

Abstract
Factors that regulate leukocyte entry and spread through CNS parenchyma during different types of CNS insults are incompletely understood. Reactive astrocytes have been implicated in restricting the spread of leukocytes from damaged into healthy parenchyma during the acute and local innate inflammatory events that follow CNS trauma, but the roles of reactive astrocytes during the chronic and widespread CNS inflammation associated with adaptive or acquired immune responses are uncertain. Here, we investigated the effects of transgenically targeted ablation of proliferating, scar-forming reactive astrocytes on the acquired immune inflammation associated with experimental autoimmune encephalitis (EAE). In wild-type mice with EAE, we found that reactive astrocytes densely surrounded perivascular clusters of leukocytes in a manner reminiscent of astrocyte scar formation after CNS trauma. Transgenically targeted ablation of proliferating astrocytes disrupted formation of these perivascular scars and was associated with a pronounced and significant increase in leukocyte entry into CNS parenchyma, including immunohistochemically identified macrophages, T lymphocytes and neutrophils. This exacerbated inflammation was associated with a substantially more severe and rapidly fulminant clinical course. These findings provide experimental evidence that reactive astrocytes form scar-like perivascular barriers that restrict the influx of leukocytes into CNS parenchyma and protect CNS function during peripherally initiated, acquired immune inflammatory responses in the CNS. The findings suggest that loss or disruption of astrocyte functions may underlie or exacerbate the inflammation and pathologies associated with autoimmune diseases of the CNS, including multiple sclerosis.
Introduction
Inflammation is a prominent feature of many disorders of the CNS. It can be initiated in numerous ways and exhibit a wide variety of different characteristics. For example, CNS inflammation triggered after trauma is locally restricted and predominantly resolves over time in the absence of secondary infection (Bush et al., 1999; Schnell et al., 1999; Donnelly and Popovich, 2008). Alternatively, CNS inflammation that is triggered peripherally by autoimmune events tends to be widespread and continuous or recurring (Raine et al., 1980; Adams et al., 1989; McFarland and Martin, 2007). Understanding the factors that regulate the entry and spread through CNS parenchyma of different types of leukocytes during different types of CNS disorders will be fundamental to developing appropriate treatment strategies.
CNS resident cells, including astrocytes, are increasingly recognized as playing central roles in regulating leukocyte trafficking and inflammation in the CNS. The ability of astrocytes in vitro to produce a wide variety of proinflammatory chemokines and cytokines, and various reactive oxygen species (ROS) suggests that they may act as proinflammatory mediators (Dong and Benveniste, 2001; Chen and Swanson, 2003; Farina et al., 2007; Nair et al., 2008). Nevertheless, equally strong in vitro evidence suggests that astrocytes may serve to mitigate or restrict inflammation by producing anti-inflammatory cytokines and ROS scavengers (Aloisi et al., 1997; Dong and Benveniste, 2001; Dringen and Hirrlinger, 2003; Nair et al., 2008). Thus, astrocytes, which are intimately associated with and signal to blood vessels (Iadecola and Nedergaard, 2007), also have the molecular machinery to play complex roles in regulating leukocyte trafficking in the CNS (John et al., 2005). This notion is further supported by additional lines of evidence, which on the one hand implicate astrocytes as key players in locally triggered innate proinflammatory responses after CNS trauma and stroke (Farina et al., 2007), and on the other hand show that scar-forming reactive astrocytes form essential barriers that restrict leukocyte migration from areas of damaged tissue into neighboring healthy tissue during acute and locally restricted innate inflammation after CNS trauma (Bush et al., 1999; Faulkner et al., 2004; Myer et al., 2006; Okada et al., 2006; Herrmann et al., 2008) or stroke (Li et al., 2008).
Reactive astrocytosis is also a prominent feature of the chronic and widespread adaptive immune inflammation of the CNS that occurs during conditions such as experimental autoimmune encephalomyelitis (EAE) and multiple sclerosis (MS) (Eng et al., 1970, 2000; Liedtke et al., 1998), but the roles of reactive astrocytes in regulating leukocyte trafficking in CNS parenchyma during peripherally initiated, adaptive or acquired immune CNS inflammation in vivo are not well understood. In this study we used a well characterized model for the transgenically targeted ablation of proliferating, scar-forming reactive astrocytes (Bush et al., 1999; Faulkner et al., 2004; Myer et al., 2006) to investigate the roles of these cells during the acquired autoimmune inflammation associated with EAE induced by peripheral inoculation with a myelin associated peptide (Liu et al., 2003; Jones et al., 2008).
Materials and Methods
Animals.
All experiments were conducted using male mice. All mice used, including GFAP-TK transgenic as well as nontransgenic, were derived from the same breeding colony backcrossed onto a C57BL/6 background for >15 generations and maintained by mating heterozygous GFAP-TK females of line 7.1 (Bush et al., 1998) with wild-type C57BL/6 males (Charles River). Mice were genotyped by PCR and immunohistochemistry for HSV-TK. Mice were housed in a 12 h light/dark cycle, allowed ad libitum food and water, and maintained in accordance with guidelines set by the National Institutes of Health and as mandated by the University of California Los Angeles Office for the Protection of Research Subjects and the Chancellor's Animal Research Committee.
EAE induction.
Active EAE was induced by subcutaneous immunization with 300 μg of myelin oligodendrocyte glycoprotein (MOG) peptide, amino acids 35–55, and 500 μg of Mycobacterium tuberculosis in complete Freund's adjuvant as previously described (Suen et al., 1997; Liu et al., 2003). Mice were also injected intraperitoneally with Bordatella pertussis toxin (Difco Laboratories) in PBS on day 0 and 2 following the first immunization. MOG 33–55 was synthesized to >98% purity by Mimotopes. Mice (n = 7 per treatment group) were monitored and scored daily for clinical disease severity according to the standard 0–5 EAE grading scale: 0, unaffected; 1, tail limpness; 2, failure to right upon attempt to roll over; 3, partial paralysis; 4, complete paralysis; and 5, moribund as described previously (Liu et al., 2003). The mean of the clinical scores of all mice within a given treatment group were determined daily, thereby yielding the mean clinical score for that treatment group.
Experimental model for transgenic astrocyte ablation.
To conditionally ablate dividing reactive astrocytes, we used GFAP-TK transgenic mice in which thymidine kinase (TK) from the herpes simplex virus (HSV) was targeted to reactive astrocytes using the mouse glial fibrillary acid protein (GFAP) promoter (Bush et al., 1998). Treatment of these GFAP-TK mice with the antiviral agent ganciclovir (GCV) selectively and specifically ablates dividing, reactive, GFAP-TK-expressing astrocytes (Bush et al., 1999) via a noninflammatory apoptotic mechanism. Cells that express transgene-derived HSV-TK, phosphorylate GCV, and pGCV disrupts DNA synthesis (Frank et al., 1984; Heyman et al., 1989), thereby triggering apoptotic death of proliferating cells (Fischer et al., 2005). The efficacy and specificity of the ablation of proliferating, scar-forming, reactive astrocytes using these mice after CNS trauma and other insults has been demonstrated and confirmed in a number of different studies (Bush et al., 1999; Faulkner et al., 2004; Myer et al., 2006).
GCV treatment.
Ganciclovir (Cytovene Hoffman-La Roche) was administered in sterile physiological saline as single subcutaneous injections given every second day at 25 mg/kg/every other day 7 d before EAE induction and continued for the duration of the study.
Measurements of peripheral immune responses.
Cytokine and chemokine profiles of immune cells were obtained as previously described (Liu et al., 2003). Briefly, spleens were harvested after terminal anesthesia before perfusion. Splenocytes were stimulated with MOG peptide 35–55 autoantigen at 25 μg/ml, and cell proliferation assessed using standard H3 incorporation assays. Supernatants were collected after 48 and 72 h, and levels of tumor necrosis factor α (TNFα), interferon γ (IFNγ), interleukin 5 (IL5), interleukin 6 (IL6), interleukin 2 (IL2), interleukin 4 (IL4), monocyte chemotactic protein-1 (MCP-1), aurine macrophage inflammatory protein 1 α (MIP1α), and regulated upon activation normal T-cell expressed and secreted (RANTES) were determined by cytometric bead array (BD Biosciences).
Histological procedures.
Mice were anesthetized by barbiturate overdose and perfused transcardially with ice-cold 0.9% saline, followed by 10% formalin. Spinal cords were postfixed for 2 h and cryoprotected in buffered 30% sucrose overnight. Some blocks of tissue were embedded in a 15% gelatin/sucrose mix and were further postfixed and stored in 30% sucrose. Thirty or forty micrometer transverse frozen sections were prepared using a cryostat microtome (Leica) and processed for either bright-field or fluorescence immunohistochemistry. The following primary antibodies were used: rabbit anti-HSV-TK (1:2000, Bush et al., 1998), rabbit anti-GFAP (1:2000 or 1:20,000, Dako), rat anti-GFAP (1:2500, Millipore Bioscience Research Reagents), mouse anti-Ki67 (1:1000, Vector Laboratories), rat anti-CD45 (1:2000, Millipore Bioscience Research Reagents), rat anti-Ag4/7 (1:100, Serotec), rabbit anti-Iba1 (1:1000, Wako Chemicals), rat anti-mouse CD-3 (1:1000, PharMingen), rat anti-myelin basic protein (MBP; 1:2000, Millipore Bioscience Research Reagents), and rabbit anti-APP (1:500, Abcam). Bright-field immunohistochemistry was performed using biotinylated secondary antibodies (Vector Laboratories), biotin–avidin–peroxidase complex (Vector Laboratories) and diaminobenzidine (DAB, Vector Laboratories) as the developing agent. Fluorescence immunohistochemistry was performed using Alexa Fluor-tagged secondary antibodies Alexa 488 (green), Alexa 568 (red), or Alexa 350 (blue) (Molecular Probes), or secondary antibodies conjugated to TRITC, FITC, and Cy5 (Vector Laboratories and Millipore Bioscience Research Reagents). The nuclear stain, 4′,6′- diamidino-2-phenylindole dihydrochloride (DAPI; 2 ng/ml; Molecular Probes), was used as a general cytological stain in fluorescence-stained sections. Stained sections were examined and photographed using bright-field and fluorescence microscopy (Zeiss), and scanning confocal laser microscopy (Leica).
Morphometric procedures and statistical evaluation.
Immune and inflammatory cell counts were determined using unbiased sampling (Gundersen et al., 1988) and stereological image analysis software (StereoInvestigator, MicroBrightField) operating a computer-driven microscope regulated in the x–z axes (Zeiss) in a manner described in detail previously (Faulkner et al., 2004). For cell density measurements, separate gray and white matter areas were traced at low magnification in three randomly selected sections per mouse in the lumbar spinal cord. In each section, sampling frames of 100 × 100 μm separated by 100 μm were selected at random within each traced region by the image analysis software and computer-driven stage. The number of positive cells per counting frame and per tissue depth was determined at high magnification according to exclusion criteria of the stereology program. The volume of the sampled tissue was calculated and final cell counts per gray or white matter per animal were expressed as cells per cubic millimeter. Axon number was determined in the dorsal column white matter using ImageJ software (NIH) analysis of particles to discriminate individual NF200-stained axons. All statistical evaluations were performed by ANOVA with post hoc, independent pairwise analysis (Prism, GraphPad).
Results
Transgenic astrocyte ablation during EAE
We began by testing whether our transgenic strategy for ablating scar-forming astrocytes in GFAP-TK mice treated with GCV would be effective during EAE. We first examined the specificity of the transgenic targeting of HSV-TK to astrocytes during EAE by using double labeling immunohistochemistry for TK and GFAP. In agreement with previous studies (Bush et al., 1999; Faulkner et al., 2004), essentially all cells that expressed detectable levels of TK also expressed GFAP in EAE-induced GFAP-TK mice (Fig. 1A,B).
Figure 1.
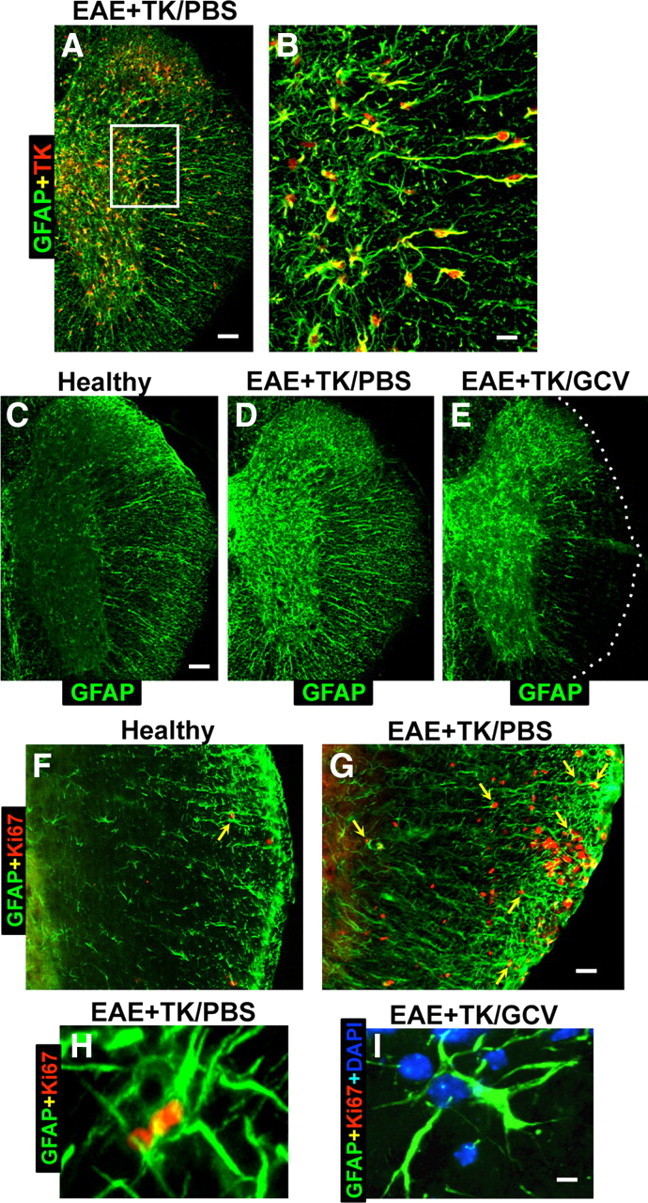
Verification of strategy for transgenic ablation of proliferating astrocytes during EAE. A–I, Single- or two-color immunofluorescence for GFAP (green, A–I) and either transgene derived-TK (red, A–E) or the cell cycle marker, Ki67 (red, F–I) in mouse spinal cord. A, B, Survey and detail images show that in EAE-induced GFAP-TK transgenic mice treated with PBS (EAE+TK/PBS), all TK-expressing cells also express GFAP. Note that TK staining is prominent in astrocyte cell bodies, whereas GFAP is distributed primarily in cell processes. C–E, Survey images of thoracic spinal cord from GFAP-TK mice that were healthy (C) or induced with EAE and given either PBS (D) or GCV (E) to ablate proliferating, transgene-expressing astrocytes (EAE+TK/GCV). Note that GFAP staining is increased in EAE+TK/PBS (D) relative to healthy (C) and reduced in a patchy manner in EAE+TK/GCV (E) relative to EAE+TK/PBS (D), particularly in white matter. F, G, Survey images show that there are few proliferating, Ki67-positive cells in a healthy mouse and that spontaneously proliferating astrocytes (arrow) are rare (F), whereas in a mouse with EAE+TK/PBS, there are many proliferating, Ki67-positive cells, including proliferating astrocytes (arrows) (H). H, I, Detail images show proliferating, Ki67-positive, perivascular astrocytes in a mouse with EAE+TK/PBS (H) and the persistence of surviving, nonproliferating perivascular astrocytes in a mouse with EAE+TK/GCV (I). Scale bars: A, 100 μm; B, 25 μm; C–E, 125 μm; F, G, 50 μm; H, I, 4 μm.
We next conducted pilot studies to identify a dose of GCV that would successfully ablate proliferating scar-forming astrocytes in GFAP-TK mice with EAE. On the basis of these pilot studies we selected an intermittent low-dose GCV treatment (25 mg/kg every other day) given to GFAP-TK mice with EAE. The efficacy of this dosing combination was evaluated by using immunohistochemistry for GFAP. In the spinal cords of healthy (non-EAE) mice, GFAP-positive astrocytes were faintly stained and evenly distributed (Fig. 1C). Twenty-one days after EAE induction in PBS-treated GFAP-TK mice, there was a robust reactive astrogliosis with a visible increase in the amount of GFAP staining in both white and gray matter (Fig. 1D) in a manner indistinguishable from the effects of EAE in wild-type C57BL/6 mice (data not shown). Twenty-one days after EAE induction in GCV-treated GFAP-TK, there was a patchy, subtotal loss of GFAP-positive astrocytes in many CNS regions, particularly in white matter (Fig. 1E).
To look for evidence of astrocyte proliferation induced by EAE we used double-staining immunofluorescence for GFAP and the cell cycle marker, Ki67 (Brown and Gatter, 1990). In healthy (non-EAE) mice, the spinal cord contained few Ki67-labeled proliferating cells and proliferating astrocytes that were double stained with Ki67 plus GFAP were rare but could occasionally be found (Fig. 1F). In PBS-treated GFAP-TK mice harvested 21 d after induction of EAE, there were many Ki67-labeled cells proliferating cells, particularly in white matter (Fig. 1G), and a subset of these cells were proliferating astrocytes that were double stained with both Ki67 and GFAP, particularly in white matter (Fig. 1G,H). In GFAP-TK mice harvested after 21 d of combined GCV treatment and EAE, there were few or no proliferating astrocytes that were double stained for Ki67 plus GFAP (Fig. 1I), and it is noteworthy that despite visible astrocyte loss these mice continued to exhibit many nondividing astrocytes throughout gray and white matter (Fig. 1I), and that astrocyte loss severe enough to be visibly detectable occurred in patches (Fig. 1E).
We next looked for potential confounds that might influence the interpretation of results obtained using the GFAP-TK plus GCV transgenic model for astrocyte ablation in EAE. Necropsy of abdominal organs showed that GFAP-TK mice given the low intermittent dosing regimen of GCV, either with or without EAE, did not exhibit any detectable visceral pathology as noted previously with an eightfold higher continuous dosing regimen of GCV (Bush et al., 1998). In agreement with previous studies, GFAP-TK mice that received the low intermittent dosing regimen of GCV treatment for 21 d, but which were not induced with EAE, exhibited no detectable effects on astrocytes stained by immunohistochemistry for GFAP, because astrocytes rarely proliferate in normal, healthy adult mice (Bush et al., 1999). Last, EAE was clinically and histopathologically indistinguishable among three control groups: (1) wild-type mice without GCV, (2) wild-type mice with GCV, and (3) GFAP-TK mice not given GCV. We therefore used this regimen of GCV treatment to examine the effects of conditional ablation of proliferating reactive astrocytes on the clinical course and histopathology of EAE.
Exacerbation of EAE clinical signs by astrocyte ablation
GFAP-TK mice given vehicle (PBS) but not given GCV exhibited a moderate EAE course indistinguishable from that in wild-type C57BL/6 mice, in which the mice reached a plateau and were clinically stable at grades 2–3. These mice did not become moribund and did not require euthanasia (Fig. 2). In contrast, GFAP-TK mice given GCV to ablate proliferating reactive astrocytes exhibited a rapid, acute EAE course that was significantly more severe (p < 0.001), in which mice reached clinical grades of 4.5 and higher, became paralyzed or moribund, and were killed by day 21 after EAE induction (Fig. 2). GFAP-TK mice given GCV but not induced with EAE exhibited no detectable clinical signs (Fig. 2).
Figure 2.

Exacerbated clinical signs of EAE in mice with transgenically targeted ablation of proliferating reactive astrocytes. Graph showing observer scored clinical rating scale (mean ± SEM) of EAE signs in GFAP-TK transgenic mice that were induced with EAE and given either PBS (EAE+TK/PBS) as a vehicle control, or GCV (EAE+TK/GCV) to ablate transgene-expressing astrocytes, or GFAP-TK mice given GCV but not induced with EAE (Non-EAE+TK/GCV). GFAP-TK mice induced with EAE and given PBS exhibited a moderately severe clinical course that reached a plateau as expected. GFAP-TK mice induced with EAE and given GCV exhibited a rapidly fulminant clinical course that was significantly more severe than that of mice given PBS. GFAP-TK mice given GCV but not induced with EAE exhibited no clinical signs. Data are representative of two separate experiments. ***p < 0.001 relative to EAE+TK/PBS, ANOVA with post hoc pairwise comparisons; n = 7 per group.
No difference in peripheral immune modulation in EAE with astrocyte ablation
Peripheral immune responses play a primary role in the induction of CNS inflammation in EAE (Mokhtarian et al., 1984). We therefore examined whether any differences observed in GCV-treated GFAP-TK mice with EAE might be due to an effect on the peripheral immune system. To do so, we assessed the immune responses of splenocytes upon ex vivo stimulation with autoantigen, MOG 35–55 peptide. We found no significant differences in autoantigen-induced production of cytokines (TNFα, IFNγ, IL5, IL6, and IL2) or chemokines (MCP-1, MIP1α, and RANTES) by splenocytes from GCV-treated GFAP-TK mice with EAE compared with splenocytes from PBS-treated GFAP-TK mice with EAE (Table 1). There was also no significant difference in splenocyte cell proliferation in PBS-treated GFAP-TK mice [19,851 ± 1784 counts per minute (cpm)] compared with GCV-treated GFAP-TK mice (21,143 ± 2410 cpm; p > 0.05).
Table 1.
Response of peripheral immune cells to stimulation with autoantigen during EAE is not altered by ablation of proliferating reactive astrocytes
| TK/PBS no Ag | TK/GCV no Ag | TK/PBS + Ag | TK/GCV + Ag | |
|---|---|---|---|---|
| TNFα | 194.4 ± 43.9 | 58.6 ± 30.3 | 553.0 ± 49.7 | 555.1 ± 30.4 |
| IFNγ | 48.6 ± 30.7 | 16.1 ± 11.0 | 4108.0 ± 472.8 | 3953.3 ± 710.6 |
| IL5 | 3.3 ± 3.3 | 1.5 ± 0.8 | 34.1 ± 7.7 | 48.9 ± 12.2 |
| IL6 | 185.2 ± 28.4 | 55.1 ± 12.0 | 1102.0 ± 64.9 | 857.5 ± 47.6 |
| IL2 | 12.1 ± 3.4 | 15.8 ± 2.9 | 271.8 ± 121.0 | 294.2 ± 60.2 |
| MCP-1 | 65.9 ± 39.1 | 43.5 ± 16.4 | 1785.1 ± 289.4 | 1752.7 ± 90.8 |
| MIP1α | 4.7 ± 0.3 | 7.6 ± 2.3 | 66.9 ± 17.0 | 87.7 ± 30.0 |
| RANTES | 31.5 ± 7.2 | 32.9 ± 4.9 | 91.2 ± 6.1 | 80.2 ± 9.1 |
Splenocytes harvested from mice at day 21 of EAE, in different experimental groups, were examined ex vivo for cytokine and chemokine production either with (+ Ag) or without (no Ag) stimulation with MOG peptide autoantigen. IL4 levels were low and at the limit of detection (data not shown). Values are expressed as mean pg/ml ± standard error. In all measures, autoantigen induced responses of splenocytes from GFAP-TK mice with EAE given GCV to ablate proliferating reactive astrocytes (TK/GCV) did not differ significantly from GFAP-TK mice with EAE given PBS (TK/PBS), p > 0.05, ANOVA with pairwise post hoc comparison.
Increased inflammation in EAE with astrocyte ablation
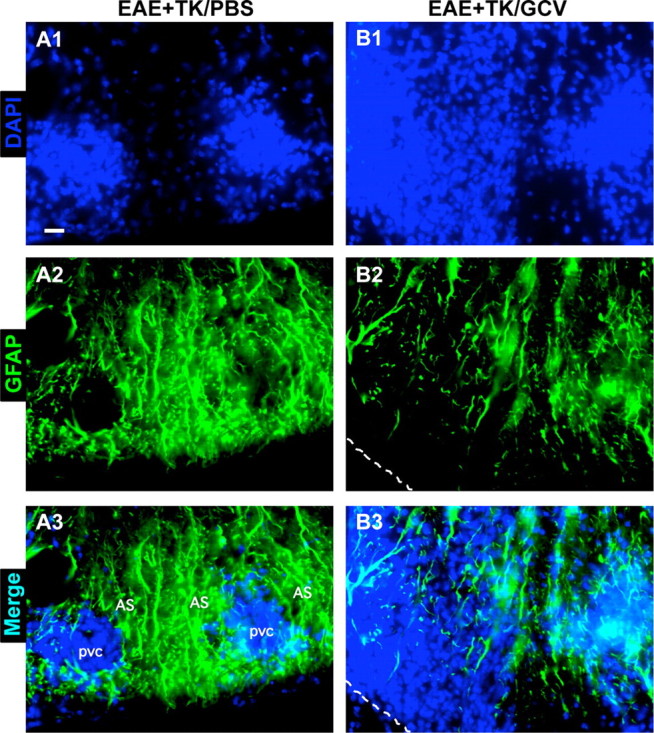
We next qualitatively and quantitatively evaluated CNS inflammation in the spinal cords of mice from different experimental groups 21 d after induction of EAE. To do so we used immunohistochemistry for markers of microglia and different types of leukocytes, alone and in various combinations with staining for astrocytes and cytological markers. Initial observations were made using fluorescent double staining with GFAP and the cytological marker, DAPI. As expected, spinal cords of mice with EAE exhibited a characteristic cytological appearance associated with autoimmune CNS inflammation (Raine et al., 1980), with widespread, randomly distributed patches of increased cellularity and many dense perivascular cell clusters (Fig. 3A1). Double-stained sections revealed that areas of increased cellularity were associated with a prominent reactive astrogliosis (Fig. 3A2) and that all perivascular cell clusters were surrounded by intensely stained GFAP-positive astrocytes and their cell processes (Fig. 3A1–A3). In GCV-treated GFAP-TK mice with EAE, there was a qualitative appearance of greater and more widespread parenchymal infiltration of DAPI-stained cells (Fig. 3B1). Double-stained sections showed an inverse relationship of DAPI and GFAP staining, such that areas of pronounced cellularity corresponded to areas of low density of GFAP-positive astrocyte processes (Fig. 3B1–B3). These findings were consistent with the notion that the inflammatory response in CNS tissue during EAE was exacerbated by ablation of proliferating reactive astrocytes.
Figure 3.
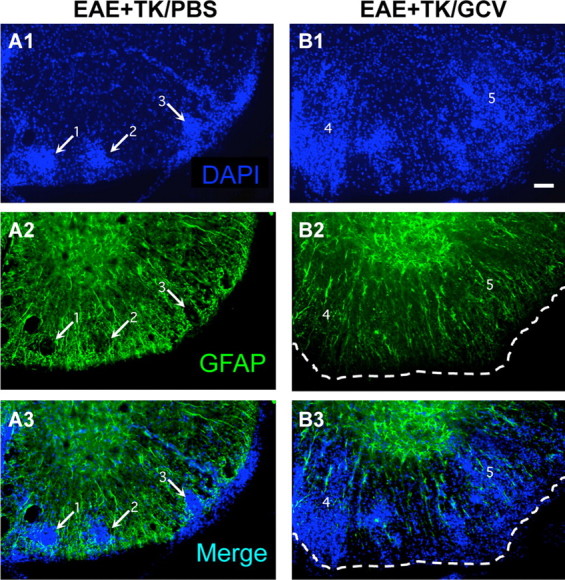
Increased spread of infiltrating inflammatory cells during EAE with ablation of proliferating reactive astrocytes. A1–A3, B1–B3, Single-channel and merged two-color fluorescence survey images of spinal cord sections stained for GFAP (green) and the nuclear counterstain, DAPI (blue), in GFAP-TK transgenic mice that were induced with EAE and given either PBS (EAE+TK/PBS) as a vehicle control (A1–A3), or GCV (EAE+TK/GCV) to ablate transgene-expressing astrocytes (B1–B3). Note that in EAE+TK/PBS, DAPI-stained infiltrating inflammatory cells (A1) are found primarily in dense perivascular clusters (e.g., 1–3) surrounded by intensely stained GFAP-positive astrocytes (A2, A3). Clusters 1 and 2 are shown at higher magnification in Figure 10A. In EAE+TK/GCV (B1–B3), inflammation is markedly increased and there is little perivascular clustering in comparison with EAE+TK/PBS (A1–A3); instead, DAPI-stained infiltrating inflammatory cells (B1) spread widely in the parenchyma of the white matter (e.g., 4, 5). GFAP staining shows that the white matter is substantially depleted of GFAP-positive astrocytes in EAE+TK/GCV (B2), and double staining reveals that the spread of DAPI-positive inflammatory cells is heaviest in areas depleted of astrocytes (B3). Region 4 is shown at higher magnification in Figure 10B. Scale bar, 50 μm.
To compare more specifically the nature and level of CNS inflammation that occurred in mice with either EAE or EAE plus astrocyte ablation, we began with a qualitative evaluation of immunohistochemistry for CD45, a global marker of all inflammatory cells including microglia and different types of leukocytes, including macrophages and T lymphocytes (Charbonneau et al., 1988). In healthy (non-EAE) mice, CD45 staining was faint and limited to small, finely branched, quiescent microglia in both gray and white matter (Fig. 4A,D,G,J). CD45-positive cells with the globoid appearance of leukocytes or microglial-derived macrophages were rare in normal, healthy CNS parenchyma. By comparison, mice with EAE exhibited a widespread increase in CD45 staining in both the gray and white matter, a large proportion of which was associated with highly branched microglia that appeared to be hypertrophied and more numerous than in normal mice (Fig. 4B,E,H). In addition, mice with EAE exhibited many randomly distributed patches of globoid CD45-immunoreactive cells resembling leukocytes or microglial-derived macrophages, either in perivascular clusters or scattered in the parenchyma (Fig. 4B,E,H,K). Mice with EAE plus astrocyte ablation exhibited an even more intense CD45 staining in both the gray and white matter, and a far greater proportion of this staining appeared associated with large globoid CD45-immunoreactive cells (Fig. 4C,F,I,L). It is noteworthy that far more globoid CD45-positive cells were distributed in the parenchyma, with fewer in dense perivascular clusters, compared with EAE mice (Fig. 4I,L). These findings provided qualitative evidence that the inflammatory response associated with EAE was exacerbated in mice with ablation of proliferating reactive astrocytes.
Figure 4.
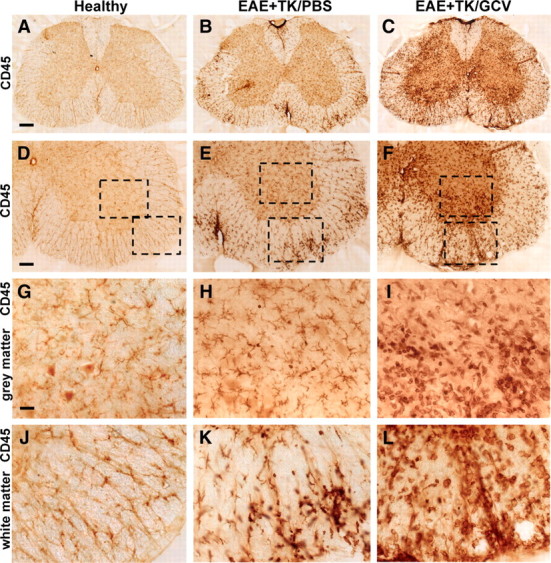
Increased upregulation of CD45-positive inflammatory cells during EAE with ablation of proliferating reactive astrocytes. A–L, Survey and detail bright-field images of lumbar spinal cord sections immunohistochemically stained for the pan leukocyte and microglial marker, CD45 in spinal cord of GFAP-TK mice that were either healthy (A, D, G, J) or had EAE+TK/PBS (B, E, H, K) or EAE+TK/GCV (C, F, I, L). Note the increase in both staining intensity and number of CD45-positive cells in EAE+TK/PBS (B, E, H, K) relative to normal (A, D, G, J), and a further increase in EAE+TK/GCV (C, F, I, L) relative to EAE+TK/PBS (B, E, H, K). Note also the marked increase in large globoid CD45-positive cells with the appearance of leukocytes in EAE+TK/GCV (C, F, I, L) relative to EAE+TK/PBS (B, E, H, K) in both gray (I, H) and white (L, K) matter. Scale bars: A–C, 170 μm; D–F, 90 μm; G–L, 27 μm.
To compare quantitatively the inflammatory changes in mice with EAE and EAE plus astrocyte ablation, we determined the relative tissue densities of CD45-positive cells with typical morphological appearances of (1) branching, ramified microglia (Fig. 5A,B) and (2) globoid inflammatory cells (Fig. 5C,D) using an automated, computer-assisted, and unbiased stereological sampling procedure. Spinal cord tissue of healthy mice exhibited many branching microglia but only rare examples of globoid inflammatory cells (Fig. 5B,D). By comparison, mice with EAE exhibited a significant doubling in the density of CD45-positive branching microglia, and a significant more than 5-fold or 10-fold increase in CD45-positive globoid inflammatory cells in gray and white matter, respectively (Fig. 5B,D). In mice with EAE plus astrocyte ablation, the number of CD45-positive branching microglia did not differ significantly from that in mice with EAE alone (Fig. 5B), but the number of CD45-positive globoid inflammatory cells was markedly and significantly increased by more than double in gray and white matter (Fig. 5D). Together these findings provided quantitative evidence that the inflammatory response triggered by EAE was significantly exacerbated in mice with ablation of proliferating reactive astrocytes.
Figure 5.
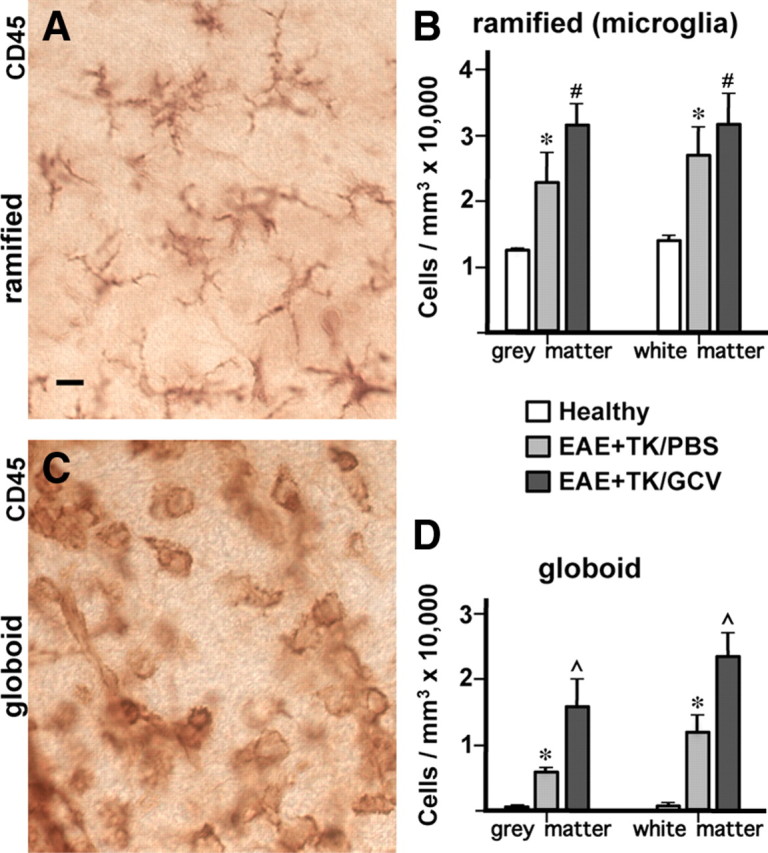
Significantly increased numbers of inflammatory cells in spinal cord during EAE with ablation of proliferating reactive astrocytes. A–D, Qualitative appearance (A, C) and quantitative stereological evaluations (B, D) of CD45-positive inflammatory cells in spinal cord of GFAP-TK mice that were either healthy or had EAE+TK/PBS or EAE+TK/GCV. A, C, Bright-field detail images of immunohistochemically stained CD45-positive cells meeting the criteria of ramified microglia (A) or globoid inflammatory cells (C) for quantitative analysis. B, D, Graphs of quantitative stereological evaluations of density of ramified (B) or globoid (D) CD-45-positive cells in spinal cord gray and white matter show that the densities of both ramified microglia (B) and globoid inflammatory cells (D) were increased significantly in EAE+TK/PBS relative to healthy, and globoid inflammatory cells were further increased significantly in EAE+ TK/GCV relative to EAE+TK/PBS (B, D). *p < 0.01 relative to healthy; #not significant relative to EAE+TK/PBS; ⋀p < 0.01 relative to EAE+TK/PBS; ANOVA with post hoc pairwise comparisons; n = 6 per group. Scale bar, 10 μm.
Increased CNS entry of multiple types of leukocytes in EAE with astrocyte ablation
To identify specific types of inflammatory cells involved in the increased inflammation in CNS parenchyma in mice with EAE plus astrocyte ablation, we used immunohistochemistry for markers of specific leukocyte populations: Iba1 for monocytes/macrophage lineage cells (including microglia) (Ahmed et al., 2007), CD3 for T lymphocytes (von Boehmer, 1988), and Ag4/7 for neutrophils (Hirsch and Gordon, 1983).
Staining with Iba1 clearly identified the widespread distribution of finely branching microglia and confirmed the essential absence of globoid monocytes or macrophages in the CNS parenchyma of normal mice (Fig. 6A,D,G). In mice with EAE, Iba1 staining confirmed a pronounced increase in the number of branched microglia as noted with CD45 staining in both gray and white matter (Fig. 6B,E,H) and showed that many of the globoid CD45-positive cells in perivascular clusters were monocytes or macrophages (Fig. 6H) derived either from leukocyte infiltration or from local microglia. In mice with EAE plus astrocyte ablation, there was a further qualitatively obvious increase in the number of cells stained with Iba1 compared with EAE mice (Fig. 6C,F,I) with a much more widespread distribution of Iba1-positive globoid macrophages in the parenchyma, particularly in white matter (Fig. 6C,I).
Figure 6.
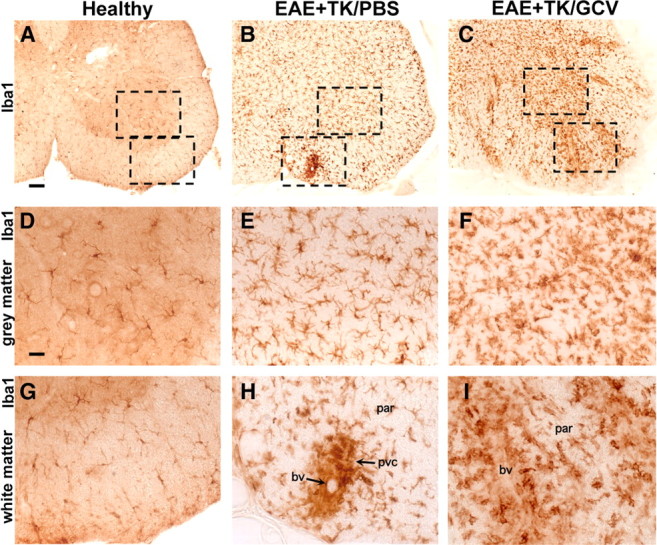
Increased upregulation of Iba1-positive microglia and spread of Iba1-positive macrophages during EAE with ablation of proliferating reactive astrocytes. A–I, Survey and detail bright-field images of lumbar spinal cord sections immunohistochemically stained for the microglial cell and macrophage marker, Iba1, in GFAP-TK mice that were either healthy (A, D, G) or had EAE+TK/PBS (B, E, H) or EAE+TK/GCV (C, F, I). Note the increase in both staining intensity and number of Iba1-positive cells in EAE+TK/PBS (B, E, H) relative to healthy (A, D, G), and a further increase in EAE+TK/GCV (C, F, I) relative to EAE+TK/PBS (B, E, H). Note also that globoid Iba1-positive macrophages are primarily in perivascular clusters (pvc) in EAE (B, E, H) and are spread widely in the parenchyma (par) in EAE+TK/GCV (C, F, I). bv, Blood vessel. Scale bars: A–C, 90 μm; D–I, 27 μm.
Staining with CD3 and quantitative evaluation using an automated, computer-assisted, and unbiased stereological sampling procedure (Fig. 7), confirmed that T lymphocytes were rare in the CNS parenchyma of healthy mice. Mice with EAE exhibited significantly more CD3-stained T lymphocytes in both gray and white matter relative to healthy mice (Fig. 7A,C), and most of these cells were located in perivascular clusters, with few cells scattered in the parenchyma, particularly in white matter (Fig. 7A). By comparison, mice with EAE plus astrocyte ablation exhibited a further pronounced and significant increase in the number of T lymphocytes in white but not gray matter relative to mice with EAE alone (Fig. 7B–D) and a much more widespread distribution of CD3-positive T lymphocytes in the white matter parenchyma (Fig. 7B,D).
Figure 7.

Increased infiltration and spread of CD3-positive T lymphocytes during EAE with ablation of proliferating reactive astrocytes. A, B, Survey bright-field images of lumbar spinal cord sections immunohistochemically stained for the T lymphocyte marker, CD3 in mice with EAE+TK/PBS (A) or EAE+TK/GCV (B). Note that in EAE+TK/PBS, CD3-positive T lymphocytes are found primarily in perivascular clusters (pvc, A), whereas in EAE+TK/GCV, CD3-positive T lymphocytes are spread widely in the parenchyma (par) of the white matter and there is little perivascular clustering (B). C, Graph of quantitative stereological evaluations of density of CD3-positive T lymphocytes in spinal cord gray and white matter, showing that CD3 cells were increased significantly in EAE+TK/PBS relative to healthy, and were further increased significantly in EAE+ TK/GCV relative to EAE+TK/PBS in white but not in gray matter. *p < 0.01 relative to healthy; #not significant relative to EAE+TK/PBS, ⋀p < 0.01 relative to EAE+TK/PBS; ANOVA with post hoc pairwise comparisons; n = 3 per group. D, Detail of boxed area in B showing CD3-positive T lymphocytes in white matter parenchyma. bv, Blood vessel. Scale bars: A, B, 20 μm; D, 5 μm.
Staining with Ag4/7 confirmed the absence of neutrophils from CNS parenchyma of healthy mice (data not shown). In mice with EAE, Ag4/7 staining showed the occasional presence of neutrophils, most commonly in perivascular clusters together with other leukocytes (Fig. 8A1,A2). In mice with EAE plus astrocyte ablation, Ag4/7-stained neutrophils were markedly more numerous and were more frequently found distributed in the parenchyma (Fig. 8B1,B2).
Figure 8.
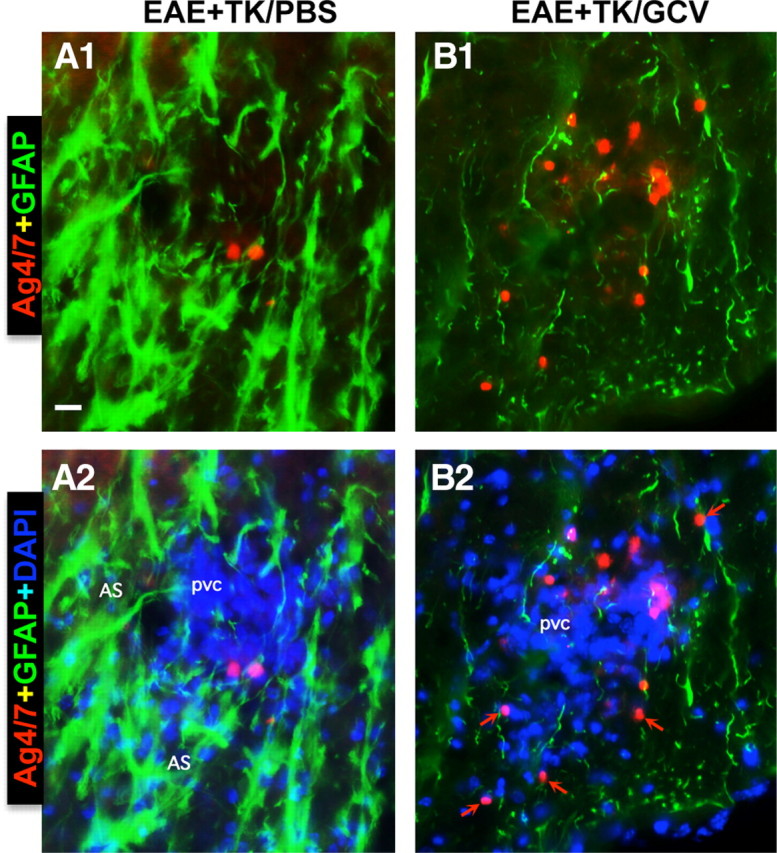
Increased spread of neutrophils during EAE with ablation of proliferating reactive astrocytes. A, B, Detail, merged two- or three-color fluorescence images of spinal cord white matter stained for the neutrophils marker, Ag4/7 (red), GFAP (green), and (DAPI blue) in EAE+TK/PBS (A1, A2) and EAE+TK/GCV (B1, B2). Note that in EAE+TK/PBS, a few positive neutrophils are present but confined to a perivascular cluster of infiltrating inflammatory cells (pvc) surrounded by tightly packed GFAP-positive astrocytes (AS) (A1, A2), whereas in EAE+TK/GCV, Ag4/7-positive neutrophils (red) are increased in number and together with other inflammatory cells (blue) spread widely in the white matter (wm) parenchyma in regions depleted of astrocytes (B1, B2). Scale bar, 35 μm.
Together these findings showed that the quantitative increase in CNS inflammation observed with CD45 staining in mice with EAE plus astrocyte ablation (Fig. 5), was due to the increased spread in CNS parenchyma of at least three different kinds of inflammatory cells and leukocytes including macrophages, T lymphocytes, and neutrophils.
Perivascular astrocytes are critical barriers to leukocytes during EAE
We next examined more closely the relationship between reactive astrocytes and leukocytes in CNS parenchyma during EAE and EAE plus astrocyte ablation, by using various combinations of multicolor fluorescence staining together with the cytological stain, DAPI, in combination with immunohistochemistry for GFAP alone or with different leukocyte markers. As noted above, mice with EAE exhibited a prominent reactive astrogliosis in areas with inflammatory cell infiltrates (Fig. 3A1–A3). On closer examination, there was a clear anatomical juxtaposition of groups of highly reactive and hypertrophied astrocytes around all perivascular clusters of inflammatory cells, such that the cell processes of reactive astrocytes appeared to surround and wall off the cluster. This observation was consistently and ubiquitously noted in all tissue stained for GFAP and DAPI (Figs. 3A1–A3, 9A1–A3) as well as for GFAP and DAPI in combination with the pan leukocyte marker CD45 (Fig. 10A) or markers for monocytes/macrophages (Fig. 10C), T lymphocytes (Fig. 10E), and neutrophils (Fig. 8A1,A2). Moreover, the groups of highly reactive astrocytes that surrounded perivascular clusters of leukocytes during EAE had many overlapping processes (Figs. 8–10) and were reminiscent in appearance of proliferating reactive astrocytes that form scars long the borders of damaged tissue after CNS trauma (Faulkner et al., 2004; Herrmann et al., 2008). In mice with EAE plus selective ablation of proliferating reactive astrocytes, despite the presence of many nonproliferating astrocytes in a scattered distribution (Figs. 1E,I, 3B2), there was a pronounced reduction in densely packed formations of astrocytes in perivascular regions and this reduction occurred in conjunction with (1) a pronounced reduction in the perivascular clustering of leukocytes and (2) a pronounced increase and widespread distribution of inflammatory cells and leukocytes in the surrounding CNS parenchyma (Figs. 8–10). Again, this observation was consistently and ubiquitously noted in all tissue stained for GFAP and DAPI (Figs. 3B1–B3, 9B1–B3) as well as for GFAP and DAPI in combination with the pan leukocyte marker CD45 (Fig. 10B) or markers for monocytes/macrophages (Fig. 10D), T lymphocytes (Fig. 10F), and neutrophils (Fig. 8B1,B2). Together, these observations suggest that reactive astrocytes form scar-like perivascular barriers to leukocytes that restrict leukocyte to perivascular clusters and limit entry into CNS parenchyma during EAE.
Figure 9.
Disruption of perivascular barriers to inflammatory cells during EAE with ablation of proliferating reactive astrocytes. A1–A3, B1–B3, Detail, single-channel and merged two-color fluorescence images of spinal cord sections stained for GFAP (green) and the nuclear counterstain, DAPI (blue), in EAE+TK/PBS (A1–A3) and EAE+TK/GCV (B1–B3). A1–A3, Detail images of perivascular inflammatory cell clusters 1 and 2 from Figure 3A. B1–B3, Detail images of region 4 from Figure 3B. Note that in EAE+TK/PBS, dense clusters of DAPI-stained infiltrating inflammatory cells (A1) are surrounded by tightly packed GFAP-positive astrocytes (A2, A3), whereas in EAE+TK/GCV, DAPI-stained inflammatory cells spread widely in the parenchyma of the white matter (B1) and this spread is heaviest in areas most depleted of astrocytes (B2, B3). Scale bar, 20 μm.
Figure 10.
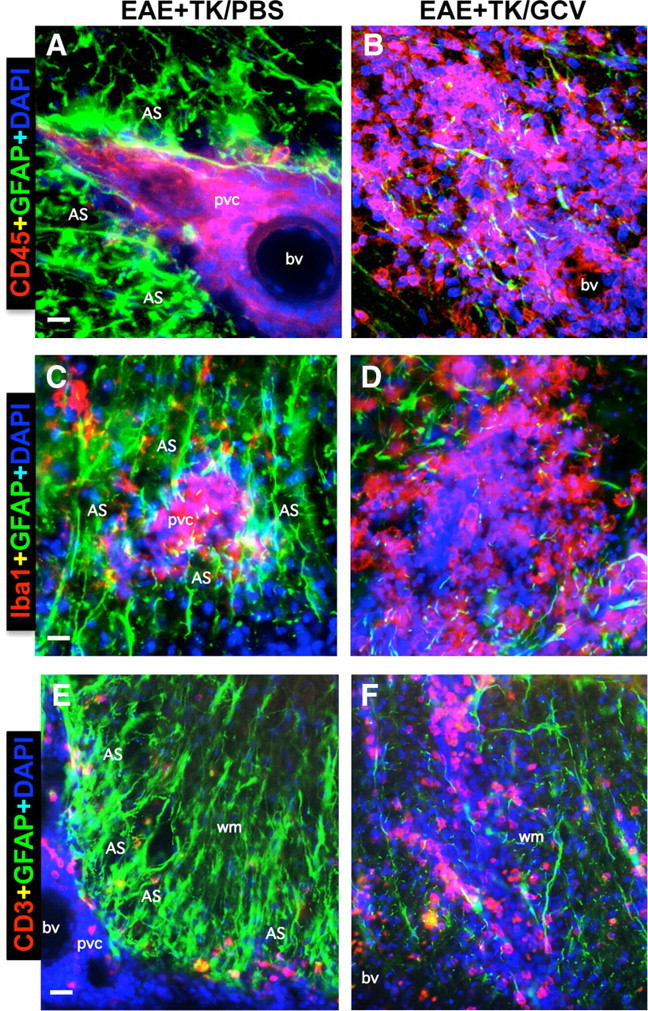
Perivascular astrocyte barriers restrain the spread of extravasting CD45-positive leukocytes, Iba1-positive macrophages, and CD3-positive T lymphocytes during EAE. A–F, Detail, merged three-color fluorescence images of spinal cord white matter stained for GFAP (green) and (DAPI blue) in combination with either the pan leukocyte marker, CD45 (red, A, B), the macrophage marker, Iba1 (red, C, D), or the T lymphocyte marker, CD3 (red, E, F), in mice with EAE+TK/PBS (A, C, E) or EAE+TK/GCV (B, D, F). Note that in EAE+TK/PBS, CD45-positive leukocytes, Iba1-positive globoid macrophages, and CD3-positive T lymphocytes are largely confined to perivascular clusters of extravasting inflammatory cells (pvc) surrounded by tightly packed GFAP-positive astrocytes (AS) (A, C, E), whereas in EAE+TK/GCV, all of these inflammatory cell types spread widely in the white matter (wm) parenchyma in regions depleted of astrocytes (B, D, F). bv, Blood vessel. Scale bars: A, B, 27 μm; C, D, 15 μm; E, F, 35 μm.
Demyelination and axon pathology in EAE with astrocyte ablation
We next looked for potential changes that might compromise neural function. Demyelination is a feature of EAE histopathology (Raine et al., 1980; Suen et al., 1997; Morales et al., 2006; Tiwari-Woodruff et al., 2007). We therefore compared the appearance of myelin in our experimental groups using histochemical staining with the myelin stain, Luxol fast blue (LFB), and immunohistochemistry for myelin basic protein (MBP). In comparison with healthy mice (Fig. 11A,D), mice with EAE exhibited demyelination throughout the white matter as shown by patchy decreases in both LFB and MBP staining (Fig. 11B,E). Mice with EAE plus astrocyte ablation also exhibited a pronounced patchy demyelination throughout the white matter (Fig. 11C,F) in a manner at least as severe or worse than that observed in with mice with EAE alone. Considerable evidence also now indicates that axonal injury is prominent in MS and EAE (Ferguson et al., 1997; Trapp et al., 1998, 1999; Wujek et al., 2002), and suggests that axonal injury may play an important part in progression of clinical signs (De Stefano et al., 2003; Sastre-Garriga et al., 2005). We therefore evaluated potential axonal pathology by using immunohistochemistry for the axonal marker NF200 and for amyloid precursor protein (APP), a prototypical marker of axonal damage in MS and EAE (Ferguson et al., 1997; Trapp et al., 1998; Wujek et al., 2002). In comparison with healthy mice (Fig. 12A1–A3), mice with EAE exhibited numerous APP-positive axons and a significant reduction in the total number of NF200-positive axonal profiles in the dorsal columns (Fig. 12B1–B3,D). In comparison with mice with EAE alone, mice with EAE plus astrocyte ablation exhibited a further significant reduction in the number of NF200-positive axonal profiles in the dorsal columns and had at least as many APP-positive axonal profiles (Fig. 12C1–C3,D). Together these findings suggest a more severe degree of neuronal pathology in mice with EAE plus astrocyte ablation.
Figure 11.
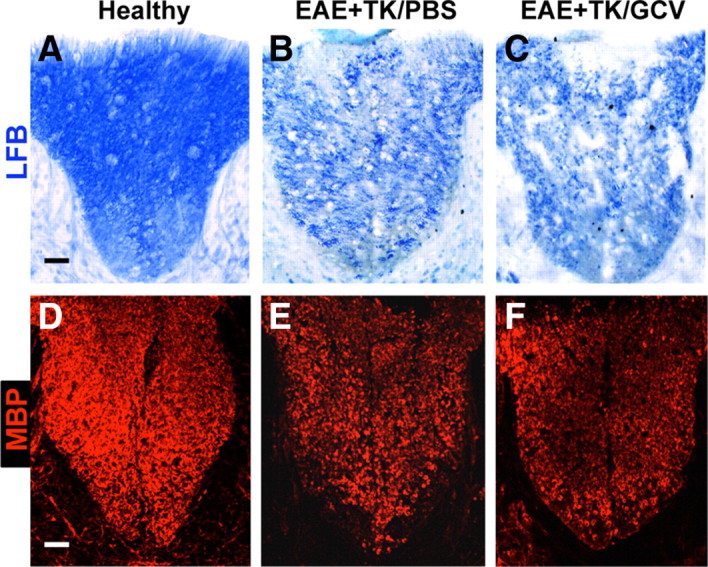
Substantial demyelination in EAE with ablation of proliferating reactive astrocytes. A–F, Survey images of dorsal columns of thoracic spinal cord of GFAP-TK mice that were either healthy (A, D) or had EAE+TK/PBS (B, E) or EAE+TK/GCV (C, F). Sections were stained either with the myelin stain Luxol fast blue (LFB, A–C), or with single-color immunofluorescence for myelin basic protein (MBP, red, D–F). Note that there is reduced staining for both LFB and MBP due to demyelination in EAE+TK/PBS (B, E) relative to healthy (A, D), and that the demyelination is as severe or worse in EAE+TK/GCV (C, F) relative to EAE+TK/PBS (B, E). Scale bars, 80 μm.
Figure 12.

Severe axonal pathology in EAE with ablation of proliferating reactive astrocytes. A–C, Single-color or merged confocal microscopic images of two-color immunofluorescence for the axonal marker, neurofilament 200 (NF200, green), and amyloid precursor protein (APP, red) in thoracic spinal cord dorsal column white matter of healthy mice (A1–A3) and in mice with EAE+TK/PBS (B1–B3) or EAE+TK/GCV (C1–C3). Note that mice with EAE+TK/PBS exhibit reduce staining of NF200 and increased staining of APP (B1–B3) relative to healthy mice (A1–A3), and that these axon pathologies are as severe or worse in mice with EAE+TK/GCV (C1–C3) relative to EAE+TK/PBS (B1–B3). D, Graph of quantitative evaluations of total number of NF200-positive axons (i.e., cross-sectional axonal profiles) in midthoracic dorsal columns of healthy mice and mice with EAE+TK/PBS or EAE+TK/GCV, showing that axon number was significantly reduced in EAE+TK/PBS relative to healthy and was further reduced significantly in EAE+TK/GCV relative to EAE+TK/PBS. *p < 0.01 relative to healthy; ⋀p < 0.01 relative to EAE+TK/PBS; ANOVA with post hoc pairwise comparisons; n = 6 per group. Scale bar, 10 μm.
Discussion
In this study we conducted a loss of function experiment based on cell deletion, to identify roles of scar-forming reactive astrocytes during peripherally initiated, adaptive or acquired immune inflammation of the CNS. To do so, we used a well characterized transgenically targeted strategy for the apoptotic ablation of proliferating reactive astrocytes, in combination with EAE, a classic model of an in vivo acquired autoimmune inflammatory challenge to the CNS. Our findings show that during EAE, reactive astrocytes form scar-like barriers that serve to restrict leukocytes to perivascular clusters and limit the infiltration of leukocytes into adjacent CNS parenchyma (Fig. 13A). Our experimental disruption of this scar-like barrier led to widespread parenchymal inflammation (Fig. 13B) and a markedly more severe clinical course. These findings demonstrate a novel barrier function for scar-forming reactive astrocytes during peripherally initiated, acquired immune CNS inflammation and suggest that the loss or disruption of this barrier function of reactive astrocytes may contribute to the neuropathology associated with autoimmune conditions such as multiple sclerosis (MS) and related disorders.
Figure 13.

Schematic summary of perivascular barrier functions of proliferating reactive astrocytes during autoimmune-mediated leukocyte infiltration in the CNS. A, In EAE, reactive astrocytes and their processes densely surround perivascular clusters of extravasated immune and inflammatory leukocytes in a manner reminiscent of scar formation after CNS injury; and there is limited spread of inflammatory leukocytes into the neighboring CNS parenchyma. B, In EAE combined with transgenically targeted ablation of proliferating scar-forming reactive astrocytes, there is a failure of astrocytes to surround leukocytes in the perivascular region and this failure is accompanied by widespread infiltration of inflammatory leukocytes (including macrophages, T lymphocytes, and neutrophils) throughout the CNS parenchyma. Together, these observations argue that perivascular reactive astrocytes are exerting an essential scar-like barrier function that restricts infiltrating leukocytes to the perivascular space and limits their spread into nearby healthy CNS parenchyma. This process provides a cellular mechanism underlying the common neuropathological observation of perivascular clustering or “cuffing” of inflammatory leukocytes. The loss or attenuation of this scar-like perivascular barrier function of reactive astrocytes has the potential to exacerbate the spread of parenchymal spread of inflammation in various CNS disorders.
Reactive astrocytes form scar-like perivascular barriers to leukocytes during EAE
The aggregation of extravasated immune and inflammatory cells into perivascular clusters (often referred to as “perivascular cuffs”) is a hallmark of primary CNS inflammatory conditions such as EAE, MS, and viral encephalitis (Raine et al., 1980; Adams et al., 1989; Gareau et al., 2002; Kurt-Jones et al., 2004), but the cellular mechanisms that lead to the formation of these perivascular leukocyte clusters and their significance are elusive. Our findings provide experimental evidence for a novel functional paradigm whereby reactive scar-forming astrocytes form perivascular barriers that serve to restrict extravasated leukocytes to perivascular clusters during adaptive or acquired immune CNS inflammatory challenges and limit their spread into adjacent CNS parenchyma.
Astrocyte scars that form around areas of damaged and inflamed tissue after traumatic injury to the CNS are well known for their inhibitory effects on axon migration (Silver and Miller, 2004) and have also been shown to limit the spread of inflammatory cells from damaged areas into neighboring healthy tissue (Bush et al., 1999; Faulkner et al., 2004; Sofroniew, 2005; Okada et al., 2006; Herrmann et al., 2008). Our findings here show (1) that dense profiles of reactive astrocytes form around perivascular clusters of extravasating inflammatory leukocytes during EAE in a manner reminiscent of astrocyte scar formation around areas of CNS trauma, and (2) that transgenically targeted ablation of these perivascular reactive astrocytes leads to widespread parenchymal infiltration of inflammatory cells. These findings provide direct experimental evidence that scar-forming reactive astrocytes form neuroprotective barriers to immune and inflammatory leukocyte infiltration not only during centrally initiated, innate CNS inflammation after trauma as shown previously, but also during peripherally initiated, adaptive or acquired immune CNS inflammation as shown here.
These findings add to the accumulating evidence that astrocytes play important roles in the regulation of leukocyte trafficking in CNS tissue and suggest that their functions are complex and likely to be context dependent, and cannot be regarded as uniformly proinflammatory or anti-inflammatory (John et al., 2005; Sofroniew, 2005; Nair et al., 2008; Qin et al., 2008). One simple model of potential astrocyte roles that is parsimonious with most available evidence, is that locally activated astrocytes can take part both in attracting inflammatory cells to specific sites and in restricting inflammatory cells to those sites by limiting their spread into adjacent healthy CNS parenchyma. Because CNS inflammation can be initiated not only by local events, but also peripherally by infection, autoimmune inoculation or molecular mimicry (Sospedra and Martin, 2005), and because, as shown here, astrocytes can exert regulatory functions in both cases, such a model has interesting implications for understanding how astrocytes might respond to a peripherally initiated autoimmune inflammatory challenge that has widespread effects; and may provide novel insights into certain disease mechanisms. For example, during autoimmune inflammation, astrocytes may be activated in a manner and time course so as to contribute to the attraction of immune and inflammatory cells to perivascular regions while at the same time attempting to restrict their infiltration into neighboring CNS parenchyma.
Methodological considerations
The GCV-mediated death of TK-expressing cells is driven by noninflammatory apoptotic pathways (Fischer et al., 2005). Numerous studies ablating different cell types using the HSV-TK+GCV transgenic strategy (Borrelli et al., 1989; Canfield et al., 1996), including oligodendrocyte progenitors in the CNS (Mathis et al., 2000; Jalabi et al., 2005), have shown that this method of cell ablation on its own does not cause inflammation in vivo. In uninjured GFAP-TK mice of the transgenic strain used in the present study, the HSV-TK+GCV-mediated ablation of proliferating GFAP-expressing adult neural stem cells does not result in inflammation (Garcia et al., 2004). Targeting HSV-TK+GCV to microglia attenuates both the clinical response to EAE and the infiltration of T lymphocytes (Heppner et al., 2005), which is the opposite effect to that observed after ablation of astrocytes during EAE in the present study and indicates that the HSV-TK+GCV ablation procedure does not simply on its own induce nonspecific inflammation. Furthermore, inhibiting reactive astrogliosis by conditional deletion of the intracellular signaling molecule, STAT3, results in increased inflammation after spinal cord injury (Okada et al., 2006; Herrmann et al., 2008) in a manner that is similar to HSV-TK+GCV-mediated astrocyte ablation (Faulkner et al., 2004) but does so by attenuating rather than ablating reactive astrocytes. Together, these observations argue strongly that the increased inflammation we observe after ablating proliferating reactive scar-forming astrocytes is due to the selective loss of these specific cells and to the consequent loss of effects that they exert, rather than to a nonspecific response to the nonspecific death of any cell type in the CNS triggered by the cell ablation strategy.
Protective role of perivascular astrocyte scar formation
The pronounced exacerbation of CNS inflammation that we observed after ablation of scar-forming astrocytes in EAE was associated with a substantially more severe clinical course as well as with evidence of severe axonal pathology in white matter. Axonal pathology is prominent in both MS and EAE, and the severity of inflammation is thought to be related to the level of axonal degeneration (Ferguson et al., 1997; Trapp et al., 1998, 1999; Wujek et al., 2002). Chronic deficits in MS patients have been reported to correlate with axonal loss (De Stefano et al., 2003; Sastre-Garriga et al., 2005), and EAE induced in transgenic mice that express yellow fluorescent protein (YFP) in neurons revealed that a loss in YFP fluorescence correlated with immune cell infiltration (Shriver and Dittel, 2006). A growing body of evidence also indicates that different kinds of inflammatory cells can directly mediate axonal damage, including lymphocytes (Shriver and Dittel, 2006; Howe et al., 2007; Howe, 2008) and macrophage/microglia lineage cells (Horn et al., 2008). Other evidence is accumulating that both astrocytes and astroglioma cells can exert direct, potent anti-inflammatory effects on microglia, macrophages, and other inflammatory cells both in vitro and in vivo, via mechanisms that at least in part involve JAK-STAT signaling (Karaosmanoglu et al., 1996; Walker et al., 2003; Min et al., 2006; Herrmann et al., 2008; Kostianovsky et al., 2008). Our observations here are compatible with, and extend these previous findings by providing experimental evidence that scar-forming reactive astrocytes protect CNS tissue and function by limiting the spread of immune and inflammatory leukocytes into CNS parenchyma during acquired autoimmune CNS inflammation.
Reactive astrocyte roles in EAE and MS
The basic process of reactive astrogliosis has long been regarded as primarily detrimental in MS and EAE, and reactive astrocytes have in some cases been proposed as potential mediators in the effector phase of EAE (Girvin et al., 2002). However, it has also been known for some time that mice developmentally lacking the astrocyte structural protein GFAP (gfap−/−) exhibit a more severe course of EAE (Liedtke et al., 1998). Recent evidence also indicates that mice lacking the heat shock protein αB-crystallin (Cryab−/−) exhibit more severe clinical EAE and increased CNS inflammation in combination with an increased apoptosis of astrocytes (Ousman et al., 2007). Although the Cryab−/− was not targeted to a specific cell type, the findings are consistent with the idea that a loss of astrocyte functions may exacerbate inflammation and clinical severity in EAE. Nevertheless, numerous findings suggest potential proinflammatory roles for reactive astrocytes (Dong and Benveniste, 2001; Chen and Swanson, 2003; Farina et al., 2007; Nair et al., 2008) and inhibition of astroglial NfkB has recently been reported to improve functional outcome in EAE (Brambilla et al., 2009). These observations are not incompatible with our observations here or with the notion that reactive astroglia may serve both to attract inflammatory cells to specific CNS sites and at the same time limit their migration away from those sites (Sofroniew, 2005). Our findings here argue that normally functioning, scar-forming reactive astrocytes exert a beneficial role in EAE and MS by acting to restrict extravasated leukocytes to perivascular clusters and thereby reduce the infiltration of immune and inflammatory leukocytes into adjacent CNS parenchyma. This idea is supported by recent clinical evidence that the majority (>90%) of patients with neuromyelitis optica, a CNS inflammatory disease with similarities to MS, have autoantibodies to aquaporin-4, a molecule expressed in the CNS only on astrocyte foot processes (Lennon et al., 2005), and that the severity of clinical signs and spinal cord lesions in such patients correlate with aquaporin-4-specific antibody titers (Takahashi et al., 2007). Together, these observations suggest that a loss or disruption of barrier functions of scar-forming reactive astrocyte may represent a pathophysiological mechanism that could precipitate or exacerbate CNS pathology in acquired immune inflammatory disorders of the CNS such as MS or related conditions.
Footnotes
This work was supported by a Collaborative Center Grant (CA1028) from the National Multiple Sclerosis Society (R.R.V.), grants from the Adelson Medical Research Foundation (R.R.V., M.V.S.), and National Institutes of Health–National Institute of Neurological Disorders and Stroke Grant NS057624 (M.V.S.). R.S.P. is a Fellow of the National Multiple Sclerosis Society. We thank D. S. Crandall for artwork.
The authors declare no competing financial interests.
References
- Adams CW, Poston RN, Buk SJ. Pathology, histochemistry and immunocytochemistry of lesions in acute multiple sclerosis. J Neurol Sci. 1989;92:291–306. doi: 10.1016/0022-510x(89)90144-5. [DOI] [PubMed] [Google Scholar]
- Ahmed Z, Shaw G, Sharma VP, Yang C, McGowan E, Dickson DW. Actin-binding proteins coronin-1a and IBA-1 are effective microglial markers for immunohistochemistry. J Histochem Cytochem. 2007;55:687–700. doi: 10.1369/jhc.6A7156.2007. [DOI] [PubMed] [Google Scholar]
- Aloisi F, Penna G, Cerase J, Menéndez Iglesias B, Adorini L. IL-12 production by central nervous system microglia is inhibited by astrocytes. J Immunol. 1997;159:1604–1612. [PubMed] [Google Scholar]
- Borrelli E, Heyman RA, Arias C, Sawchenko PE, Evans RM. Transgenic mice with inducible dwarfism. Nature. 1989;339:538–541. doi: 10.1038/339538a0. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Persaud T, Hu X, Karmally S, Shestopalov VI, Dvoriantchikova G, Ivanov D, Nathanson L, Barnum SR, Bethea JR. Transgenic inhibition of astroglial NF-kappaB improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J Immunol. 2009;182:2628–2640. doi: 10.4049/jimmunol.0802954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DC, Gatter KC. Monoclonal antibody Ki-67: its use in histopathology. Histopathology. 1990;17:489–503. doi: 10.1111/j.1365-2559.1990.tb00788.x. [DOI] [PubMed] [Google Scholar]
- Bush TG, Savidge TC, Freeman TC, Cox HJ, Campbell EA, Mucke L, Johnson MH, Sofroniew MV. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell. 1998;93:189–201. doi: 10.1016/s0092-8674(00)81571-8. [DOI] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Canfield V, West AB, Goldenring JR, Levenson R. Genetic ablation of parietal cells in transgenic mice: A new model for analyzing cell lineage relationships in the gastric mucosa. Proc Natl Acad Sci U S A. 1996;93:2431–2435. doi: 10.1073/pnas.93.6.2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbonneau H, Tonks NK, Walsh KA, Fischer EH. The leukocyte common antigen (CD45): a putative receptor-linked protein tyrosine phosphatase. Proc Natl Acad Sci U S A. 1988;85:7182–7186. doi: 10.1073/pnas.85.19.7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Swanson RA. Astrocytes and brain injury. J Cereb Blood Flow Metab. 2003;23:137–149. doi: 10.1097/01.WCB.0000044631.80210.3C. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Matthews PM, Filippi M, Agosta F, De Luca M, Bartolozzi ML, Guidi L, Ghezzi A, Montanari E, Cifelli A, Federico A, Smith SM. Evidence of early cortical atrophy in MS: relevance to white matter changes and disability. Neurology. 2003;60:1157–1162. doi: 10.1212/01.wnl.0000055926.69643.03. [DOI] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Donnelly DJ, Popovich PG. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol. 2008;209:378–388. doi: 10.1016/j.expneurol.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem. 2003;384:505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- Eng LF, Gerstl B, Vanderhaeghen JJ. A study of proteins in old multiple sclerosis plaques. Trans Am Soc Neurochem. 1970;1:42. [Google Scholar]
- Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000) Neurochem Res. 2000;25:1439–1451. doi: 10.1023/a:1007677003387. [DOI] [PubMed] [Google Scholar]
- Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120:393–399. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- Fischer U, Steffens S, Frank S, Rainov NG, Schulze-Osthoff K, Kramm CM. Mechanisms of thymidine kinase/ganciclovir and cytosine deaminase/ 5-fluorocytosine suicide gene therapy-induced cell death in glioma cells. Oncogene. 2005;24:1231–1243. doi: 10.1038/sj.onc.1208290. [DOI] [PubMed] [Google Scholar]
- Frank KB, Chiou JF, Cheng YC. Interaction of herpes simplex virus-induced DNA polymerase with 9-(1,3-dihydroxy-2-propoxymethyl)guanine triphosphate. J Biol Chem. 1984;259:1566–1569. [PubMed] [Google Scholar]
- Garcia ADR, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principle source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Gareau PJ, Wymore AC, Cofer GP, Johnson GA. Imaging inflammation: direct visualization of perivascular cuffing in EAE by magnetic resonance microscopy. J Magn Reson Imaging. 2002;16:28–36. doi: 10.1002/jmri.10136. [DOI] [PubMed] [Google Scholar]
- Girvin AM, Gordon KB, Welsh CJ, Clipstone NA, Miller SD. Differential abilities of central nervous system resident endothelial cells and astrocytes to serve as inducible antigen-presenting cells. Blood. 2002;99:3692–3701. doi: 10.1182/blood-2001-12-0229. [DOI] [PubMed] [Google Scholar]
- Gundersen HJG, Bendtsen TF, Korbo L, Marcussen N, Møller A, Nielsen K, Nyengaard JR, Pakkenberg B, Sørensen FB, Vesterby A, West MJ. Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. APMIS. 1988;96:379–394. doi: 10.1111/j.1699-0463.1988.tb05320.x. [DOI] [PubMed] [Google Scholar]
- Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hövelmeyer N, Waisman A, Rülicke T, Prinz M, Priller J, Becher B, Aguzzi A. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11:146–152. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyman RA, Borrelli E, Lesley J, Anderson D, Richman DD, Baird SM, Hyman R, Evans RM. Thymidine kinase obliteration: creation of transgenic mice with controlled immune deficiency. Proc Natl Acad Sci U S A. 1989;86:2698–2702. doi: 10.1073/pnas.86.8.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch S, Gordon S. Polymorphic expression of a neutrophil differentiation antigen revealed by monoclonal antibody 7/4. Immunogenetics. 1983;18:229–239. doi: 10.1007/BF00952962. [DOI] [PubMed] [Google Scholar]
- Horn KP, Busch SA, Hawthorne AL, van Rooijen N, Silver J. Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci. 2008;28:9330–9341. doi: 10.1523/JNEUROSCI.2488-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe CL. Immunological aspects of axon injury in multiple sclerosis. Curr Top Microbiol Immunol. 2008;318:93–131. doi: 10.1007/978-3-540-73677-6_5. [DOI] [PubMed] [Google Scholar]
- Howe CL, Ure D, Adelson JD, LaFrance-Corey R, Johnson A, Rodriguez M. CD8+ T cells directed against a viral peptide contribute to loss of motor function by disrupting axonal transport in a viral model of fulminant demyelination. J Neuroimmunol. 2007;188:13–21. doi: 10.1016/j.jneuroim.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- Jalabi W, Boehm N, Grucker D, Ghandour MS. Recovery of myelin after induction of oligodendrocyte cell death in postnatal brain. J Neurosci. 2005;25:2885–2894. doi: 10.1523/JNEUROSCI.2748-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John GR, Lee SC, Song X, Rivieccio M, Brosnan CF. IL-1-regulated responses in astrocytes: relevance to injury and recovery. Glia. 2005;49:161–176. doi: 10.1002/glia.20109. [DOI] [PubMed] [Google Scholar]
- Jones MV, Nguyen TT, Deboy CA, Griffin JW, Whartenby KA, Kerr DA, Calabresi PA. Behavioral and pathological outcomes in MOG 35–55 experimental autoimmune encephalomyelitis. J Neuroimmunol. 2008;199:83–93. doi: 10.1016/j.jneuroim.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Karaosmanoglu T, Aygun B, Wade PR, Gershon MD. Regional differences in the number of neurons in the myenteric plexus of the guinea pig small intestine and colon: an evaluation of markers used to count neurons. Anat Rec. 1996;244:470–480. doi: 10.1002/(SICI)1097-0185(199604)244:4<470::AID-AR5>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Kostianovsky AM, Maier LM, Anderson RC, Bruce JN, Anderson DE. Astrocytic regulation of human monocytic/microglial activation. J Immunol. 2008;181:5425–5432. doi: 10.4049/jimmunol.181.8.5425. [DOI] [PubMed] [Google Scholar]
- Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A. 2004;101:1315–1320. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, Nodin C, Ståhlberg A, Aprico K, Larsson K, Yabe T, Moons L, Fotheringham A, Davies I, Carmeliet P, Schwartz JP, Pekna M, Kubista M, Blomstrand F, Maragakis N. Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab. 2008;28:468–481. doi: 10.1038/sj.jcbfm.9600546. [DOI] [PubMed] [Google Scholar]
- Liedtke W, Edelmann W, Chiu FC, Kucherlapati R, Raine CS. Experimental autoimmune encephalomyelitis in mice lacking glial fibrillary acidic protein is characterized by a more severe clinical course and an infiltrative central nervous system lesion. Am J Pathol. 1998;152:251–259. [PMC free article] [PubMed] [Google Scholar]
- Liu HB, Loo KK, Palaszynski K, Ashouri J, Lubahn DB, Voskuhl RR. Estrogen receptor alpha mediates estrogen's immune protection in autoimmune disease. J Immunol. 2003;171:6936–6940. doi: 10.4049/jimmunol.171.12.6936. [DOI] [PubMed] [Google Scholar]
- Mathis C, Hindelang C, LeMeur M, Borrelli E. A transgenic mouse model for inducible and reversible dysmyelination. J Neurosci. 2000;20:7698–7705. doi: 10.1523/JNEUROSCI.20-20-07698.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8:913–919. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- Min KJ, Yang MS, Kim SU, Jou I, Joe EH. Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J Neurosci. 2006;26:1880–1887. doi: 10.1523/JNEUROSCI.3696-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokhtarian F, McFarlin DE, Raine CS. Adoptive transfer of myelin basic protein-sensitized T cells produces chronic relapsing demyelinating disease in mice. Nature. 1984;309:356–358. doi: 10.1038/309356a0. [DOI] [PubMed] [Google Scholar]
- Morales LB, Loo KK, Liu HB, Peterson C, Tiwari-Woodruff S, Voskuhl RR. Treatment with an estrogen receptor alpha ligand is neuroprotective in experimental autoimmune encephalomyelitis. J Neurosci. 2006;26:6823–6833. doi: 10.1523/JNEUROSCI.0453-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- Nair A, Frederick TJ, Miller SD. Astrocytes in multiple sclerosis: a product of their environment. Cell Mol Life Sci. 2008;65:2702–2720. doi: 10.1007/s00018-008-8059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Ousman SS, Tomooka BH, van Noort JM, Wawrousek EF, O'Connor KC, Hafler DA, Sobel RA, Robinson WH, Steinman L. Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature. 2007;448:474–479. doi: 10.1038/nature05935. [DOI] [PubMed] [Google Scholar]
- Qin H, Niyongere SA, Lee SJ, Baker BJ, Benveniste EN. Expression and functional significance of SOCS-1 and SOCS-3 in astrocytes. J Immunol. 2008;181:3167–3176. doi: 10.4049/jimmunol.181.5.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raine CS, Barnett LB, Brown A, Behar T, McFarlin DE. Neuropathology of experimental allergic encephalomyelitis in inbred strains of mice. Lab Invest. 1980;43:150–157. [PubMed] [Google Scholar]
- Sastre-Garriga J, Ingle GT, Rovaris M, Téllez N, Jasperse B, Altmann DR, Benedetti B, Stevenson VL, Cercignani M, Leary SM, Barkhof F, Brochet B, Dousset V, Filippi M, Montalban X, Kalkers NF, Polman CH, Rovira A, Miller DH, Thompson AJ. Long-term clinical outcome of primary progressive MS: predictive value of clinical and MRI data. Neurology. 2005;65:633–635. doi: 10.1212/01.wnl.0000173061.12776.1f. [DOI] [PubMed] [Google Scholar]
- Schnell L, Fearn S, Klassen H, Schwab ME, Perry VH. Acute inflammatory responses to mechanical lesions in the CNS: differences between brain and spinal cord. Eur J Neurosci. 1999;11:3648–3658. doi: 10.1046/j.1460-9568.1999.00792.x. [DOI] [PubMed] [Google Scholar]
- Shriver LP, Dittel BN. T-cell-mediated disruption of the neuronal microtubule network: correlation with early reversible axonal dysfunction in acute experimental autoimmune encephalomyelitis. Am J Pathol. 2006;169:999–1011. doi: 10.2353/ajpath.2006.050791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- Suen WE, Bergman CM, Hjelmström P, Ruddle NH. A critical role for lymphotoxin in experimental allergic encephalomyelitis. J Exp Med. 1997;186:1233–12340. doi: 10.1084/jem.186.8.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura M, Watanabe S, Shiga Y, Kanaoka C, Fujimori J, Sato S, Itoyama Y. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain. 2007;130:1235–1243. doi: 10.1093/brain/awm062. [DOI] [PubMed] [Google Scholar]
- Tiwari-Woodruff S, Morales LB, Lee R, Voskuhl RR. Differential neuroprotective and antiinflammatory effects of estrogen receptor (ER)alpha and ERbeta ligand treatment. Proc Natl Acad Sci U S A. 2007;104:14813–14818. doi: 10.1073/pnas.0703783104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Ransohoff R, Rudick R. Axonal pathology in multiple sclerosis: relationship to neurologic disability. Curr Opin Neurol. 1999;12:295–302. doi: 10.1097/00019052-199906000-00008. [DOI] [PubMed] [Google Scholar]
- von Boehmer H. The developmental biology of T lymphocytes. Annu Rev Immunol. 1988;6:309–326. doi: 10.1146/annurev.iy.06.040188.001521. [DOI] [PubMed] [Google Scholar]
- Walker PR, Calzascia T, de Tribolet N, Dietrich PY. T-cell immune responses in the brain and their relevance for cerebral malignancies. Brain Res Brain Res Rev. 2003;42:97–122. doi: 10.1016/s0165-0173(03)00141-3. [DOI] [PubMed] [Google Scholar]
- Wujek JR, Bjartmar C, Richer E, Ransohoff RM, Yu M, Tuohy VK, Trapp BD. Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. J Neuropathol Exp Neurol. 2002;61:23–32. doi: 10.1093/jnen/61.1.23. [DOI] [PubMed] [Google Scholar]