

Abstract
7-Ketocholesterol (7KC) is a cytotoxic component of oxidized low density lipoproteins (OxLDLs) and induces apoptosis in macrophages by a mechanism involving the activation of cytosolic phospholipase A2 (cPLA2). In the current study, we examined the role of ACAT in 7KC-induced and OxLDL-induced apoptosis in murine macrophages. An ACAT inhibitor, Sandoz 58-035, suppressed 7KC-induced apoptosis in P388D1 cells and both 7KC-induced and OxLDL-induced apoptosis in mouse peritoneal macrophages (MPMs). Furthermore, compared with wild-type MPMs, ACAT-1-deficient MPMs demonstrated significant resistance to both 7KC-induced and OxLDL-induced apoptosis. Macrophages treated with 7KC accumulated ACAT-derived [14C]cholesteryl and [3H]7-ketocholesteryl esters. Tandem LC-MS revealed that the 7KC esters contained primarily saturated and monounsaturated fatty acids. An inhibitor of cPLA2, arachidonyl trifluoromethyl ketone, prevented the accumulation of 7KC esters and inhibited 7KC-induced apoptosis in P388D1 cells. The decrease in 7KC ester accumulation produced by the inhibition of cPLA2 was reversed by supplementing with either oleic or arachidonic acid (AA); however, only AA supplementation restored the induction of apoptosis by 7KC. These results suggest that 7KC not only initiates the apoptosis pathway by activating cPLA2, as we have reported previously, but also participates in the downstream signaling pathway when esterified by ACAT to form 7KC-arachidonate.
Keywords: 7-ketocholesterol, oxysterol esterification, low density lipoproteins
The presence of apoptotic macrophages within atherosclerotic lesions of both animals and humans has been observed consistently during the past decade; however, the physiological significance of macrophage apoptosis in atherogenesis is not fully understood. Macrophage apoptosis has been proposed to be a mechanism by which lipids accumulate within the coronary vasculature and thereby contribute to plaque formation and progression (1, 2). Alternatively, macrophage apoptosis has been speculated to be a defense mechanism, perhaps to limit the number of lesional macrophages and the hazardous consequences associated with necrotic cell death within lesions (3). Recent results from our laboratory demonstrated that macrophage apoptosis via the mitochondrial apoptosis pathway has an overall protective role in atherogenesis (4). In this study, mice with genetically altered macrophages deficient for Bax, a proapoptotic gene, developed larger and more advanced lesions with reduced apoptosis compared with control mice with normal macrophages. This result underscores the necessity to fully elucidate the mechanisms that regulate macrophage apoptosis and potentially influence the development and progression of atherosclerosis.
Many of the pathological events associated with the development of atherosclerosis are related to the uptake and metabolism of modified LDL, such as oxidized low density lipoprotein (OxLDL). The continuous uptake of OxLDL by macrophages in the vascular intima via specific receptor-mediated mechanisms and by phagocytosis (5-7) results in the formation of lipid-laden “foam cells” that are the defining characteristic of early atherosclerotic lesions (8). The ingestion of OxLDL by macrophages has been shown to be eventually cytotoxic (9-12), and this cytotoxicity proceeds, at least in part, through the induction of the mitochondrial apoptosis pathway (13, 14).
Cholesterol oxidation products found in OxLDL, collectively referred to as oxysterols (15, 16), have been demonstrated to largely account for the apoptotic activity of OxLDL in macrophages (17, 18). Oxysterols are commonly found in foods of animal origin (19) and have been demonstrated to exist both in OxLDL (20) and in atherosclerotic plaque (21). The predominant oxysterol present in OxLDL is 7-ketocholesterol (7KC), which accounts for up to 30% of the total sterols in OxLDL (22). 7KC in the 10-25 μM range induces apoptosis in macrophages, vascular endothelial cells, and smooth muscle cells (23). 25-Hydroxycholesterol (25-OHC) is another oxysterol found in OxLDL, albeit at much lower concentrations than 7KC (22). 25-OHC has been used extensively as a model oxysterol compound in studies of both oxysterol toxicity and cellular cholesterol homeostasis. 25-OHC induces apoptosis in cultured monocyte-macrophages (13, 24, 25) and lymphoid cell lines (26, 27) in the range of 1-10 μM. The concentrations of oxysterols used to induce apoptosis in these in vitro studies are below the upper limit of the physiological concentration of oxysterols detected in human plasma after the ingestion of a test meal, which has been reported to be as high as 37 μM (28), and are within the range of oxysterols most commonly detected in plasma of individuals with hypercholesterolemia, 10-30 μM (29-31). Like OxLDL, oxysterols induce the release of cytochrome c from mitochondria (32, 33), the hallmark of the mitochondrial apoptosis pathway.
Our previous work demonstrated that the mechanism by which oxysterols (25-OHC and 7KC) induce apoptosis involves an increase in cytosolic calcium and the subsequent activation of cytosolic phospholipase A2 (cPLA2), and the loss of cPLA2 activity, either by the use of specific inhibitors or by cPLA2 gene knockout, results in resistance to oxysterol-induced apoptosis in macrophages (13, 34). We have also shown previously that macrophages treated with 5,8,11,14-eicosatetraynoic acid (ETYA), an inhibitor of arachidonic acid (AA) metabolism, are resistant to the induction of apoptosis by 25-OHC or 7KC (13, 34), indicating that metabolism of cPLA2-released AA may be required for the induction of apoptosis by these oxysterols.
OxLDL also activates cPLA2 in macrophages and the fatty acids that are released as a result of this activation have been shown to become esterified to OxLDL-derived cholesterol (35). Intracellular cholesterol esterification is catalyzed by the microsomal enzyme acyl-coenzyme A:cholesterol acyltransferase (ACAT), which uses cholesterol and fatty acyl CoA as substrates (36, 37). In addition to cholesterol, a variety of oxysterols are also ACAT substrates (38), and the oxysterols found in atherosclerotic plaques are predominantly esterified (39). This suggests that the fatty acids released by the oxysterol-induced activation of cPLA2 in macrophages may be used as substrates in esterification reactions catalyzed by ACAT and that ACAT potentially plays a role in the apoptotic signaling pathway induced by the oxysterol treatment of macrophages.
Therefore, we undertook the current study to examine the hypothesis that ACAT plays a role in OxLDL/oxysterol-induced apoptosis in murine macrophages. For this study, we used the well-studied P388D1 macrophage cell line as a model and then verified our findings in isolated mouse peritoneal macrophages (MPMs), as we have done previously to elucidate the oxysterol apoptotic signaling mechanism in macrophages (13, 34). Our results reveal that the loss of ACAT activity in macrophages, either by pharmacological inhibition or by genetic knockout of ACAT-1, results in reduced induction of apoptosis in response to 7KC or OxLDL. Furthermore, we provide evidence that the apoptotic signal generated in macrophages by ACAT may be an arachidonyl oxysterol.
MATERIALS AND METHODS
Materials
P388D1 cells were purchased from the American Tissue Type Collection (Manassas, VA). BioWhittaker cell culture media and Falcon tissue culture plates were purchased from Fisher Scientific (Pittsburgh, PA). NovaCell 1 FBS was purchased from Novatech, Inc. (Grand Island, NY). All other tissue culture supplements were purchased from Invitrogen (Carlsbad, CA). The acyl-CoA: cholesterol acyltransferase inhibitor 3-[decyldimethylsily]N-[2(4-methylphenyl)-1-phenylethyl]propanamide (58-035) was kindly provided by Novartis Pharmaceuticals Corp. (formerly Sandoz, Inc.) (East Hanover, NJ). The following were obtained from Biomol Research Laboratories, Inc. (Plymouth Meeting, PA): arachidonyl trifluoromethyl ketone (AACOCF3), ETYA, 3β-(2-diethylaminoethoxy)-androstenone (U-18666A), and caspase-3 assay substrate (Ac-DEVD-AFC) and inhibitor (Ac-DEVD-CHO). 7KC was obtained from Steraloids, Inc. (Newport, RI). All radiolabeled chemicals, [1,2,6-3H]7KC and [14C]cholesterol, were purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO). Oleoyl chloride, arachidonoyl chloride, stearoyl chloride, linoleoyl chloride, and palmitoyl chloride were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). Silicic acid (100-200 mesh) was purchased from Bio-Rad Laboratories (Richmond, CA). Organic solvents and reagents were of analytical or HPLC grade and were obtained from either Fisher Scientific or Sigma-Aldrich Chemical Co.
Mice
C57Bl/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME). ACAT-1-deficient (ACAT-1-/- ) mice were bred and fully backcrossed to C57BL/6J genetic strain at Vanderbilt University School of Medicine to allow direct comparison with wild-type C57Bl/6J mice and have been described previously (40). All animal procedures described in this study were performed in accordance with guidelines administered by the Animal Research Facility at East Tennessee State University Quillen College of Medicine or by the Institutional Animal Care and Usage Committee at Vanderbilt University School of Medicine.
Cell Culture
All cells were maintained in a humidified atmosphere of 5% CO2 and 95% air at 37°C as described previously (34). P388D1 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Preparation of OxLDL
LDL (1.019 < d < 1.063) was prepared from normal human serum by sequential ultracentrifugation, extensively oxidized by incubation with CuCl2, and evaluated as described previously (11). The OxLDL was dialyzed against PBS containing 200 μM EDTA at 4°C, stored in the dark, and used within 2 weeks.
Caspase-3 assay
Log-phase P388D1 cells were seeded at 2 × 106/well: 12-well culture plates in 1 ml of cell culture media (DMEM + 10% FBS). One hour before the addition of 7KC or OxLDL, inhibitors were diluted directly into the media from stock solutions made in DMSO, whereas controls received an equivalent volume of vehicle. 7KC (from 2 mg/ml ethanol stock solutions) or OxLDL was then added to the wells. Controls received an equivalent volume of vehicle. The cells were incubated for 16 h, harvested, and assayed for caspase-3 and caspase-3-like activity using a fluorogenic substrate (Ac-DEVD-AFC) and a spectrofluorometer equipped with a microplate reader, as described previously (34). The relative fluorescence units were normalized to the protein concentration of the sample, which was determined by μBCA assay (Pierce, Rockford, IL).
Analysis of apoptosis in MPMs
MPMs were isolated by lavage of the peritoneal cavity and cultured essentially as described previously (13). Isolated MPMs were plated at a density of 1 × 105/well on eight-well glass chamber slides (Nalge Nunc International, Naperville, IL) in DMEM + 10% FBS and allowed to attach for 24 h in a humidified 5% CO2 incubator at 37°C. Unattached cells were removed by washing twice with PBS. The MPMs were preincubated in DMEM + 10% FBS supplemented with and without inhibitors for 1 h before the addition of 7KC or OxLDL, as indicated in the figure legends. Control wells received an equivalent volume of vehicle. After incubating for 24 or 48 h, the cells were rinsed with PBS and fixed in 4% buffered paraformaldehyde for 30 min at room temperature. The cells were then rinsed with PBS, permeabilized, and subjected to DNA end labeling with isothiocyanate-dUTP using a commercially available in situ apoptosis detection kit, as described previously (13). The terminal transferase-mediated dUTP nick end labeling (TUNEL) assay was performed blind, and TUNEL-positive cells were identified by observation using a Nikon Diaphot-200. For each treatment, the percentage of TUNEL-positive cells in 10 randomly chosen fields (45-60 cells/field) was determined.
Determination of cholesteryl and oxysteryl ester formation
P388D1 cells were seeded as described above for caspase-3 assays. The culture medium was supplemented with and without inhibitors, and the controls received an equivalent volume of vehicle. After a 1 h incubation, a tracer amount of a radiolabeled oxysterol (1 μCi/ml [3H]7KC) or cholesterol (0.5 μCi/ml [14C]cholesterol) was added. At this time, unlabeled oxysterols were added (controls received an equivalent volume of vehicle) and the incubation continued for 16 h. The cells were collected, rinsed with ice-cold PBS twice, and the lipids were extracted with hexane-isopropanol (60:40, v/v). The lipids were dried under a stream of N2 gas, resuspended in hexane, and subjected to TLC on silica gel 60 plates with hexane-diethyl ether-acetic acid (80:20:1, v/v/v) as the mobile phase. Using this system, esters of 7KC and cholesterol migrate farther (relative mobilities of ∼0.41 and ∼0.7, respectively) than unesterified 7KC and cholesterol, which remain at the origin. The plates were dried, saturated with En3Hance (Perkin-Elmer Life and Analytical Sciences, Inc., Boston, MA), and subjected to autofluorography. The identity of ACAT-derived products was inferred by their inhibition with 58-035 and when possible by their comigration with authentic standards. The areas corresponding to 7KC or cholesteryl esters were scraped from the plate, and radioactivity was measured by liquid scintillation counting. The lipid-extracted cell pellets were dissolved in 0.1 N NaOH, and protein concentrations were determined by μBCA assay (Pierce). All treatments were done in triplicate and data are presented as mean dpm/mg protein ± SD.
Analysis of 7-ketocholesteryl esters by tandem LC/MS
Log-phase P388D1 cells (2.0 × 106/ml) were cultured in DMEM + 10% FBS supplemented with 7KC (20 μg/ml) for 16 h at 37°C. The cells were then collected, and rinsed with ice-cold PBS twice, and the intracellular lipids were extracted with hexane-isopropanol (60:40, v/v) in the presence of butylated hydroxyl toluene (50 μg/ml) at room temperature for 20 min. The mixture was centrifuged for 15 min at 1,000 g, the lipid extracts were placed in a clean glass vial, dried under a stream of N2 gas, resuspended in chloroform-methanol (3:1), and stored at -80°C until analyzed.
Similar to Brown, Dean, and Jessup (22), we synthesized five 7-ketocholesteryl esters (7KC esters): palmitate, stearate, oleate, linoleate, and arachidonate. The synthesis of these 7KC esters was by means of modification of the method of Piretti and Pagliuca (41). Equal molar amounts of 7KC and fatty acyl chlorides were dissolved in 0.5 ml of anhydrous pyridine and incubated at 56°C for 3 h. The reaction products were extracted into petroleum ether, the organic phase was placed into a clean glass tube, and dried under nitrogen. After evaporating the solvent, the crude residue was resuspended in hexane, and the free 7KC was separated from the esterified 7KC on silica gel columns (42). The purity of the 7KC esters was >98%, as determined by reverse-phase high-performance liquid chromatography on a Versapack C18 column (Alltech, Nicholasville, KY) using a mobile phase of acetonitrile-isopropanol-water (44:54:2, v/v/v) as described (22). The purified 7KC esters were dissolved in chloroform-methanol (3:1, v/v) and stored at -80°C. The concentration of the 7KC esters was determined using an Amplex Red Cholesterol Assay Kit (Molecular Probes, Eugene, OR) with 7KC as the standard.
Equimolar mixtures of the five 7KC ester standards (16:0, 18:0, 18:1, 18:2, and 20:4) were prepared in isopropanol to give concentrations of 0.1, 1.0, and 10.0 μM. The pentafluorophenylhydrazone derivatives were prepared by treatment of a 100 μl aliquot of each standard with 100 μl of 10 mM pentafluorophenylhydrazine in isopropanol, followed by the addition of 20 μl of glacial acetic acid. The reaction mixture was heated at 60°C for 1 h and then analyzed without further dilution by LC/MS. A 100 μl aliquot (from 1 ml total) of an extract from macrophage lipids was derivatized by this method.
The 7KC ester pentafluorophenylhydrazone derivatives were analyzed by tandem LC/MS using a ThermoElectron Corp. (San Jose, CA) Quantum Discovery triple quadrupole LC/MS instrument equipped with a Surveyor HPLC system and autosampler. Samples were ionized using the standard electrospray ion source. The electrospray ion source was operated at 4.0 kV, using nitrogen for both the nebulizer sheath gas and the auxiliary drying gas. The ion transfer capillary was operated at 325°C with a source declustering potential of 10 V. The collision cell was operated at an argon pressure of 1.5 mTorr. Survey studies in tandem MS mode showed that the most prominent product ion was derived from scission of the fatty acyl-sterol bond to give an ion at m/z 563 attributable to loss of the fatty acid as a neutral species. The optimum collision energy was 32 V for all of the 7KC esters that were examined. Quantitation was performed by selected reaction monitoring using the [M + H] precursor ions for selected C16, C18, and C20 esters and the common product ion at m/z 563. LC/MS separations were performed on a 2.1 × 150 mm Zorbax XDB-C8 5 μ silica column using isocratic elution at 300 μl/min. The elution solvent was acetonitrile-isopropanol-water (50:45:5) containing 0.5% acetic acid.
Quantitation was done by the method of external normalization of individual 7KC esters using accurately prepared complex mixtures of the five standards. Thus, 20 μl of a 0.1 μM standard (2 pmol total) was used to establish the absolute LC/MS response factor for individual standards in area counts per picomole. The absolute LC/MS response factor was used to calculate the absolute amount of individual lipid species injected into the instrument in picomoles based on the accurate extraction and dilution of total lipids in macrophages. The relative response factors for individual 7KC esters ranged from 0.14 (18:1) to 1.0 (18:0); these were used to calculate the normalized amount of individual species present in a macrophage lipid extract. Cell culture samples were analyzed in triplicate and the means reported. The absolute detection limit by this method is ∼10-50 fmol, depending on the fatty acyl species.
Statistical analysis
Data are presented as means ± SD of triplicate treatments. Student’s t-test was used to analyze the significance of differences between independent samples. P < 0.05 was considered statistically significant.
RESULTS
Pharmacological inhibition of ACAT prevents 7KC-induced apoptosis in a murine macrophage cell line
To determine whether ACAT activity plays a role in the induction of apoptosis by 7KC in macrophages, we tested the ability of a well-characterized ACAT inhibitor, Sandoz compound 58-035 (43), to effect 7KC-induced apoptosis in P388D1 cells. Control experiments were performed (data not shown) in which the incorporation of labeled oleate into cholesteryl ester was used to determine dosages of 58-035 that could produce the loss of detectable cholesterol esterification in macrophage cell lines and MPMs that has been reported by others (44). Treatment of P388D1 cells with 58-035 (10 μg/ml) effectively blocked the induction of caspase-3 activity by 7KC (Fig. 1). 58-035 also prevented the induction of apoptosis by 7KC in RAW 264.7 macrophages (data not shown). A similar result was observed when P388D1 or RAW 264.7 cells were incubated with 25-OHC in the presence of 58-035 (data not shown); however, 58-035 had no effect on the induction of apoptosis by staurosporine, a protein kinase inhibitor well known to induce apoptosis via the mitochondrial pathway (data not shown).
Fig. 1.
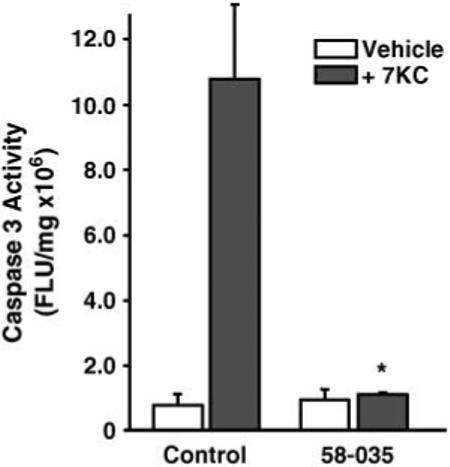
An ACAT inhibitor prevents 7-ketocholesterol (7KC)-induced apoptosis in P388D1 cells. P388D1 cells were preincubated for 1 h with an ACAT inhibitor, 58-035 (10 μg/ml), before a 16 h incubation with and without 7KC (10 μg/ml), as indicated. The cells were then assayed for caspase-3 activity as described in Materials and Methods. FLU, fluorescence light units. The data represent means ± SD of triplicate treatments. * P < 0.05 versus 7KC-treated controls.
An ACAT inhibitor attenuates 7KC- and OxLDL-induced apoptosis in isolated resident peritoneal macrophages
We also analyzed the effects of 58-035 on apoptosis induced by 7KC in isolated MPMs. Isolation of resident MPMs does not yield enough cells to accurately quantitate caspase-3 activity by our method unless we pool MPMs isolated from a large cohort of mice. Therefore, we used an in situ TUNEL method to quantitate the apoptosis. The TUNEL assay has been demonstrated to be a highly sensitive and specific method for detecting individual apoptotic cells grown on coverslips (45) and has been used previously by our laboratory to quantitate apoptosis in MPMs (13). Treatment of MPMs with 7KC (10 μg/ml) for 24 h induced an ∼5.8-fold increase in the percentage of TUNEL-positive cells compared with untreated controls (Fig. 2A). When the medium was supplemented with 58-035, the induction of TUNEL-positive MPMs by 7KC was prevented (Fig. 2A). Apoptosis was also induced in MPMs by treatment with 25-OHC, which was effectively prevented by 58-035 (data not shown).
Fig. 2.
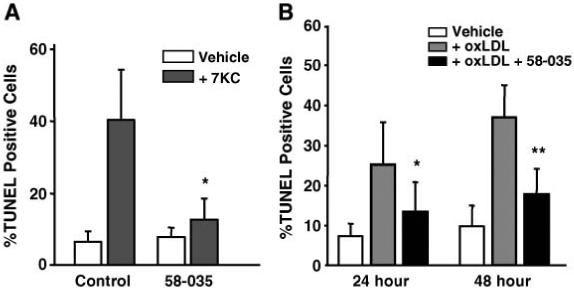
An ACAT inhibitor reduces the induction of apoptosis by 7KC and oxidized low density lipoprotein (OxLDL) in isolated peritoneal macrophages. A: The effect of 58-035 on the induction of terminal transferase-mediated dUTP nick end labeling (TUNEL)-positive cells after a 24 h treatment with 7KC treatment. Resident peritoneal macrophages were isolated, plated on glass cover slips, and allowed to attach. The media was changed and supplemented with or without 58-035 (10 μg/ml) for 1 h before supplementation with and without 7KC (10 μg/ml), as indicated. The incubations were continued for 24 h. Apoptosis was then assayed by in situ TUNEL analysis as described in Materials and Methods. B: The effect of 58-035 on the induction of TUNEL-positive cells after 24 and 48 h incubations with OxLDL. Isolated resident peritoneal macrophages were supplemented with and without 58-035 (10 μg/ml) for 1 h before the addition of OxLDL (50 μg/ml). The incubation was continued for 24 and 48 h, as indicated. The induction of apoptosis was determined by in situ TUNEL analysis. The results are presented as mean percentages of TUNEL-positive cells ± SD in 10 random fields for each condition. The results shown are representative of three independent experiments. * P < 0.01 and ** P < 0.001 versus 7KC- or OxLDL-treated controls.
Incubation with 50 μg/ml OxLDL for 24-48 h is commonly used to induce macrophage foam cell formation in vitro (35). In addition, the degree of apoptotic induction we observed using 10 μg/ml 7KC (Fig. 2A) is nearly identical to the degree of apoptotic induction we reported previously when MPMs were incubated with 50 μg/ml OxLDL for 24 h (13). Therefore, we performed TUNEL analysis of MPMs that had been incubated with 50 μg/ml OxLDL for 24 and 48 h in the presence and absence of 58-035. Treatment with OxLDL produced an ∼3.0-fold increase in TUNEL-positive cells after 24 h and an ∼4.0-fold increase after 48 h (Fig. 2B). In the presence of the ACAT inhibitor, the induction of TUNEL-positive cells by OxLDL was reduced to only ∼1.7-fold over controls at 24 h and to ∼2.0-fold over controls at 48 h.
ACAT-1-/- macrophages display significantly reduced levels of apoptosis in response to 7KC or OxLDL
ACAT-1 is the primary ACAT isoform expressed by macrophages, and peritoneal macrophages isolated from ACAT-1-/- mice display no ACAT activity (46, 47). Therefore, to directly determine whether ACAT activity was required for the induction of apoptosis by OxLDL and 7KC, we examined the ability of OxLDL and 7KC to induce apoptosis in primary macrophages isolated from ACAT-1-/- mice. Figure 3 shows a typical result from TUNEL analysis of MPMs incubated for 24 h with either 50 μg/ml OxLDL or 10 μg/ml 7KC. Compared with similarly treated wildtype MPMs, ACAT-1-/- MPMs display a significantly lower percentage of apoptotic (TUNEL-positive) cells (∼2.4-fold lower for both OxLDL- and 7KC-treated cells). Similar results were obtained with 25-OHC-treated ACAT-1-/- MPMs (data not shown). Although the absolute value for TUNEL activity varied somewhat among repeat experiments, we always found a substantially reduced level of apoptotic cells in ACAT-1-/- macrophages in response to 7KC or OxLDL compared with control macrophages. These TUNEL assay results were confirmed by performing a caspase-3 activity assay to quantitate the apoptotic induction produced by 7KC or OxLDL treatment of MPMs isolated and pooled from a large cohort (n > 6) of wildtype and ACAT-1-/- mice (data not shown).
Fig. 3.
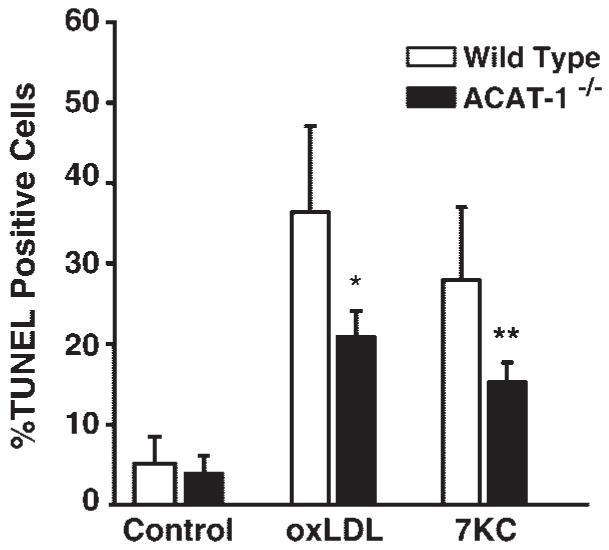
The induction of apoptosis by OxLDL and 7KC is suppressed in ACAT-1-deficient (ACAT-1-/-) peritoneal macrophages. Resident peritoneal macrophages from wild-type and ACAT-1-/- mice were isolated and treated for 24 h with OxLDL (50 μg/ml), 7KC (10 μg/ml), or vehicle, as indicated. MPMs were then subjected to in situ TUNEL analysis as described for Fig. 2. The results shown are representative of three independent experiments. * P < 0.001 and ** P < 0.0005 versus OxLDL- and 7KC-treated controls, respectively. Error bars represent standard deviation.
A cholesterol trafficking inhibitor, U18666A, preferentially inhibits cholesteryl ester accumulation, but does not inhibit 7KC-induced apoptosis
To address the issue of whether the apoptotic signal generated by ACAT might result from the production of either cholesteryl esters or 7KC esters, we took advantage of the observation that oxysterol trafficking occurs at a much faster rate than cholesterol trafficking and may involve a different trafficking mechanism (48). Amphipathic amines, such as U18666A, block cholesterol esterification by inhibiting cholesterol trafficking without having a direct effect on ACAT activity (49-52). Therefore, we determined whether U18666A could preferentially inhibit cholesteryl ester formation and, if so, what effect this inhibition would have on the 7KC-induced apoptosis. Using tracer amounts of [14C]cholesterol and [3H]7KC, the accumulation of both [14C]cholesteryl and [3H]7KC esters in P388D1 cells was observed in response to treatment with unlabeled 7KC (Fig. 4). U18666A limited the accumulation of [14C]cholesteryl esters in 7KC-treated cells to levels equivalent to those in untreated controls (Fig. 4A). However, U18666A had only a small, but significant (P = 0.048), effect on the accumulation of [3H]7KC esters (Fig. 4B). Thus, under these conditions U18666A differentially inhibited the synthesis of cholesteryl ester over 7KC esters. However, U18666A had no effect on the ability of 7KC to induce caspase-3 activity in P388D1 cells (Fig. 4C).
Fig. 4.
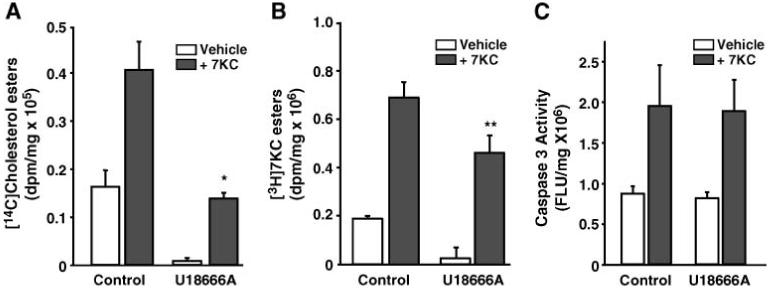
Treatment of P388D1 cells with an inhibitor of cholesterol trafficking, U18666A, preferentially inhibits cholesteryl ester accumulation, but does not inhibit 7KC-induced apoptosis. A, B: P388D1 cells were supplemented with or without U18666A (5 μM) for 1 h before supplementation with and without 7KC (10 μg/ml), as indicated. At this time, either [14C]cholesterol (0.5 μCi/ml) or [3H]7KC (1.0 °Ci/ml) was added to each well. After a 16 h incubation, total lipids were extracted, and the radioactivity incorporated into cholesteryl esters (A) and 7-ketocholesteryl esters (7KC esters; B) was determined. C: The effect of U18666A on the induction of caspase-3 activity in P388D1 cells. P388D1 cells were preincubated with U18666A (5 μM) for 1 h before a 16 h treatment with and without 7KC (10 μg/ml), as indicated. The cells were then harvested and caspase-3 activity was determined as described above. FLU, fluorescence light units. * P < 0.05 and ** P = 0.048 versus oxysterol-treated controls. Error bars represent standard deviation.
A phospholipase A2 inhibitor, a global inhibitor of arachidonate metabolism, and an ACAT inhibitor prevent esterification of 7KC and induction of apoptosis by 7KC
Our previously published results demonstrated that an inhibitor of cPLA2 (AACOCF3) and an inhibitor of AA metabolism (ETYA) prevented the induction of apoptosis by oxysterols (13, 34). Therefore, we examined the effect of AACOCF3 and ETYA on the induction of 7KC esters in P388D1 cells. In agreement with the results shown in Fig. 4, treatment of P388D1 cells with unlabeled 7KC resulted in the formation of [3H]7KC esters from [3H]7KC (Fig. 5). When supplemented with either AACOCF3 or ETYA, the stimulation of 7KC ester formation was prevented. In contrast, supplementation with the cyclooxygenase inhibitors FR-122047 and NS-398, which do not protect macrophages from 7KC-induced apoptosis (data not shown), had no effect on the induction of [3H]7KC esters (Fig. 5B). Thus, compounds capable of inhibiting 7KC-induced apoptosis in macrophages also prevent the formation of 7KC esters.
Fig. 5.
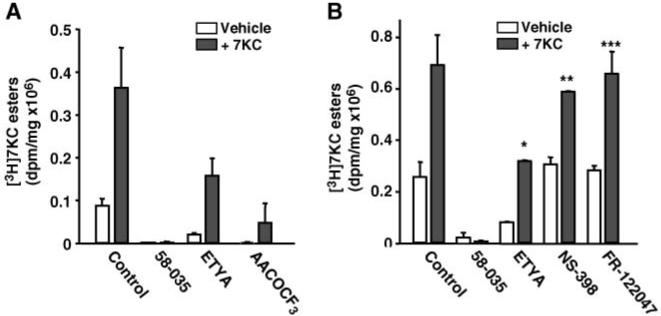
58-035, 5,8,11,14-eicosatetraynoic acid (ETYA), and arachidonyl trifluoromethyl ketone (AACOCF3) inhibit the formation of 7KC esters in P388D1 cells. A: The effects of 58-035, ETYA, and AACOCF3 on the accumulation of [3H]7KC esters in P388D1 cells. P338D1 cells were cultured in the presence of [3H]7KC (1.0 μCi/ml) in media supplemented with 58-035 (10 μg/ml), ETYA (20 μM), or AACOCF3 (20 μM), as indicated. Unlabeled 7KC (10 μg/ml) was added and the cells were incubated for 16 h. Total lipids were extracted, and the radioactivity incorporated into 7KC esters was determined. B: The effects of a cyclooxygenase-1 inhibitor, FR-122047 (10 μM), and a cyclooxygenase-2 inhibitor, NS-398 (5 μM), on the accumulation of [3H]7KC esters in P388D1 cells were compared with treatment with 58-035 and ETYA. P388D1 cells were cultured in the presence or absence of the indicated inhibitors for 1 h before supplementation with [3H]7KC or unlabeled 7KC for 16 h, as described for A. * P < 0.05, ** P = 0.12, and *** P = 0.33 versus oxysterol-treated controls. Error bars represent standard deviation.
Analysis of 7KC ester content of P388D1 cells by tandem LC/MS
A range of different oxysteryl esters have been shown to accumulate in macrophages upon uptake of modified LDL or 7KC-enriched acetylated LDL (53). To identify the distribution of 7KC esters formed in P388D1 cells, we synthesized authentic standards for the five predominant 7KC esters identified previously in J774.1 macrophages loaded with 7KC-enriched acetylated LDL (53). We then performed a semiquantitative mass spectral analysis of 7KC esters formed in P388D1 cells after treatment with 7KC for 16 h (Table 1). Macrophage 7KC esters contain primarily saturated and monounsaturated fatty acids, the majority are derived mainly from palmitate and oleate, which represented ∼21% and ∼71% of the total 7KC esters, respectively. Stearate, arachidonate, and linoleate made up <10% of the total 7KC esters combined. In addition to the five 7KC esters for which we synthesized authentic standards, we also were able to detect 7KC esters of C16:1, C18:2, C20:0, and C20:1 (Fig. 6). Because we did not have standards for these 7KC esters, we were unable to quantitate the relative amounts of each; however, C20:0 and C20:1 were probably also present in significant amounts (∼5-10% of total).
TABLE 1.
7KC ester content of P388D1 cells
| 7KC Estera | Normalized Amount | Absolute Mass |
|---|---|---|
| % | pmol/mg cell protein | |
| 7-Ketocholesteryl palmitate (16:0) | 20.98 ± 0.36 | 66.42 ± 7.15 |
| 7-Ketocholesteryl stearate (18:0) | 7.26 ± 0.88 | 22.81 ± 1.18 |
| 7-Ketocholesteryl oleate (18:1) | 71.12 ± 1.16 | 225.26 ± 25.57 |
| 7-Ketocholesteryl linoleate (18:2) | 0.22 ± 0.13 | 0.71 ± 0.51 |
| 7-Ketocholesteryl arachidonate (20:4) | 0.43 ± 0.13 | 1.35 ± 0.45 |
7KC ester, 7-ketocholesteryl ester. The MS response factors were calculated as described in Materials and Methods. P388D1 cells were incubated for 16 h in media supplemented with 20 μ/ml 7-ketocholesterol (7KC). The cells were rinsed twice with PBS and total lipids were extracted for mass spectral analysis as described in Materials and Methods. Data provided are of individual 7KC esters expressed as percentages of the total measured oxysteryl ester pool in the cell and as the absolute masses of the detected oxysteryl ester. All data are means ± SD of triplicate samples.
The numbers in parentheses refer to the length of the carbon chain and the number of double bonds of the fatty acyl chain.
Fig. 6.

Selected reaction monitoring chromatographic traces of a standard mixture (A) containing 16:0, 18:0, 18:1, 18:2, and 20:4 7KC esters, as described in Materials and Methods. Selected reaction monitoring traces of a macrophage total lipid extract (B) represent analysis of ∼1.4% of the total extract. The top panel in each set contains overlaid selected reaction monitoring traces for 16:0 and 16:1; the middle panel contains traces for 18:0, 18:1, 18:2, and 18:3; and the bottom panel contains traces for 20:0, 20:1, and 20:4. The amplification factors for each trace relative to 7KC-18:0 are shown in parentheses; the same factor was applied to each selected reaction monitoring trace in each panel.
Arachidonate supplementation can overcome the inhibitory effect of AACOCF3 on 7KC-induced apoptosis
The cPLA2 inhibitor AACOCF3 suppresses apoptosis induced by 25-OHC and 7KC (34) (data not shown) as well as the esterification of these oxysterols (Fig. 5 and data not shown). Supplementation of free fatty acids to macrophages in the presence of cPLA2 inhibition has been shown to restore OxLDL-induced cholesterol esterification (35). Therefore, we determined whether supplementation of free fatty acids in the presence of cPLA2 inhibition could restore 7KC esterification and, if so, what effect this would have on the induction of apoptosis in P388D1 cells. Figure 7A shows that supplementation of P388D1 cells with 50 μM oleic acid or AA overcame the inhibitory effect of AACOCF3 on the 7KC-induced formation of [3H]7KC esters. However, only supplementation with AA, but not oleic acid, reversed the effect of AACOCF3 on 7KC-induced caspase-3 activity (Fig. 7B). When the cells were further supplemented with the ACAT inhibitor 58-035, the stimulation of [3H]7KC ester formation produced by AA supplementation in the presence of AACOCF3 was prevented, as was the restoration of caspase-3 activity (Fig. 7A, B).
Fig. 7.
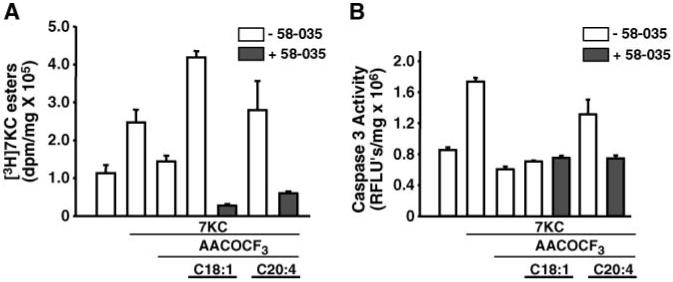
The effect of supplementing arachidonic acid (AA) and oleic acid in the presence of AACOCF3 on the formation of [3H]7KC esters and the induction of caspase-3 activity by 7KC. A: The effect of AA or oleic acid supplementation on AACOCF3 inhibition of [3H]7KC ester formation. P388D1 cells were cultured in the presence or absence of AACOCF3 (20 μM) or 58-035 (10 μg/ml) for 1 h and then supplemented with or without 7KC (10 μg/ml) and either oleic acid or AA (50 μM) complexed to fatty acid-free BSA, as indicated. [3H]7KC (1.0 μCi/ml) was added to the cells, and after a 6 h incubation, the lipids were extracted and the radioactivity incorporated into [3H]7KC esters was determined as described for Fig. 5. B: The effect of AA or oleic acid supplementation on AACOCF3 inhibition of 7KC-induced apoptosis. P388D1 cells were supplemented with or without oleic acid or AA (50 μM) in the presence of either AACOCF3 (20 μM) or 58-035 (10 μg/ml) for 1 h before the addition of unlabeled 7KC (10 μg/ml), as indicated. After a 16 h incubation, the cells were harvested, and caspase-3 activity was determined as described above. FLU, fluorescence light units. Error bars represent standard deviation.
DISCUSSION
The current study was initiated to investigate the potential role of ACAT in OxLDL/oxysterol-induced apoptosis in macrophages. The data presented demonstrate that a pharmacological inhibitor of ACAT, 58-035, produces a nearly complete loss of 7KC-induced apoptosis in a macrophage cell line, P388D1 (Fig. 1), and isolated primary macrophages (Fig. 2A). This suggests that ACAT activity is required for 7KC-induced apoptosis in macrophages. In support of this conclusion, we observed a statistically significant reduction in 7KC-induced apoptosis in ACAT-1-/- MPMs compared with wild-type MPMs (Fig. 3). These results are also consistent with our prior observation that oxysterols must be internalized by cells to be cytotoxic, even though their initial target is a cell surface calcium channel (11). Based on our previous and present observations, we hypothesize that 7KC not only initiates an apoptotic signaling pathway in macrophages via the activation of cPLA2, but also forms part of a second message in that pathway when esterified by ACAT.
There have been prior indications in the literature that ACAT may play a role in the cytotoxicity of oxysterols, albeit under somewhat different conditions. Previous reports from two laboratories have described the isolation of ACAT defective CHO-K1 mutants with selection for resistance to killing by 25-OHC (54, 55). One of these studies also described partial resistance to 25-OHC cytotoxicity in wild-type CHO-K1 cells treated with 58-035 (54). The selections for 25-OHC resistance were under different conditions than those described for the induction of apoptosis in CHO-K1 or other mammalian cells, in that the culture media was cholesterol-deficient. These conditions select for regulatory mutations in cholesterol synthesis that act to confer resistance to the downregulatory effect of 25-OHC on the transcriptional control of cholesterol biosynthetic genes (56). It was suggested in these prior studies that the selection for ACAT deficiency was a cholesterol-sparing mechanism because oxysterols are known to stimulate cholesterol esterification in whole cells. However, it is also possible that some of the resistance to killing by 25-OHC, conferred by the loss of ACAT or treatment with 58-035, could have been caused by the inhibition of oxysteryl ester formation.
Oxysterols are cytotoxic components of OxLDL (17, 18) and we present evidence that these oxysterols must be esterified to manifest their cytotoxicity. The ability of 58-035 to significantly suppress OxLDL-induced apoptosis in MPMs (Fig. 2B) and the similar observation of attenuated apoptotic induction by OxLDL in ACAT-1-/- MPMs (Fig. 3) are consistent with the hypothesis that the oxysterol components of OxLDL contribute to its cytotoxicity. The residual induction of apoptosis by OxLDL in ACAT-1-/- MPMs and MPMs treated with 58-035 probably reflects the presence of components in OxLDL that are capable of inducing apoptosis by mechanisms independent of ACAT activity (17). In prior studies demonstrating a role for cPLA2 in OxLDL-induced apoptosis, we observed a similar attenuation, rather than elimination, of apoptotic induction by OxLDL in MPMs isolated from cPLA2-/- mice (13). In these MPMs, the cPLA2-released fatty acid used for oxysterol esterification, and according to one report (35), cholesterol esterification would not be available. Furthermore, it is likely that some of the apoptosis induced by OxLDL, observed in both cPLA2-/- and ACAT-1-/- MPMs, is attributable to the accumulation of intracellular free cholesterol (57).
We cannot rule out a role for ACAT-2, in addition to ACAT-1, in the OxLDL and 7KC induction of apoptosis in ACAT-1-/- macrophages (Fig. 3). Although Sakashita et al. (58) reported that ACAT-2 is not expressed in resident MPMs, this same report shows that ACAT-2 can be induced during foam cell formation, which does occur when MPMs are incubated with OxLDL. This ambiguity does not exist for our studies with the ACAT inhibitor because it is used under conditions in which all cholesterol esterification is blocked. In these experiments, we observed a statistical 100% inhibition of 7KC-induced apoptosis in MPMs treated with 58-035 (Fig. 2A) compared with an ∼50% inhibition in the ACAT-1-/- macrophages (Fig. 3).
It has been suggested that the induction of apoptosis by oxysterols and free cholesterol loading proceed through different mechanisms (59, 60). The results of the current study agree with this assessment. The in vitro system used to induce apoptosis by free cholesterol loading in macrophages requires the inhibition of cholesterol esterification by treatment with 58-035 (61). In the current study, 58-035 proved to be a potent inhibitor of the induction of apoptosis by 7KC and OxLDL (Figs. 1, 2). Furthermore, the induction of apoptosis by free cholesterol loading is sensitive to U18666A (60, 62), whereas 7KC-induced apoptosis is insensitive to U18666A (Fig. 4).
Our prior studies have indicated that an arachidonate-derived molecule plays a role in the apoptotic response to oxysterols (13). We now hypothesize that this arachidonate derivative is an arachidonyl oxysterol. Consistent with this hypothesis, the results presented in Fig. 7 show that supplementation of macrophages in the presence of a cPLA2 inhibitor with AA, but not oleic acid, restores the induction of apoptosis by 7KC. This suggests that it is not the accumulation of 7KC esters in general, but rather the formation of a specific oxysteryl ester that produces the apoptotic response and 7KC-arachidonate may be a candidate for the apoptosis-inducing molecule. Supporting this possibility, mass spectral analysis revealed that P388D1 cells supplemented with 7KC do accumulate 7KC-arachidonate (Fig. 6, Table 1). Furthermore, Brown et al. (53) detected 7KC-arachidonate among the 7KC esters formed upon loading of MPMs and J774A.1 macrophages with OxLDL.
Our present results also provide an explanation for the observation that oxysterol toxicity can be quenched by cholesterol (63) or a mixture of nontoxic oxysterols (64). Because oxysterols as well as cholesterol are substrates for ACAT (36, 37), competition among oxysterols and cholesterol, at the level of ACAT, would be expected to diminish the levels of any specific apoptosis-inducing oxysterol arachidonate generated under these conditions. If, and by what mechanism, such an arachidonyl ester can induce apoptosis in macrophages is not known and is currently under investigation.
The data presented here have potentially important implications in atherogenesis. Our prior results (4) showed that the mitochondrial apoptosis pathway is antiatherogenic. This suggests that ACAT-1, via its ability to induce apoptosis in macrophages in response to the uptake of OxLDL, should be antiatherogenic. This hypothesis is supported by studies showing that loss of macrophage ACAT, by pharmacological inhibition (65) or by knockout of ACAT-1 (40), results in increased plaque size in animal models of atherosclerosis. In the latter study, an increase in apoptotic cells was observed in the ACAT-1-/- lesions. It was suggested that these apoptotic cells were probably macrophages undergoing cell death in response to free cholesterol toxicity. Therefore, at present, it is not possible to make a generic statement concerning the atherogenicity of apoptotic lesional macrophages.
The antiatherogenicity of macrophage ACAT-1 expression has been attributed to the ability of ACAT to limit the accumulation of free cholesterol. The data presented here suggest that another function of ACAT-1 is to produce an apoptotic signal in response to OxLDL/oxysterols. Thus, ACAT appears to play a central role in atherogenesis as a regulator of macrophage cell death by reducing the accumulation of cytotoxic free cholesterol and also promoting apoptosis in response to OxLDL/oxysterols. Further delineation of the various cell death pathways followed by in vivo testing of macrophages genetically modified in these pathways will be required to determine the precise roles of macrophage apoptosis in atherosclerosis.
Acknowledgments
The authors acknowledge the technical assistance of Theresa G. Pickle. This work was partially supported by National Institutes of Health Grant HL-65709 (S.F.).
Abbreviations
- AA
arachidonic acid
- AACOCF3
arachidonyl trifluoromethyl ketone
- ACAT
acyl-coenzyme A:cholesterol acyltransferase
- cPLA2
cytosolic phospholipase A2
- ETYA
5,8,11,14-eicosatetraynoic acid
- MPM
mouse peritoneal macrophage
- OxLDL
oxidized low density lipoprotein
- TUNEL
terminal transferase-mediated dUTP nick end labeling
- 7KC
7-ketocholesterol
- 7KC ester
7-ketocholesteryl ester
- 25-OHC
25-hydroxycholesterol
REFERENCES
- 1.Ball RY, Stowers EC, Burton JH, Cary NR, Skepper JN, Mitchinson MJ. Evidence that the death of macrophage foam cells contributes to the lipid core of atheroma. Atherosclerosis. 1995;114:45–54. doi: 10.1016/0021-9150(94)05463-s. [DOI] [PubMed] [Google Scholar]
- 2.Colles SM, Maxson JM, Carlson SG, Chisolm GM. Oxidized LDL-induced injury and apoptosis in arthrosclerosis. Potential roles for oxysterols. Trends Cardiovasc. Med. 2001;11:131–138. doi: 10.1016/s1050-1738(01)00106-2. [DOI] [PubMed] [Google Scholar]
- 3.Yao PM, Tabas I. Free cholesterol loading of macrophages induces apoptosis involving the Fas pathway. J. Biol. Chem. 2000;275:23807–23818. doi: 10.1074/jbc.M002087200. [DOI] [PubMed] [Google Scholar]
- 4.Liu J, Thewke DP, Su YR, Linton MF, Fazio S, Sinensky MS. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler. Thromb. Vasc. Biol. 2005;25:174–179. doi: 10.1161/01.ATV.0000148548.47755.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinberg D, Witztum JL. Is the oxidative modification hypothesis relevant to human atherosclerosis? Circulation. 2002;105:2107–2111. doi: 10.1161/01.cir.0000014762.06201.06. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi K, Takeya M, Sakashita N. Multifunctional roles of macrophages in the development and progression of atherosclerosis in humans and experimental animals. Med. Electron Microsc. 2002;35:179–203. doi: 10.1007/s007950200023. [DOI] [PubMed] [Google Scholar]
- 7.Steinbrecher UP. Receptors for oxidized low density lipoprotein. Biochim. Biophys. Acta. 1999;1436:279–298. doi: 10.1016/s0005-2760(98)00127-1. [DOI] [PubMed] [Google Scholar]
- 8.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr., Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler. Thromb. Vasc. Biol. 1995;15:1512–1531. doi: 10.1161/01.atv.15.9.1512. [DOI] [PubMed] [Google Scholar]
- 9.Okura Y, Brink M, Itabe H, Scheidegger KJ, Kalangos A, Delafontaine P. Oxidized low-density lipoprotein is associated with apoptosis of vascular smooth muscle cells in human atherosclerotic plaques. Circulation. 2000;102:2680–2686. doi: 10.1161/01.cir.102.22.2680. [DOI] [PubMed] [Google Scholar]
- 10.Dimmeler S, Haendeler J, Galle J, Zeiher AM. Oxidized low-density lipoprotein induces apoptosis of human endothelial cells by activation of CPP32-like proteases. A mechanistic clue to the ‘response to injury’ hypothesis. Circulation. 1997;95:1760–1763. doi: 10.1161/01.cir.95.7.1760. [DOI] [PubMed] [Google Scholar]
- 11.Rusinol AE, Yang L, Thewke D, Panini SR, Kramer MF, Sinensky MS. Isolation of a somatic cell mutant resistant to the induction of apoptosis by oxidized low-density lipoprotein. J. Biol. Chem. 2000;275:7296–7303. doi: 10.1074/jbc.275.10.7296. [DOI] [PubMed] [Google Scholar]
- 12.Hardwick SJ, Hegyi L, Clare K, Law NS, Carpenter KL, Mitchinson MJ, Skepper JN. Apoptosis in human monocyte-macrophages exposed to oxidized low density lipoprotein. J. Pathol. 1996;179:294–302. doi: 10.1002/(SICI)1096-9896(199607)179:3<294::AID-PATH590>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 13.Panini SR, Yang L, Rusinol AE, Sinensky MS, Bonventre JV, Leslie CC. Arachidonate metabolism and the signaling pathway of induction of apoptosis by oxidized LDL/oxysterol. J. Lipid Res. 2001;42:1678–1686. [PubMed] [Google Scholar]
- 14.Hegyi L, Hardwick SJ, Siow RC, Skepper JN. Macrophage death and the role of apoptosis in human atherosclerosis. J. Hematother. Stem Cell Res. 2001;10:27–42. doi: 10.1089/152581601750098192. [DOI] [PubMed] [Google Scholar]
- 15.Brown AJ, Jessup W. Oxysterols and atherosclerosis. Atherosclerosis. 1999;142:1–28. doi: 10.1016/s0021-9150(98)00196-8. [DOI] [PubMed] [Google Scholar]
- 16.Schroepfer GJ., Jr. Oxysterols: modulators of cholesterol metabolism and other processes. Physiol. Rev. 2000;80:361–554. doi: 10.1152/physrev.2000.80.1.361. [DOI] [PubMed] [Google Scholar]
- 17.Chisolm GM, Ma G, Irwin KC, Martin LL, Gunderson KG, Linberg LF, Morel DW, DiCorleto PE. 7Beta-hydroperoxycholest-5-en-3beta-ol, a component of human atherosclerotic lesions, is the primary cytotoxin of oxidized human low density lipoprotein. Proc. Natl. Acad. Sci. USA. 1994;91:11452–11456. doi: 10.1073/pnas.91.24.11452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sevanian A, Hodis HN, Hwang J, McLeod LL, Peterson H. Characterization of endothelial cell injury by cholesterol oxidation products found in oxidized LDL. J. Lipid Res. 1995;36:1971–1986. [PubMed] [Google Scholar]
- 19.Linseisen J, Wolfram G. Origin, metabolism and adverse health effects of cholesterol oxidation products. Fett/Lipid. 1998;100:211–218. [Google Scholar]
- 20.Colles SM, Irwin KC, Chisholm GM. Roles of multiple oxidized LDL lipids in cellular injury: dominance of 7β-hydroxycholesterol. J. Lipid Res. 1996;37:2018–2028. [PubMed] [Google Scholar]
- 21.Carpenter KL, Taylor SE, Ballantine JA, Fussel B, Halliwell B, Mitchinson MJ. Lipids and oxidized lipids in human atheroma and normal aorta. Biochim. Biophys. Acta. 1993;1167:121–130. doi: 10.1016/0005-2760(93)90151-x. [DOI] [PubMed] [Google Scholar]
- 22.Brown AJ, Dean RT, Jessup W. Free and esterified oxysterol: formation during copper-oxidation of low density lipoprotein and uptake by macrophages. J. Lipid Res. 1996;37:320–335. [PubMed] [Google Scholar]
- 23.Lizard G, Monier S, Cordelet C, Gesquiere L, Deckert V, Gueldry S, Lagrost L, Gambert P. Characterization and comparison of the mode of cell death, apoptosis versus necrosis, induced by 7beta-hydroxycholesterol and 7-ketocholesterol in the cells of the vascular wall. Arterioscler. Thromb. Vasc. Biol. 1999;19:1190–1200. doi: 10.1161/01.atv.19.5.1190. [DOI] [PubMed] [Google Scholar]
- 24.Aupeix K, Weltin D, Mejia JE, Christ M, Marchal J, Freyssinet JM, Bischoff P. Oxysterol-induced apoptosis in human monocytic cell lines. Immunobiology. 1995;194:415–428. doi: 10.1016/S0171-2985(11)80108-7. [DOI] [PubMed] [Google Scholar]
- 25.Harada K, Ishibashi S, Miyashita T, Osuga J, Yagyu H, Ohashi K, Yazaki Y, Yamada N. Bcl-2 protein inhibits oxysterol-induced apoptosis through suppressing CPP32-mediated pathway. FEBS Lett. 1997;411:63–66. doi: 10.1016/s0014-5793(97)00662-5. [DOI] [PubMed] [Google Scholar]
- 26.Bansal N, Houle A, Melnykovych G. Apoptosis: mode of cell death induced in T cell leukemia lines by dexamethasone and other agents. FASEB J. 1991;5:211–216. doi: 10.1096/fasebj.5.2.2004665. [DOI] [PubMed] [Google Scholar]
- 27.Christ M, Luu B, Mejia JE, Moosbrugger I, Bischoff P. Apoptosis induced by oxysterols in murine lymphoma cells and in normal thymocytes. Immunology. 1993;78:455–460. [PMC free article] [PubMed] [Google Scholar]
- 28.Emanuel HA, Hassel CA, Addis PB, Bergmann SD, Zavoral JH. Plasma cholesterol oxidation products (or sterols) in human subjects fed a meal rich in oxysterols. J. Food Sci. 1991;56:843–847. [Google Scholar]
- 29.Addis PB, Emanuel HA, Bergmann SD, Zavoral JH. Capillary quantification of cholesterol oxidation products in plasma lipoproteins of fasted humans. Free Radic. Biol. Med. 1989;7:179–182. doi: 10.1016/0891-5849(89)90011-7. [DOI] [PubMed] [Google Scholar]
- 30.Sevanian A, Seraglia R, Traldi P, Rossato P, Ursini F, Hodis H. Analysis of plasma cholesterol oxidation products using gas- and high-performance liquid chromatography/mass spectrometry. Free Radic. Biol. Med. 1994;17:397–409. doi: 10.1016/0891-5849(94)90166-x. [DOI] [PubMed] [Google Scholar]
- 31.Dzeletovic S, Breuer O, Lund E, Diczfalusy U. Determination of cholesterol oxidation products in human plasma by isotope dilution-mass spectrometry. Anal. Biochem. 1995;225:73–80. doi: 10.1006/abio.1995.1110. [DOI] [PubMed] [Google Scholar]
- 32.Yang L, Sinensky MS. 25-Hydroxycholesterol activates a cytochrome c release-mediated caspase cascade. Biochem. Biophys. Res. Commun. 2000;278:557–563. doi: 10.1006/bbrc.2000.3855. [DOI] [PubMed] [Google Scholar]
- 33.Lizard G, Gueldry S, Sordet O, Monier S, Athias A, Miguet C, Bessede G, Lemaire S, Solary E, Gambert P. Glutathione is implied in the control of 7-ketocholesterol-induced apoptosis, which is associated with radical oxygen species production. FASEB J. 1998;2:1651–1663. doi: 10.1096/fasebj.12.15.1651. [DOI] [PubMed] [Google Scholar]
- 34.Rusinol AR, Thewke D, Liu J, Freeman N, Panini SR, Sinensky MS. Akt/PKB regulation of Bcl family members during oxysterol induced apoptosis. J. Biol. Chem. 2004;279:1392–1399. doi: 10.1074/jbc.M308619200. [DOI] [PubMed] [Google Scholar]
- 35.Akiba S, Yoneda Y, Ohno S, Nemoto M, Sato T. Oxidized LDL activates phospholipase A2 to supply fatty acids required for cholesterol esterification. J. Lipid Res. 2003;44:1676–1685. doi: 10.1194/jlr.M300012-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Chang TY, Chang CC, Cheng D. Acyl-coenzyme A: cholesterol acyltransferase. Annu. Rev. Biochem. 1997;66:613–638. doi: 10.1146/annurev.biochem.66.1.613. [DOI] [PubMed] [Google Scholar]
- 37.Buhman KF, Accad M, Farese RV. Mammalian acylCoA:cholesterol acyltransferases. Biochim. Biophys. Acta. 2000;1529:142–154. doi: 10.1016/s1388-1981(00)00144-x. [DOI] [PubMed] [Google Scholar]
- 38.Cases S, Novak S, Zheng YW, Myers HM, Lear SR, Sande E, Welch CB, Lusis AJ, Spencer TA, Krause BR, et al. ACAT-2, a second mammalian acyl-CoA:cholesterol acyltransferase. Its cloning, expression, and characterization. J. Biol. Chem. 1998;273:26755–26764. doi: 10.1074/jbc.273.41.26755. [DOI] [PubMed] [Google Scholar]
- 39.Brown AJ, Leong SL, Dean RT, Jessup W. 7-Hydroperoxycholesterol and its products in oxidized low density lipoprotein and human atherosclerotic plaque. J. Lipid Res. 1997;38:1730–1745. [PubMed] [Google Scholar]
- 40.Fazio S, Major AS, Swift LL, Gleaves LA, Accad M, Linton MF, Farese RV., Jr. Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J. Clin. Invest. 2001;107:163–171. doi: 10.1172/JCI10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piretti MV, Pagliuca G. Synthesis of highly pure dolichyl and cholesteryl esters. Ital. J. Biochem. 1996;45:67–69. [PubMed] [Google Scholar]
- 42.Hirsch J, Ahrens EH. The separation of complex lipid mixtures by use of silicic acid chromatography. J. Biol. Chem. 1958;233:311–320. [PubMed] [Google Scholar]
- 43.Ross AC, Go KJ, Heider JG, Rothblat GH. Selective inhibition of acyl coenzyme A:cholesterol acytransferase by compound 58-035. J. Biol. Chem. 1984;259:815–819. [PubMed] [Google Scholar]
- 44.Warner GJ, Stoudt G, Bamberger M, Johnson WJ, Rothblat GH. Cell toxicity induced by inhibition of acyl coenzyme A:cholesterol acyltransferase and accumulation of unesterified cholesterol. J. Biol. Chem. 1995;270:5772–5778. doi: 10.1074/jbc.270.11.5772. [DOI] [PubMed] [Google Scholar]
- 45.Kelly KJ, Sandoval RM, Dunn KW, Molitoris BA, Dagher PC. A novel method to determine specificity and sensitivity of the TUNEL reaction in the quantitation of apoptosis. Am. J. Physiol. Cell Physiol. 2003;284:C1309–C1318. doi: 10.1152/ajpcell.00353.2002. [DOI] [PubMed] [Google Scholar]
- 46.Meiner V, Tam C, Gunn MD, Dong LM, Weisgraber KH, Novak S, Myers HM, Erickson SK, Farese RV., Jr. Tissue expression studies on the mouse acyl-CoA: cholesterol acyltransferase gene (ACAT): findings supporting the existence of multiple cholesterol esterification enzymes in mice. J. Lipid Res. 1997;38:1928–1933. [PubMed] [Google Scholar]
- 47.Miyazaki A, Sakashita N, Lee O, Takahashi K, Horiuchi S, Hakamata H, Morganelli PM, Chang CC, Chang TY. Expression of ACAT-1 protein in human atherosclerotic lesions and cultured human monocytes-macrophages. Arterioscler. Thromb. Vasc. Biol. 1998;18:1568–1574. doi: 10.1161/01.atv.18.10.1568. [DOI] [PubMed] [Google Scholar]
- 48.Johnson W, Mahlberg F, Rothblat G, Phillips M. Cholesterol transport between cells and high-density lipoproteins. Biochim. Biophys. Acta. 1991;1085:273–298. doi: 10.1016/0005-2760(91)90132-2. [DOI] [PubMed] [Google Scholar]
- 49.Underwood KW, Andenariam B, McWilliams GL, Liscum L. Quantitative analysis of hydrophobic amine inhibition of intracellular cholesterol transport. J. Lipid Res. 1996;37:1556–1568. [PubMed] [Google Scholar]
- 50.Lange Y, Ye J, Chin J. The fate of cholesterol existing lysosomes. J. Biol. Chem. 1997;272:17018–17022. doi: 10.1074/jbc.272.27.17018. [DOI] [PubMed] [Google Scholar]
- 51.Liscum L, Faust JR. The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-beta-[2-(diethylamino)ethoxy] androst-5-en-17-one. J. Biol. Chem. 1989;264:11796–11806. [PubMed] [Google Scholar]
- 52.Liscum L, Munn NJ. Intracellular cholesterol transport. Biochim. Biophys. Acta. 1999;1438:19–37. doi: 10.1016/s1388-1981(99)00043-8. [DOI] [PubMed] [Google Scholar]
- 53.Brown AJ, Mander EL, Gelissen IC, Kritharides L, Dean RT, Jessup W. Cholesterol and oxysterol metabolism and subcellular distribution in macrophage foam cells: accumulation of oxidized esters in lysosomes. J. Lipid Res. 2000;41:226–236. [PubMed] [Google Scholar]
- 54.Metherall JE, Ridgway ND, Dawson PA, Goldstein JL, Brown MS. A 25-hydroxycholesterol-resistant cell line deficient in acyl-CoA:cholesterol acyltransferase. J. Biol. Chem. 1991;266:12734–12740. [PubMed] [Google Scholar]
- 55.Cadigan KM, Heider JG, Chang TY. Isolation and characterization of Chinese hamster ovary cell mutants deficient in acyl-coenzyme A:cholesterol acyltransferase activity. J. Biol. Chem. 1988;263:274–282. [PubMed] [Google Scholar]
- 56.Sinensky M, Leonard S. Somatic cell genetics and the study of cholesterol metabolism. Biochim. Biophys. Acta. 1988;947:101–112. doi: 10.1016/0304-4157(88)90021-4. [DOI] [PubMed] [Google Scholar]
- 57.Accad M, Smith SJ, Newland DL, Sanan DA, King LE, Jr., Linton M, Fazio S, Farese RV., Jr. Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1. J. Clin. Invest. 2000;105:711–719. doi: 10.1172/JCI9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sakashita N, Miyazaki A, Chang CC, Chang TY, Kiyota E, Satoh M, Komohara Y, Morganelli PM, Horiuchi S, Takeya M. Acyl-coenzyme A:cholesterol acyltransferase 2 (ACAT2) is induced in monocyte-derived macrophages: in vivo and in vitro studies. Lab. Invest. 2003;83:1569–1581. doi: 10.1097/01.lab.0000095687.17383.39. [DOI] [PubMed] [Google Scholar]
- 59.Tabas I. Cholesterol and phospholipid metabolism in macrophages. Biochim. Biophys. Acta. 2000;1529:164–174. doi: 10.1016/s1388-1981(00)00146-3. [DOI] [PubMed] [Google Scholar]
- 60.Yao PM, Tabas I. Free cholesterol loading of macrophages is associated with widespread mitochondrial dysfunction and activation of the mitochondrial apoptosis pathway. J. Biol. Chem. 2001;276:42468–42476. doi: 10.1074/jbc.M101419200. [DOI] [PubMed] [Google Scholar]
- 61.Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher E, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 2003;5:781–792. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 62.Kellner-Weibel G, Geng YJ, Rothblat GH. Cytotoxic cholesterol is generated by the hydrolysis of cytoplasmic cholesteryl ester and transported to the plasma membrane. Atherosclerosis. 1999;146:309–331. doi: 10.1016/s0021-9150(99)00155-0. [DOI] [PubMed] [Google Scholar]
- 63.Clare K, Hardwick SJ, Carpenter KLH, Weeratunge N, Mitchinson MJ. Toxicity of oxysterols to human monocytemacrophages. Atherosclerosis. 1995;118:67–75. doi: 10.1016/0021-9150(95)05594-m. [DOI] [PubMed] [Google Scholar]
- 64.Biasi F, Leonarduzzi G, Vizio B, Zanetti D, Sevanian A, Sottero B, Verde V, Zingaro B, Chiarpotto E, Poli G. Oxysterol mixtures prevent proapoptotic effects of 7-ketocholesterol in macrophages: implications for proatherogenic gene modulation. FASEB J. 2004;18:693–695. doi: 10.1096/fj.03-0401fje. [DOI] [PubMed] [Google Scholar]
- 65.Perrey S, Legendre C, Matsuura A, Guffroy C, Binet J, Ohbayashi S, Tanaka T, Ortuno JC, Matsukura T, Laugel T, et al. Preferential pharmacological inhibition of macrophage ACAT increases plaque formation in mouse and rabbit models of atherogenesis. Atherosclerosis. 2001;155:359–370. doi: 10.1016/s0021-9150(00)00599-2. [DOI] [PubMed] [Google Scholar]