

Abstract
Cyclin-dependent kinase (cdk) 3, a member of the cdk family of kinases, plays a critical role in cell cycle regulation and is involved in G0-G1 and G1-S cell cycle transitions. However, the role of cdk3 in cell proliferation, as well as cell transformation, is not yet clearly understood. Here, we report that the protein expression level of cdk3 is higher in human cancer cell lines and human glioblastoma tissue compared with normal brain tissue. Furthermore, we found that cdk3 phosphorylates ATF1 at serine 63 and enhances transactivation and transcriptional activity of ATF1. Results also indicated that siRNA directed against cdk3 (si-cdk3) suppresses ATF1 activity, resulting in inhibition of cell proliferation and growth of human glioblastoma T98G cells in soft agar. Importantly, we showed that cdk3 enhances EGF-induced transformation of JB6 Cl41 cells and si-cdk3 suppresses RasG12V/cdk3/ATF1-induced foci formation in NIH3T3 cells. These results clearly demonstrated that the cdk3-ATF1 signaling axis is critical for cell proliferation and transformation.
Keywords: cell transformation, cdk3, ATF1, phosphorylation, protein-protein interaction
Introduction
Cyclin-dependent kinases (cdks) are serine/threonine protein kinases that play essential roles in control of cell cycle progression by interacting with a variety of regulators and substrates (1). In eukaryotic cells, cell cycle progression is driven by the sequential and periodic activation of cyclin/cdks, and dysregulation of the cell cycle is associated with cancer development (2). Very few mutations in cdks have been noted in human cancers with the exception of a p16Ink4a-insensitive cdk4 mutation in human familial melanoma (3). However, dysregulation of cdks’ activity is found in many human cancers because of mutations or epigenetic alterations in upstream regulators of cdks or their substrates (1, 4). Overexpression of cdk6 has been observed in lymphomas, leukemias, and melanomas due to chromosomal translocation (5, 6). When cotransfected with activated H-Ras, cdk4 displayed oncogenic potential by provoking foci formation in primary rat embryo fibroblasts (7) and generating malignant human epidermal tumorigenesis (8). Cdk4 kinase activity was shown to be required for proliferation of breast cancer cells and mammary tumorigenesis in cdk4-null mice (9, 10) and kinase-deficient cyclin D1 knock-in mice (11). Hemizygous disruption of cdc25 was shown to inhibit cell transformation and mammary tumorigenesis in mice (12). Activation of cdk2 was required for human fibroblast proliferation and foci formation induced by cyclin E/SV40 small T antigen coexpression (13). Knock-down of cdk2 was shown to inhibit proliferation and colony formation of human melanoma cells (14), and ablation of cdk2 decreased Ras/cdk4-dependent malignant progression in mouse skin tumorigensis models (15). Thus, cdks are closely associated with human cancer pathogenesis.
Cdk3 is an important regulator of cell cycle. The activity of cdk3 is first observed in early G1 phase (16) and reaches a peak in mid G1(17). A dominant-negative cdk3 was shown to induce G1 arrest, which could not be rescued by cdk2, indicating that cdk3 plays a important role for G1 exit to S entry (18, 19). Recently, cdk3 was found to form a complex with cyclin C and phosphorylate the retinoblastoma protein (pRb) at serine 807/811, which is required for G0/G1 transition (20). Furthermore, cdk3 appears to be expressed in various normal human tissues and cancer cell lines including glioblastoma and neuroblastoma cells (20-22). Ectopic overexpression of cdk3, but not cdk2, enhanced proliferation and anchorage-independent growth of Ratl cells that was associated with activation of Myc (17). The genetic locus of cdk3 was mapped to chromosome 17q22-qter region, where cdk3 was involved in a chromosome rearrangement in a breast cancer cell line possibly resulting in an alteration of cdk3 gene expression or an abnormal transcript (23). However, although cdk3 might have an effect on cell proliferation and transformation, the precise role of cdk3 in carcinogenesis has not been clearly elucidated.
The activating transcription factor 1 (ATF1) is a member of a well-known transcription factor family, the cyclic AMP response element (CRE)-binding protein (CREB) family, which includes ATF1, CREB1, and the cAMP response element modulator (CREM) (24). Both ATF1(25) and CREB1 (26) are expressed ubiquitously, whereas CREM (27) is highly expressed in neuroendocrine tissues. In response to a variety of growth factors, stress signals, neurotransmitters and other agents that elevate intracellular cAMP or Ca2+ levels, CREB family members are activated and promote the expression of a large number of cellular target genes that contain CRE elements in their promoters, including proto-oncogenes such as c-fos and c-jun, cell cycle genes such as cyclin D and cyclin A, and other genes related to cell growth, proliferation and neuronal activities (28, 29).
Phosphorylation of ATF1 at serine 63 in its kinase-inducible domain (KID) by serine/threonine kinases enhances its transactivation activity by promoting recruitment of the coactivator CREB-binding protein (CBP)/p300 (30). Furthermore, overexpression of ATF1 was found in lymphomas, transformed lymphocytes (31) and may contribute to the growth of these tumor cells. ATF1 was shown to be upregulated in metastatic melanoma cells, and inhibition of ATF1 suppressed their tumorigenicity and metastatic potential in nude mice (32). Constitutive activation of ATF1 mediates Ewing’s sarcoma protein-activating transcription factor 1 (EWS-ATF1) transforming phenotypes and unique features of clear cell sarcoma (CCS) (33). However, upstream kinases and the role of ATF1 in cell proliferation and cell transformation have not been completely identified.
In this study, we provide evidence showing that cdk3 is highly expressed in human glioblastoma and that ATF1 is a novel substrate of cdk3. The phosphorylation of ATF1 at serine 63 by cdk3 induced ATF1 transactivation and transcriptional activities. Ectopic expression of cdk3 enhanced cell transformation of JB6 Cl41 cells and knockdown of endogenous cdk3 suppressed cell proliferation and growth of glioblastoma T98G cells in soft agar. Importantly, knockdown of cdk3 with si-cdk3 suppressed foci formation in NIH3T3 cells induced by coexpression of RasG12V/cdk3/ATF1, indicating that the cdk3-ATF1 signaling axis plays an important role in cell transformation.
Materials and Methods
Reagents and antibodies
Chemical reagents, including Tris, NaCl, and SDS for molecular biology and buffer preparation were purchased from Sigma-Aldrich Corp. (St. Louis, MO). Restriction enzymes were obtained from Roche Diagnostics Corp. (Indianapolis, IN) and Taq DNA polymerase was from Qiagen, Inc. (Valencia, CA). The DNA ligation kit (v. 2.0) was purchased from TAKARA Bio, Inc. (Otsu, Shiga, Japan). The Checkmate Mammalian Two-hybrid System was obtained from Promega (Madison, WI). Cell culture medium and other supplements were from Life Technologies, Inc. (Rockville, MD). Antibodies against cdk3, ATF1 and cyclin C were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) and the antibody to detect phosphorylated ATF1 was from Cell Signaling Technology, Inc. (Beverly, MA). Anti-Flag was from Sigma-Aldrich Corp. (St. Louis, MO) and anti-HA was purchased from Roche Diagnostics Corp. (Indianapolis, IN). The Human Brain Glioblastoma Tissue Microarray was purchased from US Biomax, Inc. (Rockville, MD) and the Cy3-conjugated donkey anti-rabbit IgG antibody was purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA).
Cell culture and transfections
HEK 293 cells were cultured at 37 °C in a 5% CO2 incubator in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). T98G cells were cultured in Eagle’s minimum essential medium (MEM) with 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1.0 mM sodium pyruvate, and 10% FBS. The media for cell culture were changed every 3 days and cells were split at 80-90% confluence. Transfection of the expression plasmids was performed using jetPEI (Polyplus-transfection Inc., New York, NY) according to the manufacturer’s instructions.
RT-PCR and subcloning of transcription factors and cdk3
The cdk3 (pBIND-cdk3) and the transcription factors (pACT-TFs) cDNAs were cloned by PCR-based amplification as described previously (34). Briefly, human mRNAs were reverse transcribed with an oligo-dT primer and SuperScript II RNase H Reverse Transcriptase (Life Technologies, Grand Island, NY). ATF1 and cdk3 cDNAs were amplified by PCR and the cDNAs were introduced into pACT or pBIND vectors as required and confirmed by DNA sequencing.
Construction of expression vectors
The GST-tagged ATF1 and the deletion GST-ATF1 (residues 58-141) expression plasmids were constructed by PCR and subcloned into the SalI/NotI site of the pGEX-5X-C vector. The ATF1 cDNA was also subcloned into pcDNA4 (Invitrogen, Carlsbad, CA) to generate pcDNA4-ATF1. The plasmid constructs were confirmed by restriction mapping and DNA sequencing. The pRcCMV-cdk3 (pCMV-cdk3) was a gift from Dr. Barrett J. Rollins (Department of Medical Oncology, Harvard Medical School, Boston, Massachusetts). The expression plasmids, including pET28-ATF1, pET28-ATF1S63A, p3×Flag-ATF1 and p3×Flag-ATF1S63A, were kindly provided by Dr. Ron Pryews (Department of Biological Sciences, Columbia University). The deletion GST-ATF1 fusion vectors (1-57, 1-141 and 142-271) were gifts from Dr. Hiroshi Hanbda (Faculty of Bioscience and Biotechnology, Tokyo Institute of Technology). The mutations of cdk3 (408T/C, 411G/A, 412T/C and 414G/C) to generate a si-cdk3 resistant cdk3 expression vector (rescue cdk3 vector) were carried out using the QuikChange II Site-Directed Mutagenesis Kit (Strategene) according to recommended protocols.
Mammalian two-hybrid (M2H) assay
HEK293 cells were seeded and transfected as described previously (34). Briefly, the various DNAs, pBIND-cdk3 or pACT-TFs, and pG5-luciferease reporter plasmids were combined in the same molar ratio (1:1:1) and the total amount of DNA was not more than 100 ng/well. At 36 h after transfection, the cells were disrupted by the addition of lysis buffer (0.1 M potassium phosphate buffer pH 7.8, 2 mM EDTA, 1 mM DTT, 1% Triton X-100) and luciferase activity was measured using the Luminoskan Ascent (MTX Lab, INC, Vienna, VA, USA). The luciferase activity was calculated against the pG5-luciferase basal control and normalized against Renilla luciferase activity. The relative activity was expressed as luminescence units normalized to a negative control (value for cells transfected with only pG5-luciferase reporter vector/pBIND-cdk3 = 1).
Western blotting and immunoprecipitation
Cells were harvested at 80-90% confluence and proteins were extracted in NP-40 cell lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.5% NP-40) with freezing and thawing. Protein concentration was measured using the DC protein assay kit (Bio-Rad, Hercules, CA) for Western blotting. Proteins were transferred onto PVDF membranes, hybridized with appropriate antibodies and then visualized using the ECL detection kit (Amersham Biosciences, Piscataway, NJ). To identify potential protein interactions, the cdk3 (H-45) antibody was used for immunoprecipitation of T98G cell lysates (1 mg). Antibody binding was carried out at 4 °C overnight and proteins were visualized by Western blotting.
In vitro kinase assay
The plasmids for GST-tagged or His-tagged fusion proteins were each respectively transformed into BL21 cells and induced with 0.5 mM IPTG at 25 °C for 3 h. The soluble GST-fusion proteins were purified using glutathione Sepharose 4B beads and eluted with 10 mM reduced-glutathione in 50 mM Tris-HCl (pH 8.0) buffer. The soluble His-fusion proteins were purified with Ni-NTA agarose (QIAGEN Inc., Valencia, CA). Purified fusion proteins (200 ng) or histone H1 (2ug, Upstate Biotechnology, Inc) were used for the in vitro kinase assay with 100 ng active cdk3 (Upstate Biotechnology, Inc) or endogenous cdk3 immunoprecipitated from cell lysates (1 mg). Reactions were carried out in 1× kinase buffer containing 50 μM unlabeled ATP with or without 10 μCi of [γ-32P] ATP at 30 °C for 30 min. Reactions were stopped and then proteins resolved by 12% SDS-PAGE and visualized by autoradiography or Western blotting.
ATF1 transactivation assay
T98G cells (8 × 104) were seeded into 12-well plates for 24 h before transfection. The p5×Gal4-luciferase reporter plasmid (p5×Gal4-luc) was transfected with pCMV-cdk3 and the expression vectors for Gal4-ATF1 or Gal4-ATF1 S63A. The cells were cultured for 36 h and then disrupted for firefly luciferase activity analysis. The p5×Gal4-luc activity was normalized against Renilla luciferase activity.
Construction of si-cdk3 plasmids
The pU6pro vector (a gift from David L. Turner, University of Michigan, Ann Arbor, MI) was used to construct si-RNA against cdk3 (si-cdk3). The synthetic primers were annealed and then introduced into the pU6pro vector digested with XbaI and BbsI following the standard protocols found online (http://sitemaker.umich.edu/dlturner.vectors). The si-cdk3 construct was confirmed by DNA sequencing. An si-general scramble (si-mock) was constructed as described previously and used as a negative control (35).
MTS assay
To assess cell proliferation, psi-mock and psi-cdk3 stably transfected T98G cells (1×103) were seeded into 96-well plates in 100 μl of 10% FBS/MEM and cultured at 37 °C in a 5% CO2 incubator. After culturing for 24, 48, or 72 h, 20 μl of the CellTiter 96® Aqueous One Solution (Promega, Madison, WI) were added to each well and then incubated for 1 h at 37 °C in a 5% CO2 incubator. Absorbance was measured at 492 nm.
Anchorage-independent cell transformation assay
EGF-induced cell transformation was examined in psi-mock and psi-cdk3 stably transfected T98G cells and in mock and pCMV-cdk3 stably transfected JB6 Cl41 cells. In brief, cells (8 × 103) were exposed to EGF (10 ng/ml) in 1 ml of 0.3% Basal Medium Eagle (BME) agar/10% FBS. The cultures were maintained at 37°C for 1-2 weeks and colonies were scored using a microscope and the Image-Pro PLUS computer software program (v. 4, Media Cybernetics, Silver Spring, MD, USA) as described by Colburn et al (36).
Foci forming assay
Following standard protocols (37), NIH3T3 cells were transiently transfected with various combination of H-RasG12V (100 ng), cdk3 (500 ng), ATF1 (500 ng), ATF1 S63A (500 ng), psi-cdk3 (500 ng), or pcDNA3-mock (compensation for equal amount of DNA) and then cultured in 5% calf serum/DMEM for 2 weeks. Foci were fixed with methanol, stained with 0.5% crystal violet and then counted using a microscope and the Image-Pro PLUS (v. 4) software program.
Immunofluorescent detection
A human glioblastoma tissue array section was deparaffinized in xylene, rehydrated in alcohol, and the antigen was retrieved in 10 mM sodium citrate buffer. The tissues were first blocked in 5% normal goat serum/PBS for 1 h at room temperature and then incubated overnight at 4°C with a 1:50 dilution of anti-cdk3 in PBS containing 1% BSA. The tissues were washed and then incubated in the dark with a 1:200 dilution of a Cy3-donkey anti-rabbit antibody for 2 h at room temperature. Image stacks were captured using laser scanning confocal microscopy (NIKON C1si Confocal Spectral Imaging System, NIKON Instruments Co., Melville, NY) and analyzed with the ImageJ (version 1.37v) software program. The intensity score of fluorescence from each sample was measured and the average intensity score from duplicate scores of each case was recorded as the cdk3 expression level.
Results
Cdk3 is highly expressed in human cancer cells and glioblastoma tissue
To investigate whether cdk3 is involved in human cancer, we examined cdk3 protein expression level in 15 different human cell lines by Western blotting and cdk3 activity by immunoprecipitation kinase assay. The results indicated higher cdk3 protein levels in most of the cancer cell lines including glioblastoma T98G cells, large cell lung cancer H460 cells, colorectal carcinoma HCT116 cells, colorectal adenocarcinoma HT29 cells, prostate cancer PC-3 and DU-145 cells, osteosarcoma Saos-2 cells, epidermoid carcinoma A431 cells, and melanoma SK-MEI-28 and RPMI-1951 cells (Fig. 1A, upper panels). Furthermore, cdk3 activities in these cell lines are also accordingly higher (Fig. 1A, bottom panels). The protein levels of cyclin C were also detected in these cell lines and our data indicated that the protein expression pattern of cyclin C is not the same as cdk3 (Fig. 1A, upper panels). Others have reported high levels of cdk3 in glioblastoma cells (20-22), and our results confirmed that cdk3 is highly expressed in glioblastoma T98G cells. Thus, we then examined cdk3 protein level in a human glioblastoma cancer tissue array, which contained duplicate core samples of 35 cases of glioblastoma, 3 cases of non-neoplastic normal cerebrum tissues, and 2 cases of normal cerebrum tissues adjacent to cancer tissue (US Biomax, Inc). We found that cdk3 expression was significantly higher in glioblastoma tissues with a median expression level of 12.09 compared with expression in non-tumor tissues, which had a median score of 9.47 (Fig. 1B). These results demonstrated that high expression of cdk3 is associated with human glioblastoma.
Figure 1. Cdk3 is overexpressed in glioblastoma tissue and various cancer cell lines.

(A, top) Endogenous cdk3 and cyclin C protein levels were detected in 15 different cell lines. Cells were harvested and proteins (40 μg) were resolved by SDS-PAGE and detected by Western blotting using anti-cdk3 or anti-cyclin C. (A, bottom) Endogenous cdk3 activity was examined by immunoprecipitation with anti-cdk3 and subjected to a kinase assay using histone H1 as substrate. Kinase reactions were analyzed by SDS-PAGE followed by autoradiography. (B) Immunofluorescent staining and confocal microscopy were used to detect the expression of cdk3 in tissue from 35 cases of human brain glioblastoma, 2 cases of normal cerebrum tissue that were adjacent to cancer tissue, and 3 cases of non-neoplastic normal cerebrum tissue. Cdk3 was detected by immunofluorescent staining using anti-cdk3 as the primary antibody and a Cy3-conjugated donkey anti-rabbit antibody as the secondary. The intensity score of fluorescence from each sample was determined and the horizontal lines indicate the median of glioblastoma or non-cancer tissue samples. Significant differences were evaluated using the Mann-Whitney U test (*, p < 0.01). Pictures from representative cases are shown in the bottom panels.
ATF1 is a novel binding partner of cdk3
Cdk3 is a serine/threonine protein kinase that phosphorylates a number of substrates, including pRb (20) and Cables (38, 39). We hypothesized that cdk3 might phosphorylate additional substrates, including transcription factor(s). To examine this idea, we conducted the mammalian two-hybrid (M2H) assay using cdk3 (pBIND-cdk3) as bait for various candidate transcription factors (pACT-TFs) (34) in order to identify new binding partners of cdk3. Each individual pACT-TF and the pG5-luciferase reporter plasmid were cotransfected with pBIND-cdk3 into HEK293 cells. Each interaction activity was compared against the basal level (value = 1) of pG5-luc/pBIND-cdk3/pACT (Fig. 2A, top, lane 1). E2F1 and E2F2 (19) were used as positive controls (Fig. 2A, top, lanes 2-3). ATF1 showed more than a 15-fold increase in interacting activity with pBIND-cdk3 compared with the pG5-luc/pBIND-cdk3/pACT control (Fig. 2A, top, lane 4 vs. 1). To determine whether the interaction of ATF1 with cdk3 occurs endogenously, we used T98G cells to perform immunoprecipitation (IP) experiments. The results showed that ATF1 binds with cdk3 ex vivo (Fig. 2A, bottom panels). Then to examine whether cdk3 phosphorylates ATF1, a GST-ATF1 fusion protein was constructed, expressed, partially purified as described in “Materials and Methods”, and then subjected to a kinase assay with active cdk3. The results demonstrated that cdk3 phosphorylated ATF1 in vitro (Fig. 2B, lane 6). To determine the specific sites of ATF1 that are phosphorylated by cdk3, we used the human protein reference database (http://www.hprd.org) to compare the amino acid similarity among known cdk3 substrates. Results showed that the putative consensus motif for cdk3 is the SP amino acid motif. We then examined the amino acid sequence of ATF1 and found that serine 63 was a potential candidate for phosphorylation by cdk3 (Fig. 3C, upper panel). For confirmation, we expressed a mutant ATF1 that harbored a point mutation at serine 63 (S63A), performed an in vitro kinase assay, and detected phosphorylation of ATF1 by Western blotting with an ATF1 serine 63 specific antibody (Fig. 2C, bottom panels). The results indicated that cdk3 phosphorylates wildtype ATF1 at serine 63. When we performed the in vitro kinase assay with [γ−32p ATP, the phospho-band intensity of the ATF1S63A mutant was decreased, but was still visible (Supplementary Fig. S1A). Furthermore, in an in vitro kinase assay using deletion mutants of GST-ATF1 proteins (1-57, 1-141 or 142-271) and active cdk3, we found that another phosphorylation target amino acid exists in the N-terminal region of ATF1 spanning amino acids 1-57 (Supplementary Fig. S1B). We further confirmed that endogenous cdk3 phosphorylated ATF1 at serine 63 (Fig. 2D) using an immunoprecipitation kinase assay and T98G cells. Overall, these results indicated that ATF1 is a novel substrate of cdk3, which phosphorylates ATF1 at serine 63.
Figure 2. Cdk3 interacts with and phosphorylates ATF1 in vitro and ex vivo.
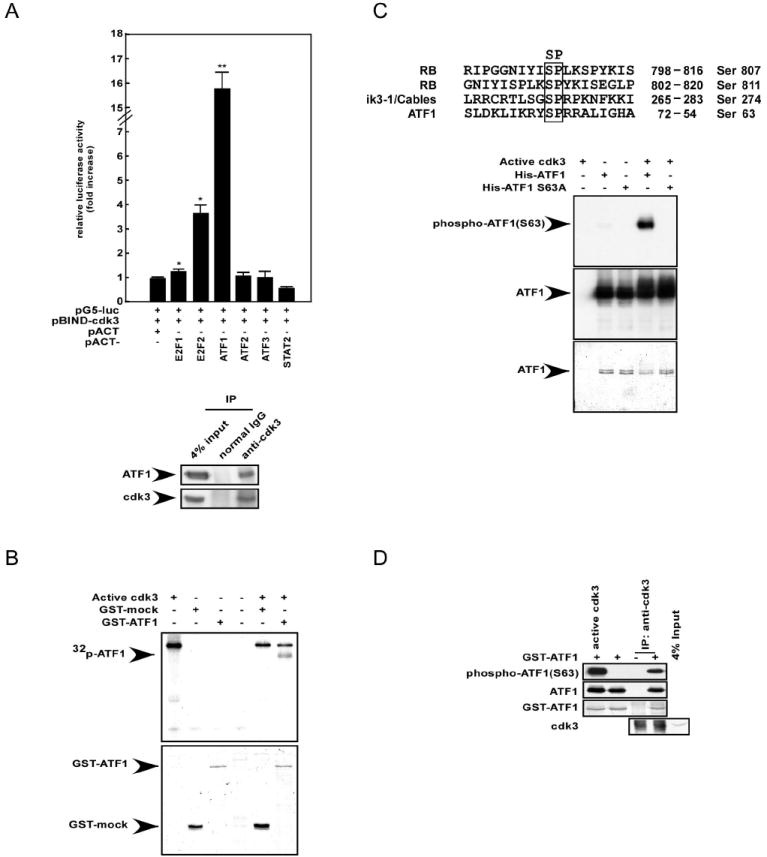
(A, top) The mammalian two-hybrid assay was performed using HEK293 cells to assess the ex vivo protein–protein interaction of pBIND–cdk3 with various pACT–transcription factors (TFs). Activity is expressed as relative luminescence units normalized to a negative control (value for cells transfected with pG5-luc/pBIND-cdk3/pACT = 1.0). The firefly luciferase activity was normalized against the Renilla luciferase activity. Data are represented as mean ± S.D. of values obtained from triplicate experiments. Significant differences were evaluated using the Student’s t–test (*, p < 0.05; **, p < 0.001). (A, bottom) Co-immunoprecipitation (co-IP) was performed using T98G cells. Cells cultured in 10% FBS/MEM were harvested and then subjected to immunoprecipitation with anti-cdk3 and subsequent Western blotting with antibodies to detect ATF1 or cdk3. (B) A GST-ATF1 fusion protein was used in an in vitro kinase assay with active cdk3 and results were visualized by autoradiography (top panel). Coomassie blue staining indicates the respective GST fusion proteins (bottom panel). (C) His-ATF1 and His-ATF1 S63A fusion proteins were partially purified and subjected to an in vitro kinase assay with active cdk3. Results were visualized by Western blotting with anti-phospho-ATF1 or anti-ATF1. Coomassie blue staining indicates the respective His fusion proteins. (D) T98G cells were harvested and subjected to immunoprecipitation with anti-cdk3. Kinase activity was assayed using GST-ATF1 as a substrate, followed by Western blotting with anti-phospho-ATF1, anti-ATF1, or anti-cdk3.
Figure 3. Cdk3 enhances transactivation activity of ATF1.

(A) A pGal4-ATF1 expression vector was constructed as indicated (left panel). The transactivation activity of ATF1 was measured in T98G cells transfected with pGal4-ATF1 or pGal4-ATF1 S63A as described in “Materials and Methods” (middle panel). The mammalian two-hybrid assay was performed in HEK293 cells to assess the interaction of pBIND cdk3 with pACT-ATF1 or pACT-ATF1 S63A (right panel). B, C, A c-jun-luciferase reporter plasmid (B) or a c-fos-luciferase reporter plasmid (C) was co-transfected with increasing amounts of a pcDNA4-ATF1 plasmid into T98G cells and then firefly luciferase activity was analyzed (left panels). The pcDNA4-ATF1 and c-jun-luciferase or c-fos-luciferase reporter plasmid were co-transfected with increasing amounts of pCMV-cdk3 into T98G cells and then firefly luciferase activity was analyzed (right panels). For all experiments (A-C), the firefly luciferase activity was analyzed at 36 h and normalized against the Renilla luciferase activity. Data are shown as the mean ± S.D. of values obtained from triplicate experiments. Significant differences were evaluated using the Student’s t–test (*, p < 0.05; **, p < 0.001).
Cdk3 enhances transactivation and transcriptional activity of ATF1
Our studies thus far have demonstrated that cdk3 phosphorylates ATF1 at serine 63. To examine the biological significance of cdk3-mediated ATF1 phosphorylation, we constructed Gal4-ATF1 and Gal4-ATF1 S63A fusion expression vectors (Fig. 3A, left panel) and analyzed ATF1 transactivation activity as described previously (40). Our results indicated that ATF1 transactivation activity was increased dose-dependently by cotransfection of increasing amounts of pCMV-cdk3 (Fig. 3A, middle panel). However, the mutant Gal4-ATF1 S63A did not exhibit any transactivation activity regardless of the amount of cdk3 present (Fig. 3A, middle panel). On the other hand, we found that cdk3 bound equally well with the mutant ATF1 (S63A) or wildtype ATF1 (Fig. 3A, right panel). These results demonstrated that cdk3 positively regulated ATF1 transactivation activity through phosphorylation at serine 63.
Because c-Jun and c-Fos expression are regulated by ATF1, we examined cdk3-mediated transcriptional activity of ATF1 using cells transfected with either a luciferase linked c-jun (c-jun-luc) or c-fos (c-fos-luc) promoter (41, 42). Results indicated that c-Jun-luciferase activity was increased dose dependently by cotransfection with increasing amounts of ATF1 (Fig. 3B, left panel) and even more by increasing amounts of cdk3 (Fig. 3B, right panel). Similar results were obtained for c-Fos (Fig. 3C). These results indicated that cdk3-mediated ATF1 phosphorylation at serine 63 is critical for ATF1 transactivation and transcriptional activity.
Knockdown of cdk3 suppresses proliferation and colony formation of T98G cells
To examine whether knockdown of cdk3 affects proliferation and colony formation in soft agar, we used glioblastoma T98G cells because of the high expression of cdk3 in these cells and human glioblastoma tissues (Fig. 1). To knockdown cdk3 expression, we designed siRNA against cdk3 (si-cdk3; Supplementary Fig. S2) and introduced it into T98G cells. A stable cell line was established and knockdown efficiency was examined by Western blotting. The results revealed that si-cdk3 suppressed the endogenous cdk3 protein level by 60% compared with si-mock control T98G cells (Fig. 4A, bottom left panels). Using these cells and the MTS assay, we estimated proliferation, and the si-cdk3 stably-transfected cell line showed significantly suppressed proliferation compared with the si-mock T98G control cells (Fig. 4A, upper left panel). Further, si-cdk3 T98G cells displayed about a 50% reduction in EGF-induced anchorage-independent colony formation in soft agar compared to mock cells (Fig. 4A, right panels). To determine whether reduction of proliferation and growth in soft agar in si-cdk3 cells is associated with ATF1 activity, we analyzed ATF1 transactivation activity and transcriptional activity using a pGl4.29/Luc2p/CRE reporter plasmid, which contains an ATF1 binding consensus sequence in the CRE motif. The results indicated that Gal4-ATF1 transactivation activity was significantly inhibited by transfection of cells with si-cdk3 (Fig. 4B, left panel). In addition, pGl4.29/Luc2p/CRE luciferase activity was also dose-dependently suppressed by introduction of si-cdk3 (Fig. 4B, right panel). These results indicated that knockdown of cdk3 by si-cdk3 inhibited T98G proliferation and colony formation in soft agar through the inhibition of ATF1 activity. To further verify whether EGF-induced ATF1 phosphorylation is mediated through cdk3, we analyzed ATF1 phosphorylation level in si-mock and si-cdk3 stable T98G cells by Western blotting. The results indicated that phosphorylation of ATF1 at serine 63 was suppressed in si-cdk3 stable cells without an effect on total ATF1 protein levels (Fig. 4C, upper left panels). By Western blotting, we found that EGF-induced ATF1 phosphorylation at serine 63 was increased at 15 min, maintained to 30 min, and decreased at 60 min in si-mock control T98G cells. However, phosphorylation of ATF1 at serine 63 was minimal in si-cdk3 stably-transfected cells (Fig. 4C, bottom left panels). Furthermore, EGF-induced transactivation activity of ATF1 was also suppressed in si-cdk3 stable cells compared with the si-mock cells (Fig. 4C, right panel), indicating that the diminished proliferation and colony formation in soft agar displayed by si-cdk3 cells occurs through a suppression of EGF-induced ATF1 activity. To further confirm a role for cdk3 in proliferation and colony formation, we constructed a mutant cdk3 expression plasmid (rescue cdk3 vector), which was not affected by si-cdk3 and then performed rescue experiments by returning the rescue cdk3 vector to si-cdk3 stably-transfected T98G cells (Fig. 4D). At 24 h after transfection of the rescue cdk3 vector, cdk3 expression was confirmed by Western blotting (Fig. 4D, bottom left) in these cells and proliferation and colony formation in soft agar were assayed. The results indicated that return of the rescue cdk3 vector reversed the si-cdk3-induced reduction of cell proliferation (Fig. 4D, upper left panel compared with Fig. 4A, left upper panel), as well as reversing the si-cdk3-induced decreased colony formation in soft agar (Fig. 4D, right panels compared with Fig. 4A, right panels).
Figure 4. Knockdown of cdk3 reduces tumor growth of T98G cells in soft agar.

(A, left) The T98G-si-cdk3 cell line was generated by stable transfection of psi-cdk3 into glioblastoma T98G cells. Cell lysates (40 μg) from T98G-si-mock and T98G-si-cdk3 cell lines were used for SDS-PAGE and subjected to Western blotting using anti-cdk3 (bottom panels). The growth curves of T98G-si-mock and T98G-si-cdk3 cells were examined by MTS assay as described in “Materials and Methods”. Data are presented as the mean ± S.D. of values obtained from four experiments. Significant differences were evaluated using the Student’s t–test (*, p < 0.05; upper panel). (A, right) Anchorage-independent cell growth in soft agar was determined using T98G-si-mock and T98G-si-cdk3 cells stimulated with EGF (10 ng/ml) as described in “Materials and Methods”. Representative photomicrographs are shown (top panels) and data (bottom panel) are presented as the mean ± S.D. of values obtained from triplicate experiments. Significant differences were evaluated using the Student’s t–test and asterisks indicate a significant suppression effect on number of colonies formed in T98G-si-cdk3 cells compared to mock (*, p < 0.05; **, p < 0.001). (B) T98G cells were used to measure transactivation activity (left panel) and the transcriptional activity of ATF1 in cells transfected with a pGL4.29-luc2P/CRE luciferase reporter plasmid (containing a CRE in its promoter; right panel) in the presence of increasing amounts of psi-cdk3. (C) Phosphorylation of ATF1 at serine 63 was detected in T98G-si-mock and T98G-si-cdk3 cell lines under normal culture conditions (top left panels) or with EGF (10 ng/ml) treatment (bottom left panels). EGF-induced transactivation activity of ATF1 in T98G-si-mock and T98G-si-cdk3 cell lines was compared (right panel). At 24 h after transfection, cells were exposed to EGF (10 ng/ml) and incubated for another 12 h and then firefly luciferase activity was analyzed. Data are presented as mean ± S.D. from triplicate experiments and asterisks indicate a significant difference between si-mock and si-cdk3 cell lines (*, p < 0.05; **, p < 0.001). (D) The rescue experiments involved the introduction of a rescue cdk3 vector, which was not affected by si-cdk3, and were performed using the T98G-si-cdk3 cell line. At 24 h after transfection, the protein level of transfected rescue cdk3 was confirmed by Western blotting using anti-HA (bottom left panels), and the growth curves and anchorage-independent cell growth were examined by MTS assay (upper left panel) or colony formation in soft agar (right panels), respectively, as described in Fig. 4A.
The cdk3-ATF1 signaling axis plays an important role in cell transformation
Next we examined whether the cdk3-ATF1 signaling axis plays an important role in cell transformation by establishing a stable JB6 Cl41 cell line that expresses high levels of cdk3. High expression of the cdk3 protein was confirmed in this cell line by Western blotting (Fig. 5A). The ATF1 protein level was not changed. Next, we examined the effect of ectopic expression of cdk3 on EGF-induced colony formation in soft agar. The results demonstrated that, compared with mock control cells, cdk3-expressing JB6 Cl41 cells displayed an approximate 8-fold increase in EGF-induced colony formation (Fig. 5B). We also analyzed EGF-induced transactivation activity of ATF1 and the results indicated that ATF1 transactivation activity was significantly increased in cdk3-expressing stable cells compared with mock cells (Fig. 5C). Importantly, we further found that RasG12V induced cell transformation (Fig. 5D, middle panel, lane 4), and overexpression of RasG12V/cdk3 or RasG12V/ATF1 induced more foci formation in NIH3T3 cells (Fig. 5D, middle panel, lanes 5-6). Moreover, cotransfection of RasG12V/cdk3/ATF1 induced even more foci formation (Fig. 5D, middle panel, lane 8). However, cotransfection of si-cdk3 with RasG12V/cdk3/ATF1 blocked foci formation in NIH3T3 cells (Fig. 5D, middle panel, lane 9) compared with RasG12V/cdk3/ATF1. In addition, cotransfection of RasG12V/cdk3/ATF1 S63A blocked foci formation to the same level of RasG12V/cdk3/ATF1/si-cdk3 compared with RasG12V/cdk3/ATF1 (Fig. 5D, middle panel, lane 10). These results indicated that cdk3-ATF1 signaling plays an important role in cell transformation.
Figure 5. Ectopic expression of cdk3 induces cell transformation through activation of ATF1.
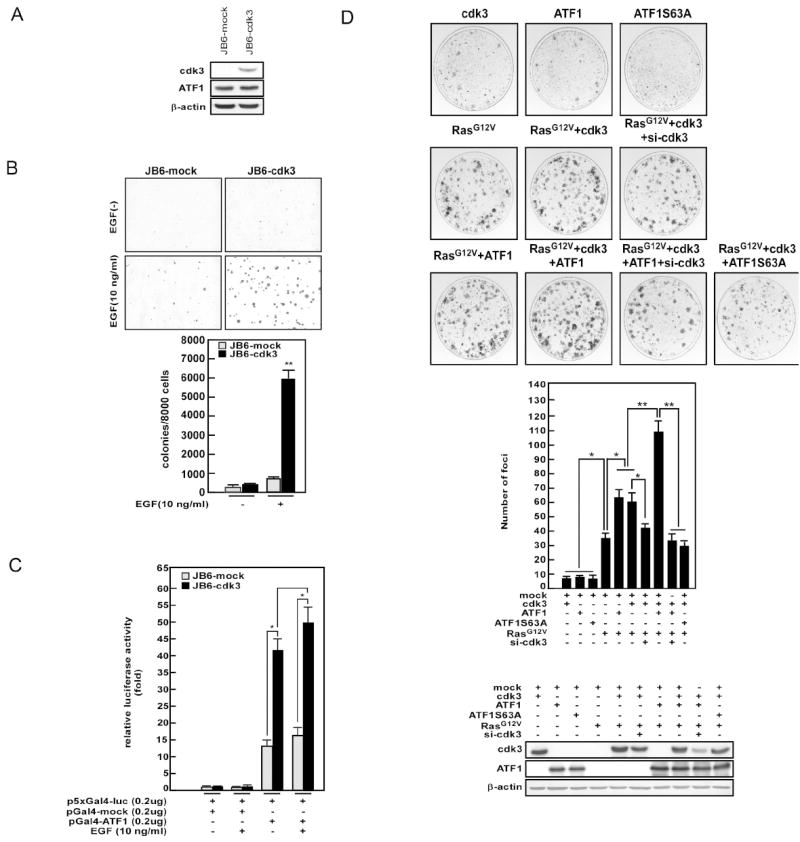
(A) The JB6-cdk3 cell line was established by stable transfection of pCMV-cdk3 into JB6 C141 cells. Proteins (60 μg) from JB6-mock and JB6-cdk3 cell lines were resolved by SDS-PAGE for Western blotting with antibodies against cdk3 or ATF1. (B) An anchorage-independent cell transformation assay was performed using JB6-cdk3 and JB6-mock cells stimulated with EGF (10 ng/ml) as described in “Materials and Methods”. Representative photomicrographs are shown (top panels). Data are presented as the mean ± S.D. of values obtained from triplicate experiments (bottom panel). Significant differences were evaluated using the Student’s t–test and the asterisks indicate a significantly higher number of colonies in cdk3-overexpressing cells compared to mock cells (**, p < 0.001). (C) EGF-induced transactivation activity of ATF1 in JB6-mock- and JB6-cdk3-transfected cell lines was examined. At 24 h after transfection, cells were exposed to EGF (10 ng/ml) in 5% FBS/MEM and firefly luciferase activity was analyzed after 12 h incubation. Data are presented as the mean ± S.D. of values obtained from triplicate experiments. Significant differences were evaluated using the Student’s t–test and the asterisk indicates a significantly increase in ATF1 transactivation in cdk3-overexpressing cells compared to mock cells (*, p < 0.05). (D) Ras-induced foci formation was examined. Various combinations of expression vectors were transfected into NIH3T3 cells as indicated and a foci formation assay was performed following standard protocols as described in “Materials and Methods”. The protein level of transfected cdk3 or ATF1 was measured with anti-HA or anti-Flag by Western blotting, respectively (bottom panels). Representative photomicrographs for each condition are shown (upper panels). Foci were counted and data are presented as mean ± S.D. of values obtained from triplicate experiments. Significant differences were evaluated using the Student’s t test and asterisks indicate a significant difference in foci formation compared to each respective control (*, p < 0.01; **, p < 0.001).
Discussion
Activation of cdks induced by overexpression of activator cyclins, inactivation of cdk inhibitors such as INK4, Cip and Kip, or inactivation of substrates such as pRb has been observed in many human cancers (1, 4). Thus cdks are important regulators in cell cycle progression and have long been regarded as targets for human cancer therapeutic intervention (43). Previous studies demonstrated that cdk3 was expressed in some human tissues and cell lines (20-22). In the present study, we found that cdk3 is overexpressed in glioblastoma tissues and in numerous human cancer cell lines, including T98G, H460, HCT116, HT29, PC-3, DU-145, Saos-2, A431, SK-MEI-28 and RPMI-1951 cells compared with the HaCaT cell line, which is an immortalized non-malignant human keratinocyte cell line (Fig. 1); and knockdown of cdk3 suppressed proliferation (Fig. 4A) and colony formation of glioblastoma T98G cells in soft agar (Fig. 4A). Furthermore, knockdown of cdk3 suppressed foci formation induced by RasG12V/cdk3/ATF1 in NIH3T3 cells (Fig. 5D). These results indicated that the cdk3-ATF1 signaling axis plays an important role in cell transformation.
The well-known substrates of cdk3 include the tumor suppressor pRb and Cables/IK3-1. Cdk3 phosphorylates pRb at serine 807/811during the G0-G1 transition, which is required for cells to exit G0 efficiently (20). Cdk3 phosphorylates Cables/IK3-1 at serine 274 (38). Cables/IK3-1 can bind with cdk5 and c-Abl and induces cdk5 phosphorylation at tyrosine 15 by Abl resulting in increased kinase activity of the p35/cdk5 complex and neurite outgrowth (44). In HeLa cells, Cables enhances cdk2 tyrosine 15 phosphorylation by Weel leading to decreased cdk2 kinase activity and inhibition of cell growth (45). Furthermore, loss of Cables expression has been found in 50 to 60% of primary colon and head and neck cancer samples (45). However, the biological significance of Cables/IK3-1 phosphorylation by cdk3 is not clear. Here, we found that cdk3 phosphorylates ATF1 at serine 63 (Fig. 2).
The expression of ATF1 is upregulated in lymphomas (31) and melanoma cells (32) and contributes to the growth of these cancers. The EWS-ATF1 fusion oncoprotein is typically associated with clear cell sarcoma (CCS) (46) because of enhanced target gene expression induced by constitutively activated ATF1 in the absence of stimuli (33). In our study, we found that transactivation and transcriptional activities of ATF1 were increased by cdk3 in a dose dependent manner (Fig. 3). Furthermore, our study demonstrated that knockdown of cdk3 suppressed cell transformation and ATF1 transactivation and transcriptional activity (Fig. 4B and C). In addition, c-jun and c-fos promoter activity was increased by ATF1 and cdk3 dose-dependently (Fig. 3B and C). ATF1 belongs to CREB transcription factor family and influences cellular physiological processes by regulating expression of downstream target genes, which are related to growth, survival and other cellular activities (24). Our data indicated that c-jun and c-fos might play an important role in cell proliferation and cell transformation induced by cdk3-ATF1 signaling.
Various stimuli, including growth factors, stress signals, neurotransmitters and other agents, induce activation of CREB family members (24). The transactivation domain of ATF1 consists of a KID domain and a glutamine-rich constitutive activation domain (Q2). Our finding that cdk3 phosphorylates ATF1 serine 63 in the KID domain is important because this phosphorylation promotes recruitment of CBP/p300, a core protein in the transcriptional machinery, leading to enhancement of transactivation and transcriptional activity (30). Furthermore, ATF1 can be phosphorylated at serine 63 by PKA (47) and calmodulin-dependent protein kinase (CaMK) I / II (47, 48). In response to EGF or TPA stimulation, ATF1 can also be phosphorylated by mitogen- and stress-activated protein kinase1 (MSK1/2) (41, 49, 50), but in MSK1/MSK2 double knockout embryonic fibroblasts, mitogen-induced phosphorylation of ATF1 at serine 63 is reduced but not totally abolished (50), suggesting phosphorylation by additional kinases. We found that ectopic expression of cdk3 in JB6 Cl41 cells stimulated EGF- induced cell transformation and ATF1 activation (Fig. 5B and C). Moreover, coexpression of cdk3 and RasG12V, a downstream kinase of the EGFR signaling pathway, induced cell transformation through cdk3-ATF1 signaling in NIH3T3 cells (Fig. 5D), indicating that cdk3 may participate as a downstream kinase of the EGFR-Ras pathway. In summary, we found that ATF1 is a novel substrate of cdk3 and the cdk3-ATF1 signaling axis plays an important role in cell proliferation and cell transformation.
Supplementary Material
Acknowledgments
We would like to thank Dr. Barrett J. Rollins (Department of Medical Oncology, Harvard Medical School, Boston, Massachusetts) for the pRcCMV-cdk3 (pCMV-cdk3); Dr. Ron Pryews (Department of Biological Sciences, Columbia University) for pET28-ATF1, pET28-ATF1S63A, p3×Flag-ATF1 and p3×Flag-ATF1S63A mutant; Dr. Hiroshi Hanbda (Faculty of Bioscience and Biotechnology, Tokyo Institute of Technology) for the deletion GST-ATF1 fusion vectors (1-57, 1-141 and 142-271); Dr. David L. Turner (University of Michigan, Ann Arbor, MI) for the pU6pro vector; and Ms. Andria Hansen for secretarial assistance.
Grant support: This work is supported in part by The Hormel Foundation, National Institutes of Health grants CA77646, CA27502, and CA111356.
Footnotes
Note: The University of Minnesota is an equal opportunity educator and employer.
References
- 1.Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001;1:222–31. doi: 10.1038/35106065. [DOI] [PubMed] [Google Scholar]
- 2.de Carcer G, de Castro IP, Malumbres M. Targeting cell cycle kinases for cancer therapy. Curr Med Chem. 2007;14:969–85. doi: 10.2174/092986707780362925. [DOI] [PubMed] [Google Scholar]
- 3.Wolfel T, Hauer M, Schneider J, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–4. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- 4.Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005;30:630–41. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Corcoran MM, Mould SJ, Orchard JA, et al. Dysregulation of cyclin dependent kinase 6 expression in splenic marginal zone lymphoma through chromosome 7q translocations. Oncogene. 1999;18:6271–7. doi: 10.1038/sj.onc.1203033. [DOI] [PubMed] [Google Scholar]
- 6.Hayette S, Tigaud I, Callet-Bauchu E, et al. In B-cell chronic lymphocytic leukemias, 7q21 translocations lead to overexpression of the CDK6 gene. Blood. 2003;102:1549–50. doi: 10.1182/blood-2003-04-1220. [DOI] [PubMed] [Google Scholar]
- 7.Haas K, Staller P, Geisen C, Bartek J, Eilers M, Moroy T. Mutual requirement of CDK4 and Myc in malignant transformation: evidence for cyclin D1/CDK4 and p16INK4A as upstream regulators of Myc. Oncogene. 1997;15:179–92. doi: 10.1038/sj.onc.1201171. [DOI] [PubMed] [Google Scholar]
- 8.Lazarov M, Kubo Y, Cai T, et al. CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat Med. 2002;8:1105–14. doi: 10.1038/nm779. [DOI] [PubMed] [Google Scholar]
- 9.Yu Q, Sicinska E, Geng Y, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9:23–32. doi: 10.1016/j.ccr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Reddy HK, Mettus RV, Rane SG, Grana X, Litvin J, Reddy EP. Cyclin-dependent kinase 4 expression is essential for neu-induced breast tumorigenesis. Cancer Res. 2005;65:10174–8. doi: 10.1158/0008-5472.CAN-05-2639. [DOI] [PubMed] [Google Scholar]
- 11.Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell. 2006;9:13–22. doi: 10.1016/j.ccr.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 12.Ray D, Terao Y, Nimbalkar D, et al. Hemizygous disruption of Cdc25A inhibits cellular transformation and mammary tumorigenesis in mice. Cancer Res. 2007;67:6605–11. doi: 10.1158/0008-5472.CAN-06-4815. [DOI] [PubMed] [Google Scholar]
- 13.Sotillo E, Garriga J, Kurimchak A, Grana X. Cyclin E and SV40 small T antigen cooperate to bypass quiescence and contribute to transformation by activating CDK2 in human fibroblasts. J Biol Chem. 2008 doi: 10.1074/jbc.M709055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du J, Widlund HR, Horstmann MA, et al. Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by MITF. Cancer Cell. 2004;6:565–76. doi: 10.1016/j.ccr.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 15.Macias E, Kim Y, Miliani de Marval PL, Klein-Szanto A, Rodriguez-Puebla ML. Cdk2 deficiency decreases ras/CDK4-dependent malignant progression, but not myc-induced tumorigenesis. Cancer Res. 2007;67:9713–20. doi: 10.1158/0008-5472.CAN-07-2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keezer SM, Gilbert DM. Evidence for a pre-restriction point Cdk3 activity. J Cell Biochem. 2002;85:545–52. doi: 10.1002/jcb.10162. [DOI] [PubMed] [Google Scholar]
- 17.Braun K, Holzl G, Soucek T, Geisen C, Moroy T, Hengstschlager M. Investigation of the cell cycle regulation of cdk3-associated kinase activity and the role of cdk3 in proliferation and transformation. Oncogene. 1998;17:2259–69. doi: 10.1038/sj.onc.1202145. [DOI] [PubMed] [Google Scholar]
- 18.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–4. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 19.Hofmann F, Livingston DM. Differential effects of cdk2 and cdk3 on the control of pRb and E2F function during G1 exit. Genes Dev. 1996;10:851–61. doi: 10.1101/gad.10.7.851. [DOI] [PubMed] [Google Scholar]
- 20.Ren S, Rollins BJ. Cyclin C/cdk3 promotes Rb-dependent G0 exit. Cell. 2004;117:239–51. doi: 10.1016/s0092-8674(04)00300-9. [DOI] [PubMed] [Google Scholar]
- 21.Meyerson M, Enders GH, Wu CL, et al. A family of human cdc2-related protein kinases. Embo J. 1992;11:2909–17. doi: 10.1002/j.1460-2075.1992.tb05360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schang LM, Bantly A, Schaffer PA. Explant-induced reactivation of herpes simplex virus occurs in neurons expressing nuclear cdk2 and cdk4. J Virol. 2002;76:7724–35. doi: 10.1128/JVI.76.15.7724-7735.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bullrich F, MacLachlan TK, Sang N, et al. Chromosomal mapping of members of the cdc2 family of protein kinases, cdk3, cdk6, PISSLRE, and PITALRE, and a cdk inhibitor, p27Kip1, to regions involved in human cancer. Cancer Res. 1995;55:1199–205. [PubMed] [Google Scholar]
- 24.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 25.Hai TW, Liu F, Coukos WJ, Green MR. Transcription factor ATF cDNA clones: an extensive family of leucine zipper proteins able to selectively form DNA-binding heterodimers. Genes Dev. 1989;3:2083–90. doi: 10.1101/gad.3.12b.2083. [DOI] [PubMed] [Google Scholar]
- 26.Hoeffler JP, Meyer TE, Yun Y, Jameson JL, Habener JF. Cyclic AMP-responsive DNA-binding protein: structure based on a cloned placental cDNA. Science. 1988;242:1430–3. doi: 10.1126/science.2974179. [DOI] [PubMed] [Google Scholar]
- 27.Foulkes NS, Borrelli E, Sassone-Corsi P. CREM gene: use of alternative DNA-binding domains generates multiple antagonists of cAMP-induced transcription. Cell. 1991;64:739–49. doi: 10.1016/0092-8674(91)90503-q. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Odom DT, Koo SH, et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 2005;102:4459–64. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Impey S, McCorkle SR, Cha-Molstad H, et al. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119:1041–54. doi: 10.1016/j.cell.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 30.Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–9. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 31.Hsueh YP, Lai MZ. Overexpression of activation transcriptional factor 1 in lymphomas and in activated lymphocytes. J Immunol. 1995;154:5675–83. [PubMed] [Google Scholar]
- 32.Jean D, Tellez C, Huang S, et al. Inhibition of tumor growth and metastasis of human melanoma by intracellular anti-ATF-1 single chain Fv fragment. Oncogene. 2000;19:2721–30. doi: 10.1038/sj.onc.1203569. [DOI] [PubMed] [Google Scholar]
- 33.Brown AD, Lopez-Terrada D, Denny C, Lee KA. Promoters containing ATF-binding sites are de-regulated in cells that express the EWS/ATF1 oncogene. Oncogene. 1995;10:1749–56. [PubMed] [Google Scholar]
- 34.Cho YY, He Z, Zhang Y, et al. The p53 protein is a novel substrate of ribosomal S6 kinase 2 and a critical intermediary for ribosomal S6 kinase 2 and histone H3 interaction. Cancer Res. 2005;65:3596–603. doi: 10.1158/0008-5472.CAN-04-3935. [DOI] [PubMed] [Google Scholar]
- 35.Cho YY, Yao K, Kim HG, et al. Ribosomal s6 kinase 2 is a key regulator in tumor promoter induced cell transformation. Cancer Res. 2007;67:8104–12. doi: 10.1158/0008-5472.CAN-06-4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colburn NH, Wendel EJ, Abruzzo G. Dissociation of mitogenesis and late-stage promotion of tumor cell phenotype by phorbol esters: mitogen-resistant variants are sensitive to promotion. Proc Natl Acad Sci U S A. 1981;78:6912–6. doi: 10.1073/pnas.78.11.6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clark GJ, Cox AD, Graham SM, Der CJ. Biological assays for Ras transformation. Methods Enzymol. 1995;255:395–412. doi: 10.1016/s0076-6879(95)55042-9. [DOI] [PubMed] [Google Scholar]
- 38.Yamochi T, Semba K, Tsuji K, et al. ik3-1/Cables is a substrate for cyclin-dependent kinase 3 (cdk 3) Eur J Biochem. 2001;268:6076–82. doi: 10.1046/j.0014-2956.2001.02555.x. [DOI] [PubMed] [Google Scholar]
- 39.Sato H, Nishimoto I, Matsuoka M. ik3-2, a relative to ik3-1/cables, is associated with cdk3, cdk5, and c-abl. Biochim Biophys Acta. 2002;1574:157–63. doi: 10.1016/s0167-4781(01)00367-0. [DOI] [PubMed] [Google Scholar]
- 40.Cho YY, Yao K, Bode AM, et al. RSK2 mediates muscle cell differentiation through regulation of NFAT3. J Biol Chem. 2007;282:8380–92. doi: 10.1074/jbc.M611322200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gupta P, Prywes R. ATF1 phosphorylation by the ERK MAPK pathway is required for epidermal growth factor-induced c-jun expression. J Biol Chem. 2002;277:50550–6. doi: 10.1074/jbc.M209799200. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Prywes R. Activation of the c-fos enhancer by the erk MAP kinase pathway through two sequence elements: the c-fos AP-1 and p62TCF sites. Oncogene. 2000;19:1379–85. doi: 10.1038/sj.onc.1203443. [DOI] [PubMed] [Google Scholar]
- 43.Knockaert M, Greengard P, Meijer L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol Sci. 2002;23:417–25. doi: 10.1016/s0165-6147(02)02071-0. [DOI] [PubMed] [Google Scholar]
- 44.Zukerberg LR, Patrick GN, Nikolic M, et al. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron. 2000;26:633–46. doi: 10.1016/s0896-6273(00)81200-3. [DOI] [PubMed] [Google Scholar]
- 45.Wu CL, Kirley SD, Xiao H, Chuang Y, Chung DC, Zukerberg LR. Cables enhances cdk2 tyrosine 15 phosphorylation by Wee1, inhibits cell growth, and is lost in many human colon and squamous cancers. Cancer Res. 2001;61:7325–32. [PubMed] [Google Scholar]
- 46.Zucman J, Delattre O, Desmaze C, et al. EWS and ATF-1 gene fusion induced by t(12;22) translocation in malignant melanoma of soft parts. Nat Genet. 1993;4:341–5. doi: 10.1038/ng0893-341. [DOI] [PubMed] [Google Scholar]
- 47.Liu F, Thompson MA, Wagner S, Greenberg ME, Green MR. Activating transcription factor-1 can mediate Ca(2+)- and cAMP-inducible transcriptional activation. J Biol Chem. 1993;268:6714–20. [PubMed] [Google Scholar]
- 48.Shimomura A, Ogawa Y, Kitani T, Fujisawa H, Hagiwara M. Calmodulin-dependent protein kinase II potentiates transcriptional activation through activating transcription factor 1 but not cAMP response element-binding protein. J Biol Chem. 1996;271:17957–60. doi: 10.1074/jbc.271.30.17957. [DOI] [PubMed] [Google Scholar]
- 49.Arthur JS, Cohen P. MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS Lett. 2000;482:44–8. doi: 10.1016/s0014-5793(00)02031-7. [DOI] [PubMed] [Google Scholar]
- 50.Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JS. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol. 2002:2871–81. doi: 10.1128/MCB.22.8.2871-2881.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.