

Abstract
Nearly half of the proteins in the complement system serve in regulation. Control at the central step of C3 activation is provided by an orchestrated interplay of membrane and plasma regulators. A model system employing Chinese hamster ovary (CHO) cells transfected with human regulators was employed to assist in making functional comparisons. Also, in this experimental setup, the pathway and magnitude of complement activation can be varied while monitoring C4b/C3b deposition and cleavage as well as cytotoxicity. This review describes lessons learned from a CHO model and the application of this model for functionally characterizing mutations in regulators associated with atypical hemolytic uremic syndrome.
1. Introduction
The complement system is a key component of innate immunity [1, 2]. It promotes the inflammatory response, opsonizes targets, and perturbs membrane integrity (Tables 1 and 2). In so doing, it instructs and facilitates the adaptive immune response, including being a “complement” to Ab.
Table 1.
Complement activation: General principles
| Site/Target | Objectives |
|---|---|
| Microbes | Surveillance via continuous turnover Destruction and an adaptive immune response |
| Tissue injury and debris* | Wound healing and clearance |
| Apoptosis and necrosis* | Clearance |
| Spermatozoa (following acrosomal reaction)* | Fertilization (?) |
These last three situations represent examples in which restricted activation is desirable and where an adaptive immune response is not indicated (in contrast to microbes).
Table 2.
Complement regulation: General principles
|
Nearly half of the complement proteins participate in regulation. They also are pivotal relative to the host’s ability to distinguish normal self from microbes and altered self [3, 4]. Normal host tissue is protected from complement activation unless deficient in control proteins. Additionally, damaged cells and surfaces targeted by lectins or antibodies are ‘attacked’. Thus, the cell membrane represents a pivotal point for containment of the complement system.
Control at the central step of C3 generation is provided by an orchestrated interplay of a family of related proteins, the Regulators of Complement Activation (RCA). The cell-expressed members of this family are membrane cofactor protein (MCP; CD46), decay accelerating factor (DAF; CD55) and complement receptors one (CR1; CD35) and two (CR2; CD21) while the plasma members are C4-binding protein (C4BP) and factor H (FH).
Complement activation generates small fragments (anaphylatoxins) that are released to serve as triggers of cell activation and to initiate the early inflammatory response. Further, the large cleavage fragments, C3b and C4b, are deposited on and thereby mark a target for destruction and antigen processing. Along with their further degradation fragments, C3b and C4b serve as ligands for interactions with complement receptors on phagocytic and antigen presenting cells. Indeed, the complement system is nature’s premier adjuvant [5].
The evolution of the complement system can be viewed as a series of steps related to activating C3. This necessitated a parallel increase in directing and controlling this process. One evolutionary direction was to specify the target and another was to increase the efficacy of cleaving C3. Lectins and Abs represent two specific target recognition schemes. Upon their respective binding to a sugar or antigen, they initiate sequential, overlapping proteolytic activation cascades resulting in the same enzyme, the classical pathway (CP) C3 convertase, which cleaves C3 to C3b and C3a. In addition to providing specificity, the subsequent partially overlapping proteolytic steps in the lectin pathway (LP) and CP represent a means to amplify and increase the efficiency of C3 cleavage.
In converting C3 to C3b via proteolysis, the thioester bond is broken. This newly generated, highly reactive and transient C3b species has only a few microseconds to become covalently bound to the target through an ester linkage or be hydrolyzed. This process, along with the target selection by lectins and Abs, restricts C3b deposition to the immediate site of activation.
The more slowly developing alternative pathway (AP) is continuously turning or ticking over secondary to an intrinsic instability of the internal thioester bond. Thus, complement activation serves as a sentry or surveillance system of the host, particularly relative to being “a guardian of the intravascular space.” Through engagement of the AP’s feedback loop, C3b deposition is amplified.
Patients lacking factor H (FH) or factor I (FI) present with nearly complete absence of C3 secondary to enhanced AP turnover [6]. Many of these individuals have membranoproliferative glomerulonephritis Type II (MPGNII) [7]. An absence on hematopoietic cells of two complement regulators, DAF and CD59 (membrane inhibitor of lysis), explains why complement lyses erythrocytes in paroxysmal nocturnal hemoglobinuria. Two more recently defined diseases have also had a major impact on our understanding of the role of complement inhibitors in human disease. Heterozygous mutations in MCP, FH and FI cause atypical hemolytic uremic syndrome (aHUS) [6, 8-10] and a polymorphism in FH predisposes to age-related macular degeneration (AMD) [11, 12].
Strict regulation is required for maintaining homeostasis of the complement system. Complement regulators at the C3 cleavage step possess cofactor activity (CA) or decay accelerating activity (DAA) (Fig. 1). CA refers to the limited proteolytic cleavage of C3b or C4b. The resulting cleavage fragment cannot participate in complement-activating enzyme complexes (convertases). This is especially important for C3b as it can engage the AP’s feedback loop. The CA cleavage reaction requires the plasma serine protease, FI, and a cofactor protein such as FH, MCP or CR1. DAA refers to the dissociation of the catalytic serine protease domain from a convertase [4]. DAF and MCP synergize to regulate convertases with DAF decaying the convertase and then MCP and FI cleaving the C4b or C3b [13].
Figure 1.
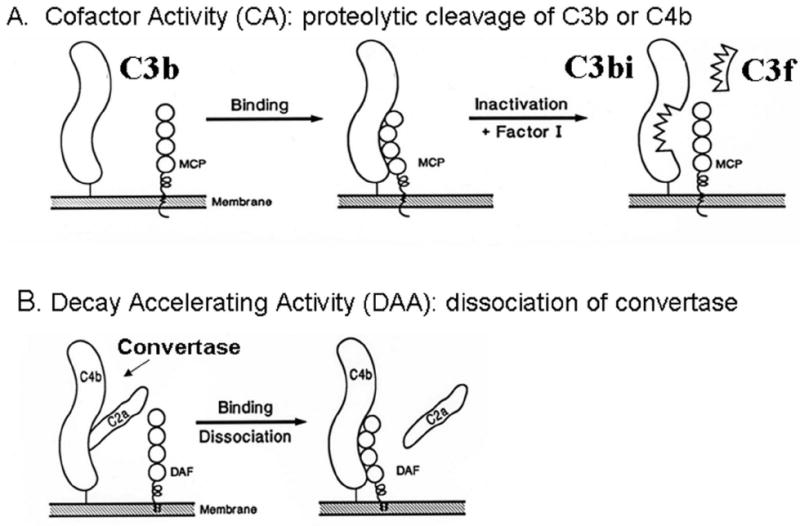
Inhibitory mechanisms for regulation of complement activation A) Cofactor activity: C3b deposits on a target and is then bound by MCP which in turn serves as a cofactor for its cleavage by the serine protease FI. B) Decay accelerating activity: the decay of convertases. This example illustrates the decay of the CP C3 convertase, C4b2a, by DAF.
2. The Chinese hamster ovary (CHO) cell model
This model was developed to facilitate comparisons among complement regulators (Fig. 2). The pathway and degree of the activation process can be varied and the outcome monitored by assessment of C4b and C3b deposition and their subsequent degradation by CA and cytotoxicity. CHO cells were chosen because they neither spontaneously activate nor express on their plasma membrane regulators of human complement. Further, they can be easily transfected and then cloned to obtain stably expressing cell lines.
Figure 2.
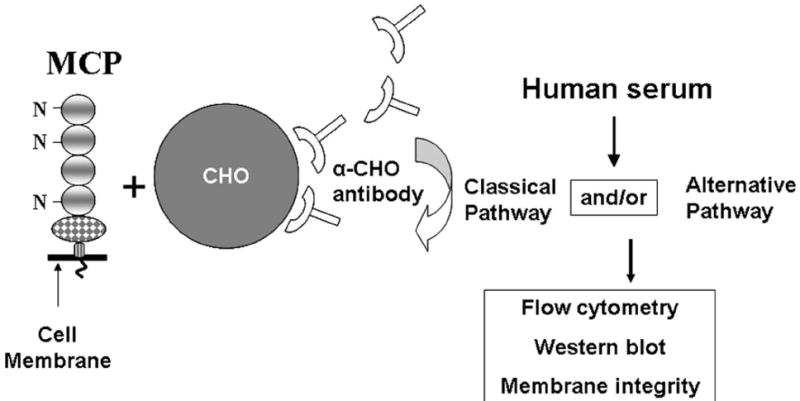
Chinese hamster ovary (CHO) cell model system. To initiate activation, MCP-transfected and wild-type CHO cells are sensitized with polyclonal Ab and then exposed to serum. To evaluate C3b/C4b deposition and their cleavage fragments on the cell surface, C7- or C8-deficient serum is used and fragment detection is with flow cytometry and Western blotting. To evaluate cytotoxity, normal human serum is used. The pathway and magnitude of activation are controlled by varying the Ab and serum concentrations and buffer conditions (e.g., Mg++EGTA blocks the CP but allows AP activation). MCP is a 65 kDa type 1 transmembrane protein consisting of four complement control protein repeats which bear three N-linked sugars, a domain with O-linked sugars, a hydrophobic transmembrane domain, a charged anchor and a cytoplasmic tail. CHO cell lines are prepared expressing different quantities of MCP. Alternative splicing of the O-linked sugar and cytoplasmic tail domains leads to expression of variable rations of four commonly expressed isoforms on most cell types.
To initiate complement activation, CHO cell lines with or without a regulator (such as MCP) are sensitized by polyclonal anti-CHO IgG and then exposed to a serum source (Fig. 2). The specificity and magnitude of complement activation can be varied by altering levels of sensitizing Ab, serum dilution and type of buffer. Flow cytometry and Western blotting are employed to monitor fragment deposition and degradation. Cytotoxicity is assessed using colorimetric indicators of viability and growth [14].
3. MCP in the CHO model system
3.1 Classical versus AP inhibition
3.1.1 CP
Sensitization of CHO cells with anti-CHO Abs engages the CP (Fig. 3A). Comparable levels of C3b deposition occur in normal compared to factor B-depleted sera, establishing that the CP is activated [14]. MCP has no effect on C3b deposition likely because its cleavage of C4b is much slower as compared to the generation C3b by the CP C3 convertase [14, 15].
Figure 3.
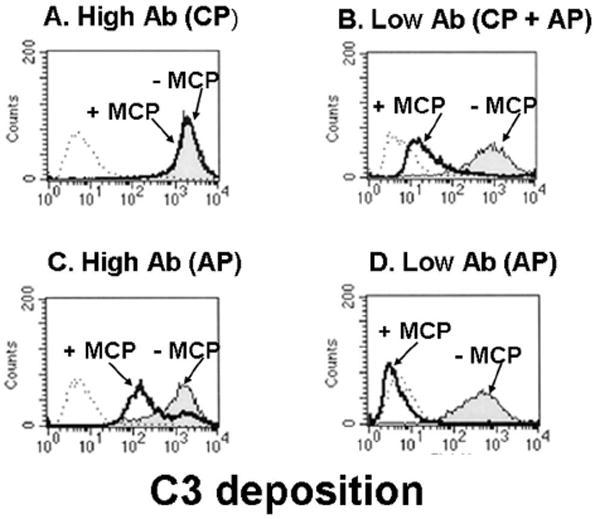
CHO model system: MCP inhibits C3b deposition by the AP but not by the CP. FACS analysis of C3b deposition using a polyclonal Ab to sensitize the CHO cells and 10% C7-deficient serum. Mg++-EGTA is used to prevent CP activation. Thick line, MCP+; thin line, MCP-; and dotted line, control. There was 8.5 times more Ab on the high vs low CHO cells. Adapted from [15].
MCP and other members of the RCA consist largely of repeating units called complement control protein (CCP) modules that consist of ~ 60 amino acids with four invariant cysteines. MCP also consists of a family of primarily four alternatively spliced isoforms that differ in the extent of O-glycosylation and in having one of two cytoplasmic tails. Although C3b deposition was not reduced by MCP, MCP isoforms with increased O-glycosylation (so-called BC isoform – see Fig. 4) inhibited cytotoxicity more efficiently than the less glycosylated C-isoform [14]. Further, deletion of two N-glycans, one in CCP-2 and one in CCP-4 of MCP, decreased the efficiency of MCP’s regulatory activity for the C5 convertase [16]. These results are consistent with CCP-2 and -4 being required for complement regulatory function and with these two N-glycans being evolutionarily conserved [16]. The data further indicate an effect of MCP on the cell bound classical pathway C5 convertase, as has been previously shown for fluid phase C5 convertases [17].
Figure 4.
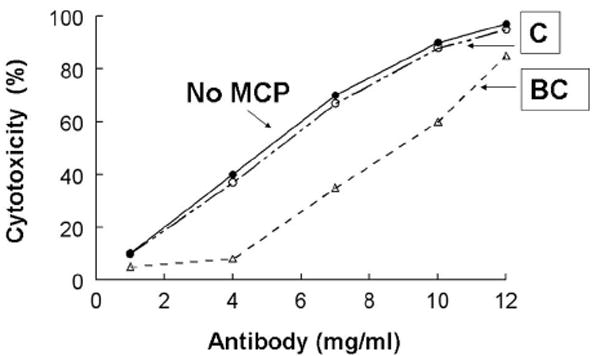
CHO model system: MCP isoforms with more O-glycosylation (BC) show less cytotoxicity and thus protect better against the CP. Transfected or wild-type CHO cells were assessed for their ability to inhibit the CP (i.e., increasing amounts of sensitizing Ab followed by incubation with 25% human serum). Cytotoxicity was measured. BC, MCP isoform with more O-linked sugars; C, MCP isoform with fewer O-linked sugars.
3.1.2. AP
The AP can be engaged independently of the CP by incubating sensitized cells in serum treated with Mg++EGTA or by utilizing serum deficient in C1q, C4 or C2. It can also be triggered by lowering the quantity of sensitizing Ab (Fig. 3B and D) [15]. Thus, in this scenario, the CP initially deposits C3b which then engages the feedback loop [14, 15].
In contrast to the CP, MCP efficiently decreases C3b deposition mediated by the AP (Fig 3B, C, and D). Using CHO clones with a four-fold difference in MCP expression levels [18], the 25,000 MCP/cell and 100,000 MCP/cell expressing clones inhibited AP-mediated C3 deposition by 82 and 95%, respectively (Fig. 5) [18]. In similar studies, we and others established that more protection against AP activation occurs as MCP copy number/cell increases [15, 19-24]. Thus, MCP regulates in a dose-dependent fashion both de novo and feedback loop engagement of the AP.
Figure 5.
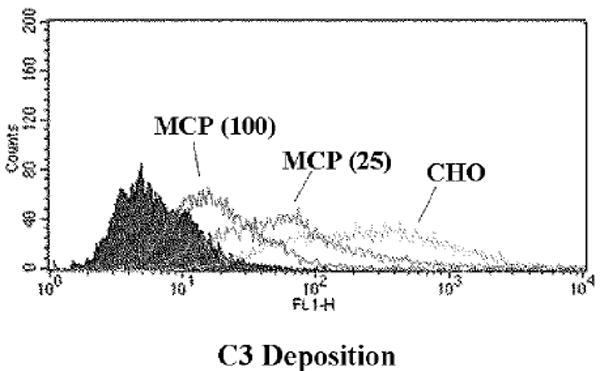
CHO model system: Dose-dependent inhibition of the AP by MCP. CHO cells expressing MCP (low: 25,000 copies per cell; and high: 100,000 copies/cell) were sensitized with (see Fig. 3) Ab and then treated with 10% C7-deficient serum. C3 fragment deposition was monitored.
3.2 Membrane versus plasma regulators
The CHO model also allows for an analysis of fluid phase regulators [14, 15]. As expected, following sensitization with Ab and incubation with C7-deficient human sera, CP-mediated C4b and C3b deposition was rapid and minimally influenced by the expression of MCP or plasma regulators C4BP and FH [15, 18]. Over the next 30 to 45 min, the majority of the deposited C4b was progressively cleaved to C4d and C4c via cofactor activity [15, 16]. The cleavage of C4b by the BC isoforms was faster than by the C isoforms [14]. Because there was no detectable cleavage of C4b on cells lacking MCP, this membrane protein and not the plasma regulator C4BP is the participating cofactor protein (Fig. 4). An increased MCP copy number/cell led to more rapid C4b cleavage, but ~ 25% of the deposited C4b remained uncleaved and was apparently not susceptible to CA.
In contrast to C4b, cleavage of the deposited C3b to C3bi and C3f was much more rapid but without a detectable effect on the quantity of C3b deposited [15]. Moreover, the C3bi fragment was generated, with or without MCP. This suggested that FH was the cofactor protein mediating C3b cleavage. Using a function-blocking mAb to FH, C3b cleavage was blocked, consistent with preceding results (Fig. 6). However, if FH was blocked, MCP could serve as the cofactor protein [15]. Thus, in this model system, MCP is the cofactor protein for deposited C4b while FH is the cofactor protein for deposited C3b. Similar overall results relative to MCP, C4BP and FH in mediating CA for cell-bound C4b and C3b have been obtained with human T cells and human endothelial cells (J.P. Atkinson, unpublished data).
Figure 6.
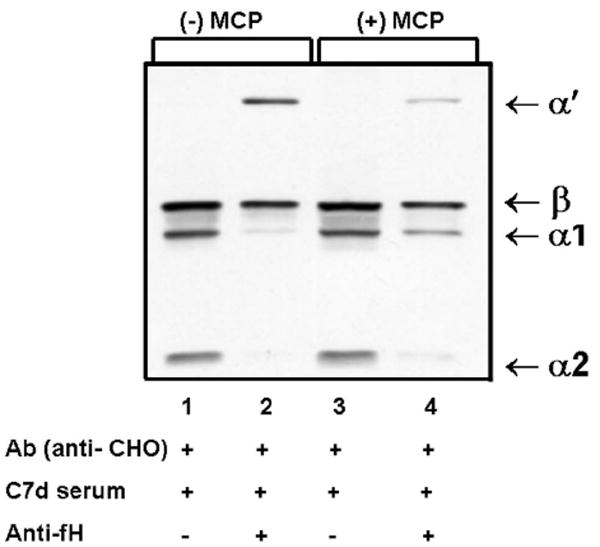
CHO model system: blocking FH inhibits cleavage of deposited C3b. Western blot profiles of C3b fragments deposited on cells incubated with high levels of sensitizing Ab and exposed to serum (10%, C7-deficient) for 5 min, in the presence or absence of anti-FH mAb (1 mg/ml). Note the presence of α’ fragment and only trace amounts of α1 and α2 fragments in the presence of anti-fH Abs. These results indicate that FH is the cofactor protein for C3b cleavage. Although minimally influencing the quantity of C3b deposition, the cleavage of C3b to C3bi by FH would provide a ligand for CR3 and CR4 to assist protection against excessive and unnecessary further complement activation. Adapted from [15].
4. Cooperation of decay accelerating factor (DAF; CD55) and MCP
DAF and MCP are widely expressed sister proteins designed to protect cells from complement attack. They consist of four CCPs as well as N- and O-linked glycans and regulate C3 and C5 convertases (see Fig. 1). While MCP is a type one transmembrane protein possessing cofactor activity, DAF is attached via a glycosylphosphatidyl (GPI) anchor and possesses DAA. This division of labor contrasts with that of plasma regulatory components, which inhibit either C3b or C4b and possess both DAA and CA for the respective protein and the convertases of which they are a part.
During AP activation on cell lines with equivalent copy numbers of MCP or DAF, MCP and DAF equivalently inhibited C3b deposition [18]. Additionally, during CP activation, even though the majority of C3b (~90%) was deposited in < 2 min, DAF but not MCP was able to partially inhibit (~70%) C3b deposition [18].
To determine if DAF and MCP cooperate on the cell surface to inhibit autologous complement attack, MCP was engineered carrying DAF’s GPI anchor and examined in the CHO model as well as on rabbit erythrocytes [13]. Quantitative studies using complement-coated rabbit erythrocyte cellular intermediates demonstrated that DAF and MCP are synergistic in preventing C3b deposition. Also, the ability of MCP to catalyze the FI-mediated cleavage of cell-bound C3b is inhibited in the presence of factors B and D but restored when DAF is incorporated into the cell. In other words, MCP is a cofactor protein for C3b but not for C3bBb [13]. These results have more recently been confirmed in a surface plasmon resonance based system in which the AP convertase is formed on a chip (D. Hourcade and J.P. Atkinson, unpublished results).
5. Heterozygous mutations in aHUS
Mutations in MCP predispose to the development of atypical hemolytic uremic syndrome (aHUS) [8-10, 18, 19, 25]. This microvascular thrombotic disorder is a pathologic triad consisting of microangiopathic hemolytic anemia, thrombocytopenia and acute renal failure. Atypical HUS is not associated with a preceding diarrheal illness. It is also commonly familial and has poor long-term prognosis [26]. Approximately 50% of aHUS patients have a mutation in FH (CFH), MCP (CD46), FI (CFI), factor B or C3 while ~5% of patients have anti-FH antibodies [10]. A key observation is that a reduction of 50% of the functional regulatory capacity (haploinsufficiency) predisposes these individuals to excessive AP activation at times of renal endothelial stress and injury.
Most aHUS patients are heterozygous for an MCP mutation and therefore possess one wild-type gene and 50% of the normal protein product and one mutated gene and variable quantities of an altered product, depending upon the nature of the mutation [10]. The other mutations (~25%) do not influence expression but the protein has decreased function. Reduced MCP function leads to a decreased ability to inhibit the AP [18, 25]. The initial family reported by Goodship et al had a S206P missense mutation which was expressed normally but had ~10% of the C3b binding and cofactor activity of wild type [18, 26, 27].
Renal transplantation is a treatment strategy for aHUS patients with MCP mutations since the transplanted kidney will possess normal levels of WT MCP on glomerular endothelial cells [6, 10, 26, 27]. Over 80% of renal transplant patients with MCP-aHUS have not had a recurrence [6, 10, 25]. In contrast, recurrence of aHUS in the transplanted kidney is common (50-90%) in patients with a deficiency of FH or FI [10].
6. The A304V mutation in MCP and new disease associations
The A304V mutation has been reported in three families with aHUS and also been associated in several other conditions that involve endothelial cell injury and a thrombotic microangiopathy [9, 28, 29]. The A304V recombinant mutant protein is expressed normally and functions comparably to wild type in in vitro assays; however, it is defective in situ in regulating complement activation (Fig. 7) [29].
Figure 7.
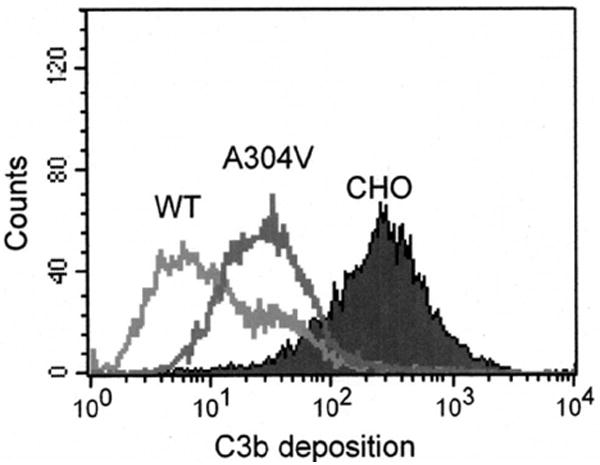
CHO model system: MCP mutant A304V is defective in its ability to control the AP. The A304V mutation is in the transmembrane domain of MCP. It has been identified in multiple aHUS families and in the HELLP syndrome [29]. A functional defect was observed in situ as shown here whereas in vitro assays failed to demonstrate a difference. Adapted from [29].
The first report of a patient with a complement regulatory protein defect in the HELLP (hemolytic anemia, elevated liver enzymes, and low platelets) syndrome had the A304V MCP mutation [29]. This patient developed HELLP during the second pregnancy and then chronic renal failure ensued one year later. She subsequently was included in a consecutive series of patients with the HELLP syndrome in which approximately one-third of the patients were shown to have a mutation in a complement regulatory protein [30].
Another example of the A304V mutation and its deleterious effect on normal cell homeostasis relates to the diarrheal-associated or epidemic HUS (D+HUS). It commonly follows an infective enteritis with a vero cytotoxin secreting bacteria, most commonly Escherichia coli O157:H7. For D+HUS, the prognosis is good with < 5% mortality during the acute phase. We reported though a child with the unlikely circumstance of D+HUS related to Shiga toxin and the A304V MCP mutation, resulting in mortality secondary to a multi-organ failure syndrome [29]. Mutations in the AP genes should be considered in patients with D+HUS if the disease process is unusually severe [29] or if renal function does not recover [31].
7. Limitations
In the CHO system, we have studied primarily Ab-mediated complement activation (Table III). In addition, this model employs a non-human cell line, provides static measurements and may not reflect local environmental conditions and pathologic influences.
Table 3.
Potential limitations of the CHO system
|
8. Conclusions
There are a number of lessons that we have learned in modeling how regulation proceeds on the cell surface using the CHO model system (Table IV). Our plans are to further mimic the clinical situation by employing human cells and mouse models of human disease.
Table 4.
Specifics of complement regulation by MCP
|
Acknowledgments
This work was supported by: National Institutes of Health grants T32AR07279 (CF) and RO1AI037618 (MKL, JPA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344(14):1058–66. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 2.Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344(15):1140–44. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 3.Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6(2):132–42. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liszewski MK, Farries TC, Lublin DM, Rooney IA, Atkinson JP. Control of the complement system. Advances in Immunology. 1996;61:201–83. doi: 10.1016/s0065-2776(08)60868-8. [DOI] [PubMed] [Google Scholar]
- 5.Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. 1996;271:348–50. doi: 10.1126/science.271.5247.348. [DOI] [PubMed] [Google Scholar]
- 6.Richards A, Kavanagh D, Atkinson JP. Inherited complement regulatory protein deficiency predisposes to human disease in acute injury and chronic inflammatory states. Advances in Immunology. 2008:141–77. doi: 10.1016/S0065-2776(07)96004-6. [DOI] [PubMed] [Google Scholar]
- 7.Smith RJ, Alexander J, Barlow PN, Botto M, Cassavant TL, Cook HT, et al. New approaches to the treatment of dense deposit disease. J Am Soc Nephrol. 2007;18(9):2447–56. doi: 10.1681/ASN.2007030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kavanagh D, Richards A, Noris M, Hauhart R, Liszewski MK, Karpman D, et al. Characterization of mutations in complement factor I (CFI) associated with hemolytic uremic syndrome. Mol Immunol. 2007;45:95–105. doi: 10.1016/j.molimm.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, et al. Genetics of HUS: the impact of MCP, CFH and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108:1267–79. doi: 10.1182/blood-2005-10-007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kavanagh D, Richards A, Atkinson JP. Complement regulatory genes and hemolytic uremic syndromes. Annual Review of Medicine. 2008:293–309. doi: 10.1146/annurev.med.59.060106.185110. [DOI] [PubMed] [Google Scholar]
- 11.Hageman GS, Hancox LS, Taiber AJ, Gehrs KM, Anderson DH, Johnson LV, et al. Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: characterization, ethnic distribution and evolutionary implications. Ann Med. 2006;38(8):592–604. [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards AO, Malek G. Molecular genetics of AMD and current animal models. Angiogenesis. 2007;10(2):119–32. doi: 10.1007/s10456-007-9064-2. [DOI] [PubMed] [Google Scholar]
- 13.Brodbeck WG, Mold C, Atkinson JP, Medof ME. Cooperation between decay-accelerating factor and membrane cofactor protein in protecting cells from autologous complement attack. J Immunol. 2000;165(7):3999–4006. doi: 10.4049/jimmunol.165.7.3999. [DOI] [PubMed] [Google Scholar]
- 14.Liszewski MK, Atkinson JP. Membrane cofactor protein (MCP; CD46). Isoforms differ in protection against the classical pathway of complement. J Immunol. 1996;156(11):4415–21. [PubMed] [Google Scholar]
- 15.Barilla-LaBarca ML, Liszewski MK, Lambris JD, Hourcade D, Atkinson JP. Role of membrane cofactor protein (CD46) in regulation of C4b and C3b deposited on cells. J Immunol. 2002;168(12):6298–304. doi: 10.4049/jimmunol.168.12.6298. [DOI] [PubMed] [Google Scholar]
- 16.Liszewski MK, Leung MK, Atkinson JP. Membrane cofactor protein (CD46): importance of N- and O-glycosylation for complement regulatory function. J Immunol. 1998;161:3711–18. [PubMed] [Google Scholar]
- 17.Seya T, Okada M, Matsumoto M, Hong K, Kinoshita T, Atkinson JP. Preferential inactivation of the C5 convertase of the alternative complement pathway by factor I and membrane cofactor protein (MCP) Mol Immunol. 1991;28:1137–47. doi: 10.1016/0161-5890(91)90029-j. [DOI] [PubMed] [Google Scholar]
- 18.Liszewski MK, Leung MK, Schraml B, Goodship TH, Atkinson JP. Modeling how CD46 deficiency predisposes to atypical hemolytic uremic syndrome. Mol Immunol. 2007;44(7):1559–68. doi: 10.1016/j.molimm.2006.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oglesby TJ, Allen CJ, Liszewski MK, White DJG, Atkinson JP. Membrane cofactor protein (MCP;CD46) protects cells from complement-mediated attack by an intrinsic mechanism. J Exp Med. 1992;175:1547–51. doi: 10.1084/jem.175.6.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loveland BE, Johnstone RW, Russell SM, Thorley BR, McKenzie IFC. Different membrane cofactor protein (CD46) isoforms protect transfected cells against antibody and complement mediated lysis. Transpl Immunol. 1993;1:101–08. doi: 10.1016/0966-3274(93)90002-p. [DOI] [PubMed] [Google Scholar]
- 21.Lublin DM, Coyne KE. Phospholipid-anchored and transmembrane versions of either decay-accelerating factor or membrane cofactor protein show equal efficiency in protection from complement-mediated cell damage. J Exp Med. 1991;174:35–44. doi: 10.1084/jem.174.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kojima A, Iwata K, Seya T, Matsumoto M, Ariga H, Atkinson JP, et al. Membrane cofactor protein (CD46) protects cells predominantly from alternative complement pathway-mediated C3-fragment deposition and cytolysis. J Immunol. 1993;151:1519–27. [PubMed] [Google Scholar]
- 23.Devaux P, Christiansen D, Fontaine M, Gerlier D. Control of C3b and C5b deposition by CD46 (membrane cofactor protein) after alternative but not classical complement activation. Eur J Immunol. 1999;29:815–22. doi: 10.1002/(SICI)1521-4141(199903)29:03<815::AID-IMMU815>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 24.Seya T, Atkinson JP. Functional properties of membrane cofactor protein of complement. Biochem J. 1989;264:581–88. doi: 10.1042/bj2640581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richards A, Liszewski MK, Kavanagh D, Fang CJ, Moulton EA, Fremeaux-Bacchi V, et al. Implications of the initial mutations in membrane cofactor protein (MCP; CD46) leading to atypical hemolytic uremic syndrome. Mol Immunol. 2007;44:111–22. doi: 10.1016/j.molimm.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 26.Jokiranta TS, Zipfel PF, Fremeaux-Bacchi V, Taylor CM, Goodship TJ, Noris M. Where next with atypical hemolytic uremic syndrome? Mol Immunol. 2007;44(16):3889–900. doi: 10.1016/j.molimm.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 27.Richards A, Kemp EJ, Liszewski MK, Goodship JA, Lampe AK, Decorte R, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci U S A. 2003;100:12966–71. doi: 10.1073/pnas.2135497100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Servais A, Fremeaux-Bacchi V, Lequintrec M, Salomon R, Blouin J, Knebelmann B, et al. Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet. 2007;44(3):193–9. doi: 10.1136/jmg.2006.045328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang CJ, Fremeaux-Bacchi V, Liszewski MK, Pianetti G, Noris M, Goodship THJ, et al. Membrane cofactor protein mutations in atypical hemolytic uremic syndrome (aHUS), fatal Stx-HUS, C3 glomerulonephritis and the HELLP syndrome. Blood. 2008;111:624–32. doi: 10.1182/blood-2007-04-084533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fakhouri F, Jablonski M, Lepercq J, Blouin J, Benachi A, Hourmant M, Factor H, et al. membrane cofactor protein and Factor I mutations in patients with HELLP syndrome. Blood. 2008 doi: 10.1182/blood-2008-03-144691. [DOI] [PubMed] [Google Scholar]
- 31.Edey MM, Mead PA, Saunders RE, Strain L, Perkins SJ, Goodship TH, et al. Association of a factor H mutation with hemolytic uremic syndrome following a diarrheal illness. Am J Kidney Dis. 2008;51(3):487–90. doi: 10.1053/j.ajkd.2007.08.030. [DOI] [PubMed] [Google Scholar]