

Abstract
The endopeptidase neprilysin (NEP) is a major amyloid-β (Aβ) degrading enzyme and has been implicated in the pathogenesis of Alzheimer's disease. Because NEP cleaves substrates other than Aβ, we investigated the potential role of NEP-mediated processing of neuropeptides in the mechanisms of neuroprotection in vivo. Overexpression of NEP at low levels in transgenic (tg) mice affected primarily the levels of neuropeptide Y (NPY) compared with other neuropeptides. Ex vivo and in vivo studies in tg mice and in mice that received lentiviral vector injections showed that NEP cleaved NPY into C-terminal fragments (CTFs), whereas silencing NEP reduced NPY processing. Immunoblot and mass spectrometry analysis showed that NPY 21–36 and 31–36 were the most abundant fragments generated by NEP activity in vivo. Infusion of these NPY CTFs into the brains of APP (amyloid precursor protein) tg mice ameliorated the neurodegenerative pathology in this model. Moreover, the amidated NPY CTFs protected human neuronal cultures from the neurotoxic effects of Aβ. This study supports the possibility that the NPY CTFs generated during NEP-mediated proteolysis might exert neuroprotective effects in vivo. This function of NEP represents a unique example of a proteolytic enzyme with dual action, namely, degradation of Aβ as well as processing of NPY.
Keywords: neuropeptide, neprilysin, Alzheimer's disease, cleavage, processing, amyloid
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder affecting the elderly and is the most common form of dementia (Ashford, 2004). A key mediator of this disease is believed to be amyloid-β (Aβ) peptides, produced by proteolytic processing of the amyloid precursor protein (APP) in the CNS (Selkoe, 1994a,b). Recent evidence supports the notion that aggregation of Aβ, resulting in the formation of oligomers rather than fibrils, might be ultimately responsible for the synaptic damage that leads to cognitive dysfunction in patients with AD (Walsh and Selkoe, 2004; Glabe, 2005; Glabe and Kayed, 2006).
Neprilysin (NEP) (also known as CD10, EC 3.4.24.11), a zinc metalloendopeptidase (Howell et al., 2005), has been identified as a critical Aβ-degrading enzyme in the brain (Iwata et al., 2000, 2001). Supporting a role for NEP in AD, previous studies have shown that NEP reduces Aβ levels in vivo (Leissring et al., 2003; Mohajeri et al., 2004; Huang et al., 2006; Farris et al., 2007; Iijima-Ando et al., 2008). These studies are also in agreement with previous experiments showing that viral vector-mediated transfer of NEP reduces the neurodegenerative and amyloid pathology in APP transgenic (tg) mice (Marr et al., 2003; Iwata et al., 2004; Hong et al., 2006; El-Amouri et al., 2008). In patients with AD, the levels of NEP in the brain are reduced (Akiyama et al., 2001; Reilly, 2001; Yasojima et al., 2001a,b; Caccamo et al., 2005), and a potential genetic linkage is currently being investigated (Sodeyama et al., 2001; Oda et al., 2002; Clarimón et al., 2003; Wood et al., 2007).
Although considerable effort has been focused on investigating the effects of NEP on Aβ pathology, less is known about alternative effects of NEP in the CNS. Previous studies have shown that NEP is capable of cleaving a wide range of neuropeptides, including substance P (SP), enkephalin (ENK), and neuropeptide Y (NPY) (Skidgel and Erdös, 2004). Among them, NPY is of interest because in AD pathology, levels of this neuropeptide are abnormal (Minthon et al., 1990; Ramos et al., 2006) and in APP tg mice the alterations in the NPY network in the hippocampus have been linked to epileptic activity (Palop et al., 2007). NPY is a 36-aa-long protein, is one of the most abundant peptide transmitter in the CNS, and has been shown to play a role in appetite regulation (Sokolowski, 2003), behavior (Albers and Ferris, 1984), seizure activity (Vezzani et al., 1999), and memory (Redrobe et al., 1999). NEP-mediated proteolysis of NPY has been traditionally considered a terminal event; however, it is possible that in the CNS some of these fragments might have neuroprotective effects relevant to AD. In this context, for the present study we show that C-terminal fragments (CTFs) of NPY derived from NEP processing might have neuroprotective effects in models of AD pathology. Together, our studies suggest that NEP might have a unique dual role by processing NPY into neuroactive fragments and reducing amyloid load in the CNS by degrading Aβ.
Materials and Methods
Mouse lines and generation of NEP, APP, and double tg mice.
For these experiments, tg mice expressing high levels of human NEP and APP were used. Transgenic mice expressing human NEP under the regulatory control of the platelet-derived growth factor-β (PDGFβ) promoter were generated as previously described (Masliah et al., 2000). These mice were screened by PCR analysis of genomic DNA extracted from tail biopsies and screened for RNA and for levels of protein expression by Western blot. Three lines of mice were generated, and one line displaying the most stable levels of NEP expression was selected for crosses with the APP tg mice as previously described (Rockenstein et al., 2002b). Transgenic lines were maintained by crossing heterozygous tg mice with non-tg C57BL/6 × DBA/2 F1 breeders. All mice were heterozygous with respect to the transgene. The APP tg mice express mutated (London V717I and Swedish K670M/N671L) human APP751 under the control of the murine Thy1 promoter (Thy1-hAPP, line 41) (Rockenstein et al., 2001). This tg model was selected because these mice produce high levels of Aβ1–42 and exhibit performance deficits in the water maze, synaptic damage, and plaque formation at an early age (beginning at 3 months) (Rockenstein et al., 2001, 2002a). Additional experiments were performed using homozygous NEP-knock-out (KO) mice (generously provided by Dr. Bao Lu, Harvard Medical School, Boston, MA) (Lu et al., 1995). Mice from all lines used were maintained until 6 months of age, followed by biochemical and neuropathological studies.
Infusion of NPY CTFs into the brains of APP tg mice.
Briefly, as previously described (Veinbergs et al., 2001), to evaluate the neuroprotective effects of NPY CTFs, groups of APP tg mice (6 months of age) received intraventricular infusions with a cannula implanted into the skull and connected to osmotic minipumps delivering solutions of vehicle alone, amidated NPY CTFs (21–36, 31–36), or nonamidated NPY CTFs (21–36, 31–36). A total of 30 APP tg mice (n = 6 per group) were used for these experiments. Mice were treated for 28 d; the compound was dissolved in 0.9% NaCl/dimethylsulfoxide (90/10) at a concentration of 120 μm. Then, 200 μl of this solution was filled into the osmotic minipump (Alzet; Charles River Laboratories) ensuring constant delivery (0.2 μl/h). The minipump was implanted subcutaneously on the back under light anesthesia. An additional group of control non-tg (n = 6) mice were implanted with minipumps filled with vehicle only. All experiments were approved by the animal subjects committee at the University of California, San Diego (UCSD), and were performed according to National Institutes of Health (NIH) recommendations for animal use.
Tissue processing.
In accordance with NIH guidelines for the humane treatment of animals, mice were killed by deep anesthesia with chloral hydrate. Brains were removed and divided sagitally. One hemibrain was postfixed in phosphate-buffered 4% paraformaldehyde, pH 7.4, at 4°C for 48 h and sectioned at 40 μm with a Vibratome 2000 (Leica), whereas the other hemibrain was snap frozen and stored at −70°C for RNA and protein analysis.
Analysis of NEP activity.
The proteolytic activity of NEP was measured as previously described (Hemming et al., 2007) using the substrate 3-dansyl-d-Ala-Gly-p-(nitro)-Phe-Gly (DAGNPG) (Sigma Pharmaceuticals). Cell lysates from the neocortex, hippocampus, caudate, and cerebellum were incubated with 50 μm DAGNPG and 1 μm captopril [to inhibit any ACE (angiotensin converting enzyme) cleavage of DAGNPG] in a volume of 200 μl at 37°C. Reactions were stopped by heating samples to 100°C for 5 min, followed by centrifugation. The supernatant was diluted into 50 mm Tris, pH 7.4, and fluorescence was determined using a Victor2 multilabel plate reader (excitation, 342 nm; emission, 562 nm).
Immunoblot analysis.
Western blot analysis was performed essentially as previously described (Rockenstein et al., 2001, 2005b). Briefly, 20 μg per lane of cytosolic and particulate fractions, assayed by the BCA method (Pierce Biotechnology), were loaded into 4–12% SDS-PAGE gels and blotted onto polyvinylidene fluoride (PVDF) membranes. Blots were incubated with antibodies against total FL-APP (mouse monoclonal; clone 22C11; 1:500; Millipore Bioscience Research Reagents), human APP (mouse monoclonal; 1:500; 8E5 clone; Elan Pharmaceuticals), Aβ (mouse monoclonal; clone 6E10; 1:1000; Signet Laboratories), NEP (mouse monoclonal; clone CD10; 1:1000; Abcam), brain-derived neurotrophic factor (BDNF) (mouse monoclonal; clone 35928.11; 1:1000; Calbiochem), nerve growth factor (NGF) (mouse monoclonal; clone 25623.1; 1:1000; Calbiochem), neurotrophin-3 (NT3) (mouse polyclonal; 1:300; Promega), NT4 (mouse monoclonal; clone 36507; 1:1000; R&D Systems), and NPY (mouse polyclonal; 1:1000; Peninsula Laboratories) followed by secondary antibodies tagged with HRP (1:5000; Santa Cruz Biotechnology) and visualized by enhanced chemiluminescence and analyzed with a Versadoc XL imaging apparatus (Bio-Rad). Analysis of actin levels was used as a loading control.
Determination of Aβ, SP, ENK, and NPY levels by ELISA, and mass spectrometry analysis.
Brain samples from the frontal cortex were homogenized in ice-cold buffer (5 m guanidine-HCl and PBS, pH 8.0) with 1× protease inhibitor mixture (Calbiochem). Homogenates were then mixed overnight at room temperature and subsequently diluted 10-fold in Dulbecco's PBS, pH 7.4, containing 5% bovine serum albumin and 0.03% Tween 20. Samples were then centrifuged at 16,000 × g for 20 min at 4°C. The resulting supernatants were subjected to quantification with commercially available ELISA kits for NPY (Phoenix Pharmaceuticals), SP (Assay Designs), and Met-ENK (Peninsula Laboratories).
To confirm the presence of NPY CTFs in mouse brain homogenates by an independent method, briefly as previously described (Medeiros Mdos and Turner, 1996), mass spectrometry was performed by HT Laboratories (San Diego, CA). Samples of non-tg and tg mouse brains were normalized to 18 mg/ml and homogenized in 100 mm Tris-HCl, pH 7.4, 1.0% Tween 20, 1 m Thiorphan, and protease inhibitors (Calbiochem). Peaks matching the NPY 21–36 and 31–36 standards were found in the tissue samples and are depicted in the mass chromatogram given in supplemental Figure 3G (available at www.jneurosci.org as supplemental material).
Immunocytochemical analysis of neurodegeneration.
To evaluate the integrity of the neuronal structure, briefly as previously described (Rockenstein et al., 2005a,b), blind-coded, 40-μm-thick vibratome sections from mouse brains fixed in 4% paraformaldehyde were immunolabeled with the mouse monoclonal antibodies against synaptophysin (synaptic marker; 1:20; Millipore Bioscience Research Reagents), microtubule-associated protein-2 (MAP2) (dendritic marker; 1:40; Millipore Bioscience Research Reagents), NeuN (neuronal marker; 1:1000; Millipore Bioscience Research Reagents), or glial fibrillary acidic protein (GFAP) (astroglial marker; 1:500; Millipore Bioscience Research Reagents) (Mucke et al., 1995). Additional sections were immunostained with the polyclonal antibody against NPY (1:200; Peninsula Laboratories). After overnight incubation with the primary antibodies, sections were incubated with fluorescein isothiocyanate (FITC)-conjugated horse anti-mouse IgG secondary antibody (1:75; Vector Laboratories), transferred to SuperFrost slides (Thermo Fisher Scientific) and mounted under glass coverslips with anti-fading media (Vector Laboratories). All sections were processed under the same standardized conditions. The immunolabeled blind-coded sections were serially imaged with the laser-scanning confocal microscope (LSCM) (MRC1024; Bio-Rad) and analyzed with the NIH Image 1.43 program, as previously described (Toggas et al., 1994; Mucke et al., 1995). For each mouse, a total of three sections were analyzed, and for each section, four fields in the frontal cortex and hippocampus were examined. For synaptophysin and MAP2, results were expressed as percentage area of the neuropil occupied by immunoreactive terminals and dendrites; for GFAP immmunostaining, levels were expressed as pixel intensity, and for NeuN and NPY, the mean neuronal density was estimated using the disector method as previously described (Chana et al., 2003). Briefly, for stereology, immunolabeled sections were counterstained with 1% cresyl violet and analyzed with the optical disector. From each case, four 100-μm-wide fields from at least three sections (180 μm interval) per animal were analyzed and results were averaged and expressed as total number per cubic millimeter.
Analysis of FL-NPY and NPY CTFs by immunoblot and immunocytochemistry.
To determine the expression levels of NPY, brain homogenates from control and tg mice were homogenized and fractioned as previously described into soluble and insoluble fractions (Kawahara et al., 2008). Briefly, tissues were sonicated in HEPES buffer with 1% Triton X-100 plus 1× thiorphan, phosphatase, and protease inhibitors. Samples were centrifuged for 1 h at 100,000 rpm in a Beckman TL100 rotor. Samples were run on a 4–12% Bis-Tris gel (Invitrogen) and transferred onto 0.2 μm PVDF membranes with a 20% MeOH transfer buffer. The membranes were then blocked in 3% BSA and incubated with the rabbit polyclonal antibodies against FL-NPY (Peninsula Laboratories) or NPY CTFs. The antibody against NPY CTFs was prepared at Invitrogen by immunizing rabbits with the peptide corresponding to the amidated 31–36 aa of NPY with an extra Cys added to the N terminus necessary for conjugation with BSA. Additional analysis was performed with a commercial rabbit polyclonal antibody specific for NPY CTFs (20–36) (Santa Cruz).
Because of the inherent difficulty in Western blot detection of peptides as small as the NPY CTFs, some studies were performed using an immunoblot protocol refined for the detection of very small proteins. For this purpose, ∼0.1 g of tissue was obtained and sonicated in 100 mm Tris-HCl, pH 7.4, 1% Tween 20, containing protease, phosphatase, and thiorphan inhibitors (Calbiochem). The homogenate was then fractionated by centrifugation at 4°C for 1 h at 100,000 rpm. For Western blot analysis, both the soluble and insoluble fractions were separated on 10–20% Tricine gels (Invitrogen) first at 125 V for 10 min, and then at 180 V until the gel ran to completion. Before transfer, a 0.1 μm nitrocellulose membrane was incubated in 0.5% gelatin for 30 min at 37°C, and then allowed to dry for 1 h at 56°C. The transfer was performed for 15 min at 10 V, and then the membrane was removed and placed into an air-tight container with towels soaked in 37% formaldehyde for 2.5 h at 37°C. After this fixation process, the membrane was reactivated quickly with ddH2O and PBS. Immunoblots were then probed with antibodies against NPY as described above.
Double immunocytochemical and laser-scanning confocal analysis.
To evaluate the colocalization between APP and NEP and NPY and NEP, double immunocytochemical analysis was performed as previously described (Masliah et al., 2000). For this purpose, vibratome sections were immunolabeled with a monoclonal antibody against NEP (1:10,000; Abcam) detected with the Tyramide Signal Amplification-Direct (Red) system (1:100; PerkinElmer Life and Analytical Sciences), and the mouse monoclonal antibody against human APP (1:500; 8E5 clone; Elan Pharmaceuticals) or the rabbit polyclonal antibodies against FL-NPY (Peninsula Laboratories) or NPY CTFs, detected with FITC-conjugated secondary antibodies (1:75; Vector Laboratories) (Masliah et al., 2000). All sections were processed simultaneously under the same conditions, and experiments were performed twice to assess reproducibility. Sections were imaged with a Zeiss 63× (numerical aperture, 1.4) objective on an Axiovert 35 microscope (Zeiss) with an attached MRC1024 LSCM system (Bio-Rad) (Masliah et al., 2000). To confirm the specificity of primary antibodies, control experiments were performed in which sections were incubated overnight in the absence of primary antibody (deleted) or preimmune serum and primary antibody alone.
Ex vivo studies of NPY processing by NEP.
The relative rate of NPY processing by NEP was determined, and the resultant products were visualized by both cell-free and ex vivo processing with homogenates from the tg mouse brains using N-terminal FITC-tagged human NPY (1–36; AnaSpec). For the cell-free assays, 200 ng of human recombinant NEP (R&D Systems) was incubated with 100 mm Tris, pH 7.4, 10 μm ZnCl2, and 50 μm FITC-labeled NPY in a final volume of 50 μl. For the ex vivo experiments with tissue homogenates, 1.2 mg of fresh murine brain tissue (from non-tg, APP tg, NEP tg, and APP/NEP tg mice) was sonicated in lysis buffer (100 mm Tris, pH 7.4, and 1% Tween 20), and centrifuged at 5000 rpm for 5 min at 4°C. The resulting supernatant was then analyzed by the BCA method (Pierce) to determine protein concentration, after which 40 μg of protein was incubated with 100 mm Tris, pH 7.4, and 50 μm NPY-FITC in a final volume of 50 μl. Thiorphan (1 mm; Calbiochem) was used as a NEP-specific protease inhibitor. A time course was then performed for both cell-free and tissue-based assays. Aliquots were taken at varying time points, stopped with an equal volume of 8 m urea, run on 12% SDS-PAGE gels with MES buffer (Invitrogen), and analyzed with a Versadoc XL imaging apparatus (Bio-Rad).
Lentivirus NEP preparation and intracerebral injections in mice.
To verify the effects of NEP in cleaving NPY using an alternative system, the ex vivo assay for NPY-FITC was performed with brain homogenates from mice that received intrahippocampal injections with a lentiviral vector (LV) expressing either NEP, mutant (E585V) inactive NEP (NEP X), or green fluorescent protein (GFP). The effects of silencing NEP were studied by injecting a LV expressing either short hairpin RNA (shRNA) (a 19-mer construct with a sequence of GCACGTGGTTGAAGACTTG; designed and cloned by Dr. O. Singer, The Salk Institute, La Jolla, CA) for NEP or a control scrambled shRNA. Briefly, as previously described (Marr et al., 2003; Singer et al., 2005), 293T cells were transfected with vector and packaging plasmids, and the supernatants were collected and vectors concentrated by centrifugation. The LV titers were estimated by measuring the amount of HIV p24 gag antigen with an ELISA kit (100,000 TU/ng p24; PerkinElmer Life and Analytical Sciences) or by flow cytometry using an anti-NEP specific antibody (56C6; Research Diagnostics).
In total, 24 non-tg (n = 4 mice per group; 6 months of age) (C57) mice were injected with 2 μl of the lentiviral preparations (1.5 × 109 TU) into the frontal cortex and hippocampus (using a 5 μl Hamilton syringe; 0.25 μl/min). Mice received bilateral injections with either LV control (empty vector), LV-NEP, LV-NEP X, LV-GFP, LV-shRNA NEP, or LV-shRNA scrambled. Four weeks after injection, mice were killed, and the brains were removed and prepared for immunoblot and ex vivo NPY-FITC analysis.
Effects of NPY fragments in primary human neurons treated with Aβ.
To investigate the potential neuroprotective effects of NEP-derived NPY fragments, cultures of primary human neurons were pretreated with NPY fragments followed by Aβ exposure. For this purpose, primary fetal human neurons (generously provided by G. Chana, UCSD, La Jolla, CA) were plated onto a 48-well tissue culture plate at 5 × 105 cells/well in DMEM/F12 media (10% FBS, 1% sodium pyruvate, 0.1% nonessential amino acids, 0.75 g sodium bicarbonate in 500 ml). Cells were then incubated for 24 h in minimal media containing either FL-NPY (AnaSpec) or amidated or nonamidated NPY CTFs (21–36 and 31–36; Invitrogen) at dilutions ranging from 1 nm to 10 μm in 10% DMSO. After NPY incubation, 10 μm freshly solubilized Aβ1–42 (American Peptide) was added to each well, followed by fixation with 4% paraformaldehyde and immunocytochemical analysis with antibodies against synaptophysin and MAP2 as described above. In another set of experiments, the primary cultured neurons were pretreated with (S)-N2-[[1-[2-[4-[(R,S)-5,11-dihydro-6(6h)-oxodibenz[b,e]azepin-11-yl]-1-piperazinyl]-2-oxoethyl]cyclopentyl]acetyl]-N-[2-[1,2-dihydro-3,5(4H)-dioxo-1,2-diphenyl-3H-1,2,4-triazol-4-yl]ethyl]-argininamid (BIIE0246) (Y2 receptor inhibitor; Tocris Bioscience) or R-N2-(diphenylacetyl)-N-(4-hydroxyphenyl)-methyl argininamide (BIBP3226) (Y1 receptor inhibitor; Sigma-Aldrich) or vehicle control at 1 μm for 24 h followed by incubation with the amidated NPY 21–36 (10 nm) and challenge with 10 μm freshly solubilized Aβ1–42 for 24 h. Coverslips were prepared in triplicate and analyzed with the MRC1024 LSCM system (Bio-Rad) to determine the levels of synaptophysin immunoreactivity.
Statistical analyses.
Analyses were performed with the StatView 5.0 program (SAS Institute). Differences among means were assessed by one-way ANOVA with post hoc Dunnett's or Tukey–Kramer's tests. All values in the figures are expressed as means ± SEM. Comparisons between two groups were done with the unpaired two-tailed Student's t test. Correlation studies were performed by simple linear regression analysis, and the null hypothesis was rejected at the 0.05 level.
Results
Levels of neurotrophic factors and neuropeptides in NEP tg mice
Because NEP cleaves substrates other than Aβ, it is important to investigate the potential degradative effects of this metalloprotease on neuropeptides and trophic factors. For these studies, tg mice expressing NEP under the PDGFβ promoter were generated. These tg mice express NEP ∼1- to 1.5-fold above endogenous levels (Fig. 1A,B), whereas NEP KO mice show very low levels of NEP expression (supplemental Fig. 1A–C, available at www.jneurosci.org as supplemental material). The activity assay confirmed similar low levels of NEP activity in NEP KO mice (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material), whereas in NEP tg mice there was a 50–60% increase in NEP activity in the neocortex and hippocampus, and an approximate fourfold increase in the caudate, compared with the cerebellum (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material). The increased levels of NEP activity were similar in APP/NEP double tg and NEP single tg mice (Fig. 1C). The NEP tg mice were crossed with APP tg mice because NEP has been shown to play an important role in the pathogenesis of AD and has been considered to be a potential therapeutic target. Levels of total (including murine) APP and human APP were comparable between single APP tg mice and APP/NEP double tg mice. Levels of murine APP were comparable between non-tg and NEP tg mice (Fig. 1A,B). Double labeling experiments confirmed that, compared with controls (Fig. 1D–F), the transgene-driven human APP and NEP colocalized in neuronal populations in the neocortex (Fig. 1G–I) and hippocampus. To investigate the effects of NEP overexpression on neuropeptides other than Aβ, levels of BDNF, NT3, NT4, SP, Met-ENK, and NPY were analyzed by immunoblot. Remarkably, only levels of total FL-NPY were significantly affected in the APP/NEP and NEP tg groups compared with APP and non-tg controls (Fig. 2A,B). Levels of the other neurotransmitter peptides and neurotrophic factors were not different among the four groups (Fig. 3A–C). Levels of Met-ENK were slightly elevated in the APP tg mice compared with non-tg animals (Fig. 3D). Because previous studies have shown that NEP cleaves NPY at the C terminus (Medeiros and Turner, 1994), the reduction in the levels of FL-NPY in the total homogenates of the NEP tg might be related to the proteolytic processing of this neuropeptide and the concomitant generation of potentially bioactive fragments rather than being attributable to decreased NPY expression. In support of this possibility, quantitative reverse transcription-PCR showed no differences in the levels of NPY mRNA among the four groups of mice (Fig. 2C).
Figure 1.

Characterization of NEP expression and activity in tg mouse models. A, Representative immunoblot analysis illustrating increased expression levels of FL-tAPP, FL-hAPP, and NEP in the APP/NEP tg mice compared with non-tg mice. B, Image analysis of the levels of immunoreactivity of FL-tAPP, APP-CTFs, and NEP in the frontal cortex of non-tg, APP tg, APP/NEP, and NEP tg mice (n = 6 mice per group; 6 months of age). C, Levels of NEP activity in the frontal cortex of non-tg, APP tg, APP/NEP, and NEP tg mice (n = 6 mice per group; 6 months of age). Error bars indicate SEM. D–I, Double immunocytochemical analysis and laser-scanning confocal microscopy analysis of the patterns of colocalization between APP and NEP. NEP immunoreactivity is in the FITC (green) channel, whereas hAPP is in the red channel. D–F, Non-tg control. G–I, Double tg expressing APP and NEP. Scale bar, 10 μm.
Figure 2.

Levels of NPY expression in NEP and APP/NEP tg mice. A, Representative immunoblot analysis of NPY immunoreactivity in soluble fractions from the frontal cortex of non-tg, APP tg, APP/NEP tg, and NEP tg mice. B, Analysis of the levels of FL-NPY by immunoblot with total homogenates from the frontal cortex. C, Levels of NPY mRNA expression by real-time quantitative PCR analysis. D, Immunoblot analysis of levels of NPY CTFs in the frontal cortex of non-tg, APP tg, APP/NEP tg, and NEP tg mice using an antibody raised with a peptide corresponding to the NPY 20–36 region (Santa Cruz). E, Analysis of the levels of NPY CTFs by immunoblot in the frontal cortex. Error bars indicate SEM. F–I, Patterns of NPY CTFs immunoreactivity comparing non-tg and tg mice. H, I, Increased NPY CTFs immunoreactivity in APP/NEP and NEP tg mice compared with controls (*p < 0.05 compared with non-tg controls by one-way ANOVA with post hoc Dunnett's; n = 6 mice per group; 6 months of age). Scale bar, 50 μm.
Figure 3.

Expression levels of neurotrophic factors and neurotransmitter peptides in APP/NEP tg mice. A, Representative immunoblot analysis of BDNF, NT3, and NT4 in homogenates from the frontal cortex of non-tg, APP, APP/NEP, and NEP tg mice. B, Analysis of the immunoblot levels of BDNF, NT3, and NT4 immunoreactivity. C, Levels of SP in the frontal cortex determined by ELISA. D, Levels of Met-Enk in the frontal cortex determined by ELISA (*p < 0.05 compared with non-tg controls by one-way ANOVA with post hoc Dunnett's; n = 6 mice per group; 6 months of age). Error bars indicate SEM.
Identification of NPY CTFs in the brains of NEP tg mice
To search for NPY fragments consistent with NEP processing, immunoblot studies with antibodies against NPY CTFs, mass spectrometry, and ex vivo proteolysis analyses were performed in the brains of NEP tg and APP/NEP tg mice. To begin investigating whether NEP promotes the formation of NPY fragments, Western blot analysis of synthetic NPY peptides was performed with an antibody generated against the C terminus of NPY (obtained from Santa Cruz Biotechnology). This antibody recognized the 0.8 and 2 kDa bands corresponding to the 31–36 and 21–36 fragments of NPY (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). Immunoblot analysis with this NPY CTFs antibody using mouse brain homogenates and gelatin-coated, formaldehyde-treated membranes demonstrated the presence in vivo of the 0.8 and 2 kDa bands corresponding to the NPY CTFs in the insoluble fraction (Fig. 2D). Compared with the non-tg and APP tg mice, in the NEP and APP/NEP tg mice levels of immunoreactivity for the lower molecular weight (MW) bands corresponding to NPY CTFs were increased in the insoluble fraction (Fig. 2D,E). To confirm the results obtained with the commercial NPY CTFs antibody, we generated a polyclonal antibody with the peptide corresponding to amino acids 31–36 of NPY. Immunoblot analysis with synthetic NPY peptides showed that this antibody recognized a band at a MW of 2 kDa consistent with the 21–36 CTF, and a band at ∼0.8 kDa consistent with the 31–36 NPY CTF (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). Similar to the commercial NPY CTFs antibody, by immunoblot analysis our antibody (generated with the 31–36 peptide of NPY) detected bands at a MW of 0.8 and 2 kDa in the insoluble fraction of the brain homogenates (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material). Consistent with this result, in the NEP and APP/NEP tg mice, levels of immunoreactivity with our antibody were increased for the lower MW bands corresponding to NPY CTFs (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material).
Immunocytochemical analysis with the commercial NPY CTFs antibody (Fig. 2F–I) showed immunoreactivity associated with neurons similar to those recognized by the antibody against FL-NPY but also immunolabeled a subset of pyramidal neurons (Fig. 2H,I; supplemental Fig. 2B,C, available at www.jneurosci.org as supplemental material). Our NPY CTFs antibody displayed similar patterns of immunoreactivity (supplemental Figs. 2D, 3C–F, available at www.jneurosci.org as supplemental material). Compared with non-tg and APP tg mouse brains, the antibody from the commercial source (Fig. 2F–I) and our NPY CTFs antibody (supplemental Fig. 3C–F, available at www.jneurosci.org as supplemental material) showed more intense immunoreactivity in the NEP and APP/NEP tg mice. In contrast, in NEP KO mice, levels of NPY CTFs were reduced compared with non-tg and NEP tg animals (supplemental Fig. 1E–G,K, available at www.jneurosci.org as supplemental material), whereas FL-NPY levels were not modified (supplemental Fig. 1H–J,L, available at www.jneurosci.org as supplemental material).
To provide additional confirmation of the presence of NPY CTFs in vivo, mass spectrometry analysis was performed with mouse brain homogenates. This study showed that FL-NPY (supplemental Fig. 3G, available at www.jneurosci.org as supplemental material) and two distinct fragments of NPY consistent with NEP processing (21–36 and 31–36) (supplemental Fig. 3G, available at www.jneurosci.org as supplemental material) could be detected in the brains of non-tg and NEP tg mice.
Additional analysis by laser-scanning confocal microscopy of double-labeled sections confirmed that NEP was present in neurons displaying NPY CTFs (Fig. 4A–C) and FL-NPY (Fig. 4D–F) immunoreactivity in NEP and APP/NEP tg mice. NPY was present in ∼8% of the NeuN-positive neurons (Fig. 4G–I). Abundant NPY-positive axons were identified in the neuropil, and the varicosities of these axons colocalized with the nerve terminal marker, synaptophysin (Fig. 4J–L). Together, these studies suggest that NEP mediates the processing of NPY into CTFs that can be detected at higher concentrations in the brains of NEP tg mice.
Figure 4.
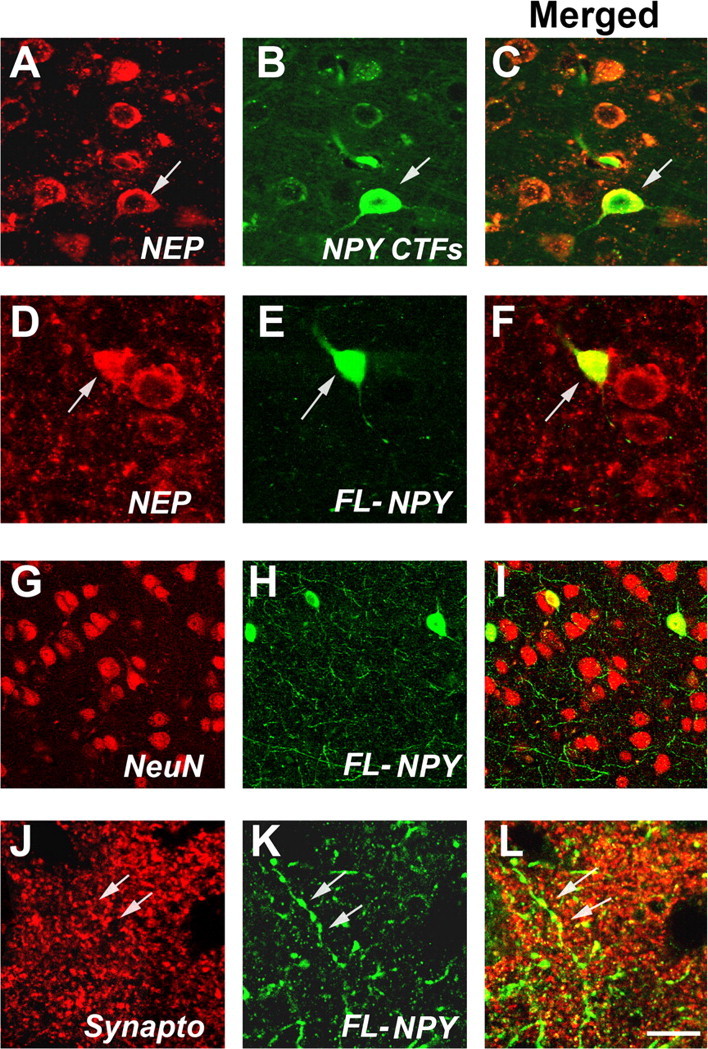
Colocalization of NEP and NPY in the tg mice. All images were obtained by laser-scanning confocal microscopy analysis of sections from the APP/NEP tg mice (frontal cortex, 6 months old). A–C, Colocalization of NEP and NPY CTFs in non-pyramidal (arrow) and pyramidal neurons. D–F, Colocalization of NEP and FL-NPY to non-pyramidal neurons (arrows). G–I, FL-NPY colocalizes with some NeuN positive neurons, and in the neuropil abundant NPY-positive processes are present. J–L, Colocalization of axonal varicosities immunolabeled with FL-NPY to synaptophysin-immunoreactive nerve terminals (arrows). Scale bar: (in L) A–F, 10 μm; G–I, 30 μm; J–L, 5 μm.
Analysis of NPY processing in vivo in tg mice and mice treated with lentiviral vectors expressing NEP or NEP shRNA
Because the increased levels of NPY fragments in NEP tg mice and colocalization of NEP and NPY in neurons suggest that these two proteins might interact in the same subcellular compartments, we developed an ex vivo detection system to further investigate the interactions of NEP and NPY and the ability of NEP to generate NPY fragments. For this purpose, FITC-tagged (at the N terminus site) NPY was incubated with homogenates from non-tg and tg mice. After 4 h of incubation of the tagged peptide with the brain homogenates, gel electrophoresis and image analysis of the resulting bands were performed. This study showed that levels of FITC-tagged NPY were reduced in all four groups (Fig. 5A). Moreover, brain homogenates cleaved the FITC-tagged NPY, resulting in the generation of two lower molecular-weight bands, one at ∼3.5 kDa, consistent with the N-terminal 1–30 fragment (corresponding to the C-terminal 31–36), and a fainter and more diffuse band at 2.2 kDa, representing the N-terminal 1–20 fragment (corresponding to the C-terminal 21–36) (Fig. 5A,B). The brain homogenates from the NEP and APP/NEP tg mice generated on average 50% more N-terminal NPY fragments compared with APP tg and non-tg controls (Fig. 5C–E). The effects of the brain homogenates on NPY cleavage were blocked by the NEP inhibitor thiorphan (Fig. 5A) but not by other protease inhibitors (data not shown). Incubation of the FITC-tagged NPY with recombinant human NEP resulted in similar patterns of cleavage that were inhibited by thiorphan (Fig. 5A).
Figure 5.

Patterns of NPY processing and immunoreactivity in APP/NEP tg mice. A, Representative gel of the patterns of FITC-tagged NPY (1–36) proteolysis at time 0 and after 4 h exposure to brain homogenates from non-tg, APP, APP/NEP, and NEP tg mice. B, Schematic representation of NPY metabolism and generation of NPY fragments with FITC-tagged NPY. C–E, Analysis of the levels of FL and N-terminal NPY fragments (1–30 and 1–20) at time 0 and after 4 h exposure to brain homogenates from non-tg, APP, APP/NEP, and NEP tg mice. Error bars indicate SEM.
To verify the ex vivo effects of NEP with an alternative model, levels of NPY-FITC cleavage were analyzed in mice that received intracerebral injections with a LV expressing NEP or an shRNA to silence NEP (Fig. 6A). Four weeks after infection, the levels of NEP expression in the area of the injection were analyzed by immunoblot. Whereas, with LV-NEP, levels of expression were increased by 45% (Fig. 6B), with LV-NEP shRNA, there was a 90% decrease (Fig. 6B). When NPY-FITC was incubated with homogenates from the brains of mice that received LV-NEP, levels of FL-NPY were reduced (Fig. 6C), whereas levels of NPY CTFs were increased (Fig. 6D). In contrast, NPY-FITC processing was reduced when incubated with brain homogenates from mice that were injected with LV-NEP shRNA (Fig. 6E,F). No significant effects were observed when NPY-FITC was incubated with brain samples from mice inoculated with a mutant, inactive NEP (LV-NEP X) (Fig. 6C,D) or with a scrambled LV-shRNA (Fig. 6E,F). Together, these studies indicate that most of the NEP activity in the mouse brain cleaves NPY between amino acids 20 and 21, resulting in the generation of the 21–36 and 31–36 aa CTFs.
Figure 6.

Processing of NPY-FITC in mice that received intracerebral injections with LV-NEP or NEP shRNA. A, Schematic representation of the LV vectors used to increase or silence NEP expression. Controls included no LV, an empty LV vector, and an LV expressing GFP. Non-tg mice received intrahippocampal injections and brains were processed after 4 weeks for Western blot (B) and the ex vivo NPY-FITC assay (C–F). B, Immunoblot analysis of levels of NEP immunoreactivity in mice treated with LV- or LV-NEP shRNA. C, Levels of un-degraded FL-NPY were lower in mice treated with LV-NEP compared with controls and LV-NEP X. D, Levels of NPY CTFs were higher in mice treated LV-NEP compared with controls and LV-NEP X. E, Levels of un-degraded FL-NPY were higher in mice treated with LV-NEP shRNA compared with controls and LV-scrambled shRNA. F, Levels of NPY CTFs were lower in mice treated LV-NEP with LV-NEP shRNA compared with controls and LV-scrambled shRNA (*p < 0.05 compared with LV-vector controls by one-way ANOVA with post hoc Dunnett's; n = 4 mice per group; 6 months of age). Error bars indicate SEM.
NPY CTFs are neuroprotective in in vitro and in vivo models of AD
To investigate whether the NPY CTFs generated by NEP processing might be neuroprotective or represent a pathway to terminate the effects of NPY in the CNS, amidated and nonamidated peptides of 21–36 and 31–36 NPY CTFs (Fig. 7A) were infused into the brains of non-tg and APP tg mice. After 4 weeks, the brains of mice were analyzed with antibodies against NPY CTFs and MAP2. Confocal microscopy showed that, compared with vehicle controls (Fig. 7B,C), mice that were infused with NPY 21–36 (Fig. 7D,E) and 31–36 (data not shown) peptides displayed increased levels of NPY CTFs immunoreactivity in the neocortex and hippocampus. Compared with vehicle-infused non-tg controls (Fig. 7F,J), vehicle-infused APP tg mice displayed a significant decrease in the area occupied by MAP2-immunoreactive dendrites in the neocortex (Fig. 7G,J). In contrast, infusion of amidated NPY 21–36 or NPY 31–36 ameliorated the neurodegenerative pathology in the APP tg mice (Fig. 7H–J). However, infusion of the nonamidated NPY CTFs did not revert the alterations in MAP2 immunoreactivity in the APP tg mice (Fig. 7J).
Figure 7.

Neuroprotective effects of NPY fragments infused into the brains of APP tg mice. A, Peptide sequences and diagram of amidated (ami) NPY 31–36 and 21–36 CTFs. B, C, Low-power (20×) (B) and high-power (200×) (C) images of a section from an APP tg mouse infused with vehicle and immunolabeled with the polyclonal antibody against NPY CTFs. D, E, Low-power (D) and high-power (E) images of a section from an APP tg mouse infused with amidated (ami) NPY 21–36 and immunolabeled with the polyclonal antibody against NPY CTFs. F–I, MAP2 immunoreactivity in the frontal cortex of non-tg or APP tg mice treated with vehicle control or amidated NPY fragments. J, Levels of MAP2 immunoreactivity in the frontal cortex of non-tg or APP tg mice treated with vehicle control or amidated NPY fragments (*p < 0.05 compared with non-tg controls by one-way ANOVA with post hoc Dunnett's; n = 6 mice per group; 6 months of age). Error bars indicate SEM. Scale bars: (in E) B, D, 100 μm; C, E, 40 μm; (in I) F–I, 10 μm.
To confirm whether the NPY CTFs generated by NEP processing might be neuroprotective, primary human cortical neurons were pretreated with amidated and nonamidated FL-NPY, NPY 21–36, NPY 31–36, a scrambled peptide, or vehicle alone (DMSO) for 24 h followed by challenge with Aβ1–42 (Fig. 8). Laser-scanning confocal analysis of cells double-immunolabeled with antibodies against synaptophysin and MAP2 showed that, compared with vehicle-treated cells (Fig. 8A,E), Aβ1–42 treatment alone resulted in an average 40% decrease in synaptophysin immunoreactivity after 24 h of exposure (Fig. 8B,E). In contrast, pretreatment with the amidated NPY peptides protected neurons from the Aβ1–42-mediated decrease in synaptophysin immunoreactivity compared with controls (Fig. 8C,E). The amidated FL-NPY had similar neuroprotective effects to the 21–36 and 31–36 NPY CTFs. No significant differences were noted between amidated 21–36, 31–36, or FL-NPY. The scrambled peptide (data not shown) and the nonamidated fragments had no protective effects (Fig. 8D,E). To verify the specificity of the effects of the NPY fragments, primary neuronal cells were pretreated with a Y1 (BIBP3226) or Y2 receptor (BIIE0246) inhibitor followed by NPY and Aβ exposure. This study showed that the Y2 receptor inhibitor was able to block the protective effects of the amidated 21–36 NPY CTF in primary neuronal cells challenged with Aβ1–42 (Fig. 8F). In contrast, the Y1 inhibitor and a control had no effects in blocking the neuroprotective effects of the 21–36 NPY CTF (Fig. 8F).
Figure 8.

Neuroprotective effects of NPY fragments in primary human neurons. Primary cultures were treated with Aβ for 24 h in the presence or absence of amidated (ami) NPY 21–36 peptide or nonamidated NPY 21–36 peptide. Fixed cells were double-immunolabeled with antibodies against synaptophysin (red) and MAP2 (green) and imaged with the laser-scanning confocal microscope. A, Vehicle treatment (1% DMSO). B, Reduced synaptophysin and MAP2 immunoreactivity in primary cultures treated with 10 μm freshly solubilized Aβ for 24 h. C, Pretreatment with amidated 21–36 CTF of NPY (10 nm) followed by Aβ challenge. D, Control experiment preincubating the cells with nonamidated 21–36 CTF of NPY (10 nm) followed by Aβ treatment. E, Effects of the amidated and nonamidated NPY CTFs on synaptophysin immunoreactivity in primary neurons treated with Aβ as assessed by computer-aided image analysis. F, Effects of pretreatment with Y1 (BIBP3226) or Y2 receptor (BIIE0246) inhibitors followed by incubation with 21–36 NPY CTF on synaptophysin immunoreactivity in primary neurons challenged with Aβ (*p < 0.05 compared with vehicle-treated controls by one-way ANOVA with post hoc Dunnett's; experiments were performed in triplicate). Error bars indicate SEM. Scale bar, 20 μm.
Together, the in vitro and in vivo studies support the contention that NPY CTFs resulting from NEP activity have neuroprotective abilities in AD-related models.
Discussion
Most studies on NEP activity in AD models have focused on the ability of NEP to degrade Aβ (Iwata et al., 2001; Leissring et al., 2003; Mohajeri et al., 2004; Carter et al., 2006; Huang et al., 2006; Farris et al., 2007; El-Amouri et al., 2008; Iijima-Ando et al., 2008); however, NEP is known to cleave other neuropeptides and processing of these alternative substrates might contribute to the neuroprotective effects. For this reason, we investigated the effects of NEP on neurotrophic factors and neuropeptides. Our study showed that expression of NEP at moderate levels in tg mice and in crosses with an APP tg model results in increased generation of NPY CTFs that displayed neuroprotective activity both in primary neurons and APP tg mice.
The mature NPY protein, one of the most abundant neuropeptides in the CNS, is a 36 aa protein that possesses an amidated C-terminal residue (Medeiros Mdos and Turner, 1996; Silva et al., 2005). The FL-NPY can be cleaved by dipeptidyl peptidase IV and aminopeptidase P resulting in NPY 3–36 and NPY 2–36, respectively, and these fragments are agonists of the Y2/Y5 receptors (Silva et al., 2003). Although considerable attention has been devoted to the NPY 2–36 and NPY 3–36 because of their ability to regulate food intake (Naveilhan et al., 1999), less is known about the intriguing role of alternative, shorter NPY CTFs (21–36, 31–36).
In the present study, we show that NPY CTFs consistent with NEP processing can be found in the brains of NEP and NEP/APP tg mice. This is in agreement with other in vitro analysis that NEP can generate such CTF products (Medeiros Mdos and Turner, 1996). FL-NPY can be cleaved by NEP at four different places in the C-terminal region; the primary sites are between Tyr20–Tyr21 and Leu30–Ile31. This cleavage results in the production of the N-terminal fragments of NPY 1–20 and NPY 1–30 and the complementary C-terminal portions of NPY 21–36 and NPY 31–36. In general, it has been thought that NEP hydrolyzes and terminates the activity of a variety of neuropeptides, including enkephalins, tachykinins, brain natriuretic peptides, and somatostatin (Skidgel and Erdös, 2004); however, the present study suggests that the NPY CTFs resulting from NEP processing might play an active role in neuroprotection. This hypothesis is consistent with a previous report that has proposed that such fragments might bind the Y receptors (Kaga et al., 2001) and that NPY protects hippocampal neurons from glutamate excitotoxicity in a receptor-dependent manner (Silva et al., 2003). Previous studies have shown that binding of NPY to the Y receptors protects CA1, and binding to Y1, Y2, and Y5 protects CA3 and the dentate gyrus (Silva et al., 2003). The direct detection and binding to the Y receptors by the putative C-terminal NPY fragments derived from NEP processing awaits future investigation.
NPY fragments consistent with NEP processing have been also detected by matrix-assisted laser desorption ionization–mass spectrometry in the CSF of control and AD patients (Nilsson et al., 2001); and here we confirmed their presence in the brains of non-tg and tg mice by mass spectrometry. However, it has been difficult to detect these products in the brain. For this reason, we generated a new NPY CTFs antibody (with a coupled 21–36 peptide) and used FITC-tagged FL-NPY to show ex vivo that NEP in brain homogenates generates NPY fragments that, by infusion into APP tg mice and in neuronal cultures, provide protection from the toxic effects of Aβ. The difficulty in directly detecting NPY fragments in the brain might be related to the possibility that these products are generated at low levels in vivo and that they have short half-lives. Thus, isolating and characterizing the bioactivity of these fragments will require more detailed investigation. Nonetheless, it is reasonable to postulate that NPY CTFs might be neuroactive, because the intact C terminus is necessary for binding to Y2, whereas the N terminus is necessary for binding to the Y1 receptor. Moreover, whereas Y1 is mostly postsynaptic and has been linked to food intake and anxiety (Silva et al., 2005), the Y2 receptor is mainly presynaptic and activation of the Y2 receptor in the hippocampus has been implicated in learning and memory (Redrobe et al., 2004). Thus, it is possible that, rather than simply representing terminal products of hydrolysis, the amidated shorter NPY CTFs generated by NEP might protect from neurotoxicity by activating Y2 receptors in the hippocampus. In the present study, we showed that direct delivery of NPY CTFs is neuroprotective in a mouse model of AD. Consistent with these findings, a recent study showed that AAV (adenoassociated virus)-mediated expression of NPY CTFs is neuroprotective in a rat seizure model (Foti et al., 2007).
This is also of interest because a recent study by Palop et al. (2007) showed that abnormalities in the NPY network in the hippocampus of APP tg mice might be responsible for the seizure activity that is characteristic in these mice and that has also been reported in patients with advanced AD (Silva et al., 2005; Amatniek et al., 2006). This suggests that therapy with NEP or alternatively with NPY CTFs might play a role in remodeling and controlling seizure activity in the APP tg model. Future studies will be necessary to investigate this possibility.
NPY is colocalized with somatostatin and GABA in interneurons in the cerebral cortex and subcortical white matter (Jinno and Kosaka, 2003). Moreover, we have shown that NPY also colocalizes with NEP in the brains of tg mice. During the aging process (Cha et al., 1997; Cadacio et al., 2003) and in AD, NPY neurons are susceptible to degeneration and previous studies have shown decreased levels of these neuropeptide-positive neurons in the neocortex and hippocampus (Chan-Palay et al., 1985; Davies et al., 1990). In APP×PS1 tg mice, the levels of NPY mRNA and the numbers of NPY/somatostatin interneurons are significantly reduced (Ramos et al., 2006). In PDAPP (Diez et al., 2000) and APP23 tg mouse lines, NPY fibers in the hippocampus are prominent (Palop et al., 2007) and dystrophic neurites around plaques show abundant NPY, galanin, ENK, and cholecystokinin immunoreactivity (Diez et al., 2003).
Finally, our study also showed that at moderate levels NEP overexpression had no adverse effects on neurotrophic factors and other neurotransmitter pathways. NEP did not alter the expression or proteolysis of BDNF, NGF, NT4, NT3, or other NEP substrates, such as SP. This finding is important because, based on its anti-amyloidogenic and neuroprotective effects, NEP has been considered as a potential therapeutic target for AD. In conclusion, this study suggests that NEP might also have beneficial effects by generating protective neuropeptides. This function of NEP represents a unique example of a proteolytic enzyme with dual action, namely, degradation of Aβ as well as processing of NPY.
Footnotes
This work was supported by National Institutes of Health Grants AG10435, AG022074, AG18440, and AG5131.
References
- Akiyama H, Kondo H, Ikeda K, Kato M, McGeer PL. Immunohistochemical localization of neprilysin in the human cerebral cortex: inverse association with vulnerability to amyloid β-protein (Aβ) deposition. Brain Res. 2001;902:277–281. doi: 10.1016/s0006-8993(01)02390-3. [DOI] [PubMed] [Google Scholar]
- Albers HE, Ferris CF. Neuropeptide Y: role in light-dark cycle entrainment of hamster circadian rhythms. Neurosci Lett. 1984;50:163–168. doi: 10.1016/0304-3940(84)90480-4. [DOI] [PubMed] [Google Scholar]
- Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, Albert M, Brandt J, Stern Y. Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia. 2006;47:867–872. doi: 10.1111/j.1528-1167.2006.00554.x. [DOI] [PubMed] [Google Scholar]
- Ashford JW. APOE genotype effects on Alzheimer's disease onset and epidemiology. J Mol Neurosci. 2004;23:157–165. doi: 10.1385/JMN:23:3:157. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Oddo S, Sugarman MC, Akbari Y, LaFerla FM. Age- and region-dependent alterations in Aβ-degrading enzymes: implications for Aβ-induced disorders. Neurobiol Aging. 2005;26:645–654. doi: 10.1016/j.neurobiolaging.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Cadacio CL, Milner TA, Gallagher M, Pierce JP. Hilar neuropeptide Y interneuron loss in the aged rat hippocampal formation. Exp Neurol. 2003;183:147–158. doi: 10.1016/s0014-4886(03)00126-2. [DOI] [PubMed] [Google Scholar]
- Carter TL, Pedrini S, Ghiso J, Ehrlich ME, Gandy S. Brain neprilysin activity and susceptibility to transgene-induced Alzheimer amyloidosis. Neurosci Lett. 2006;392:235–239. doi: 10.1016/j.neulet.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Cha CI, Lee YI, Lee EY, Park KH, Baik SH. Age-related changes of VIP, NPY and somatostatin-immunoreactive neurons in the cerebral cortex of aged rats. Brain Res. 1997;753:235–244. doi: 10.1016/s0006-8993(97)00009-7. [DOI] [PubMed] [Google Scholar]
- Chana G, Landau S, Beasley C, Everall IP, Cotter D. Two-dimensional assessment of cytoarchitecture in the anterior cingulate cortex in major depressive disorder, bipolar disorder, and schizophrenia: evidence for decreased neuronal somal size and increased neuronal density. Biol Psychiatry. 2003;53:1086–1098. doi: 10.1016/s0006-3223(03)00114-8. [DOI] [PubMed] [Google Scholar]
- Chan-Palay V, Lang W, Allen YS, Haesler U, Polak JM. Cortical neurons immunoreactive with antisera against neuropeptide Y are altered in Alzheimer's-type dementia. J Comp Neurol. 1985;238:390–400. doi: 10.1002/cne.902380404. [DOI] [PubMed] [Google Scholar]
- Clarimón J, Muñoz FJ, Boada M, Tàrraga L, Sunyer J, Bertranpetit J, Comas D. Possible increased risk for Alzheimer's disease associated with neprilysin gene. J Neural Transm. 2003;110:651–657. doi: 10.1007/s00702-002-0807-3. [DOI] [PubMed] [Google Scholar]
- Davies CA, Morroll DR, Prinja D, Mann DM, Gibbs A. A quantitative assessment of somatostatin-like and neuropeptide Y-like immunostained cells in the frontal and temporal cortex of patients with Alzheimer's disease. J Neurol Sci. 1990;96:59–73. doi: 10.1016/0022-510x(90)90057-t. [DOI] [PubMed] [Google Scholar]
- Diez M, Koistinaho J, Kahn K, Games D, Hökfelt T. Neuropeptides in hippocampus and cortex in transgenic mice overexpressing V717F β-amyloid precursor protein—initial observations. Neuroscience. 2000;100:259–286. doi: 10.1016/s0306-4522(00)00261-x. [DOI] [PubMed] [Google Scholar]
- Diez M, Danner S, Frey P, Sommer B, Staufenbiel M, Wiederhold KH, Hökfelt T. Neuropeptide alterations in the hippocampal formation and cortex of transgenic mice overexpressing β-amyloid precursor protein (APP) with the Swedish double mutation (APP23) Neurobiol Dis. 2003;14:579–594. doi: 10.1016/j.nbd.2003.08.003. [DOI] [PubMed] [Google Scholar]
- El-Amouri SS, Zhu H, Yu J, Marr R, Verma IM, Kindy MS. Neprilysin: an enzyme candidate to slow the progression of Alzheimer's disease. Am J Pathol. 2008;172:1342–1354. doi: 10.2353/ajpath.2008.070620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris W, Schütz SG, Cirrito JR, Shankar GM, Sun X, George A, Leissring MA, Walsh DM, Qiu WQ, Holtzman DM, Selkoe DJ. Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am J Pathol. 2007;171:241–251. doi: 10.2353/ajpath.2007.070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foti S, Haberman RP, Samulski RJ, McCown TJ. Adeno-associated virus-mediated expression and constitutive secretion of NPY or NPY13–36 suppresses seizure activity in vivo. Gene Ther. 2007;14:1534–1536. doi: 10.1038/sj.gt.3303013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glabe CC. Amyloid accumulation and pathogensis of Alzheimer's disease: significance of monomeric, oligomeric and fibrillar Aβ. Subcell Biochem. 2005;38:167–177. doi: 10.1007/0-387-23226-5_8. [DOI] [PubMed] [Google Scholar]
- Glabe CG, Kayed R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology. 2006;66:S74–S78. doi: 10.1212/01.wnl.0000192103.24796.42. [DOI] [PubMed] [Google Scholar]
- Hemming ML, Patterson M, Reske-Nielsen C, Lin L, Isacson O, Selkoe DJ. Reducing amyloid plaque burden via ex vivo gene delivery of an Aβ-degrading protease: a novel therapeutic approach to Alzheimer disease. PLoS Med. 2007;4:e262. doi: 10.1371/journal.pmed.0040262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong CS, Goins WF, Goss JR, Burton EA, Glorioso JC. Herpes simplex virus RNAi and neprilysin gene transfer vectors reduce accumulation of Alzheimer's disease-related amyloid-β peptide in vivo. Gene Ther. 2006;13:1068–1079. doi: 10.1038/sj.gt.3302719. [DOI] [PubMed] [Google Scholar]
- Howell OW, Doyle K, Goodman JH, Scharfman HE, Herzog H, Pringle A, Beck-Sickinger AG, Gray WP. Neuropeptide Y stimulates neuronal precursor proliferation in the post-natal and adult dentate gyrus. J Neurochem. 2005;93:560–570. doi: 10.1111/j.1471-4159.2005.03057.x. [DOI] [PubMed] [Google Scholar]
- Huang SM, Mouri A, Kokubo H, Nakajima R, Suemoto T, Higuchi M, Staufenbiel M, Noda Y, Yamaguchi H, Nabeshima T, Saido TC, Iwata N. Neprilysin-sensitive synapse-associated amyloid-β peptide oligomers impair neuronal plasticity and cognitive function. J Biol Chem. 2006;281:17941–17951. doi: 10.1074/jbc.M601372200. [DOI] [PubMed] [Google Scholar]
- Iijima-Ando K, Hearn SA, Granger L, Shenton C, Gatt A, Chiang HC, Hakker I, Zhong Y, Iijima K. Overexpression of neprilysin reduces alzheimer amyloid-β42 (Aβ42)-induced neuron loss and intraneuronal Aβ42 deposits but causes a reduction in cAMP-responsive element-binding protein-mediated transcription, age-dependent axon pathology, and premature death in Drosophila. J Biol Chem. 2008;283:19066–19076. doi: 10.1074/jbc.M710509200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee HJ, Hama E, Sekine-Aizawa Y, Saido TC. Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–150. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee HJ, Saido TC. Metabolic regulation of brain Aβ by neprilysin. Science. 2001;292:1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- Iwata N, Mizukami H, Shirotani K, Takaki Y, Muramatsu S, Lu B, Gerard NP, Gerard C, Ozawa K, Saido TC. Presynaptic localization of neprilysin contributes to efficient clearance of amyloid-β peptide in mouse brain. J Neurosci. 2004;24:991–998. doi: 10.1523/JNEUROSCI.4792-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinno S, Kosaka T. Patterns of expression of neuropeptides in GABAergic nonprincipal neurons in the mouse hippocampus: quantitative analysis with optical disector. J Comp Neurol. 2003;461:333–349. doi: 10.1002/cne.10700. [DOI] [PubMed] [Google Scholar]
- Kaga T, Fujimiya M, Inui A. Emerging functions of neuropeptide Y Y(2) receptors in the brain. Peptides. 2001;22:501–506. doi: 10.1016/s0196-9781(01)00362-x. [DOI] [PubMed] [Google Scholar]
- Kawahara K, Hashimoto M, Bar-On P, Ho GJ, Crews L, Mizuno H, Rockenstein E, Imam SZ, Masliah E. α-Synuclein aggregates interfere with Parkin solubility and distribution: role in the pathogenesis of Parkinson disease. J Biol Chem. 2008;283:6979–6987. doi: 10.1074/jbc.M710418200. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ. Enhanced proteolysis of β-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–1093. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- Lu B, Gerard NP, Kolakowski LF, Jr, Bozza M, Zurakowski D, Finco O, Carroll MC, Gerard C. Neutral endopeptidase modulation of septic shock. J Exp Med. 1995;181:2271–2275. doi: 10.1084/jem.181.6.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr RA, Rockenstein E, Mukherjee A, Kindy MS, Hersh LB, Gage FH, Verma IM, Masliah E. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J Neurosci. 2003;23:1992–1996. doi: 10.1523/JNEUROSCI.23-06-01992.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Medeiros MS, Turner AJ. Post-secretory processing of regulatory peptides: the pancreatic polypeptide family as a model example. Biochimie. 1994;76:283–287. doi: 10.1016/0300-9084(94)90159-7. [DOI] [PubMed] [Google Scholar]
- Medeiros Mdos S, Turner AJ. Metabolism and functions of neuropeptide Y. Neurochem Res. 1996;21:1125–1132. doi: 10.1007/BF02532423. [DOI] [PubMed] [Google Scholar]
- Minthon L, Edvinsson L, Ekman R, Gustafson L. Neuropeptide levels in Alzheimer's disease and dementia with frontotemporal degeneration. J Neural Transm Suppl. 1990;30:57–67. doi: 10.1007/978-3-7091-3345-3_6. [DOI] [PubMed] [Google Scholar]
- Mohajeri MH, Kuehnle K, Li H, Poirier R, Tracy J, Nitsch RM. Anti-amyloid activity of neprilysin in plaque-bearing mouse models of Alzheimer's disease. FEBS Lett. 2004;562:16–21. doi: 10.1016/S0014-5793(04)00169-3. [DOI] [PubMed] [Google Scholar]
- Mucke L, Abraham CR, Ruppe MD, Rockenstein EM, Toggas SM, Mallory M, Alford M, Masliah E. Protection against HIV-1 gp120-induced brain damage by neuronal overexpression of human amyloid precursor protein (hAPP) J Exp Med. 1995;181:1551–1556. doi: 10.1084/jem.181.4.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveilhan P, Hassani H, Canals JM, Ekstrand AJ, Larefalk A, Chhajlani V, Arenas E, Gedda K, Svensson L, Thoren P, Ernfors P. Normal feeding behavior, body weight and leptin response require the neuropeptide Y Y2 receptor. Nat Med. 1999;5:1188–1193. doi: 10.1038/13514. [DOI] [PubMed] [Google Scholar]
- Nilsson CL, Brinkmalm A, Minthon L, Blennow K, Ekman R. Processing of neuropeptide Y, galanin, and somatostatin in the cerebrospinal fluid of patients with Alzheimer's disease and frontotemporal dementia. Peptides. 2001;22:2105–2112. doi: 10.1016/s0196-9781(01)00571-x. [DOI] [PubMed] [Google Scholar]
- Oda M, Morino H, Maruyama H, Terasawa H, Izumi Y, Torii T, Sasaki K, Nakamura S, Kawakami H. Dinucleotide repeat polymorphisms in the neprilysin gene are not associated with sporadic Alzheimer's disease. Neurosci Lett. 2002;320:105–107. doi: 10.1016/s0304-3940(02)00057-5. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos B, Baglietto-Vargas D, del Rio JC, Moreno-Gonzalez I, Santa-Maria C, Jimenez S, Caballero C, Lopez-Tellez JF, Khan ZU, Ruano D, Gutierrez A, Vitorica J. Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1×APP transgenic model of Alzheimer's disease. Neurobiol Aging. 2006;27:1658–1672. doi: 10.1016/j.neurobiolaging.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Redrobe JP, Dumont Y, St-Pierre JA, Quirion R. Multiple receptors for neuropeptide Y in the hippocampus: putative roles in seizures and cognition. Brain Res. 1999;848:153–166. doi: 10.1016/s0006-8993(99)02119-8. [DOI] [PubMed] [Google Scholar]
- Redrobe JP, Dumont Y, Herzog H, Quirion R. Characterization of neuropeptide Y, Y(2) receptor knockout mice in two animal models of learning and memory processing. J Mol Neurosci. 2004;22:159–166. doi: 10.1385/JMN:22:3:159. [DOI] [PubMed] [Google Scholar]
- Reilly CE. Neprilysin content is reduced in Alzheimer brain areas. J Neurol. 2001;248:159–160. doi: 10.1007/s004150170259. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Mante M, Sisk A, Masliaha E. Early formation of mature amyloid-b proteins deposits in a mutant APP transgenic model depends on levels of Aβ1–42. J Neurosci Res. 2001;66:573–582. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Mante M, Alford M, Windisch M, Moessler H, Masliah E. Effects of Cerebrolysin on amyloid-β deposition in a transgenic model of Alzheimer's disease. J Neural Transm Suppl. 2002a;2002:327–336. [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002b;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Schwach G, Ingolic E, Adame A, Crews L, Mante M, Pfragner R, Schreiner E, Windisch M, Masliah E. Lysosomal pathology associated with alpha-synuclein accumulation in transgenic models using an eGFP fusion protein. J Neurosci Res. 2005a;80:247–259. doi: 10.1002/jnr.20446. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mante M, Alford M, Adame A, Crews L, Hashimoto M, Esposito L, Mucke L, Masliah E. High β-secretase activity elicits neurodegeneration in transgenic mice despite reductions in amyloid-β levels: implications for the treatment of Alzheimer disease. J Biol Chem. 2005b;280:32957–32967. doi: 10.1074/jbc.M507016200. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Cell biology of the amyloid β-protein precursor and the mechanisms of Alzheimer's disease. Annu Rev Cell Biol. 1994a;10:373–403. doi: 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: a central role for amyloid. J Neuropathol Exp Neurol. 1994b;53:438–447. doi: 10.1097/00005072-199409000-00003. [DOI] [PubMed] [Google Scholar]
- Silva AP, Pinheiro PS, Carvalho AP, Carvalho CM, Jakobsen B, Zimmer J, Malva JO. Activation of neuropeptide Y receptors is neuroprotective against excitotoxicity in organotypic hippocampal slice cultures. FASEB J. 2003;17:1118–1120. doi: 10.1096/fj.02-0885fje. [DOI] [PubMed] [Google Scholar]
- Silva AP, Xapelli S, Grouzmann E, Cavadas C. The putative neuroprotective role of neuropeptide Y in the central nervous system. Curr Drug Targets CNS Neurol Disord. 2005;4:331–347. doi: 10.2174/1568007054546153. [DOI] [PubMed] [Google Scholar]
- Singer O, Marr RA, Rockenstein E, Crews L, Coufal NG, Gage FH, Verma IM, Masliah E. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat Neurosci. 2005;8:1343–1349. doi: 10.1038/nn1531. [DOI] [PubMed] [Google Scholar]
- Skidgel RA, Erdös EG. Angiotensin converting enzyme (ACE) and neprilysin hydrolyze neuropeptides: a brief history, the beginning and follow-ups to early studies. Peptides. 2004;25:521–525. doi: 10.1016/j.peptides.2003.12.010. [DOI] [PubMed] [Google Scholar]
- Sodeyama N, Mizusawa H, Yamada M, Itoh Y, Otomo E, Matsushita M. Lack of association of neprilysin polymorphism with Alzheimer's disease and Alzheimer's disease-type neuropathological changes. J Neurol Neurosurg Psychiatry. 2001;71:817–818. doi: 10.1136/jnnp.71.6.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolowski MB. NPY and the regulation of behavioral development. Neuron. 2003;39:6–8. doi: 10.1016/s0896-6273(03)00398-2. [DOI] [PubMed] [Google Scholar]
- Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- Veinbergs I, Van Uden E, Mallory M, Alford M, McGiffert C, DeTeresa R, Orlando R, Masliah E. Role of apolipoprotein E receptors in regulating the differential in vivo neurotrophic effects of apolipoprotein E. Exp Neurol. 2001;170:15–26. doi: 10.1006/exnr.2001.7684. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Sperk G, Colmers WF. Neuropeptide Y: emerging evidence for a functional role in seizure modulation. Trends Neurosci. 1999;22:25–30. doi: 10.1016/s0166-2236(98)01284-3. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett. 2004;11:213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- Wood LS, Pickering EH, McHale D, Dechairo BM. Association between neprilysin polymorphisms and sporadic Alzheimer's disease. Neurosci Lett. 2007;427:103–106. doi: 10.1016/j.neulet.2007.09.019. [DOI] [PubMed] [Google Scholar]
- Yasojima K, Akiyama H, McGeer EG, McGeer PL. Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of β-amyloid peptide. Neurosci Lett. 2001a;297:97–100. doi: 10.1016/s0304-3940(00)01675-x. [DOI] [PubMed] [Google Scholar]
- Yasojima K, McGeer EG, McGeer PL. Relationship between β amyloid peptide generating molecules and neprilysin in Alzheimer disease and normal brain. Brain Res. 2001b;919:115–121. doi: 10.1016/s0006-8993(01)03008-6. [DOI] [PubMed] [Google Scholar]