

Abstract
Gold (Au) nanoshells were grown on silica nanoparticles with differing average diameters, ranging from 30 nm to 120 nm. Au nanoshells were also formed on silica spheres encapsulating 5 nm diameter magnetic iron oxide nanocrystals. The optical absorbance spectra of these Au nanoshells are reported. The plasmon resonance wavelengths of the smaller diameter nanoshells were significantly less tunable than those of the larger diameter nanoshells. This is due to a reduced range of accessible core-shell ratio—the geometric factor that determines the plasmon peak position—as the silica core diameter shrinks. The smaller diameter nanoshells were also found to be highly prone to aggregation, which broadens the plasmon absorption peak. Model calculations of dispersion stability as a function of silica core diameter reveal that smaller diameter Au shells exhibit more aggregation because of the size-dependence of the electrostatic double-layer potential.
Introduction
Gold (Au) nanoshells are plasmonic materials that strongly absorb and scatter visible and near-IR light with size-tunable color.1–6 Au nanoshells are typically made by coating a nanoparticle of a low dielectric constant material like silica with Au; the core/shell size ratio determines the nanoshell plasmon resonance frequency.2,3,7 Metal nanoshells can be conjugated with biological molecules using relatively straightforward chemistry,8–12 and since nanoshells can be made with plasmon resonances at wavelengths that are particularly transparent for tissue (between 700 nm and 1000 nm),13,14 they have been explored for use as optical contrast agents for tumor cell imaging.8,15 Additionally, medical therapeutics might also be combined with imaging, as Au nanoshells could serve as vehicles for drug delivery or for minimally-invasive photothermal therapy since light excitation leads to local heating that can be used as a trigger for targeted cell death.14,16–19
Au nanoshells are typically synthesized between 80 and 150 nm in diameter.1–6 For in vivo medical applications, Au nanoshells in this size range are restricted to direct delivery to the site of interest,6,14,20,21 as there are problems with systemic delivery associated with quick clearance from the blood by liver and spleen22–26 and limited diffusion of such big particles in tissue.27–29 Considering also that the various molecular coatings, such as polyethylene glycol (PEG) to suppress immune response and recognition molecules like antibodies, aptamers or peptide fragments needed to bind biological targets,16,30- can add another 40 nm or so to the bare particle diameter,34,35 nanoshells with smaller diameters, of 10 to 60 nm prior to bioconjugation, are better suited for in vivo medical applications.
There are only a few reports of nanoshells with diameters of 10 to 60 nm. 36–39 Xia, et al.39 recently reported Au nanoshells on 32 nm diameter silica-coated Au particles. The optical absorption peak was relatively broad with a maximum wavelength of 681 nm. Their transmission electron microscopy (TEM) showed that the nanoshells were not spherical, and the limited optical tunability was attributed in part to rough surfaces and agglomeration. Alternatives to silica core particles for making smaller diameter Au nanoshells have also been reported; for instance, Au nanoshells ranging from 45 to 60 nm in diameter have been made by depositing Au on 9 nm diameter iron oxide nanoparticles.37,38 The plasmon wavelength range for these nanoshells was again relatively limited, from 535 and 540 nm—comparable to the plasmon resonance wavelengths of solid Au nanoparticles—because of their small core/shell thickness ratios. Hollow Au nanoshells have also recently been made by galvanic displacement of silver or cobalt nanospheres. 5,36 The nanoshells grown from cobalt spheres ranged from 58 nm to 60 nm in diameter with plasmon peak wavelengths ranging from 530 to 630 nm and the nanoshells produced from silver spheres were 10–20 nm in diameter with a plasmon resonances that could be tuned between 550 and 800 nm. These are promising results, but the structural stability of such unsupported nanoshells remains to be tested and may present a problem for in vivo medical applications. Furthermore, a silica core enables the incorporation of smaller nanocrystals of different materials, like magnets or luminescent semiconductors within the nanoshell that can provide added functionality, for multimodal medical imaging contrast with both optical and magnetic signatures for example. 20,40
Considering the stability, biocompatibility, and potential for development into a multi- functional material by manipulating the core material composition of the silica-supported Au nanoshells, we aimed to understand the limitations of this system for obtaining small diameter (20 to 60 nm diameter) Au nanoshells with size-tunable optical resonances from visible to NIR frequencies. Herein, we demonstrate the formation of Au nanoshells on silica spheres as small as 30 nm in diameter. We also demonstrate the formation of Au nanoshells on 40 nm diameter silica spheres encapsulating 5 nm diameter magnetic iron oxide nanocrystals. However, as the nanoshell diameter decreased, the plasmon peak could only be tuned to increasingly shorter wavelengths. With decreasing silica core size, the geometric ratio of the Au shell thickness to the diameter becomes increasingly limited by the size of the ~2 nm diameter Au nanocrystals used to seed shell growth; the gold shell simply cannot be made thin enough relative to the core diameter to shift the plasmon to NIR wavelengths. The smaller diameter nanoshells were also found to be highly susceptible to aggregation—to a much greater extent than the larger diameter nanoshells. With model calculations of the size-dependent interparticle potential, we reveal that larger nanoshells are more stable than smaller nanoshells during synthesis because of a more effective charge double-layer repulsion. These factors lead to fundamental challenges in the synthesis of smaller diameter Au nanoshells with NIR plasmon resonances.
Experimental Details
Chemicals
Octyl ether (99%) and oleic acid (≥99%) were purchased from Fluka. Iron pentacarbonyl (Fe(CO)5, 99.999%), tetraethylorthosilicate (TEOS, 98%), IgePal CO-520 (Mn ~ 441), tetrakis(hydroxymethyl)phosphonium chloride (THPC, 80% solution in water), sodium hydroxide pellets (97+%), tetrachloroauric acid (HAuCl4·3H2O, 99.9+%), tris(2′2-bipyridyl)dichlororuthenium(II) hexahydrate (Rubpy, 99.95%), n-hexanol (anhydrous, ≥99%), Triton X-100 (97+%), and 3-aminopropyltrimethoxysilane (APTMS, 97%) were purchased from Sigma Aldrich. Cyclohexane (99.9%), potassium carbonate (anhydrous, 99.8%), and formaldehyde solution (37% w/w in water) were obtained from Fisher Scientific. Ammonium hydroxide (NH4OH, 28% in water) was purchased from EM Science. Colloidal silica particles (product name Organosilicasol™ IPA-ST-ZL, 30% w/w SiO2 in isopropanol, 70–100 nm particle diameter) were obtained from Nissan Chemical Company. All chemicals were used as received. Doubly-distilled, deionized (18 M Ω resistance), 0.2-micron filtered water (DI-H2O) was used for all preparations.
Silica-coated Iron Oxide Nanocrystal Synthesis
Iron oxide nanocrystal synthesis and subsequent silica coating was performed using published procedures.41,42
Hydrophobic 5 nm diameter iron oxide nanocrystals were first prepared on a Schlenk line under nitrogen atmosphere. 10 mL octyl ether and 0.9588 mL oleic acid were added to a 3-neck flask fitted with a cooling water condenser and two rubber septa. A thermocouple was fed through one septum and allowed to contact the solution, and the condenser was connected to a stopcock with Teflon valve. The system was purged of oxygen by connecting the stopcock to the Schlenk line, closing the stopcock valve and pulling vacuum, switching to nitrogen flow and opening the stopcock valve for a few minutes, and repeating these steps for three additional cycles. Subsequently, the solution was heated to 100°C under magnetic stirring using a heating mantle, which was powered by a variable autotransformer linked to the thermocouple by an electronic temperature controller. As soon as the solution temperature stabilized at 100°C for five minutes, 0.2 mL of iron pentacarbonyl was then injected into the flask and the mixture was heated to 297°C.43 During heating, the reaction mixture proceeds through a series of color changes from orange to clear yellow, and finally black at the final temperature setting. The heating rate is important: the autotransformer was set to 70% power output and the solution reached its temperature set point after 30–40 minutes. The mixture was refluxed at 297°C for one hour, then the heating mantel was removed and the mixture was allowed to cool under nitrogen by equilibrating the flask with room temperature air. When the solution temperature reached 70°C, the reaction mixture was exposed to oxygen by disconnecting the open stopcock from the Schlenk line. Once the reaction mixture cooled to room temperature, it was poured into a centrifuge tube and centrifuged with ethanol antisolvent (4 mL per 1 mL of reaction product) at 8000 rpm for 5 minutes at 20°C. The black precipitate was suspended in 10 mL hexane and re-precipitated with ethanol as just described at least four times, then finally dispersed in cyclohexane and stored in a parafilm-sealed glass vial. The iron oxide nanocrystals had a mean diameter of 5.0 ± 0.8 nm, determined by TEM.
To coat the iron oxide nanocrystals with silica, 16.2 mL of cyclohexane was first added to a 20 mL screwcap glass vial. With magnetic stirring, 0.800 mL Igepal, 37.7 μL iron oxide nanocrystals (15.5 mg/mL in cyclohexane), 0.130 mL of ammonium hydroxide (28% in water), and 0.100 mL of TEOS were added to the vial in precisely that order. The microemulsions were covered and stirred for 2 days. The nanoparticles were extracted with about 5 mL of methanol. The nanoparticles were purified by adding 20 mL of hexane per 5 mL of extracted methanol dispersion. The solutions were centrifuged at 2500 rpm for 6 hours. The precipitate was collected and re-dispersed in 5 mL ethanol. Hexane was added as an antisolvent to re-precipitate the nanoparticles. This dispersion was centrifuged again to isolate the precipitated nanoparticles. This purification procedure was repeated three times to remove unreacted TEOS and excess Igepal. The final nanoparticle product was re-dispersed in 5 mL of ethanol. High centrifugation speeds, complete drying (rotary evaporation), and an extensive number of centrifugation cycles were all seen to make the silica particles very difficult to redisperse in any solvent, so these actions should be avoided with all of the silica nanoparticle preparations.
Microemulsion-Assisted Synthesis of Colloidal Silica Particles
30 nm diameter silica particles were synthesized in microemulsion media using the published procedures.44 Two mL of Igepal, 0.325 mL of NH4OH (28% w/w) and 0.250 mL of TEOS (0.250 mL) were added sequentially to 41 mL of cyclohexane. After stirring this cyclohexane solution for two days, the silica particles were extracted from the organic phase with ~5 mL methanol. The particles were isolated and purified as described for the silica-coated iron oxide.
Larger diameter (70 nm) silica particles were also made by a microemulsion method, but required a different surfactant combination.45 The published procedures had incorporated a fluorescent dye, Rubpy, and therefore this procedure was followed exactly as described, including the addition of Rubpy, since the silica chemistry is sensitive to any chemical changes. First, 7.5 mL cyclohexane, 1.8 mL n-hexanol, 1.77 mL Triton X-100, 0.48 mL Rubpy (20 mM, aq), and 0.20 mL of TEOS were placed under magnetic stirring for 20 minutes in a conical flask. Next, 0.060 mL of ammonium hydroxide was added and the reaction was stirred for 24 hours. Subsequently, an equal volume (11.65 mL) of acetone was added to the completed reaction with vortexing to break the microemulsion. The dye-doped silica nanoparticles were collected by centrifuging at 3000 rpm for 30 minutes, the supernatant was discarded, and the particles were then suspended in 5 mL of ethanol. 20 mL of hexane was added, the particles were centrifuged again at the same parameters, and the supernatant was discarded. The particles were suspended in 5 mL ethanol, and the centrifugation cycle was repeated two more times.
Commercial Silica Preparation
The commercial silica colloid IPA-ST-ZL is supplied as an isopropanol dispersion. Prior to gold shell growth, these silica particles need to be purified by size selective precipitation and suspended in ethanol for surface modification. 20 mL of hexane was added to 5 mL of ST-ZL isopropanol-based colloid and centrifuged for 1 hour at 2500 rpm. The supernatant was discarded and the white precipitate was re-dispersed in 5 mL of ethanol with mild sonication. The concentrated sample was further purified through size selective precipitation to collect the largest silica spheres. Hexane was added dropwise to the nanoparticles until observing a slight change in turbidity (typically after adding ~0.5 mL), the particles were centrifuged for 1 hour at 2500 rpm and the precipitate was collected by pouring off the supernatant. The precipitated nanoparticles were dispersed in 5 mL ethanol, and the observed average particle diameter was 118 ± 5 nm from TEM measurement.
Silica Surface Modification with APTS
Prior to Au shell deposition, the silica particles were functionalized with amines (APTS) using a modification of the procedures described by Lee for attaching octyltrimethoxysilane to silica spheres.42 This method was more reproducible than previously reported methods of refluxing silica spheres in APTS,39 and thus more effective for reproducibly achieving high surface coverage of Au nanocrystals on the amine-terminated silica particles in the next step. 0.10 mL of 30% NH4OH (aq) was added to 10 mL of a 1 mg/mL dispersion of purified silica particles under magnetic stirring.46 0.5 mL of 10% v/v of APTS in ethanol was added dropwise to the dispersion. The dispersion was stirred for 24 hours. The nanoparticles were then precipitated by adding 40 mL of hexane and centrifuging at 3000 rpm for one hour. After discarding the supernatant, the particles were suspended in 5 mL ethanol with sonication. This precipitation was repeated 2 times, after which the particles were re-dispersed with sonication in 10 mL DI-H2O (1 mg/mL APTS-silica). The pH of this suspension is typically ~10, which is above the pKa of the amine. Therefore, to protonate the amine for better dispersibility, the pH was reduced to ~3 by adding a few drops of concentrated HCl and sonicating the suspension.
Au Nanoshell Formation
Au nanoshells were prepared following published methods.39 Shell growth is carried out on silica nanoparticles surface-modified with exposed amines. A gold nanoparticle seed layer is first adsorbed onto the silica particle surface, followed then by the addition of a gold salt shell growth solution. Note that shell growth is extremely sensitive to the details of the procedures and need to be carefully followed.
A few days in advance of shell growth, Au seeds were prepared as previously described.47 45 mL of DI-H 2O was poured into a conical flask and stirred. 0.5 mL of 1M NaOH was added, followed by 1 mL of a THPC solution (12 μL of 80% THPC per mL of DI-H2O). The mixture was stirred for 5 minutes before adding 2 mL of a fresh 1% w/w HAuCl4 (aq) solution. After 15 minutes, the solution turned dark brown, indicating the formation of small (2–3 nm) gold seed particles. This colloidal Au dispersion was stored at 4°C for a week until use, as suggested in the literature.14,16,48
The gold hydroxide solution for shell deposition was prepared one day prior to shell deposition. 25 mg of potassium carbonate was dissolved in 100 mL DI-H2O, followed by the addition of 1.5 mL of a 1.0 % w/w HAuCl 4 solution. After stirring for ~30 minutes, the solution appears colorless, indicating that hydrolysis of AuCl4− has occurred.47 This gold hydroxide solution was then stored for 24 hours in the dark and used immediately. Storing the gold hydroxide solution for more than 2–3 days leads to a faint pink color and a solution that is no longer suitable for gold shell deposition, as evidenced by formation of a nanoshell reaction product having low optical tunability, and this is confirmed by other sources.48
Prior to shell growth, Au seed particles were adsorbed to silica particles.39 The APTS-treated silica particles were mildly sonicated for 20 minutes to ensure uniform particle dispersion and then 2.5 mL of the APTS-silica dispersion (~100 cm2 silica surface area per mL solution) was added to a 15 mL centrifuge tube, followed immediately by the addition of 5.0 mL of THPC Au colloid.49 The pH was re-adjusted (if necessary) to 3.0 with a few drops of concentrated HCl, and the dispersion was gently shaken for 2 minutes before storing in the dark overnight at 4°C. The resulting nanoparticle product was isolated by centrifugation at 2500 rpm for 3 hours. The supernatant was discarded and the precipitate was re-dispersed with sonication in 2.5 mL DI- H 2O. This centrifugation and re-dispering cycle was repeated two more times to remove any unattached gold remaining, as indicated by a colorless supernatant above the precipitate.
For Au nanoshell growth, 4 mL of the as-prepared gold hydroxide reactant solution was added to a 20 mL glass vial and placed under magnetic stirring.39 After sonicating for 20 minutes, Au-decorated silica was then added to this reactant solution with amounts that depended on the desired Au shell thickness, from 0.01 mL to 1.5 mL. The pH was adjusted from ~6.0 to 8.0 by adding 1–2 μL of NH4OH (aq, 30% w/w), as recommended by ref 6 to inhibit homogeneous Au particle nucleation. 10 μL of formaldehyde was then immediately added. The dispersion changes from colorless to blue over the course of about 10 minutes. The nanoshell product was isolated by centrifugation at 2500 rpm for 1 hour. After discarding the supernatant, the precipitate of Au nanoshells was redispersed in 4 mL of DI-H2O with sonication. The nanoshell dispersions varied in color from blue to red, with the red color corresponding to the thickest Au shells. The larger diameter nanoshells remained dispersed without settling for much longer than the smaller diameter nanoshells.
Materials Characterization
The nanoparticles were characterized using transmission electron microscopy (TEM) and UV-visible-NIR optical absorbance spectroscopy. TEM samples were prepared by drop-casting 5 μL of a nanocrystal dispersion on 200 mesh carbon-coated copper TME grids (Electron Microscopy Sciences) held at their edge with anti-capillary tweezers. To create a thin nanocrystal film on the grid, the droplet was drawn off slowly by touching a clean piece of filter paper to the edge of the grid. Low-resolution TEM images were taken using a Philips 208 TEM, equipped with an AMT Advantage HR model CCD camera, at 80 kV accelerating voltage. High resolution TEM images were acquired with a JEOL 2010F TEM at 200 kV using a Gatan multipole scanning CCD camera. Absorbance spectra were acquired with a Cary 500 spectrophotometer using quartz cuvettes with 10 mm optical path lengths.
Results and Discussion
Au Nanoshell Formation and Morphology
Figure 1 shows TEM images of Au nanoshells on silica particles with average diameters decreasing from 118 nm to 28 nm. The Au nanoshells became rougher as the silica particle diameter decreased. There was also much more aggregation of the smaller diameter nanoshells. A variety of different synthetic parameters were modified to try to eliminate the aggregation and nanoshell roughness, 50 but it could not be avoided. Aggregation in particular was found to be a major problem associated with the smaller diameter particles as discussed in more detail below.
Figure 1.

TEM images of silica particles with varying size: (A) 118 ± 5 nm; (B) 74 ± 3 nm; (C) 38 ± 1 nm with 5.0 ± 0.8 nm diameter iron oxide core; (D) 28 ± 1 nm—coated with Au nanoshells. Each frame shows the nanoshells at different stages of the deposition process: (i) bare silica; (ii) Au nanocrystal-decorated silica; (iii) partial Au shell growth; (iv) complete Au shell formation.
Figure 2 illustrates the nanoshell growth process. Silica spheres are first decorated with Au nanoparticles that seed the deposition of the Au shell. TEM images of silica spheres coated with Au seed layers prior to nanoshell deposition are shown in Figure 1. The seed layer coverage on the silica surface is only 20–30 % based on TEM images, which is consistent with previous reports.1,51 This sparse coating of the silica particles is most likely due to the charge double-layer repulsion between neighboring Au particles.52,53 The Au seed particle surface coverage did not vary noticeably with silica particle diameter.
Figure 2.

Illustration of Au nanoshell (yellow) deposition on a silica particle (gray) or silica-coated iron oxide (brown) particle. Formaldehyde reduces gold hydroxide to Au, which deposits heterogeneously on the nanoparticle (NP) seeds on the silica particle surface. Nanoshell growth is complete when sufficient gold hydroxide has been added so that the seeds grow and coalesce into a continuous shell (final step).
In the initial stages of shell growth, Au deposits isotropically on each spherical seed particle, which eventually becomes large enough to fuse with neighboring Au seeds into a semicontinuous film on the silica surface (Figure 1, proceeding from panels (ii) to (iii)). Additional Au deposits on the surface and fills the voids in the Au film, ultimately forming a continuous Au shell (Figure 1, panels (iv)). In practice, the amount of Au deposited on the silica particles is manipulated by changing the ratio of Au-primed silica cores (Figure 1(ii)) to the amount of the gold reactant solution added for nanoshell growth. There was no apparent dependence of the Au deposition process on the core silica particle size, although the smaller diameter Au nanoshells were found to be rougher than the larger diameter shells. The Au shell roughness depends on the size distribution of the Au seeds and any Au clustering that might occur on the silica surface. The variation in shell thickness from the seed layer should be independent of silica particle size, and therefore, the roughness is simply more pronounced and apparently rougher when the silica cores have smaller diameter.
Au Nanoshell Optical Properties
Figure 3 shows experimental UV-vis-NIR optical absorbance spectra of aqueous dispersions of Au nanoshells grown on silica particles of varying diameter. Prior to shell growth, each sample exhibits a plasmon peak at 513 nm that corresponds to the plasmon resonance of the 2 nm diameter Au seed particles. The plasmon peak than shifts to longer wavelength as Au deposits on the particles and fills out the shell. The absorbance wavelength reaches a maximum value when shell growth comes to completion to form a uniform coating on the silica cores. As the Au shell thickness continues to increase, the plasmon peak wavelength also decreases. This is consistent with expectations, as the plasmon absorbance peak of a Au nanoshell shifts to shorter wavelength as the ratio of core radius to shell thickness, decreases.2,3,7
Figure 3.

Optical absorbance spectra of Au nanoshells on silica spheres of varying diameter and shell thickness. Nanoshells with the most red-shifted plasmon peak are labeled as “100% complete.” Incomplete nanoshells exhibit plasmon peaks at lower wavelength than the complete shells and the plasmon peak shifts back to the blue as the nanoshell thickness is further increased. The absorbance axis scale is linear.
Table 1 summarizes the lowest wavelength plasmon peak of each nanoshell sample. The largest silica core particles exhibited the maximum red-shift of the plasmon absorbance peak of the samples at 733 nm, and the maximum plasmon peak wavelength was found to decrease as the core diameter was decreased. Part of the reason for this observed size dependence of the longest wavelength plasmon peak is that the ratio of the core radius to shell thickness is limited by the the seed particle diameter—the shell thickness cannot be less than the seed particle size. Therefore, the maximum core radius/shell thickness ratio possible for each sample depends on the silica core size. For instance, 28 nm diameter silica spheres have a minimum possible core radius/shell thickness ratio of 7 when 2 nm diameter Au seeds are used, which yields a plasmon peak wavelength of 790 nm.54 In practice, the minimum shell thickness is always slightly greater than the seed diameter; the minimum Au shell thickness observed in any of these samples was 10 nm. The thinnest Au shells obtained on 118 nm diameter silica particles had a thickness of 11 nm (from TEM), which corresponds to a core radius/shell thickness ratio of 5.4. Based on Mie theory, nanoshells need a core radius/shell thickness ratio of at least 4.5 to exhibit a plasmon resonance of 700 nm or greater. To obtain core/shell ratios of 4.5, Au shell thicknesses of 3.1 and 4.2 nm are needed for 28 and 38 nm diameter silica spheres, respectively, which are not experimentally accessible and the reddest plasmon peak wavelengths for these nanoshells was 653 and 644 nm, respectively.
Table 1.
The lowest wavelength plasmon peak position (λmax) observed for each nanoshell as a function of silica particle size.
| Silica Diameter (nm) | Silica Particle Concentration during Growth* (×1010NP/mL) | Total Silica Surface Area during Growth* (cm2/mL) | Nanoshell Extinction Peak, λmax (nm) | Core/shell Ratio Calculated from λmax† | Core/shell Ratio Estimate from TEM † |
|---|---|---|---|---|---|
| 28 | 100 | 25 | 653 | 3.1 | 1.1±0.4 |
| 38 | 30 | 14 | 644 | 1.9 | 1.0±0.3 |
| 74 | 9 | 15 | 697 | N/A‡ | 3.6±0.7 |
| 118 | 2 | 9 | 733 | 6.6 | 5.4±1.8 |
Refers to the Au-decorated silica spheres added to the Au shell growth solution.
Ratio of silica core radius to the Au shell thickness. Mie theory calculations assumed the nanoshells to be smooth and non-aggregated.54
The plasmon peak for this sample was not within range of λmax predicted by Mie theory.
The nanoshell thickness was estimated using the experimentally measured plasmon peak wavelength with Mie theory,54 and compared to the thickness measured by TEM. Table 1 lists the core/shell size ratios determined from the two measures. The shell thicknesses estimated from the using Mie theory were generally thinner than those measured by TEM and for the nanoshells smaller than 100 nm, the deviation was significant. Part of the deviation is due to the fact that the experimental absorbance peaks are generally broader than those predicted using Mie theory, as a result of the size distribution in the sample, surface roughness and aggregation. In the case of the 74 nm diameter nanoshells, Mie theory does not predict a single plasmon resonance peak at 697 nm for any cores/shell ratio, suggesting that the Au shells may still be incomplete for this sample (See Supporting Information for more discussion about comparison between Mie theory calculations and the experimental data).
Model for Size-Dependent Nanoshell Aggregation
After many experimental trials to deposit Au nanoshells on silica particles less than 60 nm in diameter, it became clear that these smaller diameter Au-coated silica particles were much more prone to aggregation than the larger diameter nanoshells. Calculation of the interparticle potential as a function of nanoshell diameter reveals why this is the case. Due to their higher surface curvature, the smaller diameter nanoshells have a more diffuse charge double-layer and as a result are not as well stabilized from aggregation as the larger diameter particles, despite their weaker van der Waals attraction compared to the larger diameter nanoshells.
The pair interparticle potential Vnet, can be approximated as the sum of an (attractive) van der Waals VA, potential and a (repulsive) double-layer potential VE:55
| (1) |
The magnetic dipole coupling between silica-coated iron oxide nanoparticles was not included in the calculations because the interaction is of the order of 0.001 kT or less and is insignificant (Supporting Information). VE between two particles of equal radius r, separated by a distance x, (c.f. Figure 4) is55,56
Figure 4.

Model geometry of silica particles (material, p; radius, r) covered by a sheath layer of Au nanoparticles (material, s; thickness, δ) that was used to determine the pair interparticle potentials. Material s is a composite layer made of Au nanoparticles covering 30% of the silica surface and separated by solvent. The nanoparticle surfaces are separated by a distance x across the water medium (material, w). The overall nanoparticle radius is r + δ.
| (2) |
where κ is the inverse Debye length and Y is a function,
| (3) |
| (4) |
Table 2 provides a description of the parameters. The solvent ionic strength I, is estimated to be ≤5 mM during Au nanoshell growth, and also ~5 mM during Au nanoparticle attachment to the APTS-silica (see Supporting Information for a further discussion).
Table 2.
Parameters used to model nanoshell aggregation.
| Parameter | Description | Value | Reference |
|---|---|---|---|
| kT | Thermal energy | 4.04 × 10−21 J | 57 |
| e | Fundamental charge | 1.60 × 10−19 C | 57 |
| ε0 | Vacuum permittivity | 8.85 × 10−12 C2/N-m2 | 57 |
| εrel | Solvent relative dielectric constant | 80.1 | 57 |
| L | Avogadro’s number | 6.02 × 1023 mol−1 | 57 |
| I | Solvent ionic strength | ~5 mM | See supporting info. |
| φ | Au nanoparticle surface coverage | 30% | 6, 39 |
| ρg | Au bulk density | 19.3 g/cm3 | 57 |
| ρw | Water bulk density | 0.997 g/cm3 | 57 |
| Mg | Au molar mass | 196.97 g/mol | 57 |
| Mw | Water molar mass | 18.02 g/mol | 57 |
| ηw | Water viscosity | 0.89 cP | 57 |
| δ | Thickness of Au nanocrystal layer | 3 nm | TEM measurement |
| Ag | Au Hamaker constant in vacuo | 48 × 10−20 J | See supporting info. |
| Aw | Water Hamaker constant in vacuo | 3.7 × 10−20 J | 58 |
| Ap | Silica Hamaker constant in vacuo | 14.7 × 10−20 J | 58 |
VA can be determined for silica particles coated with Au seed particles, like those illustrated in Figure 4. The Hamaker constant As, of such a typical sheath layer composed of Au and water is59
| (5) |
Nw0 and Ng0 are the number densities of pure water (w) and Au (g), and Nw and Ng are the number densities of water and Au in the sheath layer. The number n of Au nanoparticles on each silica particle prior to Au deposition is
| (6) |
It follows that the Au and water concentrations in the sheath layer are60
| (7) |
| (8) |
The Hamaker attractive potentialVA is59
| (9) |
The Hamaker constants (Aw, Ap, As) are listed in Table 2. The Hamaker functions for the sheath-sheath (Hs), particle-particle (Hp), and particle-sheath (Hps) interactions used for the calculations are provided in the Supporting Information.
Calculations of Vnet as a function of nanoshell core diameter under conditions that reflect those at the onset of Au nanoparticle attachment and Au nanoshell growth are plotted in Figures 5 and 6. For all of the nanoshell diameters, there is a repulsive peak in Vnet (x), but it is weaker for nanoshells with decreased diameter. The peak repulsion for the 30 nm diameter nanoshells is ~10 kT (260 meV) versus ~25 kT for the 120 nm diameter nanoshells. Since this barrier represents an activation energy for particle aggregation (W ∝ exp (− Vnet/kT)), the 30 nm net diameter nanoshells are approximately 107 times more likely to aggregate with each interparticle collision in solution.61 The Au-decorated silica spheres also exhibit this dramatic difference in stability with differing particle diameter, though the silica spheres of all the sizes studied are expected to become more stable after the Au is adsorbed.
Figure 5.
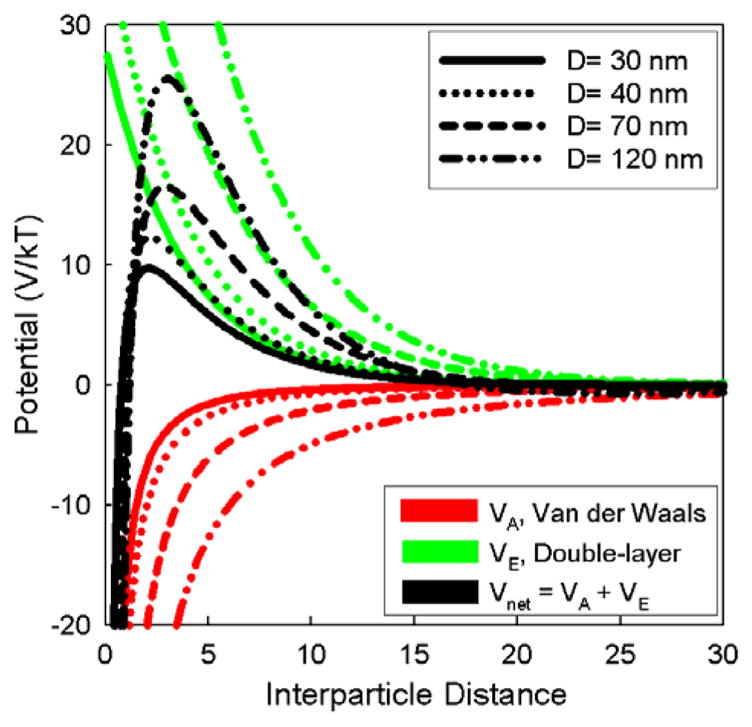
Pair interparticle potential (black curves), van der Waals attraction (red curves) and electrostatic double-layer repulsion (green) calculated using Eqns 1–8 for nanoshells with silica core diameters noted in the legend. Calculations were performed for silica spheres coated with APTMS at pH of 3: ζ=42 mV.62,63
Figure 6.
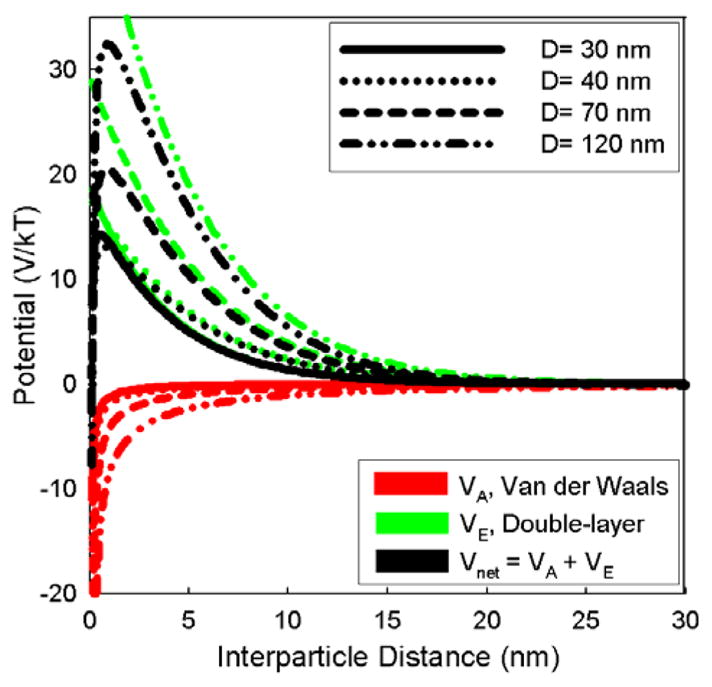
Pair interparticle potential (black curves), van der Waals attraction (red lines), and electrostatic double-layer repulsion (green) calculated using Eqns 1–8 for nanoshells with silica core diameters shown in the legend. The calculations are for silica spheres decorated with 3 nm Au nanoparticles (30% coverage) at pH of 7: ζ= −32.5 mV.62,63
Considering that the aggregation rate z (per unit volume of two spherical particles of equal size is64
| (10) |
where ηw is the solvent viscosity, and N is the number concentration of colliding particles, perhaps the extent of aggregation could be reduced by decreasing the concentration of the smaller diameter silica particles. In practice, however, this is not possible. As noted in Table 1, smaller diameter nanoshells required higher silica particle concentrations than the larger diameter nanoshells. The nanoshells were deposited using the same Au concentration in the growth solutions, as diluting the nanoshell growth solution was found to be less effective at producing nanoshells with red-shifted plasmon resonance peaks.50 Therefore, because deposition of the same amount of Au on a smaller sphere results in a larger Au shell thickness than for a bigger silica particle, a much higher number concentration of the small silica spheres is needed to obtain the same Au thickness as on the larger silica particles. Figures 7 and 8 show the calculated colloid stability coefficient and aggregation rate for different particle sizes.61 The calculated aggregation rate increased by several orders of magnitude as the silica core size was reduced from 130 nm to 20 nm. Under identical solution conditions, the 30 and 40 nm diameter silica spheres are significantly less stable than the larger diameter silica spheres, and therefore are much more prone to aggregation. The aggregation happens prior to Au deposition, which begins after adding the formaldehyde reducing agent and fuses the silica aggregates together with a common layer of Au.
Figure 7.
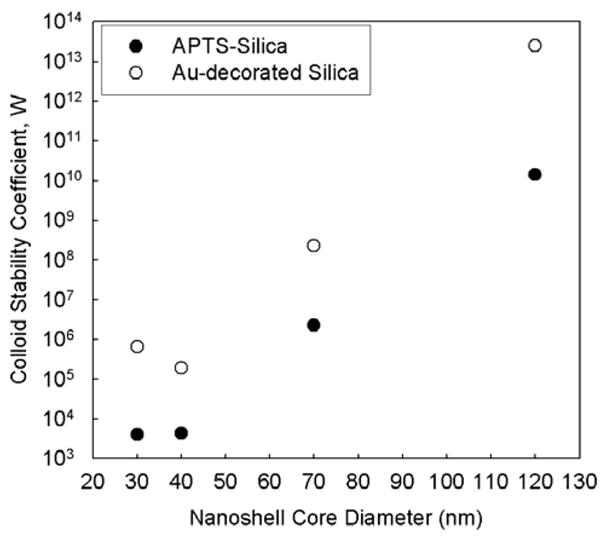
Calculated colloid stability coefficient for different size Au nanoshells at the onset of Au-decoration (APTS-Silica) and Au shell deposition (Au-decorated Silica).
Figure 8.

Calculated aggregation rate for different size Au nanoshells at the onset of Au-decoration (APTS-Silica) and Au shell deposition (Au-decorated Silica).
Conclusions
As their diameter decreases from 120 nm to 30 nm, Au nanoshells are more prone to aggregation during the Au shell deposition step. Rather extensive aggregation is observed by TEM and is apparent from their absorbance spectra. Model calculations of the interparticle potential revealed that the significant aggregation of smaller diameter Au nanoshells is related to the more diffuse charge double layer of the smaller particles. The energy barrier inhibiting aggregation decreases as the particle diameter decreases; for example, 30 nm diameter nanoshells are 107 times more likely to aggregate than the 120 nm diameter nanoshells with each Brownian collision.
The smaller diameter nanoshells also exhibit less optical tunability of their plasmon absorbance peak to the red than larger diameter nanoshells. The core/shell size ratio dictates the plasmon peak energy and the size of the gold seeds provides a lower limit to nanoshell thickness. In practice, the shells are thicker than this as a result of the shell deposition process, with a minimum thickness of ~10 nm. This limits the plasmon resonance frequency well into the visible wavelength range for the smallest diameter 30 nm silica spheres.
To obtain small diameter Au nanoshells with strong absorption near the NIR wavelengths, smaller Au nanoparticle seeds are needed in order to obtain higher core/shell size ratios. This is a challenge, but perhaps more significantly, a high nanoshell aggregation rate would still be a problem. Perhaps one approach that might enable stable small diameter Au nanoshells (20–60 nm in diameter) to be synthesized would be the use of a surfactant- or microemulsion-based reaction medica that provides steric stabilization of the growing shells, as opposed to charge stabilization. This chemistry has yet to be developed, but should be possible.
Supplementary Material
Acknowledgments
We acknowledge the National Institutes of Health (CA10383) and the Robert A. Welch Foundation for partial financial support of this research, and the Nissan Chemical Company for donating the silica colloid.
Footnotes
Supporting Information Available: Additional nanoshell synthesis details, TEM images and EDS data, methods for calculating nanoparticle concentrations, Mie theory calculations of absorbance spectra compared to experimental spectra, calculations of the dispersion ionic strength, magnetic dipole interactions between nanoshells with iron oxide cores, and Hamaker functions,.. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Oldenburg SJ, Averitt RD, Westcott SL, Halas NJ. Chem Phys Lett. 1998;288:243–247. [Google Scholar]
- 2.Oldenburg SJ, Jackson JB, Westcott SL, Halas NJ. Appl Phys Lett. 1999;75:2897–2899. [Google Scholar]
- 3.Prodan E, Radloff C, Halas NJ, Nordlander P. Science. 2003;302:419–422. doi: 10.1126/science.1089171. [DOI] [PubMed] [Google Scholar]
- 4.Nehl CL, Grady NK, Goodrich GP, Tam F, Halas NJ, Hafner JH. Nano Lett. 2004;4:2355–2359. [Google Scholar]
- 5.Liang H, Wan L, Bai C, Jiang L. J Phys Chem B. 2005;109:7795–7800. doi: 10.1021/jp045006f. [DOI] [PubMed] [Google Scholar]
- 6.Yong KT, Sahoo Y, Swihart MT, Prasad PN. Colloids Surf A. 2006;290:89–105. [Google Scholar]
- 7.Jain PK, Lee KS, El-Sayed IH, El-Sayed MA. J Phys Chem B. 2006;110:7238–7248. doi: 10.1021/jp057170o. [DOI] [PubMed] [Google Scholar]
- 8.Loo C, Lin A, Hirsch L, Lee M, Barton J, Halas NJ, West J, Drezek R. Technol Cancer Res Treat. 2004;3:33–40. doi: 10.1177/153303460400300104. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Qian W, Tan Y, Ding S. Biosensors and Bioelectronics. 2008;23:1166–1170. doi: 10.1016/j.bios.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 10.Slocik JM, Tam F, Halas NJ, Naik RR. Nano Lett. 2007;7:1054–1058. doi: 10.1021/nl070267x. [DOI] [PubMed] [Google Scholar]
- 11.Levin CS, Bishnoi SW, Grady NK, Halas NJ. Anal Chem. 2006;78:3277–3281. doi: 10.1021/ac060041z. [DOI] [PubMed] [Google Scholar]
- 12.Levin CS, Kundu J, Janesko BG, Scuseria GE, Raphael RM, Halas NJ. J Phys Chem B. 2008;112:14168–14175. doi: 10.1021/jp804374e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cubeddu R, Pifferi A, Taroni P, Torricelli A, Valentini G. Appl Phys Lett. 1999;74:874–876. [Google Scholar]
- 14.Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL. Proc Natl Acad Sci USA. 2003;100:13549–13554. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loo C, Lowery A, Halas N, West J, Drezek R. Nano Lett. 2005;5:709–711. doi: 10.1021/nl050127s. [DOI] [PubMed] [Google Scholar]
- 16.O’Neal DP, Hirsch LR, Halas NJ, Payne JD, West JL. Cancer Lett. 2004;209:171–176. doi: 10.1016/j.canlet.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 17.Halas NJ. MRS Bulletin. 2005;30:362–367. [Google Scholar]
- 18.Liao H, Nehl CL, Hafner JH. Nanomedicine. 2006;1:201–208. doi: 10.2217/17435889.1.2.201. [DOI] [PubMed] [Google Scholar]
- 19.Fortina P, Kricka LJ, Graves DJ, Park J, Hyslop T, Tam F, Halas N, Surrey S, Waldman SA. TRENDS in Biotech. 2007;25:145–152. doi: 10.1016/j.tibtech.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Ji X, Shao R, Elliott AM, Stafford RJ, Esparaza-Coss E, Bankson JA, Liang G, Luo Z, Park K, Markert JT, Li C. J Phys Chem C. 2007;111:6245–6251. doi: 10.1021/jp0702245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Qian W, Tan Y, Ding S, Zhang H. Talanta. 2007;72:1134–1140. doi: 10.1016/j.talanta.2007.01.026. [DOI] [PubMed] [Google Scholar]
- 22.Arruebo M, Fernandes-Pacheko R, Ibarra MR, Santamaria J. Nano Today. 2007;2:22–32. [Google Scholar]
- 23.Schipper ML, Iyer G, Koh AL, Cheng Z, Ebenstein Y, Aharoni A, Keren S, Bentolila LA, Li J, Rao J, Chen X, Banin U, Wu AM, Sinclair R, Weiss S, Gambhir SS. Small. 2009;5:126–134. doi: 10.1002/smll.200800003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Ipe BI, Bawendi MG, Frangioni JV. Nature Biotech. 2007;25:1165–1170. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadauskas E, Wallin H, Stoltenberg M, Vogel U, Doering P, Larsen A, Danscher G. Part Fibre Toxicol. 2007;4:10. doi: 10.1186/1743-8977-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balogh L, Nigavekar SS, Nair BM, Lesniak W, Zhang C, Sung LY, Kariapper MST, El-Jawahri A, Llanes M, Bolton B, Mamou F, Tan W, Hutson A, Minc L, Khan MK. Nanomedicine. 2007;3:281–296. doi: 10.1016/j.nano.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 27.Buzea C, Blandino IIP, Robbie K. Biointerphases. 2007;2:MR17–MR172. doi: 10.1116/1.2815690. [DOI] [PubMed] [Google Scholar]
- 28.Chan VSW. Reg Tox Pharm. 2006;46:218–224. doi: 10.1016/j.yrtph.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 29.Banerjee R. Nanomedicine. 2006;1:481–485. doi: 10.2217/17435889.1.4.481. [DOI] [PubMed] [Google Scholar]
- 30.Katz E, Willner I. Angew Chem Int Ed. 2004;43:6042–6108. doi: 10.1002/anie.200400651. [DOI] [PubMed] [Google Scholar]
- 31.Chu TC, Shieh F, Lavery LA, Levy M, Richards-Kortum R, Korgel BA, Ellington AD. Biosensors and Bioelectronics. 2006;21:1859–1866. doi: 10.1016/j.bios.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 32.Winter JO, Liu TY, Korgel BA, Schmidt CE. Adv Mater. 2001;13:1673–1677. [Google Scholar]
- 33.Winter JO, Schmidt CE, Korgel BA. Mat Res Soc Symp Proc. 2004;789:N6.2.1–4. [Google Scholar]
- 34.McCarthy JR, Kelly KA, Sun EY, Weissleder R. Nanomedicine. 2007;2:153–167. doi: 10.2217/17435889.2.2.153. [DOI] [PubMed] [Google Scholar]
- 35.Duguet E, Vasseur S, Mornet S, Devoisselle J. Nanomedicine. 2006;1:157–168. doi: 10.2217/17435889.1.2.157. [DOI] [PubMed] [Google Scholar]
- 36.Liu Z, Song H, Yu L, Yang L. Appl Phys Lett. 2005;86:113109. [Google Scholar]
- 37.Lyon JL, Fleming DA, Stone MB, Schiffer P, Williams ME. Nano Lett. 2004;4:719–723. [Google Scholar]
- 38.Larson TA, Bankson J, Aaron J, Sokolov K. Nanotechnology. 2007;18:325101. [Google Scholar]
- 39.Xia X, Liu Y, Backman V, Ameer GA. Nanotechnology. 2006;17:5435–5440. [Google Scholar]
- 40.Heitsch AT, Smith DK, Patel RN, Ress D, Korgel BA. J Solid State Chem. 2008;181:1590–1599. doi: 10.1016/j.jssc.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hyeon T, Lee SS, Park J, Chung Y, Na HB. J Am Chem Soc. 2001;123:12798–12801. doi: 10.1021/ja016812s. [DOI] [PubMed] [Google Scholar]
- 42.Lee DC, Mikulec FV, Pelaez JM, Koo B, Korgel BA. J Phys Chem B. 2006;110:11160–11166. doi: 10.1021/jp060974z. [DOI] [PubMed] [Google Scholar]
- 43.Warning: Iron pentacarbonyl is a highly flammable, very toxic chemical that should be handled with caution.
- 44.Arriagada FJ, Osseo-Asare K. J Coll Int Sci. 1999;211:210–220. doi: 10.1006/jcis.1998.5985. [DOI] [PubMed] [Google Scholar]
- 45.Lian W, Litherland SA, Badrane H, Tan W, Wu D, Baker HV, Gulig PA, Lim DV, Jin S. Anal Biochem. 2004;334:135–144. doi: 10.1016/j.ab.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 46.The APTS coating was performed with 1 mg/mL silica for all sizes of silica particles, despite the variation in total silica surface area. The referenced procedure for the OTMOS coating provided about 2.8 μmol OTMOS per mm2 of silica. For the 30 nm silica spheres (10 mL, 1 mg/mL), the given reactant ratios provide 3.2 μmol of APTS per mm2 of silica. At the same mass concentration (1 mg/mL), the larger silica spheres then have extra APTS per mm2 relative to the 30 nm silica spheres. This did not appear to give the larger spheres any advantage over the smaller-sized silica for attachment of Au nanoparticles.
- 47.Duff DG, Baiker A. Langmuir. 1993;9:2301–2309. [Google Scholar]
- 48.Brinson BE, Lassiter JB, Levin CS, Bardhan R, Mirin N, Halas NJ. Langmuir. 2008;24:14166–14171. doi: 10.1021/la802049p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.The Au nanoparticle seeds were exposed to the same ATPS-silica surface area during their attachment, regardless of the silica particle size. This was to ensure consistent Au attachment during this step. Calculation of nanoparticle concentration and surface area are described in the Supporting Information. A large excess of Au nanoparticles are used for attachment, and silica surface area concentrations in the range of 100–500 cm2/mL provided about the same Au surface coverage. Concentrations of silica surface area lower than this range did not improve the Au surface coverage.
- 50.We tried diluting the nanoshell growth solution to reduce the aggregation that occurred during Au shell deposition. The experimental details and results are provided in the Supporting Information. First, we tested how changes to the shell growth solution parameters affected Au deposition onto the largest silica spheres (118 nm diameter). The pH (~8.0, adjusted with dilute NH4OH), concentration of reducing agent, and ratio of Au ion concentration to silica particles were held constant, and the shell growth medium was diluted using one of three different solutions: 1) DI water (adjusted to pH~8.0 with concentrated NaOH), 2) aqueous potassium carbonate (1.8 mM, the same concentration for preparing the gold hydroxide solution), and 3) an aqueous HEPES buffer (50 mM, pH~8.0). For the 118 nm nanoshells, only dilution with 1) did not affect the wavelength of the plasmon absorbance peak. Unfortunately, applying this dilution strategy with 1) to the small silica spheres (30 nm diameter) yielded a nanoshell product with a very weak, blue-shifted plasmon absorbance peak relative to the case with no dilution. We also tried diluting the growth solution with 118 nm silica spheres using DI water at pH=5.5, allowing the pH of the growth solution to drop away from the amine pKa, though this did not offer any improvement in the broadness or position of the plasmon absorbance peak.
- 51.Grabar KC, Smith PC, Musick MD, Davis JA, Walter DG, Jackson MA, Guthrie AP, Natan MJ. J Am Chem Soc. 1996;118:1148–1153. [Google Scholar]
- 52.Sudhir B, Kumar S. Resonance. 2002;7:67–81. [Google Scholar]
- 53.Westcott SL, Oldenburg SJ, Lee TR, Halas NJ. Langmuir. 1998;14:5396–5401. [Google Scholar]
- 54.Plasmon peak wavelength calculations are described in Supporting Information, along with specific comparisons to the experimental absorbance spectra.
- 55.Kim T, Lee K, Gong M, Joo S. Langmuir. 2005;21:9524–9528. doi: 10.1021/la0504560. [DOI] [PubMed] [Google Scholar]
- 56.Lee K, Sathyagal AN, McCormick AV. Colloids Surf A. 1998;144:115–125. [Google Scholar]
- 57.CRC Handbook of Chemistry and Physics. 89. CRC Press/Taylor and Francis; Boca Raton, FL: (Internet Version 2009) [Google Scholar]
- 58.Vincent B. J Coll Interf Sci. 1973;42:270–285. [Google Scholar]
- 59.Suhara T, Shimano F, Sato Y, Fukui H, Yamaguchi M. Colloids Surf A. 1996;119:105–114. [Google Scholar]
- 60.If n Au seeds on each silica particle grow symmetrically and independent of one another, equations 7 and 8 can apply to the sheath layer during shell growth by increasing δ until the seed particles form a closely-packed lattice. This tight arrangement of n small spheres on the surface of a larger sphere relates to the Tammes problem, which has only been solved for n up to about 80.65,66 For a large number of tightly-packed spheres (50 < n < 80), the maximum fraction of spherical surface covered remains constant at about 0.83 independent of n.66 Applying this result to each of the four silica core sizes, we found that the seed particles attain optimum packing on each core surface after growing to a diameter of 5.0 nm. Thus, the Au nanoparticles will definitely have to fuse when the shell thickness exceeds 5.0 nm, and equations 7 and 8 will no longer be valid beyond this point (the sheath layer will essentially be all Au anyway). The TEM images in Figure 2iii suggest that the Au seeds probably do not grow uniformly, so we focused our analysis of the sheath layer on the 2–3 nm Au-decorated silica spheres at the onset of Au shell growth.
-
61.The colloidal stability coefficient W, or inverse aggregation probability, is64,67
The stability coefficients were evaluated using trapezoidal numerical integration and the results are plotted in Figure 7. For the undecorated silica spheres, W increases with particle size, and the results are well within the range of calculated values reported by Speilman and coworkers.67(11) - 62.Wu Z, Xiang H, Kim T, Chun M, Lee K. J Coll Int Sci. 2006;304:119–124. doi: 10.1016/j.jcis.2006.08.055. [DOI] [PubMed] [Google Scholar]
- 63.Park S, Park M, Han P, Lee S. Bull Korean Chem Soc. 2006;27:1341–1345. [Google Scholar]
- 64.Valioulis FA, List JE. Adv Colloid Interface Sci. 1984;20:1–20. [Google Scholar]
- 65.Clare BW, Kepert DL. Proc R Soc Lond A. 1986;405:329–344. [Google Scholar]
- 66.Bruinsma RF, Gelbart WM, Regurea D, Rudnick J, Zandi R. Phys Rev Lett. 2003;90:248101. doi: 10.1103/PhysRevLett.90.248101. [DOI] [PubMed] [Google Scholar]
- 67.Spielman LA. J Coll Interf Sci. 1970;33:562–571. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.