

Abstract
New antiviral drugs are needed for the treatment of cytomegalovirus (CMV) infections, particularly in immunocompromised patients. These studies evaluated the in vitro and in vivo activity of the non-nucleosidic CMV inhibitor, BAY 38-4766, against guinea pig cytomegalovirus (GPCMV). Plaque reduction assays indicated that BAY 38-4766 was active against GPCMV, with an IC50 of 0.5 μM. Yield reduction assays demonstrated an ED90 and ED99 of 0.4 and 0.6 μM, respectively, of BAY 38-4766 against GPCMV. Guinea pigs tolerated oral administration of 50 mg/kg/day of BAY 38-4766 without evidence of biochemical or hematologic toxicity. Plasma concentrations of BAY 38-4766 were high following oral dosing, with a mean peak level at 1-h post-dose of 26.7 mg/ml (n = 6; range, 17.8-35.4). Treatment with BAY 38-4766 reduced both viremia and DNAemia, as determined by a real-time PCR assay, following GPCMV infection of cyclophosphamide-immunosuppressed strain 2 guinea pigs (p < 0.05, Mann-Whitney test). BAY 38-4766 also reduced mortality following lethal GPCMV challenge in immunosuppressed Hartley guinea pigs, from 83% (20/24) in placebo-treated guinea pigs, to 17% (4/24) in BAY 38-4766-treated animals (p < 0.0001, Fisher’s exact test). Mortality differences were accompanied by reduction in DNAemia in Hartley guinea pigs. Based upon its favorable safety, pharmacokinetic, and therapeutic profiles, BAY 38-4766 warrants further investigation in the GPCMV model.
Keywords: Guinea pig, Cytomegalovirus, Antiviral therapy, CMV infection, Immunocompromised animal model, Real-time PCR, BAY 38-4766, Placenta
1. Introduction
Infection with human cytomegalovirus (HCMV) is ubiquitous, and generally asymptomatic (Britt and Alford, 1996). Certain patient populations, however, may develop life- and sight-threatening HCMV disease, usually in association with immunosuppression. Patients at risk for HCMV disease include immunocompromised solid organ and bone marrow transplant patients, and individuals with HIV infection (Schleiss, 2002). Congenital infection with HCMV also produces significant morbidity and mortality, and can cause long-term sequelae, including deafness (Demmler, 1996). Although vaccination may ultimately offer promise for protection against HCMV disease, there are as yet no licensed vaccines (Plotkin, 1999). Therefore, antiviral therapy represents the only intervention currently available for prophylaxis and therapy of HCMV in high-risk patient populations.
The first antiviral licensed for HCMV infection was ganciclovir. This nucleoside agent inhibits the viral polymerase, and is efficacious both following intravenous administration, or oral administration as the prodrug, val-ganciclovir (Kimberlin, 2002; Nichols and Boeckh, 2000; Griffiths, 2002). Unfortunately, both drug toxicities (particularly neutropenia) and the emergence of antiviral resistance limit the clinical utility of this agent (Chou, 1999). Alternative therapies, such as foscarnet and cidofovir, are available, but fraught with toxicities, in particular renal toxicity, limiting their usefulness (Schleiss and McVoy, 2004).
Recently, a novel class of non-nucleosidic antivirals has been developed which interfere with cleavage and packaging of viral DNA concatemers during the DNA replication process. One such agent, BAY 38-4766, has been shown to be well-absorbed and safe following oral administration, and is highly active against HCMV in vitro. Based on its favorable pharmacokinetic profile following oral dosing, its activity against HCMV, and its low toxicity, this agent may represent a useful candidate for further clinical trial evaluation in high-risk patients with HCMV disease (Reefschlaeger et al., 2001; McSharry et al., 2001; Weber et al., 2001; Buerger et al., 2001; Evers et al., 2002).
Ideally, candidate antiviral therapies for HCMV should be evaluated in animal models prior to human clinical trials. However, since CMVs are species-specific, preclinical efficacy evaluation of candidate HCMV antivirals unfortunately cannot easily be performed in animal models. This necessitates the study of CMVs of other species, in order to identify promising treatments in animal models which might prove to be of value for HCMV disease. The pathogenesis of guinea pig cytomegalovirus (GPCMV) infection shares many similarities with HCMV, including the clinical manifestations of pneumonitis and disseminated visceral disease, particularly evident in immunocompromised animals. Additionally, in contrast to the CMVs of most small animals, the guinea pig CMV (GPCMV) crosses the placenta, causing infection in utero, thus making the GPCMV model uniquely useful for antiviral evaluations which target the fetus (Bia et al., 1983). These features of the GPCMV model have proven to be very useful in the study of a number of experimental antivirals (Aquino-de Jesus and Griffith, 1989; Li et al., 1990; Feng et al., 1992; Bourne et al., 2000). In view of the antiviral activity which BAY 38-4766 exhibits against HCMV, and in light of the efficacy of the agent observed against disseminated disease in mice experimentally infected with murine cytomegalovirus (MCMV; Weber et al., 2001), these studies were undertaken to evaluate the in vitro and in vivo activity of BAY 38-4766 against GPCMV.
2. Materials and methods
2.1. Virus and cells
GPCMV (strain no. 22122, ATCC VR682) was propagated on guinea pig fibroblast lung cells (GPL, ATCC CCL 158) and maintained in F-12 medium supplemented with 10% fetal calf serum (FCS, HyClone Laboratories), 10,000 IU/I penicillin, 10 mg/l streptomycin (Gibco-BRL) and 7.5% NaHCO3 (Gibco-BRL). An eGFP-expressing recombinant GPCMV, with replication kinetics identical to wild-type (ATCC) virus, was prepared as described elsewhere for in vitro antiviral testing (McGregor and Schleiss, 2001). Salivary gland workpools of GPCMV were prepared for in vivo studies as described elsewhere (Bia et al., 1983).
2.2. Analysis of in vitro activity of BAY 38-4766 against GPCMV
The compound, BAY 38-4766, was provided by Bayer® Pharmaceuticals (Tubigen, Germany). Compound was solubilized in DMSO, and diluted in F-12 media for in vitro testing, and in tylose (as described below) for in vivo administration. For in vitro testing, twofold dilutions of drug were made in F-12 media for determination of IC50 by plaque reduction assay, and EC90 and ED99 by yield reduction assay. The drug concentration determined to produce a 50% reduction in plaques was defined as the IC50. Briefly, GPL cells were seeded in 24-well plates. Confluent cell monolayers were infected with 80-100 plaque-forming units (pfu) per well of eGFP-expressing GPCMV. After a 1.5-h adsorption period, a 0.5% methylcellulose-F12 media overlay containing dilutions of drug was added. Plaques were counted by fluorescence microscopy after 72-96 h of incubation. The number of plaques in treated wells was expressed as a percentage of untreated virus control and plotted against drug concentration (Fig. 1). Yield reduction assays were performed as described elsewhere (Hu and Hsiung, 1989). Briefly, the definitions employed were based on the drug concentration required to reduce 90% (1 log10 reduction) or 99% (2 log10 reduction) of the virus yield, compared to the virus control. This was determined from dose-response curves and expressed as the ED90 or ED99 of the compound in a virus yield reduction assay (Hu and Hsiung, 1989).
Fig. 1.
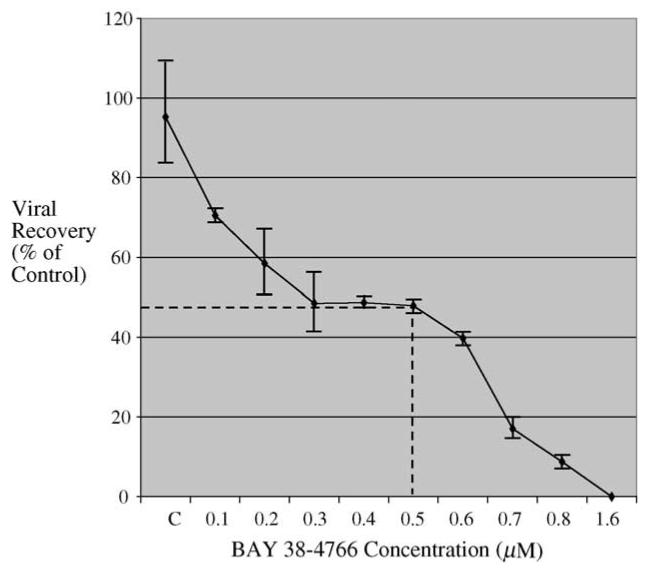
Antiviral activity in vitro of BAY 38-4766 against GPCMV. Graphical depiction of IC50 determination (plaque reduction assay), as defined in Section 2. Range of concentrations (μM concentrations), horizontal axis. C, control inoculum of eGFP-expressing GPCMV. Data points represent mean (±S.D.) of plaque counts from four independent wells. Data are representative of four independent experiments. IC50 for BAY 38-4766 against GPCMV was determined to be 0.5 μM.
2.3. Animal studies and experimental design
GPCMV seronegative Hartley guinea pigs were obtained from Harlan Laboratories (Indianapolis, IN). Inbred adult strain-2 guinea pigs were purchased from the Children’s Hospital Research Foundation (Cincinnati, OH). Young adult animals were used for these studies (mean weight, ∼600 g). Immunosuppression and GPCMV challenges were carried out as previously described (Bourne et al., 2000), except that strain 2 guinea pigs were treated with a single dose of cyclophosphamide, 200 mg/kg, 1 day prior to GPCMV infection. For experiments with Hartley guinea pigs, a second dose of cyclophosphamide, 50 mg/kg, was administered on day 6 following viral challenge. Beginning on day 1 following challenge with 4 × 105 pfu of salivary gland-passaged GPCMV by subcutaneous route, animals were either treated with tylose (vehicle control) or BAY 38-4766, at a dose of 50 mg/kg/day divided twice daily. Tylose (Tylose MH 400P2) was obtained from Clariant Corporation, and the formulation of the vehicle was as a 0.5% solution in phosphate-buffered saline. Strain 2 animals were treated for 7 days post-viral challenge, with quantitative viral blood culture and PCR analysis performed on day 10. Hartley strain animals were treated for a total of 14 days with drug or placebo. For Hartley studies, blood was obtained by toe clip for viral culture and quantitative PCR analysis of viral load at days 7 and 10 post-inoculation. A subset of animals was sacrificed at day 7, for evaluation of organ pathology (not included in mortality analysis). Animals were housed under American Association of Accreditation of Laboratory Animal Care policies, with approval from the Institutional Animal Use Committee.
2.4. Viral culture and real-time PCR analyses
Blood samples were obtained by toe clip at various times post-infection in sham-treated and BAY 38-4766-treated animals. Heparinized blood was cultured on GPL cells and a separate aliquot of blood (100 μl volume) was purified by Qiagen® column extraction, using the QIAamp® DNA mini kit, according to the manufacturer’s specifications. Before the samples were subjected to real-time PCR, they were analyzed by spectrophotometry to determine A260 DNA concentrations. The primer pair UL83 F6 (5′-CGACGACGACGATGACGAAAAC-3′) and UL83 B11 (5′-TCCTCGGTCTCAACGAAGGGTC-3′) amplifies a 225 base pair (bp) region, corresponding to Asp402 through Ser473 of the GP83 protein (Schleiss et al., 1999, 2003), and was used for real-time PCR. The PCR was performed in a volume of 20 μl as specified by the LightCycler® FastStart DNA Master SYBR Green I “Ready to Use” hot start reaction mix, and PCR was performed using the LightCycler® instrument (Roche Diagnostics). Each reaction mix contained approximately 25 pmol of UL-83 F6 forward and UL-83 B11 reverse primer, and either unknown sample or, for positive controls, 2 μl of plasmid, pKTS 502, which contains the 225 bp GP83 target sequence, at a concentration of 1 × 103 genome copies/μl. For unknown samples, 4 μl of sample at an approximate concentration of 0.1 μg was used. PCR was performed using the Roche LightCycler® 1.0 instrument under the following conditions: initial denature at 95 °C for 10 min, followed by 95 °C for 3 s, 64 °C for 5 s, 72 °C for 8 s for a total of 45 cycles, followed by melting curve analysis starting at 64 °C and ending at 95 °C, then a final hold at 40 °C. The data were analyzed with LightCycler® Data Analysis (LCDA) Software 1.0 (Roche Diagnostics). For quantification, standard curves were generated using dilutions of plasmid and viral DNA at known concentrations. The sensitivity of the assay was consistently between 1 and 10 copies/reaction. Viral load was expressed as copy numbers/μg total DNA extracted from the sample. For confirmation of positive results, gel electrophoresis was performed, on a 2.25% PCR-grade agarose gel.
2.5. Statistical analyses
Incidence data (mortality rates) were compared using Fisher’s exact test. Nonparametric data, including the comparisons of viral load and viral titers in treated versus untreated animals, were analyzed using the Mann-Whitney test. All comparisons were two-tailed. Statistical analyses were performed with the InStat® software package (GraphPad Software, San Diego).
3. Results
3.1. In vitro antiviral activity of BAY 38-4766 against GPCMV
Previous analyses of BAY 38-4766 had identified antiviral activity against both human and rodent CMVs, with IC50 values in the 0.04-0.3 μM range (Reefschlaeger et al., 2001). BAY 38-4766 was assessed for activity against GPCMV, using standard plaque reduction assays. Plaque reduction assay established an IC50 for BAY 38-4766 of 0.5 μM (Fig. 1), similar to the level of antiviral activity which had previously been described for this compound against HCMV (Reefschlaeger et al., 2001; McSharry et al., 2001). Yield reduction assays determined for BAY 38-4766 against GPCMV determined an ED90 and ED99 of 0.4 and 0.6 μM, respectively. Similar values were observed by using both an eGFP-tagged version of GPCMV (McGregor and Schleiss, 2001), as well as a tissue culture-passaged stock, derived from the ATCC strain of GPCMV (strain no. 22122, ATCC VR682). BAY 38-4766 exhibited no evidence of cytotoxicity to monolayers of GPL cells at concentrations up to 0.4 mM.
3.2. Pharmacokinetic and safety analyses of BAY 38-4766 in guinea pigs
To assess the tolerability and pharmacokinetics of BAY 38-4766 in guinea pigs, blood counts and serum chemistries were obtained following oral dosing, along with drug levels. Animals tolerated oral administration of 50 mg/kg/day of BAY 38-4766 without evidence of biochemical or hematologic toxicity (data not shown). Treated animals also had similar weight gain during therapy, compared to placebo recipients. Pharmacokinetic analyses indicated that plasma concentrations of BAY 38-4766 were high following oral dosing in nonpregnant animals, with a mean peak level 1-h post-dose of 26.7 μg/ml (0.06 mM; n = 3; range, 17.8-35.6). In all animals, drug concentration was lower on day 3 of oral dosing, with a mean level 1-h post-dose of 16.8 μg/ml (11.4-21.6 μg/ml range), and was further reduced on day 5 to a mean level of 6.5 μg/ml (0.015 mM; 4.9-8.1 μg/ml), an effect possibly compatible with cytochrome p450 induction. Blood levels were also obtained from pregnant dams (n = 3), and from pups sacrificed in utero. Plasma concentrations in dams were high, ranging from 14.3 to 36.3 μg/ml (mean, 25.6 μg/ml), 1-h post-dose on day 1 of oral dosing. On day 3 of oral dosing, the 1-h post-dose levels remained high in pregnant animals, with a mean of 23.9 μg/ml (range, 13.2-30.3 μg/ml). On day 3 of oral dosing, the 4-h post-dose level was 6.5 μg/ml (range, 4502-7441). Following this dose, dams were sacrificed, and fetal plasma obtained (n =9 pups). The mean BAY 38-4766 level in fetal plasma was 1.6 μg/ml (0.004 mM). These analyses indicated that BAY 38-4766 crossed the placenta in pregnant guinea pigs, with fetal plasma concentrations approximately 25% of maternal levels.
3.3. Reduction in GPCMV viremia and DNAemia in strain 2 guinea pigs
To assess the efficacy of BAY 38-4766 in an inbred strain of guinea pigs, strain 2 guinea pigs, GPCMV viral loads were monitored by blood culture and qcPCR following subcutaneous challenge with salivary gland-passaged virus. These animals were immunosuppressed with a single dose of cyclophosphamide, and then treated with either BAY 38-4766 for 7 days, or placebo, beginning on day 1 post-viral inoculation. Viral load assessment of whole blood obtained on day 10 indicated significant reductions in the mean titer of GPCMV in quantitative blood culture, and in viral load assessed by real-time PCR. The 50% tissue culture infectious dose endpoint (TCID50) was determined, according to the methodology of Reed and Muench (1938). The mean viral titer was 3.1 ± 0.4 log10 TCID50/ml in control animals (n = 7), and 2.0 ± 0.6 log10 TCID50/ml in BAY 38-4766-treated animals (n =6; p < 0.02 versus control, Mann-Whitney test). By real-time PCR, viral load in control animals was 2.9 ± 0.6 log10 genomes/μg, compared to 2.3 ± 0.2 log10 genomes/μg in BAY 38-4766-treated animals (p < 0.04 versus control, Mann-Whitney test; Table 1). No GPCMV-related mortality was observed in strain 2 guinea pigs under these experimental conditions.
Table 1.
GPCMV dissemination in cyclophosphamide-treated strain 2 guinea pigs: impact of BAY 38-4766 on viral load on day 10 post-GPCMV challenge
| Group | No. of animals | Mean titer (TCID50/ml) | Viral load (log10 genomes/μg DNA) |
|---|---|---|---|
| Control (tylose) | 7 | 3.1 ± 0.4 log10 | 2.9 ± 0.6 |
| BAY 38-4766 | 6 | 2.0 ± 0.6 log10* | 2.3 ± 0.2** |
p < 0.02 vs. control, Mann-Whitney test.
p < 0.04 vs. control, Mann-Whitney test.
3.4. Reduction of GPCMV mortality following lethal challenge in Hartley guinea pigs
The effect of BAY 38-4766 on mortality due to disseminated GPCMV in immunocompromised Hartley guinea pigs (two doses of cyclophosphamide) was next analyzed. For these studies, BAY 38-4766 was given as described above for strain 2 animals, but antiviral therapy was extended to a total of 14 days, and Hartley guinea pigs were more heavily immunosuppressed, via the administration of an additional dose of cyclophosphamide, as noted above. Under these experimental conditions of immunosuppression, a high rate of mortality (50-75%) is typically observed (Bourne et al., 2000). These experiments identified a dramatic impact of BAY 38-4766 on guinea pig mortality (Fig. 2). At the conclusion of the 14-day treatment regimen, the total guinea pig mortality in the control (tylose vehicle) group was 20/24 (83%), compared to only 4/24 (17%) mortality in the treatment group (p < 0.0001, Fisher’s exact test). Most mortality occurred between days 10 and 14 following GPCMV infection (Fig. 2).
Fig. 2.
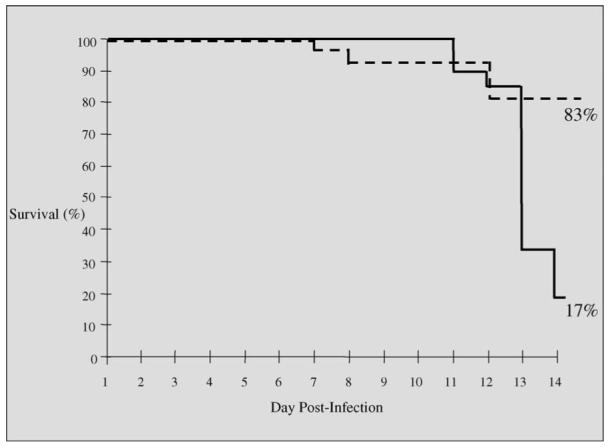
Effect of BAY 38-4766 on mortality in Hartley guinea pigs following GPCMV challenge. Animals were immunosuppressed with cyclophosphamide as described in text and challenged with GPCMV. Guinea pigs received either tylose placebo, or BAY 38-4766, 50 mg/kg/day. Treatments were initiated 24 h after viral challenge, and were administered in two divided doses per day, for a total of 14 days. Guinea pig mortality in the control (tylose vehicle) group was 20/24 (83%). In contrast, in BAY 38-4766-treated animals, mortality was 4/24 (17%; p < 0.0001, Fisher’s exact test).
To assess the impact of BAY 38-4766 on viral replication in immunosuppressed Hartley guinea pigs, quantitative viral load assessments in the blood were made by real-time PCR at days 7 and 10. In addition, a subset of animals was sacrificed (n = 6/group; not included in mortality analysis above) at day 7 post-viral challenge, for quantitative viral load assessment in solid organs (lung, liver, spleen) by viral culture and PCR, and for histopathology. Histopathologic evaluation of organs recovered at the day 7 time point revealed the presence of viral inclusions and inflammatory infiltrates in the visceral organs of animals from both groups, particularly in the lung. No differences were observed in the extent of tissue histopathology in the two groups (data not shown). Similarly, there were no differences in virus recovery by culture or PCR in visceral organ samples obtained at this time point (data not shown). However, when systemic viral load was examined in blood from guinea pigs at days 7 and 10 post-viral inoculation, total genome copy number was decreased by treatment with BAY 38-4766 by real-time PCR, compared to controls (Fig. 3). The mean genome copy numbers were 1.4 ± 0.3 log10 genomes/μg at day 7 in blood from the BAY 38-4766-treated animals, compared to 1.7 ± 0.2 log10 genomes/μg at day 7 in the tylose controls (p < 0.04 versus BAY-treated animals, Mann-Whitney test). The mean genome copy numbers were similarly decreased in the treatment group when day 10 samples were assessed, with a viral load of 2.7 ± 0.6 log10 genomes/μg in blood from BAY 38-4766-treated animals, compared to 3.0 ± 0.5 log10 genomes/μg in tylose controls (p < 0.02 versus BAY-treated animals, Mann-Whitney test).
Fig. 3.
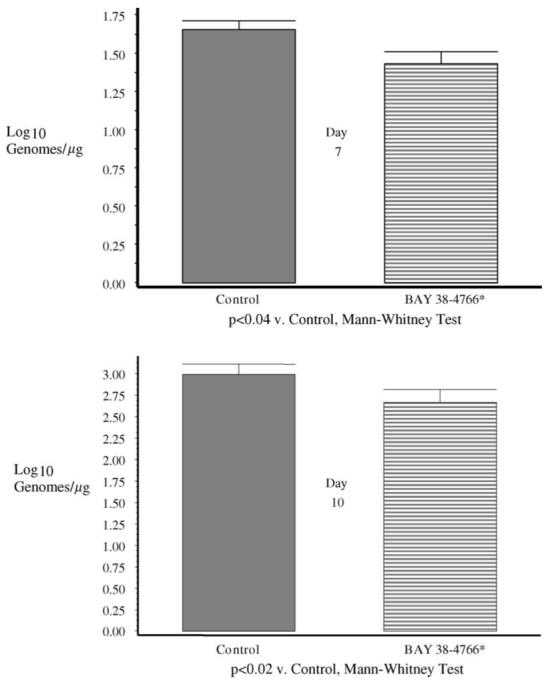
Comparison of viral loads in blood samples from BAY 38-4766 (cross-hatched bars) and tylose control groups (solid gray bars), compared at days 7 and 10 post-infection. Genome copy number was determined by real-time PCR, as described in Section 2. Significantly lower viral load was present in BAY 38-4766-treated vs. control animals at both the day 7 (upper panel; p < 0.04) and day 10 (lower panel; p < 0.02) time points (Mann-Whitney test).
3.5. BAY 38-4766-resistance in GPCMV
DNA sequencing of viruses resistant to BAY 38-4766 initially identified candidate mutations in the terminase proteins, HCMV UL56 and UL89, and MCMV M56 and M89 (Reefschlaeger et al., 2001). The amino acid changes P202A and I208N in MCMV M56 and M360I in MCMV M89 were subsequently confirmed to confer resistance (Buerger et al., 2001). To further characterize the molecular targets of BAY 38-4766 in GPCMV, a BAY 38-4766-resistant GPCMV (BAYR-GPCMV) was derived by serial passage of GPCMV in incrementally increasing concentrations of compound. Starting at a concentration of 0.01 μM, virus was maintained over multiple passages over a period of several months in twofold increasing concentrations of compound, until CPE was readily observed at concentrations of >8 μM. Viral growth curves confirmed that replication of BAYR-GPCMV was not impaired by 8 μM BAY 38-4766, a dose sufficient to reduce wild type GPCMV titers by 3-4 logs (Fig. 4). Furthermore, in the absence of BAY 38-4766, replication of BAYR-GPCMV was similar to wild type virus, indicating that the mutations that confer resistance do not result in any loss of replicative fitness (Fig. 4).
Fig. 4.
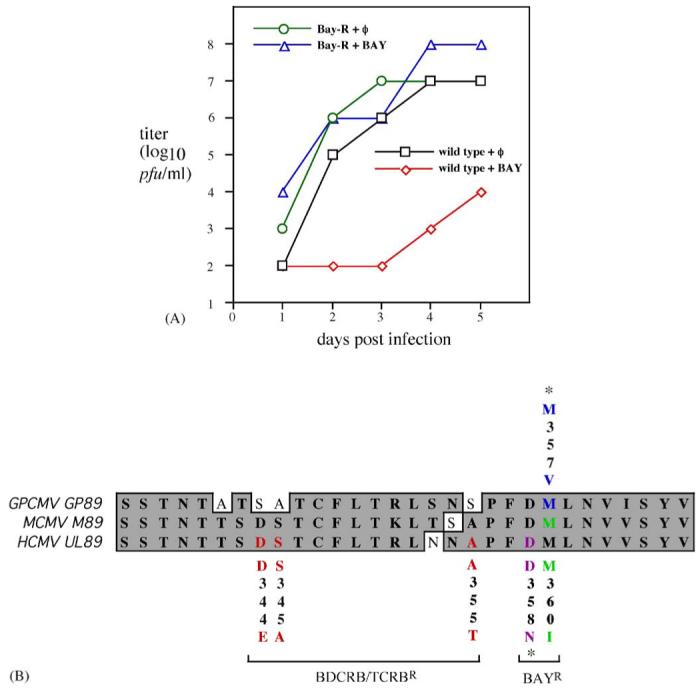
Characterization of resistance phenotype and sequence analysis of BAY 38-4766-resistant isolate of GPCMV. (A) Replication of wild-type and BAY 38-4766-resistant mutant GPCMV in the presence and absence of BAY 38-4766. Cells were infected with wild-type GPCMV or BAYR-GPCMV in the absence or presence of 8 μM BAY 38-4766 (BAY) and the culture supernatants were titrated daily for 5 days. (B) Alignment of terminase amino acid sequences from GPCMV GP89 (residues 335-364), MCMV M89 (residues 338-367), and HCMV UL89 (residues 337-366). Mutations associated with resistance are color-coded: red, BDCRB/TCRB-resistance in HCMV (Krosky et al., 1998; Underwood et al., 1998); purple, BAY 38-4766-resistance in HCMV (Reefschlaeger et al., 2001); green, BAY 38-4766-resistance in MCMV (Buerger et al., 2001); and blue, BAY 38-4766-resistance in GPCMV (this study). Note that residue numbers are specific to each virus. Mutations indicated with ‘*’ have been identified in resistant viruses by sequencing but have not been confirmed to confer resistance.
To explore the mechanism of GPCMV resistance, sequencing of BAYR-GPCMV DNA was performed, and detected a mutation resulting in the amino acid change M357V within the GPCMV GP89 terminase protein (Fig. 4). Alignment of GP89, HCMV UL89, and MCMV M89 amino acid sequences revealed that M357 in GP89 lies within a highly conserved region and corresponds to M360 in M89, the precise residue that when changed to I confers resistance BAY 38-4766 in MCMV (Buerger et al., 2001; Fig. 4). Although genetic confirmation that this mutation confers resistance in GPCMV requires a more comprehensive genetic analysis, this result strongly suggests that GP89 is the molecular target of BAY 38-4766. To confirm that observed viral replication in tissues obtained for cultures from guinea pigs treated with BAY 38-4766 did not represent the selection of resistant virus, selected viral isolates obtained from BAY 38-4766-treated animals were examined by PCR and DNA sequence analysis, and found to be of wild-type sequence in the GP89 ORF (data not shown).
4. Discussion
Of the newer compounds in development for the treatment of HCMV infection, the BAY compounds represent a class of potentially useful agents, because of their high oral bioavailability and low toxicity. The non-nucleoside compound BAY 38-4766 has a mode of anti-CMV activity consisting of inhibition of DNA cleavage and packaging. The drug acts through the viral terminase, an enzymatic complex that both packages the DNA into capsids and cleaves the DNA to form mature genomes. The precise molecular targets of BAY 38-4766 are the UL89 and UL56 gene products, which are components of the terminase complex (Reefschlaeger et al., 2001). Importantly, viruses which are resistant to foscarnet, ganciclovir, and cidofovir are susceptible to BAY 38-4766 (McSharry et al., 2001).
The mechanism by which BAY 38-4766 inhibits DNA cleavage and packaging appears to be quite similar to that of another class of maturational inhibitors, the benzimidazole ribosides, which include 2-bromo-5,6-dichloro-1-β-d-riborfuranosyl benzimidazole riboside (BDCRB) and 2,5,6-trichloro-1-β-d-riborfuranosyl benzimidazole riboside (TCRB; Krosky et al., 1998; Underwood et al., 1998). Despite pronounced dissimilarities in structure, amino acid changes that confer resistance to these compounds lie very close together in both terminase subunits (UL56 and UL89 in HCMV, and M56 and M89 in MCMV). This proximity suggests that these two groups of compounds interact with terminase regions that are adjacent if not overlapping. Even so, the fact that benzimidazole-resistant viruses are not cross-resistant to BAY 38-4766 (Evers et al., 2002) suggests subtle differences in how these compounds interact with terminase that likely reflect their differences in structure.
The M357V amino acid change that we detected in GP89 of BAYR-GPCMV precisely corresponds with the M360I change in MCMV M89, which has been shown to confer BAY 38-4766-resistance (Buerger et al., 2001). Thus, between HCMV, MCMV, and GPCMV, mutations associated with or known to confer BAY 38-4766-resistance lie within two adjacent residues (Fig. 4B). This strongly suggests that at the molecular level the inhibitory effects of BAY 38-4766 are essentially the same for all three viruses. Consequently, both MCMV and GPCMV are likely to provide models that closely emulate HCMV with respect to BAY 38-4766 efficacy and resistance.
In vivo, BAY 38-4766 has been found to have encouraging safety and pharmacokinetic properties. The IC50 we observed for BAY 38-4766 against GPCMV, 0.5 μM, was similar to that reported for HCMV, but an order of magnitude lower than that observed against MCMV (Reefschlaeger et al., 2001). In mice the drug was highly efficacious against MCMV infection (Weber et al., 2001). In animal studies, and in human volunteers, BAY 38-4766 has been found to have an exceptional safety profile. In healthy male human volunteers single oral doses of up to 2000 mg were safe and well tolerated (Nagelschmitz et al., 1999). Similarly, a highly favorable safety profile was similarly observed in guinea pigs in this study. At a dose of 50 mg/kg/day, no evidence of hematologic or biochemical toxicity (monitored by complete blood count, renal profile, and liver function testing) was observed. Comparison of weight gain between groups of animals receiving BAY 38-4766 or the placebo (tylose vehicle) indicated no adverse effect of drug on weights. In addition to its favorable safety profile, we found that BAY 38-4766 had an effect on experimental GPCMV infection. Following GPCMV challenge in immunocompromised guinea pigs, therapy with BAY 38-4766 was found to reduce systemic viral load, as assessed by real-time PCR and quantitative viral blood culture, in strain 2 guinea pigs. The drug also markedly improved survival in more heavily immunocompromised Hartley guinea pigs following GPCMV challenge, and was associated with reductions in DNAemia at both 7 and 10 days post-infection in these animals (Fig. 3). However, no differences were noted in end-organ viral load, or in histopathology of solid organs, in Hartley guinea pigs. At the dosage we used in this study, this compound may not accumulate to sufficient levels in all tissue compartments in vivo to result in a discernable reduction in viral load in visceral organs, which could explain the lack of any differences in tissue histopathology. The magnitude of the reduction of viral load was greater in strain 2 animals than in Hartley guinea pigs, suggesting that there may be differences in the pharmacokinetics of the compound, which vary depending upon the strain of guinea pigs used. Future studies of more frequent or higher doses of BAY 38-4766, or of drug administered in combination with other antiviral agents, may result in an even greater impact on viral replication in vivo in the guinea pig model.
It was of interest that BAY 38-4766 crossed the guinea pig placenta, with fetal blood levels approximating 20-30% of those of maternal blood. These observations may be germane to evaluation of treatment of HCMV infection in pregnancy (Schleiss and McVoy, 2004). Antiviral compounds which cross the placenta could conceivably be of value in management of HCMV infections acquired in utero. Given its apparent lack of toxicity, BAY 38-4766 could warrant consideration for this clinical scenario. Ongoing study and eventual licensure of this agent for HCMV disease in immunocompromised patients could prove to be of great clinical benefit, particularly in the setting of documented antiviral resistance to the traditional nucleoside antivirals (Blackman et al., 2004). Evaluation of BAY 38-4766, and related compounds, in guinea pig models of CMV infection, including both immunocompromised models and pregnancy/congenital infection models, may provide useful insights into treatment strategies for HCMV disease.
Acknowledgements
The excellent technical assistance of Amber-Renee Hickson, Kelly Pogorzelski, and Edward Tillitz is acknowledged. We also thank Rhonda Cardin for a critical review of the manuscript. This work was supported by National Institute of Health AI-65289 and HD38416-01, and by March of Dimes Basic Research Grants 6-FY98/99-0416 and FY01-22.
References
- Aquino-de Jesus MJ, Griffith BP. Cytomegalovirus infection in immunocompromised guinea pigs: a model for testing antiviral agents in vivo. Antiviral Res. 1989;12:181–193. doi: 10.1016/0166-3542(89)90028-4. [DOI] [PubMed] [Google Scholar]
- Bia FJ, Griffith BP, Fong CKY, Hsuing GD. Cytomegaloviral infections in the guinea pig: experimental models for human disease. Rev. Infect. Dis. 1983;5:177–195. doi: 10.1093/clinids/5.2.177. [DOI] [PubMed] [Google Scholar]
- Blackman SC, Lurain NS, Witte DP, Filipovich AH, Groen P, Schleiss MR. Emergence and compartmentalization of fatal multi-drug-resistant cytomegalovirus infection in a patient with autosomal-recessive severe combined immune deficiency. J. Pediatr. Hematol. Oncol. 2004;26:601–605. doi: 10.1097/01.mph.0000135283.77668.6a. [DOI] [PubMed] [Google Scholar]
- Bourne N, Bravo FJ, Bernstein DI. Cyclic HPMPC is safe and effective against systemic guinea pig cytomegalovirus infection in immune compromised animals. Antiviral Res. 2000;47:103–109. doi: 10.1016/s0166-3542(00)00100-5. [DOI] [PubMed] [Google Scholar]
- Britt WJ, Alford CA. Cytomegalovirus. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. Lippincott-Raven; Philadelphia: 1996. pp. 2493–2523. [Google Scholar]
- Buerger I, Reefschlaeger J, Bender W, Eckenberg P, Popp A, Weber O, Graeper S, Klenk HD, Ruebsamen-Waigmann H, Hallenberger S. A novel nonnucleoside inhibitor specifically targets cytomegalovirus DNA maturation via the UL89 and UL56 gene products. J. Virol. 2001;75:9077–9086. doi: 10.1128/JVI.75.19.9077-9086.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S. Antiviral drug resistance in human cytomegalovirus. Transpl. Infect. Dis. 1999;1:105–114. doi: 10.1034/j.1399-3062.1999.010204.x. [DOI] [PubMed] [Google Scholar]
- Demmler GJ. Congenital cytomegalovirus infection and disease. Adv. Pediatr. Infect. Dis. 1996;11:135–162. [PubMed] [Google Scholar]
- Evers DL, Komazin G, Shin D, Hwang DD, Townsend LB, Drach JC. Interactions among antiviral drugs acting late in the replication cycle of human cytomegalovirus. Antiviral Res. 2002;56:61–72. doi: 10.1016/s0166-3542(02)00094-3. [DOI] [PubMed] [Google Scholar]
- Feng JS, Crouch JY, Tolman RL, Lucia HL, Hsiung GD. Combined treatment with 2′-nor-cGMP and ganciclovir against cytomegalovirus infection in a guinea pig model. Antiviral Res. 1992;19:193–206. doi: 10.1016/0166-3542(92)90079-k. [DOI] [PubMed] [Google Scholar]
- Griffiths PD. The 2001 Garrod lecture. The treatment of cytomegalovirus infection. J. Antimicrob. Chemother. 2002;49:243–253. doi: 10.1093/jac/49.2.243. [DOI] [PubMed] [Google Scholar]
- Hu JM, Hsiung GD. Evaluation of new antiviral agents: I. In vitro perspectives. Antiviral Res. 1989;11:217–232. doi: 10.1016/0166-3542(89)90032-6. [DOI] [PubMed] [Google Scholar]
- Kimberlin DW. Antiviral therapy for cytomegalovirus infections in pediatric patients. Semin. Pediatr. Infect. Dis. 2002;13:22–30. doi: 10.1053/spid.2002.29754. [DOI] [PubMed] [Google Scholar]
- Krosky PM, Underwood MR, Turk SR, Feng KW, Jain RK, Ptak RG, Westerman AC, Biron KK, Townsend LB, Drach JC. Resistance of human cytomegalovirus to benzimidazole ribonucleosides maps to two open reading frames: UL89 and UL56. J. Virol. 1998;72:4721–4728. doi: 10.1128/jvi.72.6.4721-4728.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SB, Yang ZH, Feng JS, Fong CK, Lucia HL, Hsiung GD. Activity of (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine (HPMPC) against guinea pig cytomegalovirus infection in cultured cells and in guinea pigs. Antiviral Res. 1990;13:237–252. doi: 10.1016/0166-3542(90)90069-j. [DOI] [PubMed] [Google Scholar]
- McGregor A, Schleiss MR. Molecular cloning of the guinea pig cytomegalovirus (GPCMV) genome as an infectious bacterial artificial chromosome (BAC) in Escherichia coli. Mol. Gen. Metab. 2001;72:15–26. doi: 10.1006/mgme.2000.3102. [DOI] [PubMed] [Google Scholar]
- McSharry JJ, McDonough A, Olson B, Hallenberger S, Reefschlaeger J, Bender W, Drusano GL. Susceptibilities of human cytomegalovirus clinical isolates to BAY38-4766, BAY43-9695, and ganciclovir. Antimicrob. Agents Chemother. 2001;45:2925–2927. doi: 10.1128/AAC.45.10.2925-2927.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelschmitz J, Moeller JG, Stass H, et al. Safety tolerability and pharmacokinetics of single oral doses of BAY 38-4766 in healthy male subjects; Proceedings of the 39th ICAAC; San Francisco. 1999.Sep 26-29, [Google Scholar]
- Nichols WG, Boeckh M. Recent advances in the therapy and prevention of CMV infection. J. Clin. Virol. 2000;16:25–40. doi: 10.1016/s1386-6532(99)00065-7. [DOI] [PubMed] [Google Scholar]
- Plotkin SA. Vaccination against cytomegalovirus, the changeling demon. Pediatr. Infect. Dis. J. 1999;18:313–325. doi: 10.1097/00006454-199904000-00002. [DOI] [PubMed] [Google Scholar]
- Reed LJ, Muench H. A simple method of estimating 50 percent endpoints (TCID50) Am. J. Hyg. 1938;23:493–497. [Google Scholar]
- Reefschlaeger J, Bender W, Hallenberger S, Weber O, Eckenberg P, Goldmann S, Haerter M, Buerger I, Trappe J, Herrington JA, Haebich D, Ruebsamen-Waigmann H. Novel non-nucleoside inhibitors of cytomegaloviruses (BAY 38-4766): in vitro and in vivo antiviral activity and mechanism of action. J. Antimicrob. Chemother. 2001;48:757–767. doi: 10.1093/jac/48.6.757. [DOI] [PubMed] [Google Scholar]
- Schleiss MR. Cytomegalovirus in immunocompromised persons. Curr. Treat. Options Inf. Dis. 2002;4:43–50. [Google Scholar]
- Schleiss MR, McVoy MA. Overview of congenitally and perinatally acquired cytomegalovirus infections: recent advances in antiviral therapy. Expert. Rev. Anti-Infect. Ther. 2004;2(3):389–403. doi: 10.1586/14787210.2.3.389. [DOI] [PubMed] [Google Scholar]
- Schleiss MR, Bourne N, Bravo F, Jensen NJ, Bernstein DI. Quantitative competitive PCR (qcPCR) analysis of viral load following experiment guinea pig cytomegalovirus (GPCMV) infection. J. Virol. Methods. 2003;108:103–110. doi: 10.1016/s0166-0934(02)00265-3. [DOI] [PubMed] [Google Scholar]
- Schleiss MR, McGregor A, Jensen NJ, Erdem G, Aktan L. Molecular characterization of the guinea pig cytomegalovirus UL83 (pp65) protein homolog. Virus Genes. 1999;19:205–221. doi: 10.1023/a:1008136714136. [DOI] [PubMed] [Google Scholar]
- Underwood MR, Harvey RJ, Stanat SC, Hemphill ML, Miller T, Drach JC, Townsend LB, Biron KK. Inhibition of human cytomegalovirus DNA maturation by a benzimidazole ribonucleoside is mediated through the UL89 gene product. J. Virol. 1998;72:717–725. doi: 10.1128/jvi.72.1.717-725.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber O, Bender W, Eckenberg P, Goldmann S, Haerter M, Hallenberger S, Henninger K, Reefschlaeger J, Trappe J, Witt-Laido A, Ruebsamen-Waigmann H. Inhibition of murine cytomegalovirus and human cytomegalovirus by a novel non-nucleosidic compound in vivo. Antiviral Res. 2001;49:179–189. doi: 10.1016/s0166-3542(01)00127-9. [DOI] [PubMed] [Google Scholar]