

Abstract
Chronic postnatal hypoxia causes an apparent loss of cortical neurons that is reversed during recovery (Fagel et al., 2006). The cellular and molecular mechanisms underlying this plasticity are not understood. Here, we show that chronic hypoxia from postnatal days 3 (P3) to 10 causes a 30% decrease in cortical neurons and a 24% decrease in cortical volume. T-brain-1 (Tbr1)+ and SMI-32+ excitatory neuron numbers were completely recovered 1 month after the insult, but the mice showed a residual deficit in Parvalbumin+ and Calretinin+ GABAergic interneurons. In contrast, hypoxic mice carrying a disrupted fibroblast growth factor receptor-1 (Fgfr1) gene in GFAP+ cells [Fgfr1 conditional knock-out (cKO)], demonstrated a persistent loss of excitatory cortical neurons and a worsening of the interneuron defect. Labeling proliferating progenitors at P17 revealed increased generation of cortical NeuN+ and Tbr1+ excitatory neurons in wild-type mice subjected to hypoxic insult, whereas Fgfr1 cKO failed to mount a cortical neurogenetic response. Hypoxic wild-type mice also demonstrated a twofold increase in cell proliferation in the subventricular zone (SVZ) at P17 and a threefold increase in neurogenesis in the olfactory bulb (OB) at P48, compared with normoxic mice. In contrast, Fgfr1 cKO mice had decreased SVZ cell proliferation and curtailed reactive neurogenesis in the OB. Thus, the activation of FGFR-1 in GFAP+ cells is required for neuronal recovery after neonatal hypoxic injury, which is attributable in part to enhanced cortical and OB neurogenesis. In contrast, there is incomplete recovery of inhibitory neurons after injury, which may account for persistent behavioral deficits.
Keywords: Fgf, cerebral cortex, neurogenesis, mouse, repair, progenitor
Introduction
Oxygen deprivation is a major cause of neurodevelopmental impairments in premature children and leads to cortical gray and white matter loss (Hack et al., 2002; Ment et al., 2005). Serial evaluations have determined that there is progressive improvement in language measures through late childhood and early adolescence (Ment et al., 2003; Saigal and Doyle, 2008) and significant recovery of cerebral cortical volume measures at young adulthood (Allin et al., 2004; Fearon et al., 2004). Similar to neuropathologic findings in preterm children, our mouse model of chronic sublethal hypoxic injury is characterized by cortical neuron loss, decreased cortical volume, atrophy of subcortical white matter and ventriculomegaly (Turner et al., 2003; Fagel et al., 2006). Remarkably, 4 weeks after hypoxia, total neuron number in the cerebral cortex is indistinguishable from that of normal mice (Fagel et al., 2006). Such data support the idea that in mice and in some children, the immature cerebral cortex is able to repair its cellular structure by unknown developmental processes occurring after the insult.
The nature of injury may play a determinant role in recovery. For example, adult rodents that suffer infarct-producing ischemic events can regenerate striatal and hippocampal neurons, but not cortical neurons (Yoshimura et al., 2001; Arvidsson et al., 2002; Parent et al., 2002; Hoehn et al., 2005), whereas diffuse neuronal apoptosis stimulates the genesis of cortical projection neurons from endogenous progenitors (Magavi and Macklis, 2002). Furthermore, the immature brain may have a better capacity to recover. For example, we detected newly generated neurons in the juvenile mouse cortex by tagging neural precursors either by 2-bromodeoxyuridine (BrdU) incorporation or by cre-induced genetic recombination (Fagel et al., 2006; Ganat et al., 2006). Others have reported the generation of cortical inhibitory interneurons after perinatal hypoxia-ischemia, although many of these cells appear to be transient (Yang et al., 2007). In contrast, no neurogenesis has been observed in adult rodent or macaque cerebral cortex (Kornack and Rakic, 2001). Although these data suggest that the immature cortex is capable of neurogenesis, its mechanisms and physiological significance remain unclear.
Neurogenesis in the embryonic and perinatal periods is regulated by fibroblast growth factors (FGF) (Tao et al., 1996; Vaccarino et al., 1999a, 2001). FGF can induce neural fate in uncommitted stem cells (Stavridis et al., 2007) and increases progenitor proliferation (Vaccarino et al., 1999b; Wagner et al., 1999). In the embryonic cortex, FGF-2 increases the number of neural progenitors (Raballo et al., 2000) and the differentiation of inhibitory and excitatory neurons (Dono et al., 1998; Ortega et al., 1998; Vaccarino et al., 1999b; Shin et al., 2004). Furthermore, both endogenous FGF-2 (Yoshimura et al., 2001) and exogenously administered FGF-2 (Nakatomi et al., 2002; Androutsellis-Theotokis et al., 2006; Monfils et al., 2006) promote hippocampal neurogenesis and behavioral recovery after a number of injuries in adult rodents, including global hypoxia-ischemia, local cortical ischemic lesions and mechanical motor cortex injury.
Using mice with a disrupted Fgfr1 gene in GFAP+ cells, we assessed whether this FGF receptor may be responsible for reconstituting brain structure after chronic sublethal hypoxic injury and the role of neurogenesis in this process. An unbiased stereological analysis of the number of newly generated cortical and olfactory bulb neurons was performed in parallel with an assessment of the total number of excitatory and inhibitory neurons several weeks after the insult. The data suggest that FGFR-1 signaling is required not only for cell proliferation and neurogenesis after an insult, but also for indirect effects leading to enhanced neuronal survival or maturation in the posthypoxic environment.
Materials and Methods
Mouse lines.
To conditionally knockout the Fgfr1 gene, mice carrying the Fgfr1f allele (Trokovic et al., 2003) were crossed with GFAPCre mice, which express Cre under the control of the human GFAP promoter (Zhuo et al., 2001). The Fgfr1f/f;hGFAPCre mice lack Fgfr1 gene expression in telencephalic precursors, as described previously (Ohkubo et al., 2004; Smith et al., 2006). Cre negative littermate mice were used as controls. The genetic background of the animals is a mixed 129/Sv, C57/BL, FVB and CD1.
All experimental procedures were performed in accordance with the Yale Animal Resources Center and Institutional Animal Care and Use Committee policies.
Hypoxic rearing.
Mice were placed in a chamber maintaining a 9.5–10.5% O2 concentration by displacement with N2 as described previously (Turner et al., 2003; Weiss et al., 2004; Fagel et al., 2006). Hypoxia began at postnatal day 3 (P3) for 7 d until P10. A separate group of control (normoxic) mice were matched for strain and age. Mice were killed after cessation of hypoxia at P10 or 38 d later at P48. Mice were perfused transcardially with 20 ml of PBS followed by 35 ml of 4% paraformaldehyde (PFA). Brains were postfixed overnight in PFA, cryoprotected in a 20% sucrose solution overnight, and stored at −80°C after embedding in Tissue-Tek O.C.T. Compound (Electron Microscopy Sciences) medium.
2-Bromodeoxyuridine birthdating assay.
BrdU (4 mg/ml in 0.007 N HCl/0.9% NaCl, Sigma) was administered intraperitoneally (50 mg/kg) every 4 h at P17 (at 0, 4, 8 and 12 h), and animals were killed at 14 h or 31 d later. In both cases, the total labeling time was ∼14 h, since BrdU availability time is estimated to last for 4–5 h after injection (Hayes and Nowakowski, 2000). The reason for this cumulative labeling protocol is that it should label all constitutively proliferating cells of the subventricular zone (SVZ), as their cell cycle is ∼13 h (Zheng et al., 2004). Based on double labeling of BrdU and 3H thymidine, this dosage of BrdU has been shown to label S-phase cells (Nowakowski et al., 1989; Hayes and Nowakowski, 2000).
Immunohistochemistry.
Brains were serially sectioned at 50 μm thickness, and sections were stored in 0.04% sodium azide (NaN3)/PBS at +4°C and immunostained as described previously (Fagel et al., 2006; Ganat et al., 2006). The following primary antibodies were used: mouse anti-NeuN (1:500, Millipore), rabbit anti-Tbr1 (1:1000) (gift from Dr. Hevner, University of Washington School of Medicine, Seattle, WA), mouse anti-SMI-32 (1:1000; Covance), mouse anti-parvalbumin (PV) (1:2500, Sigma), rabbit anti-calretinin (CR) (1:2000, Millipore), rat anti-BrdU (1:500, Accurate Chemical), mouse anti-Fgfr1 (1:500, Upstate), rabbit anti-Sox2 (1:3000, Millipore), rabbit anti-Pax6 (1:1000, University of Iowa, Iowa City, IA), rabbit anti-Tbr2 (1:2000, gift from Dr. Hevner), and rabbit anti-caspase-3 (1:500, Cell Signaling). The following secondary reagents were also used: anti-mouse Alexa 488 (1:500, Invitrogen), anti-mouse Alexa 488 cross-absorbed (1:500, Invitrogen), anti-rabbit Alexa 594 (1:1000, Invitrogen), anti-rabbit Alexa 488 (1:1000, Invitrogen) and anti-rat Alexa 594 (1:500, Invitrogen).
Morphometry and cell counting.
Unbiased estimates for volumes and cell numbers were obtained using a computer coupled to a Zeiss Axioskope 2 Mot Plus, running the StereoInvestigator software with Neurolucida (Microbrightfield). For the cortex, one in every twenty serial sections were selected for analyses; for the SVZ and OB, one in every ten sections were selected. Contours were drawn around the cerebral cortex including the infralimbic, orbital, cingulate, entorhinal and retrosplenial cortices, and cell counting was performed as described previously (Korada et al., 2002; Fagel et al., 2006). To estimate the total number of newly generated neurons in the cortex, NeuN+/BrdU+ cells as well as Tbr1+/BrdU+ and SMI-32+/Tbr1+ cells were simultaneously visualized by a 594 and 488 nm double-exposure filter and counted using the optical fractionator method in a 3-dimensional counting frame of 100 × 100 × 5 μm using a sampling grid of 1500 × 1500 μm for NeuN, 750 × 750 μm for Tbr1 and 1000 × 1000 μm for SMI-32, PV and CR. At P10, 5–6 coronal sections were sampled per brain, whereas at P48, 6–7 sections were sampled. Approximately 40 NeuN, 180 Tbr1 and 120 SMI-32, PV and CR sampling sites were counted for each cortex. The contours of the granular layer of the OB were drawn based on its increased cellular density in DAPI-stained sections. To estimate the total number of Neun+/BrdU+ cells in the OB, cells were counted in a counting frame of 50 × 50 × 5 μm, using a sampling grid of 500 × 500 μm. At P48, 5–6 coronal sections and ∼50 sampling sites were counted for each OB. Cells were classified as double-stained only if both colors were in sharp focus within a narrow (<2 μm) focal plane, using the z-axis encoder. The contours of the SVZ were drawn based on its increased cellular density in DAPI-stained sections and included the entire extent of the SVZ from its rostral end until the beginning of the hippocampal commissure. To estimate the total number of BrdU+ cells in the SVZ, cells were counted in a counting frame of 50 × 50 × 5 μm, using a sampling grid of 200 × 200 μm. Between 3 and 4 coronal sections and ∼25 sampling sites were counted for each SVZ. The average Schaffer coefficient of error for cell counting was 0.04–0.07 for cortex, 0.06–0.10 for OB and 0.15–0.17 for SVZ.
Statistical analyses.
Data were analyzed by ANOVA or Student's t test using the DataDesk statistical program. The number of samples is specified in each case in the figure legends. Post hoc analyses were performed via the Sheffe post hoc test.
Results
The recovery of cortical excitatory neurons after hypoxia requires Fgfr1
Mice were exposed to chronic sublethal hypoxia (9.5–10.5% O2) for 7 d, from P3 to P10. After injury on P10, the hypoxic mice exhibited a decrease in cortical thickness (Fig. 1A,B) and a significant 24% decrease in cortical volume (70.57 ± 2.54 mm3 for normoxic vs 53.67 ± 2.21 mm3 for hypoxic; p < 0.05). The number of NeuN+ neurons in the cerebral cortex, quantified by stereological analysis, declined from 10.2 ± 0.4 × 106 to 7.3 ± 0.8 × 106 cells (p < 0.05), revealing a 29% decrease compared with age-matched normoxic controls (Fig. 1I, blue bars). To determine whether hypoxia decreased the number of excitatory neurons, sections were immunostained using antibodies to the transcription factor Tbr1 and the neurofilament epitope SMI-32, which label subsets of cortical pyramidal neurons (Campbell et al., 1991; Hevner et al., 2006). Tbr1 is a transcription factor that regulates the differentiation of early born glutamatergic cortical neurons (Hevner et al., 2001). In the immature cortex, Tbr1 is expressed by pyramidal cells, in the subplate and in layer 1 Reelin+ cells. In adulthood, Tbr1 becomes progressively restricted to layer 6 pyramidal cells, although in rostral cortex it is expressed in layers 5 and 2/3 as well (Bulfone et al., 1995; Hevner et al., 2001, 2006). At P10, the number of Tbr1+ neurons in the cortex of hypoxic mice declined from 6.3 ± 0.4 × 106 to 4.4 ± 0.3 × 106 cells (p < 0.05), revealing a 31% statistically significant deficit in Tbr1+ cells throughout the supra- and infra-granular layers (Fig. 1A,B,J, blue bars). The total loss of Tbr1+ excitatory neurons (1.95 × 106 neurons) accounted for 67% of the NeuN+ neurons that were lost (2.92 × 106) (Fig. 1, compare I, J). There was an even greater decrease (66%) in SMI-32+ neurons (Fig. 1E,F,K, blue bars).
Figure 1.
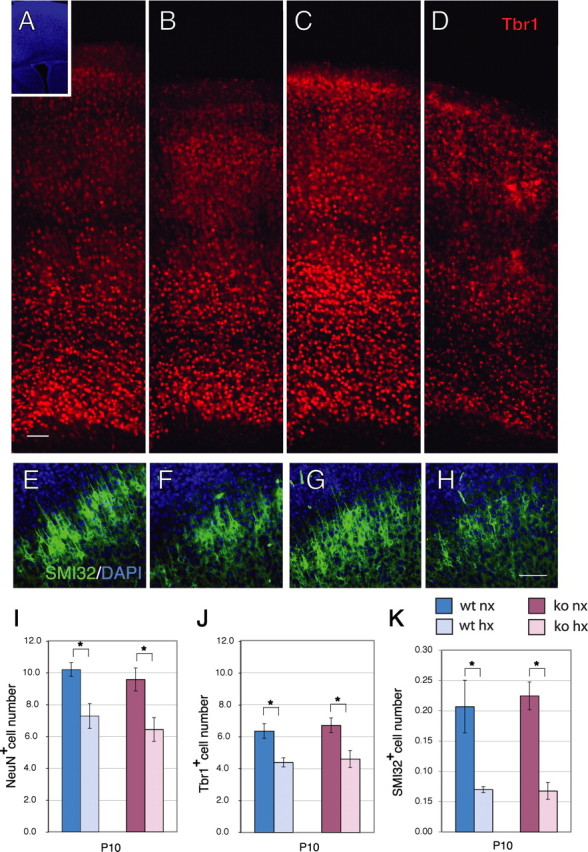
Cortical neuron number and thickness are decreased after exposure to hypoxia. A–D, Immunostaining for Tbr1 (red) in the cerebral cortex of wild-type (A, B) or Fgfr1 cKO mice (C, D) at P10 under normoxia (A, C) or after hypoxia from P3 to P10 (B, D). Inset shows DAPI low magnification. Each panel is the composite of four 20× images demonstrating the entire extent of the cerebral cortex. E–H, SMI-32 (green) and DAPI (blue) stained cortices of wild-type (E, F) or Fgfr1 cKO mice (G, H) at P10 under normoxia (E, G) or after hypoxia from P3 to P10 (F, H). Scale bars, 100 μm. I–K, Total number of NeuN (I), Tbr1 (J) and SMI-32 (K) immunoreactive neurons in the cerebral cortex by stereological analyses in wild-type (blue bars) and Fgfr1 cKO (red bars) mice. Values are expressed in 106 units. N = 3 for each group. *p < 0.05 by ANOVA with Sheffe post hoc test.
Previous findings link Fgf gene expression to the development of excitatory cortical neurons (Vaccarino et al., 1999b; Korada et al., 2002) and particularly Tbr1+ neurons (Shin et al., 2004) during embryogenesis. Fgfr1 continues to be expressed by cells of the postnatal SVZ and astrocytes throughout the brain, as assessed by immunostaining (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), as well as by an Fgfr1 BAC-EGFP transgenic mouse (Gensat Project, http://www.gensat.org/index.html). To investigate whether Fgfr1 plays a role in neuroprotection or recovery after injury, the Fgfr1 cKO mice (which lack the intact Fgfr1 gene in GFAP+ cells) (Ohkubo et al., 2004) were exposed to the chronic hypoxia protocol. Notably, the Fgfr1 cKO mice did not exhibit any baseline defect in NeuN+, Tbr1+ or SMI-32+ cortical neuron number (Fig. 1A,C,I–K), suggesting that in the absence of injury, Fgfr1 does not play a major role in cortical morphogenesis. Similar to wild-type mice, hypoxic Fgfr1 cKO mice suffered a 33, 31, and 70% loss of NeuN+, Tbr1+ and SMI-32+ cortical neurons, respectively (Fig. 1C,D,G,H,I–K). Hence, the absence of Fgfr1 did not worsen the neuronal loss caused by hypoxia.
Although there was a residual 17% decrease in cortical volume (103.49 ± 4.16 mm3 for normoxic vs 85.75 ± 1.29 mm3 for hypoxic; p < 0.05), the initial deficit in NeuN+ neurons was no longer present in the hypoxic wild-type mice at P48, after 4 weeks of recovery (Fig. 2A,B,I), confirming our previous results in mice of pure C57BL background (Fagel et al., 2006). To determine the proportion of neuronal subtypes in normoxic and hypoxic mice at this stage, we estimated the number of cortical excitatory neurons expressing Tbr1 and SMI-32 and that of inhibitory neurons expressing PV and CR by stereological analyses. Hypoxic mice showed no significant difference in the total number of Tbr1+ and SMI-32+ cortical neurons compared with normoxic controls at P48 (Fig. 2A,B,E,F,J,K, blue bars). In contrast, the total number of PV+ and CR+ interneurons was significantly decreased by 32 and 43%, respectively, in hypoxia-reared mice (p < 0.01) (Fig. 3A,B,E,F,M,N, blue bars). Given that PV and CR cells comprise a much smaller population, compared with Tbr1+ excitatory neurons, no changes in the total number of cortical neurons were detected in hypoxic mice, as shown by the analysis of NeuN (Fig. 2I). However, the proportion of excitatory and inhibitory neurons changed substantially in the cerebral cortex at 4 weeks postinjury. The ratio between Tbr1+ and the sum of PV+ and CR+ neurons in the normoxic cortex was ∼7 to 1, whereas this ratio was 12 to 1 in the hypoxic cortex. In sum, the data show no evidence for long-term neuronal loss but a substantial imbalance in the proportion of excitatory versus inhibitory neurons after hypoxia.
Figure 2.
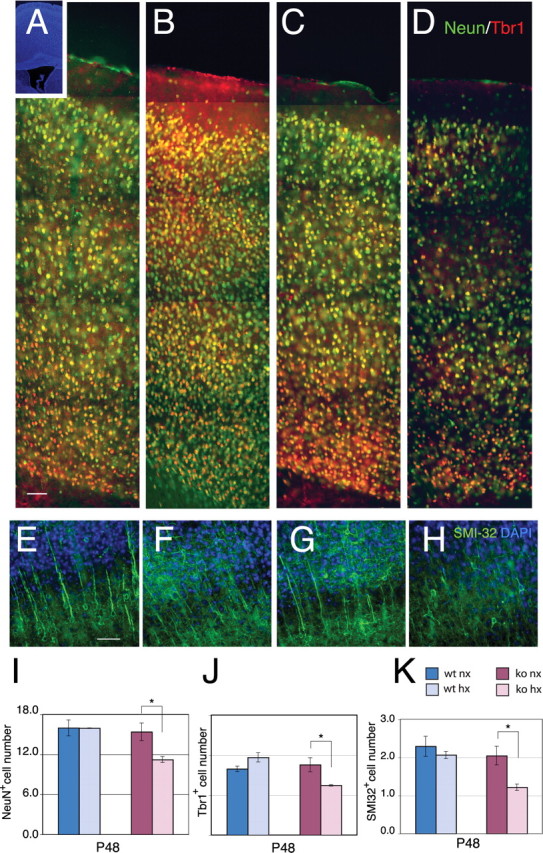
Neuron numbers recover in hypoxic wild-type mice but not in Fgfr1 cKO mice. A–D, Double immunostaining for NeuN (green) and Tbr1 (red) in the P48 cerebral cortex of wild-type (A, B) or Fgfr1 cKO mice (C, D) under normoxia (A, C) or after hypoxia (B, D). Inset shows DAPI low magnification. Each panel is the composite of five 20× images. E–H, SMI-32- (green) and DAPI- (blue) stained cortices of wild-type (E, F) or Fgfr1 cKO mice (G, H) at P48 under normoxia (E, G) or after hypoxia from P3 to P10 (F, H). Scale bars, 100 μm. I–K, Total number of NeuN (I), Tbr1 (J) and SMI-32 (K) immunoreactive neurons in the cerebral cortex by stereological analyses in wild-type (blue bars) and Fgfr1 cKO (red bars) mice. Values are expressed in 106 units. N = 3 for each group. *p < 0.05 by ANOVA with Sheffe post hoc test.
Figure 3.
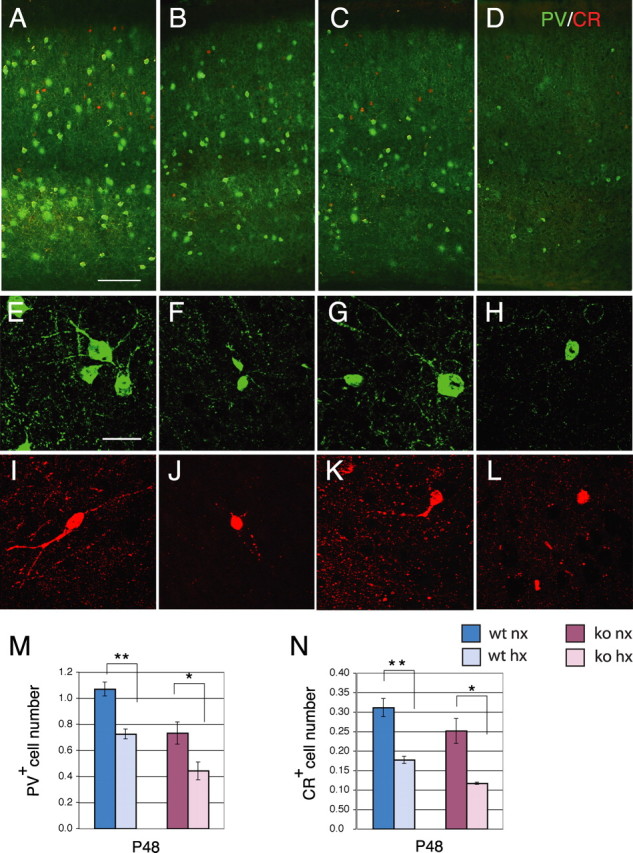
Cortical interneuron number is decreased at P48 in both hypoxic wild-type and Fgfr1 cKO mice. A–D, Double immunostaining for PV (green) and CR (red) in the P48 cerebral cortex of wild-type (A, B) or Fgfr1 cKO mice (C, D) under normoxia (A, C) or after hypoxia (B, D). Each panel is the composite of two images to show all layers of the cerebral cortex. E–L, Individual PV+ and CR+ neurons in hypoxia-reared mice (F, J, H, L) and their normoxic counterparts (E, I, G, K). Scale bars: (A–D) 100 μm; (E–L) 10 μm. M, N, Total number of PV (M) and CR (N) immunoreactive neurons by stereological analyses in wild-type (blue bars) and Fgfr1 cKO (red bars) mice. Values are expressed in 106 units. N = 3 for each group. *p < 0.05 and **p < 0.01 by ANOVA with Sheffe post hoc test.
In normoxic conditions, no difference in NeuN+, Tbr1+ or SMI-32+ neuron number was detected between Fgfr1 cKO mice and wild-type mice (Fig. 2I–K); however, these mice exhibited a basal 32% deficit of PV+ interneurons (Fig. 3M,N) as described previously (Müller Smith et al., 2008). This 32% loss of interneurons was not accompanied by a detectable decrease in total neurons, presumably because PV+ interneurons comprise a relatively small subset of total cortical neurons (i.e., 1.07 ± 0.06 million cells for PV+ neurons vs 15.98 ± 1.19 million cells for NeuN+ neurons).
The long-term response to hypoxia was significantly different in Fgfr1 cKO and wild-type mice. First, hypoxic Fgfr1 cKO mice continued to show statistically significant deficits of 27, 29 and 37% in NeuN+, Tbr1+ and SMI-32+ cortical neurons, respectively (p < 0.05) (Fig. 2C,D,G,H,I–K, red bars). Second, the Fgfr1 cKO mice, which had a baseline 32% decrease in PV+ interneurons, showed an additional decrease of 39 and a 53% in PV+ and CR+ interneurons, respectively, after hypoxic rearing (Fig. 3C,D,M,N).
Hypoxia-induced cortical and olfactory bulb neurogenesis requires Fgfr1
We next investigated the possible mechanisms underlying the recovery of Tbr1+ cortical neuron number after hypoxia in wild-type mice and the absence of this recovery in Fgfr1 cKO mice. We first examined whether progenitors, labeled with BrdU 1 week after hypoxia, can differentiate into Tbr1+ neurons. Proliferative cells were labeled with four cumulative BrdU injections administered at P17 to incorporate BrdU in the whole population of constitutively proliferating cells and their fate was analyzed 31 d later at P48. Our estimate suggested the presence of 54,800 NeuN+/BrdU+ and 19,200 Tbr1+/BrdU+ cortical neurons in normoxic cortices. These cells had a relatively small nucleus and were often present in doublets, suggesting that they were the product of a recent cell division (Fig. 4A–Q). In hypoxic mice, NeuN+/BrdU+ cortical neurons at P48 were estimated to be 114,040, while Tbr1+/BrdU+ neurons were estimated to be 72,225, which represent a 108 and a 280% increase, respectively, compared with normoxic controls (Fig. 4R,S, blue bars). Tbr1+/BrdU+ neurons accounted for just 35% of all NeuN+/BrdU+ neurons in normoxic mice, whereas they accounted for 63% of all NeuN+/BrdU+ neurons in hypoxic mice. The data suggest that proliferative progenitors give rise to new neurons in the immature cerebral cortex, that this phenomenon is increased by previous exposure to hypoxic insult, and that the proportion of progenitors giving rise to Tbr1+ neurons is higher in hypoxic-reared mice.
Figure 4.
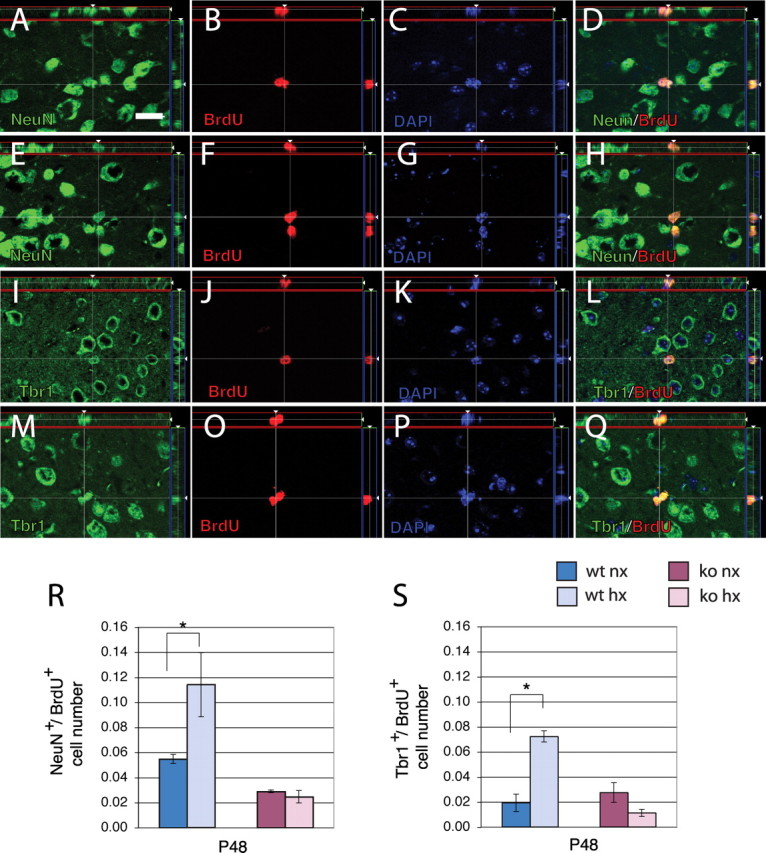
Newly generated NeuN+ and Tbr1+ cortical neurons are increased at P48 in hypoxic wild-type but not in Fgfr1 cKO mice. A–Q, Apotome 1 μm single slices of BrdU staining in the P48 cerebral cortex double-immunostained with NeuN (A–H) or Tbr1 (I–Q). BrdU was injected at P17 and analysis performed at P48. The image analyses on the z-axis are shown in the side panels. NeuN (A, E), Tbr1 (I, M), BrdU (B, F, J, O), DAPI (C, G, K, P), and merged images (D, H, L, Q). BrdU colocalization is observed in neurons born at P17 in normoxic (A–D, I–L) and hypoxic cortex (E–H, M–Q). R, S, Total number of NeuN/Brdu (R) and Tbr1/Brdu (S) double-labeled neurons in the cerebral cortex by stereological analyses in wild-type (blue bars) and Fgfr1 cKO (red bars). Scale bar, 10 μm. Values are expressed in 106 units. N = 3 for each group. *p < 0.05 by ANOVA with Sheffe post hoc test.
To clarify whether Fgfr1 cKO mice differed in their reaction to hypoxia, these mutants were injected with BrdU at P17 and assessed 31 d later, according to the protocol described previously. The results indicate that hypoxic Fgfr1 cKO mice were unable to upregulate the rate of cortical neurogenesis 1 week after the insult (Fig. 4R,S, red bars). Hypoxic Fgfr1 cKO showed the same amount of newly generated neurons as their normoxic counterparts, although it remains possible that the Fgfr1 cKO have a smaller neurogenetic response that reaches completion before P17, that is insufficient to reverse the deficits. We also noticed that in the absence of injury, the Fgfr1 cKO mice showed a 48% basal deficit in NeuN+/BrdU+ cortical neurons, but not in Tbr1+/BrdU+ neurons, compared with wild-type littermates (Fig. 4R,S). Hence, whereas Tbr1+/BrdU+ neurons accounted for just 35% of NeuN+/BrdU+ neurons in normoxic wild-type mice, the Tbr1+/BrdU+ cells accounted for 95% of NeuN+/BrdU+ neurons in normoxic Fgfr1 cKO mice (Fig. 4R,S). The data indicate that Fgfr1 is required for the production of non-Tbr1+ neurons under normoxic conditions, as well as for the production of both non-Tbr1+ and Tbr1+ neurons after injury.
Neurogenesis was also assessed in the OB at P48, after the cumulative BrdU injections at P17. The results indicate that while the total density of NeuN+/BrdU+ cells in the granular layer of the OB increased by 335% in hypoxic wild-type mice (p < 0.005), the hypoxic cKO did not show any significant increase compared with their normoxic counterparts (Table 1). The proportion of neurons that were newly generated from these BrdU-labeled cells with respect to all neurons in the OB was 30% in hypoxic wild-type compared with 10% in the normoxic mice, a difference which was highly significant (p < 0.005). Interestingly, the hypoxic cKO did show a modest increase in percentage of OB neurons that arose from BrdU-labeled precursors compared with their normoxic counterparts (from 8 to 16%, p < 0.05) albeit smaller than the wild type (Table 1).
Table 1.
Fgfr1 is required for the hypoxia-induced increase in OB neurogenesis
| Condition | NeuN density (cells/mm3) | BrdU/NeuN density (cells/mm3) | Ratio BrdU/ NeuN to total NeuN |
|---|---|---|---|
| Normoxic WT | 547,795 ± 62,476 | 56,388 ± 9895 | 0.102 ± 0.01 |
| Hypoxic WT | 634,300 ± 58,263a | 189,575 ± 20,981** | 0.303 ± 0.04** |
| Normoxic Fgfr1 cKO | 620,820 ± 206,132 | 61,181 ± 30,414 | 0.088 ± 0.02 |
| Hypoxic Fgfr1 cKO | 658,110 ± 30,250a | 104,210 ± 4605a | 0.160 ± 0.01* |
Mice were exposed to chronic hypoxia from P3 to P10 or were kept under normoxic conditions. Proliferative cells were labeled by in vivo BrdU incorporation for 14 h at P17. The density of NeuN+ neurons and that of newly generated BrdU+/NeuN+ labeled neurons was estimated in the granular layer of the OB at P48 by unbiased stereological analyses. The ratio between newly generated BrdU+/NeuN+ neurons to total NeuN+ neurons is shown in the last column. N = 3 control and 4 hypoxic animals. *p < 0.05 and **p < 0.005 by Student's t test comparing hypoxic versus normoxic mice. WT, Wild type.
aNot statistically different from normoxic mice.
Fgfr1-dependent proliferation of neural progenitors in the reactive period
Confirming data obtained in rat tissue (Ganat et al., 2002), we observed that after cessation of the hypoxic insult, there was a 60% increase in FGFR-1 immunoreactivity in the SVZ of wild-type mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), along with an increase in immunoreactivity for Sox2, Pax6 and Tbr2 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), which are transcription factors expressed by neural stem and precursor cells in the SVZ. As expected, in Fgfr1 cKO mice, the FGFR-1 protein was not detected in the SVZ neuroepithelium, and the upregulation in Sox2, Pax6 and Tbr2+ cells induced by chronic hypoxia was attenuated (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
To assess whether FGFR-1 expression in neural progenitors played a role in their proliferation or survival, BrdU was administered shortly before killing at both P10 and P17. At P10, the hypoxic wild-type and Fgfr1 cKO showed a 30 and 45% loss of BrdU+ cells in the SVZ, respectively, compared with their normoxic counterparts (Fig. 5I). To understand whether some of the stem/progenitor cells may have undergone apoptosis during the exposure to hypoxia, we analyzed the SVZ for activated Caspase-3, a marker for cells undergoing apoptosis, immediately after cessation of injury. In both wild-type and Fgfr1 cKO mice, hypoxia caused a near doubling in number of apoptotic cells in the SVZ at P10 (Fig. 5E–H,J). These data suggest that hypoxia decreases cell proliferation in the SVZ by promoting progenitor apoptosis, and that Fgfr1 does not play a major role in stem/progenitor cell proliferation or survival in normal development, neither it changes their susceptibility to die in response to injury.
Figure 5.
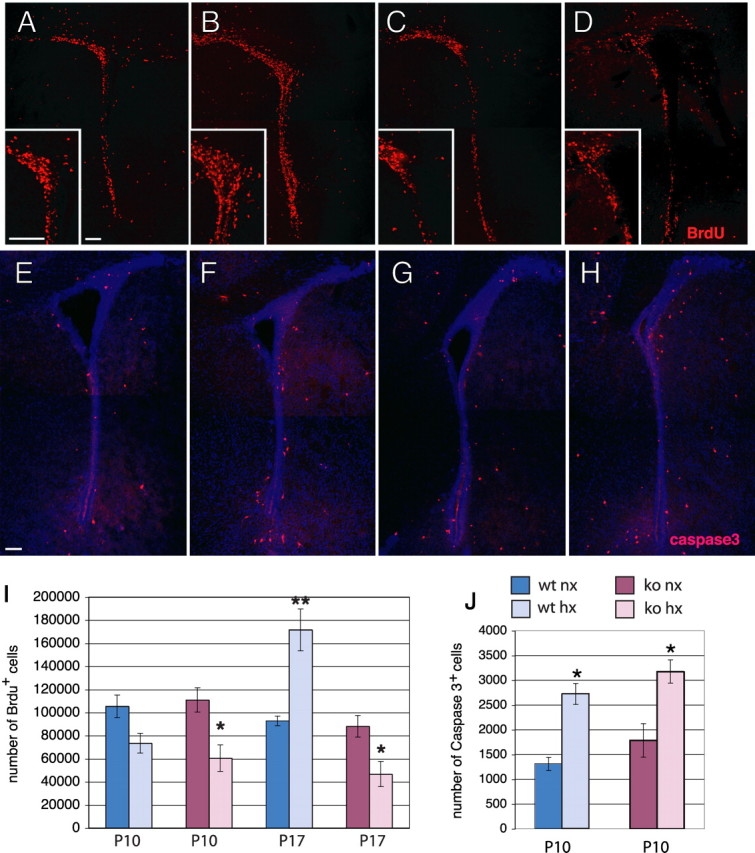
Increase in proliferation in the SVZ during recovery requires Fgfr1. A–D, BrdU immunostaining in the SVZ at P17 of wild-type (A, B) or Fgfr1 cKO mice (C, D) under normoxia (A, C) or after hypoxia (B, D). Insets show high magnifications. E–H, Caspase-3 immunostaining in the SVZ at P10 of wild-type (E, F) or Fgfr1 cKO (G, H) mice under normoxia (E, G) or after hypoxia (F, H). I, J, Total number of BrdU+ cells (I) at P10 and P17 and caspase-3+ cells (J) at P10 in the SVZ by stereological analyses in wild-type (blue bars) and Fgfr1 cKO (red bars). Scale bar, 100 μm. N = 3 for each group. *p < 0.05 and **p < 0.01 by ANOVA with Sheffe post hoc test.
Short-term BrdU incorporation assays were then performed 1 week later, at P17, to assess cell proliferation in the posthypoxia reactive phase in both wild-type and Fgfr1 cKO mice. In the absence of injury, the Fgfr1 cKO mice did not show any difference in cell proliferation compared with the wild-type littermates. After injury, the wild-type hypoxic mice showed an 85% increase in BrdU-positive cells in the SVZ compared with their normoxic counterparts (Fig. 5A,B,I). In Fgfr1 cKO mice, however, the number of BrdU+ cells at P17 further declined to 47% below that of their normoxic counterparts (Fig. 5C,D,I). Our data demonstrate that while basal cell proliferation in nonhypoxic mice was not dependent upon FGFR-1 functioning, FGFR-1 was clearly required for the surge in SVZ cell proliferation after hypoxia, suggesting that this receptor may be an essential component of the hypoxia-induced proliferative response. Combined, these data indicate that after exposure to hypoxia, FGFR-1 may be critical for inducing cell proliferation in normally quiescent astroglial stem/progenitor cells in the SVZ.
Discussion
We report that chronic sublethal hypoxia in newborn mice produces an initial 30% deficit in cortical neurons, two-thirds of which are excitatory neurons expressing the transcription factor Tbr1. Over the ensuing 4 weeks in normoxic conditions, the deficit in neuron number recovers, such that neither NeuN+, Tbr1+ nor SMI-32+ neurons are decreased in the cortex of hypoxic-reared mice at P48. However, there is an enduring loss in PV+ and CR+ inhibitory interneurons in the hypoxic mice, which creates an imbalance between excitatory and inhibitory neurons in the hypoxic cortex. Disrupting the Fgfr1 gene in GFAP+ cells of the developing dorsal telencephalon (including cortical radial glial cells and all their progeny) does not alter the initial loss of cortical neurons but precludes the recovery of NeuN+, Tbr1+ and SMI-32+ neuron number in the cerebral cortex. Hypoxic exposure increases cell proliferation and the generation of Tbr1+ cortical excitatory neurons and that of OB granular neurons, processes that are absent or greatly attenuated in Fgfr1 cKO mice. Thus the juvenile cerebral cortex possesses an unsuspected capacity to recover from a postnatal hypoxic insult, which depends on the functioning of critical signaling systems, i.e., FGFR-1. As rodents are born earlier in the developmental program compared with primates, this hypoxic protocol may model injury during late human gestation and the neonatal period, as it typically occurs in prematurely born infants, as well as ensuing recovery processes in human childhood.
The reconstitution of excitatory cortical neuron number after the hypoxic insult is surprising, as excitatory neurons were previously thought to be generated only during embryogenesis. It is possible that the initial loss may be less profound than what is revealed by NeuN and Tbr1 neuron counts, since some of the damaged neurons may not express neuronal markers and may subsequently recover. We cannot exclude that our assessment of the initial extent of neuron loss may be an over-estimate due to such a process. Nevertheless, we consistently observed corresponding decreases in neuron number, brain weight and cortical thickness in the hypoxia-reared mice, in at least two genetic backgrounds (Fig. 1A,B) (Fagel et al., 2006). Furthermore, we show that cell death is twofold higher the hypoxic brain, indicating probable cell losses.
We noticed that in the current mixed strain of mice, the total number of cortical neurons increases by almost 6 million cells between P10 and P48 in normoxic mice (compare Figs. 1 and 2), suggesting a high degree of cellular plasticity even in the normal juvenile mouse cerebral cortex. This is consistent with recent unbiased stereological studies suggesting that total neuron number progressively increases in the early postnatal rodent cerebral cortex, albeit to varying degrees in different mouse strains, and that there is delayed acquisition of cortical neuron features over the postnatal period (Lyck et al., 2007). Thus, part of the postnatal increase in the number of cortical neurons may be attributable to a progressive accumulation of neuronal antigens, such as NeuN and SMI-32. Nonetheless, using BrdU birthdating assays, we report that part of the increase in cortical neuron number is attributable to the addition of new cortical neurons in the third postnatal week after birth. These BrdU+ neurons are unlikely to be the result of neuronal repair or impending neuronal death (Kuan et al., 2004), because apoptotic cells would have been already eliminated 4 weeks after BrdU incorporation. Furthermore, we have previously shown (Zheng et al., 2004) that neurons strongly immunolabeled with BrdU do not reflect ongoing DNA repair at these dosages of the nucleotide. Also supporting the idea of a hypoxia-induced proliferative effect as opposed to DNA repair are the increase in total number of cells and volume of the SVZ observed by us (Fagel et al., 2006) and others (Plane et al., 2004) and the absence of a correlation between apoptotic cell number and BrdU+ cells in the SVZ at different survival times after the hypoxic insult (Fagel et al., 2006). Finally, we have detected postnatal addition of neurons to the immature cerebral cortex using genetic fate mapping, which allowed us to mark with β-galactosidase GFAP+ neural stem cells in vivo (Ganat et al., 2006).
In wild-type hypoxic mice, BrdU/NeuN and BrdU/Tbr1 double-positive neurons increased twofold and threefold, respectively, compared with age-matched normoxic mice. This can be attributed to hypoxia increasing the proliferation or survival of progenitors, particularly those committed to a Tbr1+ phenotype. Alternatively, hypoxia may increase the capacity of newly born, postmitotic neurons to differentiate and survive in the cortical environment. The first hypothesis is supported by a nearly twofold increase in cell proliferation in the SVZ in hypoxia-exposed mice compared with age-matched controls (this study) (Fagel et al., 2006), and by previous reports suggesting that hypoxia-ischemia increases cell proliferation in the brain (Plane et al., 2004; Ong et al., 2005; Park et al., 2006; Zhao et al., 2008). Furthermore, neural precursor cells expressing the neurogenic transcription factors Sox2, Pax6 and Tbr2 in the SVZ appear to increase after hypoxia. In contrast, in the hypoxic Fgfr1 cKO mice, the SVZ proliferative response is absent, and the increase in Sox2-, Pax6- and Tbr2-immunoreactive cells is attenuated. These data highlight the central role of FGFR-1, whose expression is restricted to glial cells (Belluardo et al., 1997; Vaccarino et al., 2001; Ganat et al., 2002) (Gensat Project: http://www.gensat.org/index.html) in promoting neuronal progenitor proliferation after postnatal hypoxic injury.
Dividing precursors in the SVZ and rostral migratory stream generate neurons for the OB (Luskin, 1993; Alvarez-Buylla and Garcia-Verdugo, 2002; Aguirre and Gallo, 2004; Hack et al., 2005) and FGF-2 (a ligand for FGFR-1) is required for the maintenance of this proliferating cell pool (Zheng et al., 2004). Consistently, Fgfr1 cKO mice are unable to properly upregulate OB neurogenesis in response to hypoxia. The failure to observe a statistically significant increase in density of NeuN+/BrdU+ cells in the OB of Fgfr1 cKO mice may be attributable in part to the large variability between animals; however, the mean increase in NeuN+/BrdU+ cells from normoxic to hypoxic cKO (70%) is much less than that observed wild-type mice (236%), suggesting that if there was an increase in OB neurogenesis in the hypoxic cKO, it was more modest. Thus, the absence of a posthypoxic proliferative response in the SVZ of Fgfr1 cKO mice likely underlies their decreased ability to regenerate OB neurons, although other events unrelated to Fgfr1, such as cell survival, may play a compensatory role in regulating OB neurogenesis.
Fgfr1 cKO mice exposed to hypoxia suffer the same level of apoptosis and initial cortical cell loss as wild-type mice, but fail to recover, exhibiting a persistent 31–33% deficit in total neurons and excitatory cortical neurons 5 weeks later. Furthermore, in the absence of Fgfr1, the generation of cortical neurons from BrdU-labeled cells is not upregulated in the recovery period. These data suggest that Fgfr1 is not involved in neuroprotection after injury, but rather, in the regenerative and reactive response. At present, we do not understand whether the unresponsiveness of SVZ progenitors underlies the failure to boost cortical neurogenesis in the Fgfr1 cKO mice, as BrdU birthdating cannot reveal whether the progenitors that generate cortical neurons arise from the SVZ. It is possible that increased generation of NeuN+ and Tbr1+ cortical neurons occurs from proliferative progenitors in the cortex itself, as mature cortical astrocytes may re-enter the cell cycle and acquire multipotency in vitro after cortical injury (Buffo et al., 2008). Furthermore, although Fgfr1 is clearly required for the hypoxia-induced increase in proliferation in the SVZ, it may play an additional role in fostering neuronal repair in the brain parenchyma, and thus contribute to the recovery of neurons damaged by hypoxia. For example, the loss of FGFR-1 may alter the ability of astroglial cells to regulate the exchange of molecules involved in energy regulation, or secrete trophic factors that might affect neurons or neuronal progenitors in a paracrine manner.
In contrast to the recovery of Tbr1+ neuron number, hypoxia caused an enduring deficit in inhibitory cortical PV+ and CR+ interneurons (32 and 43%, respectively). We could not estimate the extent of the initial loss of PV+ and CR+ inhibitory neurons after hypoxia, since at P11 the maturation of calcium-binding protein immunoreactivities is not yet complete. Hence, we cannot ascertain whether the significant decreases in PV+ and CR+ cells present in hypoxia-reared mice represent an enduring loss, a failure to properly mature, or a progressive demise.
In conclusion, chronic neonatal hypoxia induces a chain of linked events, beginning with the upregulation of FGFR-1 expression in neural stem cells, which in turn may increase the generation of Tbr2+ and Pax6+ proliferative neuronal progenitors. This work provides the first demonstration that, after neonatal chronic hypoxic injury, the initial loss of SMI-32- and Tbr1-expressing excitatory cortical neurons spontaneously recovers in the juvenile cerebral cortex. Furthermore, glial FGFR-1 plays a central role in driving this recovery, as both the increased neurogenesis and reconstitution of normal cortical neuron number are not seen in mice that lack Fgfr1 in GFAP+ cells. This reconstituted neuron number is stable 9 weeks after neonatal hypoxic injury (Fagel et al., 2006). In contrast, we show that PV and CR inhibitory interneurons do not show a comparable degree of recovery, resulting in marked imbalance in excitatory/inhibitory neurons in the hypoxic cerebral cortex.
Footnotes
This work was supported by National Institutes of Health Grant P01 NS 35476. We thank Dr. Paul Lombroso for making available the ApoTome system for this work, Drs. Stewart and Schwartz for the maintenance of the hypoxia apparatus, and Suzanna Luft and Lauren Provini for technical assistance. We are grateful to Dr. Hevner for gift of antibodies and to members of the Vaccarino laboratory for helpful discussions.
References
- Aguirre A, Gallo V. Postnatal neurogenesis and gliogenesis in the olfactory bulb from NG2-expressing progenitors of the subventricular zone. J Neurosci. 2004;24:10530–10541. doi: 10.1523/JNEUROSCI.3572-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allin M, Henderson M, Suckling J, Nosarti C, Rushe T, Fearon P, Stewart AL, Bullmore ET, Rifkin L, Murray R. Effects of very low birthweight on brain structure in adulthood. Dev Med Child Neurol. 2004;46:46–53. doi: 10.1017/s0012162204000088. [DOI] [PubMed] [Google Scholar]
- Alvarez-Buylla A, Garcia-Verdugo JM. Neurogenesis in adult subventricular zone. J Neurosci. 2002;22:629–634. doi: 10.1523/JNEUROSCI.22-03-00629.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, Rueger MA, Bae SK, Kittappa R, McKay RD. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442:823–826. doi: 10.1038/nature04940. [DOI] [PubMed] [Google Scholar]
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8:963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- Belluardo N, Wu G, Mudo G, Hansson AC, Pettersson R, Fuxe K. Comparative localization of fibroblast growth factor receptor-1, -2, and -3 mRNAs in the rat brain: in situ hybridization analysis. J Comp Neurol. 1997;379:226–246. [PubMed] [Google Scholar]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Götz M. Origin and progeny of reactive gliosis: a source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A. 2008;105:3581–3586. doi: 10.1073/pnas.0709002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulfone A, Smiga SM, Shimamura K, Peterson A, Puelles L, Rubenstein JLR. T-brain-1: a homolog of brachyury whose expression defines molecularly distinct domains within the cerebral cortex. Neuron. 1995;15:63–78. doi: 10.1016/0896-6273(95)90065-9. [DOI] [PubMed] [Google Scholar]
- Campbell MJ, Hof PR, Morrison JH. A subpopulation of corticocortical neurons is distinguished by somatodendritic distribution of neurofilament protein. Brain Res. 1991;539:133–136. doi: 10.1016/0006-8993(91)90695-r. [DOI] [PubMed] [Google Scholar]
- Dono R, Texido G, Dussel R, Ehmke H, Zeller R. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J. 1998;17:4213–4225. doi: 10.1093/emboj/17.15.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagel DM, Ganat Y, Silbereis J, Ebbitt T, Stewart W, Zhang H, Ment LR, Vaccarino FM. Cortical neurogenesis enhanced by chronic perinatal hypoxia. Exp Neurol. 2006;199:77–91. doi: 10.1016/j.expneurol.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Fearon P, O'Connell P, Frangou S, Aquino P, Nosarti C, Allin M, Taylor M, Stewart A, Rifkin L, Murray R. Brain volumes in adult survivors of very low birth weight: a sibling-controlled study. Pediatrics. 2004;114:367–371. doi: 10.1542/peds.114.2.367. [DOI] [PubMed] [Google Scholar]
- Ganat Y, Soni S, Chacon M, Schwartz ML, Vaccarino FM. Chronic hypoxia up-regulates fibroblast growth factor ligands in the perinatal brain and induces fibroblast growth factor-responsive radial glial cells in the sub-ependymal zone. Neuroscience. 2002;112:977–991. doi: 10.1016/s0306-4522(02)00060-x. [DOI] [PubMed] [Google Scholar]
- Ganat YM, Silbereis J, Cave C, Ngu H, Anderson GM, Ohkubo Y, Ment LR, Vaccarino FM. Early postnatal astroglial cells produce multilineage precursors and neural stem cells in vivo. J Neurosci. 2006;26:8609–8621. doi: 10.1523/JNEUROSCI.2532-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack M, Flannery DJ, Schluchter M, Cartar L, Borawski E, Klein N. Outcomes in young adulthood for very-low-birth-weight infants. N Engl J Med. 2002;346:149–157. doi: 10.1056/NEJMoa010856. [DOI] [PubMed] [Google Scholar]
- Hack MA, Saghatelyan A, de Chevigny A, Pfeifer A, Ashery-Padan R, Lledo PM, Götz M. Neuronal fate determinants of adult olfactory bulb neurogenesis. Nat Neurosci. 2005;8:865–872. doi: 10.1038/nn1479. [DOI] [PubMed] [Google Scholar]
- Hayes NL, Nowakowski RS. Exploiting the dynamics of S-phase tracers in developing brain: interkinetic nuclear migration for cells entering versus leaving the S-phase. Dev Neurosci. 2000;22:44–55. doi: 10.1159/000017426. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Shi L, Justice N, Hsueh Y, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, Rubenstein JL. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–366. doi: 10.1016/s0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Hodge RD, Daza RA, Englund C. Transcription factors in glutamatergic neurogenesis: conserved programs in neocortex, cerebellum, and adult hippocampus. Neurosci Res. 2006;55:223–233. doi: 10.1016/j.neures.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Hoehn BD, Palmer TD, Steinberg GK. Neurogenesis in rats after focal cerebral ischemia is enhanced by indomethacin. Stroke. 2005;36:2718–2724. doi: 10.1161/01.STR.0000190020.30282.cc. [DOI] [PubMed] [Google Scholar]
- Korada S, Zheng W, Basilico C, Schwartz ML, Vaccarino FM. Fgf2 is necessary for the growth of glutamate projection neurons in the anterior neocortex. J Neurosci. 2002;22:863–875. doi: 10.1523/JNEUROSCI.22-03-00863.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornack DR, Rakic P. Cell proliferation without neurogenesis in adult primate neocortex. Science. 2001;294:2127–2130. doi: 10.1126/science.1065467. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Schloemer AJ, Lu A, Burns KA, Weng WL, Williams MT, Strauss KI, Vorhees CV, Flavell RA, Davis RJ, Sharp FR, Rakic P. Hypoxia-ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J Neurosci. 2004;24:10763–10772. doi: 10.1523/JNEUROSCI.3883-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luskin MB. Restricted proliferation and migration of postnatally generated neurons derived from the forebrain ventricular zone. Neuron. 1993;11:173–189. doi: 10.1016/0896-6273(93)90281-u. [DOI] [PubMed] [Google Scholar]
- Lyck L, Krøigård T, Finsen B. Unbiased cell quantification reveals a continued increase in the number of neocortical neurones during early post-natal development in mice. Eur J Neurosci. 2007;26:1749–1764. doi: 10.1111/j.1460-9568.2007.05763.x. [DOI] [PubMed] [Google Scholar]
- Magavi SS, Macklis JD. Induction of neuronal type-specific neurogenesis in the cerebral cortex of adult mice: manipulation of neural precursors in situ. Brain Res Dev Brain Res. 2002;134:57–76. doi: 10.1016/s0165-3806(01)00316-9. [DOI] [PubMed] [Google Scholar]
- Ment LR, Vohr B, Allan W, Katz KH, Schneider KC, Westerveld M, Duncan CC, Makuch RW. Change in cognitive function over time in very low-birth-weight infants. Jama. 2003;289:705–711. doi: 10.1001/jama.289.6.705. [DOI] [PubMed] [Google Scholar]
- Ment LR, Allan WC, Makuch RW, Vohr B. Grade 3 to 4 intraventricular hemorrhage and Bayley scores predict outcome. Pediatrics. 2005;116:1597–1598. doi: 10.1542/peds.2005-2020. author reply 1598. [DOI] [PubMed] [Google Scholar]
- Monfils MH, Driscoll I, Kamitakahara H, Wilson B, Flynn C, Teskey GC, Kleim JA, Kolb B. FGF-2-induced cell proliferation stimulates anatomical, neurophysiological and functional recovery from neonatal motor cortex injury. Eur J Neurosci. 2006;24:739–749. doi: 10.1111/j.1460-9568.2006.04939.x. [DOI] [PubMed] [Google Scholar]
- Müller Smith K, Fagel DM, Stevens HE, Rabenstein RL, Maragnoli ME, Ohkubo Y, Picciotto MR, Schwartz ML, Vaccarino FM. Deficiency in inhibitory cortical interneurons associates with hyperactivity in fibroblast growth factor receptor 1 mutant mice. Biol Psychiatry. 2008;63:953–962. doi: 10.1016/j.biopsych.2007.09.020. [DOI] [PubMed] [Google Scholar]
- Nakatomi H, Kuriu T, Okabe S, Yamamoto S, Hatano O, Kawahara N, Tamura A, Kirino T, Nakafuku M. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural precursors. Cell. 2002;110:429–441. doi: 10.1016/s0092-8674(02)00862-0. [DOI] [PubMed] [Google Scholar]
- Nowakowski RS, Lewin SB, Miller MW. Bromodeoxyuridine immunohistochemical determination of the lengths of the cell cycle and the DNA-synthetic phase for an anatomically defined population. J Neurocytol. 1989;18:311–318. doi: 10.1007/BF01190834. [DOI] [PubMed] [Google Scholar]
- Ohkubo Y, Uchida AO, Shin D, Partanen J, Vaccarino FM. Fibroblast growth factor receptor 1 is required for the proliferation of hippocampal progenitor cells and for hippocampal growth in mouse. J Neurosci. 2004;24:6057–6069. doi: 10.1523/JNEUROSCI.1140-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong J, Plane JM, Parent JM, Silverstein FS. Hypoxic-ischemic injury stimulates subventricular zone proliferation and neurogenesis in the neonatal rat. Pediatr Res. 2005;58:600–606. doi: 10.1203/01.PDR.0000179381.86809.02. [DOI] [PubMed] [Google Scholar]
- Ortega S, Ittmann M, Tsang SH, Ehrlich M, Basilico C. Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc Natl Acad Sci U S A. 1998;95:5672–5677. doi: 10.1073/pnas.95.10.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM. Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol. 2002;52:802–813. doi: 10.1002/ana.10393. [DOI] [PubMed] [Google Scholar]
- Park KI, Hack MA, Ourednik J, Yandava B, Flax JD, Stieg PE, Gullans S, Jensen FE, Sidman RL, Ourednik V, Snyder EY. Acute injury directs the migration, proliferation, and differentiation of solid organ stem cells: evidence from the effect of hypoxia-ischemia in the CNS on clonal “reporter” neural stem cells. Exp Neurol. 2006;199:156–178. doi: 10.1016/j.expneurol.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Plane JM, Liu R, Wang TW, Silverstein FS, Parent JM. Neonatal hypoxic-ischemic injury increases forebrain subventricular zone neurogenesis in the mouse. Neurobiol Dis. 2004;16:585–595. doi: 10.1016/j.nbd.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Raballo R, Rhee J, Lyn-Cook R, Leckman JF, Schwartz ML, Vaccarino FM. Basic fibroblast growth factor (Fgf2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J Neurosci. 2000;20:5012–5023. doi: 10.1523/JNEUROSCI.20-13-05012.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saigal S, Doyle LW. An overview of mortality and sequelae of preterm birth from infancy to adulthood. Lancet. 2008;371:261–269. doi: 10.1016/S0140-6736(08)60136-1. [DOI] [PubMed] [Google Scholar]
- Shin DM, Korada S, Raballo R, Shashikant CS, Simeone A, Taylor JR, Vaccarino F. Loss of glutamatergic pyramidal neurons in frontal and temporal cortex resulting from attenuation of FGFR1 signaling is associated with spontaneous hyperactivity in mice. J Neurosci. 2004;24:2247–2258. doi: 10.1523/JNEUROSCI.5285-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KM, Ohkubo Y, Maragnoli ME, Rasin MR, Schwartz ML, Sestan N, Vaccarino FM. Midline radial glia translocation and corpus callosum formation require FGF signaling. Nat Neurosci. 2006;9:787–797. doi: 10.1038/nn1705. [DOI] [PubMed] [Google Scholar]
- Stavridis MP, Lunn JS, Collins BJ, Storey KG. A discrete period of FGF-induced Erk1/2 signalling is required for vertebrate neural specification. Development. 2007;134:2889–2894. doi: 10.1242/dev.02858. [DOI] [PubMed] [Google Scholar]
- Tao Y, Black IB, DiCicco-Bloom E. Neurogenesis in neonatal rat brain is regulated by peripheral injection of basic fibroblast growth factor (bFGF) J Comp Neurol. 1996;376:653–663. doi: 10.1002/(SICI)1096-9861(19961223)376:4<653::AID-CNE11>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Trokovic R, Trokovic N, Hernesniemi S, Pirvola U, Vogt Weisenhorn DM, Rossant J, McMahon AP, Wurst W, Partanen J. FGFR1 is independently required in both developing mid- and hindbrain for sustained response to isthmic signals. EMBO J. 2003;22:1811–1823. doi: 10.1093/emboj/cdg169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CP, Seli M, Ment L, Stewart W, Yan H, Johansson B, Fredholm BB, Blackburn M, Rivkees SA. A1 adenosine receptors mediate hypoxia-induced ventriculomegaly. Proc Natl Acad Sci U S A. 2003;100:11718–11722. doi: 10.1073/pnas.1931975100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccarino FM, Schwartz ML, Raballo R, Rhee J, Lyn-Cook R. Fibroblast growth factor signaling regulates growth and morphogenesis at multiple steps during brain development. In: Pedersen RA, Shatten G, editors. Current topics in developmental biology. San Diego: Academic; 1999a. pp. 179–200. [DOI] [PubMed] [Google Scholar]
- Vaccarino FM, Schwartz ML, Raballo R, Nilsen J, Rhee J, Zhou M, Doetschman T, Coffin JD, Wyland JJ, Hung YT. Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat Neurosci. 1999b;2:246–253. doi: 10.1038/6350. [DOI] [PubMed] [Google Scholar]
- Vaccarino FM, Ganat Y, Zhang Y, Zheng W. Stem cells in neurodevelopment and plasticity. Neuropsychopharmacology. 2001;25:805–815. doi: 10.1016/S0893-133X(01)00349-9. [DOI] [PubMed] [Google Scholar]
- Wagner JP, Black IB, DiCicco-Bloom E. Stimulation of neonatal and adult brain neurogenesis by subcutaneous injection of basic fibroblast growth factor. J Neurosci. 1999;19:6006–6016. doi: 10.1523/JNEUROSCI.19-14-06006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J, Takizawa B, McGee A, Stewart WB, Zhang H, Ment L, Schwartz M, Strittmatter S. Neonatal hypoxia suppresses oligodendrocyte Nogo-A and increases axonal sprouting in a rodent model for human prematurity. Exp Neurol. 2004;189:141–149. doi: 10.1016/j.expneurol.2004.05.018. [DOI] [PubMed] [Google Scholar]
- Yang Z, Covey MV, Bitel CL, Ni L, Jonakait GM, Levison SW. Sustained neocortical neurogenesis after neonatal hypoxic/ischemic injury. Ann Neurol. 2007;61:199–208. doi: 10.1002/ana.21068. [DOI] [PubMed] [Google Scholar]
- Yoshimura S, Takagi Y, Harada J, Teramoto T, Thomas SS, Waeber C, Bakowska JC, Breakefield XO, Moskowitz MA. FGF-2 regulation of neurogenesis in adult hippocampus after brain injury. Proc Natl Acad Sci U S A. 2001;98:5874–5879. doi: 10.1073/pnas.101034998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Zhang CP, Liu ZH, Wu LY, Huang X, Wu HT, Xiong L, Wang X, Wang XM, Zhu LL, Fan M. Hypoxia-driven proliferation of embryonic neural stem/progenitor cells–role of hypoxia-inducible transcription factor-1alpha. FEBS J. 2008;275:1824–1834. doi: 10.1111/j.1742-4658.2008.06340.x. [DOI] [PubMed] [Google Scholar]
- Zheng W, Nowakowski RS, Vaccarino FM. Fibroblast growth factor 2 is required for maintaining the neural stem cell pool in the mouse brain subventricular zone. Dev Neurosci. 2004;26:181–196. doi: 10.1159/000082136. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]