shapes are identical) but rather situations where two or more P matrices yield nearly indistinguishable state curves such that a single model is no longer identifiable. Prior to our development of the approach described in this manuscript, assessing this issue in systems with more than two intermediates was computationally demanding. Different curve shapes and separations will make the problem of identifying a single model more or less challenging. We therefore set out to determine the general applicability of our method to larger systems.
3.2. Three Intermediate Systems
To evaluate possible degeneracy in larger systems we generated 15 independent three-intermediate data sets using random combinations of P and K. We wanted to produce C⃗E(t) that had characteristics similar to what could be obtained by current ·OH footprinting technology. We therefore constrained our choice of values in the K matrix in the following way: 1.) We generated a random K matrix with values between 0 and 1, and the reverse rates (r) were multiplied by 10−3 while the forward rates (k) were multiplied by a factor of 10 for 8 of the theoretical data sets. We solved Equation 5 numerically to obtain x⃗ (t) for each K matrix. 2.) For the other 7 theoretical data sets we chose rates such that the path U→I1→I2→I3→F was favored by a factor 10 while the other rates were generated as in 1. 3.) We visually inspected the resulting x⃗ (t) curves to confirm that this approach generated a data set that is consistent current ·OH footprinting technology. 4.) A random P matrix was then used to generate the C⃗E(t) curves using Equation 6. 5.) We applied our algorithm to recover the K and P matrix for the time progress curves generated C⃗E(t).
Table 1 summarizes our results from this analysis and reveals that in a majority of the cases, we are indeed able to identify a single model that fits the data with a RMSE at least one order of magnitude lower than the second best fitting model (difference between first and second RMSE column in Table 1.) In the three cases where this is not the case (Test Number 1, 3, and 6), the degeneracy is a result of different P matrices leading to curves with identicial shape (as illustrated in Figure 3), such that the two top models are in fact equivalent, but the association of state curves to intermediates is different. (This equivalence arises from an exchange of columns in the P matrix however it is still unclear why in some cases a row exchange can result in an equivalent model while in others it does not.) Furthermore, our non-linear least squares optimization approach accurately finds the correct rate constants (k) from the curves, with errors in general between 5 and 15%. These results illustrate that for a wide variety of systems with three intermediates, it is generally possible to identify a single kinetic model both accurately and reproducibly. These results also suggest the method will be robust with respect to experimental noise, as the RMSE differences between the models that need to be tested are large (between one and four orders of magnitude).
Table 1.
RMSE errors for 15 different random data sets to determine accuracy and reproducibility on three intermediate systems.
| data set # | # of non-negative models found | mean % difference of k values | lowest RMSE values for top three models | ||
|---|---|---|---|---|---|
| 1 | 9 | 5.3 | 7.4×10−6 | 1.0×10−5 | 1.8×10−4 |
| 2 | 7 | 2.6 | 2.0×10−5 | 2.8×10−3 | 4.1×10−3 |
| 3 | 4 | 23.6 | 7.8×10−6 | 9.1×10−6 | 9.5×10−6 |
| 4 | 2 | 1.1 | 8.3×10−6 | 2.4×10−2 | - |
| 5 | 3 | 2.7 | 1.9×10−5 | 1.0×10−3 | 8.56×10−2 |
| 6 | 3 | 6.7 | 1.1×10−5 | 1.2×10−5 | 1.0×10−1 |
| 7 | 3 | 7.1 | 1.2×10−4 | 7.8×10−2 | 2.2×10−1 |
| 8 | 3 | 28.5 | 2.3×10−4 | 1.1×10−3 | 1.2×10−3 |
| 9 | 6 | 1.6 | 1.8×10−5 | 1.4×10−2 | 1.6×10−2 |
| 10 | 2 | 5.0 | 8.2×10−5 | 5.0×10−1 | - |
| 11 | 1 | 16.3 | 3.5×10−4 | - | - |
| 12 | 2 | 2.2 | 3.5×10−5 | 4.2×10−1 | - |
| 13 | 3 | 10.1 | 3.1×10−3 | 2.9×10−2 | 6.3×10−1 |
| 14 | 1 | 5.3 | 1.1×10−5 | - | - |
| 15 | 6 | 1.8 | 3.9×10−3 | 4.5×10−1 | 5.2×10−1 |
Three intermediate systems are the largest that are currently experimentally tractable. Indeed, the folding of the 5′ domain of the 16S ribosome generates four progress curves (which requires a three intermediate system) for an RNA molecule that is approximately 600 nucleotides in length [20]. Comparatively, the L-21 T. thermophila group I intron has only 388 nucleotides [24]. Recently, however, data on the entire 16S (1500 nucleotides) Ribosomal molecule has been collected, which will yield many more time-progress curves [21]. We therefore decided to evaluate the potential computational cost and tractability of four and five intermediate systems using our new approach.
3.3. Large Systems
As can be seen in Figure 2, the number of possible P matrices increases factorially with the number of intermediates. For systems with four and five intermediates, very large numbers of P matrices must thus be tested. Fortunately, our new approach allows us to test all these combinations efficiently first, to then determine which combinations of P and K require further non-linear least-squares optimization. We therefore used the same approach as described for the three intermediate case above to generate 100 sets of progress curves for four and five intermediate systems. We then applied Equation 9 to determine the subset of P matrices that produce only positive state curves. We report the average number of P matrices for each I that generate only positive state curves in Figure 2 (black line).
For all I, it is clear that our approach offers a significant reduction in the number of models that need to be tested by non-linear least-squares optimization, making this approach computationally tractable for systems with large numbers of intermediates like the Ribosome. Interestingly, the number of models that need to be tested is highly dependent on the curves, as evidenced by the large standard deviation over our 100 models (and the even larger spread in the min and max values, gray shadow Figure 2). It is therefore difficult to a priori predict the total number of P matrices that will produce only positive curves for a given data set. Our ability to identify a single kinetic model that best fits the experimental data will ultimately depend on the experimental data. Our approach offers a computationally simple solution to determining any potential degeneracy or lack thereof.
4. Discussion
In this paper we propose an improved algorithm for identifying the underlying kinetic model that best describes the folding of an RNA molecule from ·OH footprinting data. This new algorithm yields dramatic improvements in performance allowing us to tackle even the largest of RNA molecules. We achieved this significant improvement in efficiency by developing novel criteria for testing whether a particular kinetic model (in the form of a matrix P, Equation 8) produces curves that drop below zero. Since a majority of P matrices produce physically non-realistic state curves, we can eliminate most models without having to perform the computationally expensive non-linear least-squares optimization to determine K. These improvements have allowed us to test 15 three intermediate systems, and establish that in a majority of cases, it is possible to identify a single, non-degenerate kinetic model from ·OH time-progress curves. Furthermore, we were able to show that the method remains robust to experimental noise, as we were able to reproduce a previously published kinetic model (Figure 4). Finally, we were able to apply our approach to very large systems (with four and five intermediates) and we determined that this approach will work and remain computationally tractable in these cases.
Eliminating the need for a distributed computing solution to determine the underlying kinetic model from ·OH footprinting data for medium sized systems will greatly reduce the barrier for experimental labs to performing detailed computational kinetic analysis. Fundamentally, our method offers an automated approach to determining the kinetic parameters of an RNA folding reaction, allowing an experimentalist to effectively test all possible models against their data. The kinetic model defines the identity and ultimately the structure of kinetic folding intermediates. Time-progress curves correspond to specific subdomains of the RNA (Figure 1) and intermediates are often composed of several of these domains (e.g. I2 in the L-21 T. thermophila is composed of the red and green curves, corresponding to the P4P6 and peripheral elements of the RNA). The structure of the I2 intermediate has both the peripheral and P4P6 subdomains formed (third column, Equation 8), while the catalytic core (blue in Figure 1) is unstructured. Furthermore, once the kinetic model is known, we can compute the relative flux through the different pathways to determine the dominant folding mechanism. In the case of the L-21 T. thermophila grroup I intron we determined that three major pathways dominate the flux (U→I1→F, U→I2→F and U→I1→I2→F) [9]. Analysis of the folding of this molecule under multiple counter-ion conditions, established the relative contributions of electrostatic shielding and initial conditions on flux repartitioning in RNA folding [8]. With our new algorithm, the analysis we performed in these two papers is now feasible on a desktop computer. Our approach still tests all P matrices against the experimental data and this provides an equivalent level of confidence in the result as our previous implementation. It however does this in a far more computationally efficient manner.
Our analysis of larger systems with up to five intermediates shows that our approach will scale and remain tractable even for the largest experimentally known systems. These results also illustrate one fundamental limitation of the approach, in that it is likely that for these large systems, it will not always be possible to identify a single combination of P and K that fits the data better than all others. This suggests that the information content of the data is not sufficient, and that other sources of data will be required. In the case of Ribosomal assembly, methods like Pulse-Chase Mass Spectrometry [4] reveal the protein’s perspective on the RNA folding reaction and can provide the additional kinetic information to identify a single model. Furthermore, time-resolved Small Angle X-ray Scattering can provide global compaction measures [25, 26], while catalytic activity measurements indicate the rate of appearance of the native molecule[7]. Taken together, these varied sources of experimental data have the potential to accurately describe the folding reaction of very large RNA molecules. Kinetic modeling, such as the approach we describe here, will be critical in ultimately determining the rules that govern RNA folding reactions.
Supplementary Material
Acknowledgments
We thank Michael Brenowitz and Joerg Schlatterer for their insightful discussions and comments during the preparation of this manuscript. This work is supported by the US National Institutes of Health, NIGMS R00 079953 grant to A.L. The work also benefited from participation in the RNA Ontology Consortium, funded by the US National Science Foundation grant 0443508. Source code and example data sets can be downloaded from https://simtk.org/home/kinfold.
Footnotes
This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
References and Notes
- 1.Woodson SA. Folding mechanisms of group i ribozymes: role of stability and contact order. Biochem Soc Trans. 2002;30:1166–1169. doi: 10.1042/bst0301166. [DOI] [PubMed] [Google Scholar]
- 2.Vicens Q, Gooding AR, Laederach A, Cech TR. Local RNA structural changes induced by crystallization are revealed by shape. RNA. 2007;13:536–548. doi: 10.1261/rna.400207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thirumalai D, Hyeon C. RNA and protein folding: common themes and variations. Biochemistry. 2005;44:4957–4970. doi: 10.1021/bi047314+. [DOI] [PubMed] [Google Scholar]
- 4.Talkington MW, Siuzdak G, Williamson JR. An assembly landscape for the 30s ribosomal subunit. Nature. 2005;438:628–632. doi: 10.1038/nature04261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takamoto K, Das R, He Q, Doniach S, Brenowitz M, Herschlag D, Chance MR. Principles of RNA compaction: insights from the equilibrium folding pathway of the p4-p6 RNA domain in monovalent cations. J Mol Biol. 2004;343:1195–1206. doi: 10.1016/j.jmb.2004.08.080. [DOI] [PubMed] [Google Scholar]
- 6.Russell R, Herschlag D. Probing the folding landscape of the tetrahymena ribozyme: commitment to form the native conformation is late in the folding pathway. J Mol Biol. 2001;308:839–851. doi: 10.1006/jmbi.2001.4751. [DOI] [PubMed] [Google Scholar]
- 7.Russell R, Das R, Suh H, Travers KJ, Laederach A, Engelhardt MA, Herschlag D. The paradoxical behavior of a highly structured misfolded intermediate in RNA folding. J Mol Biol. 2006;363:531–544. doi: 10.1016/j.jmb.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 8.Laederach A, Shcherbakova I, Jonikas MA, Altman RB, Brenowitz M. Distinct contribution of electrostatics, initial conformational ensemble, and macromolecular stability in RNA folding. Proc Natl Acad Sci USA. 2007;104:7045–7050. doi: 10.1073/pnas.0608765104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laederach A, Shcherbakova I, Liang M, Brenowitz M, Altman RB. Local kinetic measures of macromolecular structure reveal partitioning among multiple parallel pathways from the earliest steps in the folding of a large RNA molecule. J Mol Biol. 2006;358:1179–1190. doi: 10.1016/j.jmb.2006.02.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tucker BJ, Breaker RR. Riboswitches as versatile gene control elements. Curr Opin Struct Biol. 2005;15:342–348. doi: 10.1016/j.sbi.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM. High-throughput shape analysis reveals structures in hiv-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol. 2008;6:e96. doi: 10.1371/journal.pbio.0060096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilkinson KA, Merino EJ, Weeks KM. RNA shape chemistry reveals nonhierarchical interactions dominate equilibrium structural transitions in TRNA(asp) transcripts. J Am Chem Soc. 2005;127:4659–4667. doi: 10.1021/ja0436749. [DOI] [PubMed] [Google Scholar]
- 13.Mitra S, Shcherbakova IV, Altman RB, Brenowitz M, Laederach A. High-throughput single-nucleotide structural mapping by capillary automated footprinting analysis. Nucleic Acids Res. 2008;36:e63. doi: 10.1093/nar/gkn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shcherbakova I, Mitra S, Beer RH, Brenowitz M. Fast fenton footprinting: a laboratory-based method for the time-resolved analysis of DNA, RNA and proteins. Nucleic Acids Res. 2006;34:e48. doi: 10.1093/nar/gkl055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woodson SA. Recent insights on RNA folding mechanisms from catalytic RNA. Cell Mol Life Sci. 2000;57:796–808. doi: 10.1007/s000180050042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan J, Thirumalai D, Woodson SA. Folding of RNA involves parallel pathways. J Mol Biol. 1997;273:7–13. doi: 10.1006/jmbi.1997.1311. [DOI] [PubMed] [Google Scholar]
- 17.Heilman-Miller SL, Thirumalai D, Woodson SA. Role of counterion condensation in folding of the tetrahymena ribozyme. i. equilibrium stabilization by cations. J Mol Biol. 2001;306:1157–1166. doi: 10.1006/jmbi.2001.4437. [DOI] [PubMed] [Google Scholar]
- 18.Brenowitz M, Senear DF, Shea MA, Ackers GK. “footprint” titrations yield valid thermodynamic isotherms. Proc Natl Acad Sci USA. 1986;83:8462–8466. doi: 10.1073/pnas.83.22.8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brenowitz M, Chance MR, Dhavan G, Takamoto K. Probing the structural dynamics of nucleic acids by quantitative time-resolved and equilibrium hydroxyl radical ”footprinting”. Curr Opin Struct Biol. 2002;12:648–653. doi: 10.1016/s0959-440x(02)00366-4. [DOI] [PubMed] [Google Scholar]
- 20.Adilakshmi T, Ramaswamy P, Woodson SA. Protein-independent folding pathway of the 16s RRNA 5′ domain. J Mol Biol. 2005;351:508–519. doi: 10.1016/j.jmb.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 21.Adilakshmi T, Bellur DL, Woodson SA. Concurrent nucleation of 16s folding and induced fit in 30s ribosome assembly. Nature. 2008;455:1268–1272. doi: 10.1038/nature07298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Latham JA, Cech TR. Defining the inside and outside of a catalytic RNA molecule. Science. 1989;245:276–282. doi: 10.1126/science.2501870. [DOI] [PubMed] [Google Scholar]
- 23.Das R, Laederach A, Pearlman SM, Herschlag D, Altman RB. Safa: semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. RNA. 2005;11:344–354. doi: 10.1261/rna.7214405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cech TR. Self-splicing of group i introns. Annu Rev Biochem. 1990;59:543–568. doi: 10.1146/annurev.bi.59.070190.002551. [DOI] [PubMed] [Google Scholar]
- 25.Pollack L, Tate MW, Finnefrock AC, Kalidas C, Trotter S, Darnton NC, Lurio L, Austin RH, Batt CA, Gruner SM, Mochrie SG. Time resolved collapse of a folding protein observed with small angle x-ray scattering. Phys Rev Lett. 2001;86:4962–4965. doi: 10.1103/PhysRevLett.86.4962. [DOI] [PubMed] [Google Scholar]
- 26.Russell R, Millett IS, Tate MW, Kwok LW, Nakatani B, Gruner SM, Mochrie SG, Pande V, Doniach S, Herschlag D, Pollack L. Rapid compaction during RNA folding. Proc Natl Acad Sci USA. 2002;99:4266–4271. doi: 10.1073/pnas.072589599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.