

Abstract
Transgenic mice with macrophage-specific expression of human (hu) lipoprotein lipase (LPL) were generated to determine the contribution of macrophage LPL to atherogenesis. Macrophage specificity was accomplished with the scavenger receptor A promoter. Complete characterization demonstrated that macrophages from these mice expressed huLPL mRNA and secreted enzymatically active huLPL protein. Expression of huLPL was macrophage specific, because total RNA isolated from heart, thymus, lung, liver, muscle, and adipose tissues was devoid of huLPL mRNA. Macrophage-specific expression of huLPL did not exacerbate lesions in aortas of C57BL/6 mice even after 32 weeks on an atherosclerotic diet. However, when expressed in apolipoprotein E knockout background, the extent of occlusion in the aortic sinus region of male huLPL + mice increased 51% (n= 9 to 11, P < 0.002) compared with huLPL − mice after they had been fed a Western diet for 8 weeks. The proatherogenic effect of macrophage LPL was confirmed in serial sections of the aorta obtained after mice had been fed a Western diet for 3 weeks. By immunohistochemical analysis, huLPL protein was detected in the lesions of huLPL + mice but not in huLPL − mice. Our results establish that macrophage LPL accelerates atherosclerosis in male apolipoprotein E knockout mice.
Keywords: lipoprotein lipase, macrophages, atherosclerosis, apoE knockout mice, transgenic mice
Lipoprotein lipase (LPL)1 is a 55-kDa heparin-binding glycoprotein secreted by adipocytes, muscle cells, and macrophages.1 After secretion, it anchors to the luminal side of the capillary endothelium, where it interacts with circulating lipoprotein particles and is responsible for the hydrolysis of lipoprotein triglycerides. It is believed that LPL also functions as an apoprotein (apo) by associating with remnant particles and providing the recognition signal for the rapid hepatic clearance of remnant particles (Chappell and Medh2 and references therein).
There is substantial evidence to suggest that plasma LPL is antiatherogenic because of its role in clearing the plasma of chylomicrons and VLDL particles.2,3 However, macrophages also secrete significant amounts of LPL. Several reports have implicated macrophage-derived LPL in atherogenesis.4,5 Northern blot analysis and in situ hybridization show that macrophages are the most abundant source of LPL mRNA in rabbit and human atherosclerotic plaques. Corey and Zilversmit6 demonstrated a direct correlation between local LPL protein in the aorta and the extent of aortic cholesterol deposits. Murine susceptibility to atherosclerosis is known to vary substantially among strains and correlates only weakly with plasma cholesterol levels.7 However, peritoneal macrophages from atherosclerosis-susceptible C57BL/6J mice synthesize significantly higher amounts of LPL mass, activity, and mRNA than atherosclerosis-resistant C3H/HeN mice.8 Crossbreeding between C57BL/6J and atherosclerosis-resistant A/J mice revealed that atherosclerosis susceptibility is coinherited with expression of high levels of macrophage LPL.8 Babaev et al9 recently generated mice lacking macrophage LPL by fetal liver cell transplantation techniques. Removal of macrophage LPL resulted in a 55% reduction in aortic lesion size compared with controls. In a recent report, Clee et al10 directly addressed the opposing effects of plasma and macrophage LPL on atherogenesis. They compared lipid levels and lesion size in LPL+/+ and LPL+/− mice that express, respectively, high and low levels of LPL in both the plasma and macrophages and transgenic mice that overexpress human LPL (huLPL) in most tissues except the macrophages. Plasma LPL levels were highest in huLPL transgenic mice and lowest in LPL+/− mice. Plasma triglyceride and cholesterol levels were inversely correlated with plasma LPL levels. However, lesions were largest in LPL+/+ mice and smallest in huLPL transgenic mice. Thus, Clee et al10 demonstrated that in LPL+/− mice, reduced macrophage LPL levels protected against atherosclerosis despite the dyslipidemia of reduced plasma LPL.
Given these data, we hypothesized that macrophage LPL may promote atherosclerosis. To investigate this possibility, we developed transgenic mice with macrophage-specific expression of huLPL under the control of the scavenger receptor A (SRA) promoter. When huLPL was expressed in macrophages of apoE knockout (KO) mice, there was a significant increase (50%) in lesion area compared with control huLPL −/apoE KO mice. Female mice exhibited larger lesions than males even in the absence of huLPL, and in females, huLPL expression did not significantly increase lesion area. Our data suggest that macrophage LPL is proatherogenic in male apoE KO mice.
Methods
Preparation of the huLPL Gene Construct
A construct designated as JC46 was obtained as a generous gift from Dr Chris Glass (University of California, San Diego). It consisted of a Bluescript (pBSK II–) backbone with a chimeric human SRA enhancer-promoter region (−4.5 to −4.1 kb enhancer and −245 to +46 of the promoter). A 1.65-kb EcoRI fragment containing the entire LPL cDNA was inserted into the multiple cloning site of the JC46 construct.
Production of Transgenic Mice
The JC46/LPL construct described above was microinjected into single-cell fertilized mouse embryos, which were then implanted into surrogate mothers. Founder mice were classified for the presence of transgene by huLPL-specific polymerase chain reaction (PCR) amplification of genomic DNA isolated from tail biopsy samples. For this, we used an upstream sense primer (CTCGTGCTGACTCTGGCC) from the signal sequence region (positions 19 to 36 from translation-initiating ATG), which has only 66% identity to the mouse sequence.11 The downstream antisense primer (GGTCCAGCTGGATCGAGG) was from positions 542 to 560. This set of primers was specific for huLPL, because no amplification was obtained with either mouse genomic DNA or mouse RNA as templates. huLPL+ founders were used to establish transgenic lines by breeding to C57BL/6J mice. The studies reported here were done after the fourth to sixth backcross. Each mouse line presented offspring that were either positive or negative for the transgene. Thus, all mice used in the study were genotyped by PCR analysis as described above, and huLPL− littermates served as age- and sex-matched controls.
Expression of huLPL transgene was also obtained in apoE KO mice by crossbreeding hemizygous huLPL+ mice with apoE KO mice. The first-generation offspring were apoE+/− and huLPL+ or huLPL−. Next, huLPL+/apoE+/− offspring were crossed with each other or with apoE KO mice. The apoE genotype of resulting offspring was determined by a combination of 2 PCRs. An apoE reverse primer from exon 4 (TGTCTTCCACTATTGGCTCG) was paired with a sense primer (TGGCGGACCGCTATCAGGAC) from the neocassette (to identify a recombination and apoE KO allele) or with a primer (GCGAAGATGAAGGCTCTGTGG) from exon 2 (to identify a wild-type apoE allele). This set of PCR amplifications helped determine the apoE genotype and aided in the selection of mice that were apoE−/−.
Isolation of Mouse Macrophages
All animal procedures followed were in accordance with institutional animal care and use guidelines. Resident mouse macrophages were harvested by washing the peritoneal cavity with RPMI 1640. For isolation of stimulated macrophages, the peritoneal cavity was washed 4 days after mice were injected with 2 mL of 3% thioglycollate broth intraperitoneally. We routinely obtained 0.8 to 1.2×106 resident or 10 to 20×106 stimulated macrophages from each mouse.
Isolation of RNA and Northern Blotting
Cultured mouse macrophages were harvested in Tri reagent (Sigma Chemical Co; 5 × 106 cells/mL reagent) and stored at −70°C. Tissue samples were frozen in liquid nitrogen immediately after dissection, then powdered while still frozen and suspended in Tri reagent. Total RNA was isolated as per instructions provided with the reagent. The RNA was resolved on 1% agarose gels containing 0.6 mol/L formaldehyde and transferred to Nytran membrane by capillary action. Hybridization was done with specific DNA probes radiolabeled with α-[32P]-dCTP (100 mCi per 20 to 50 ng of cDNA per reaction) and a random primed DNA labeling kit (BMB). A 1.65-kb EcoRI full-length huLPL cDNA fragment that recognized both a 2.4-kb huLPL and a 4.0-kB endogenous mouse LPL (moLPL) mRNA was used as a probe for LPL. The huLPL transgene does not contain the entire 3′ untranslated region,12,13 and thus it is smaller than the moLPL. To ascertain equal loading of RNA, membranes were probed for 18S RNA or GAPDH mRNA.
Detection of LPL Protein and Activity
Anti-LPL chicken IgY-sepharose was used to immunoprecipitate LPL secreted by peritoneal macrophages from transgenic mice. Adsorbed proteins were eluted from IgY-sepharose with 50 μL of 1× Lammeli’s sample buffer and resolved by SDS-PAGE. After electrophoretic transfer to Immobilon membrane, huLPL was detected with IgG-5D2, a monoclonal antibody specific to huLPL (IgG-5D2 was obtained from Dr John Brunzell, University of Washington, Seattle). The macrophage culture medium was assayed for LPL lipolytic activity with Triolein-labeled Intralipid emulsion as a substrate.14
Phenotypic Analysis of Transgenic Mice
C57BL/6 mice were fed a regular chow (Harlan Teklad 7012C) containing 5.7% total fat or a high-fat atherosclerotic diet (Harlan Teklad 88051) containing 7.5% cocoa butter and 1.25% cholesterol. Mice with the apoE KO genetic background were fed a Western-type diet (Harlan Teklad 88137) containing 0.15% cholesterol and 21% milk fat. After 32 weeks on the atherosclerotic diet or 3 to 17 weeks on the Western diet, blood was drawn by cardiac puncture and the animals were killed. EDTA plasma was analyzed for total cholesterol (cholesterol oxidase method) and triglyceride (GPO-Trinder method) with diagnostic kits from Sigma. The hearts were removed, fixed, and infiltrated with paraffin. They were trimmed until the aortic sinus region was reached, as identified by the presence of valve cusps, and then 8-μm-thick transverse serial sections were cut. These were subjected to histochemical analysis by the Verhoeff-van Gieson technique for detection of atherosclerotic lesions. Sections were photographed by light microscopy at a magnification of 250×. Lesion and lumen areas were calculated with V-Trace software of the University of Iowa Image Analysis Facility.
Immunohistochemical Detection of huLPL in Aortic Lesions
apoE KO mice with macrophage-specific expression of huLPL were maintained on a Western diet for 17 weeks. Mice were killed, and their hearts were removed and immediately frozen on dry ice in tissue-freezing medium. A cryostat was used to obtain 10-μm sections from the aortic sinus region. These were processed for immunohistochemistry with monoclonal antibody IgG-5D2 as the primary antibody and goat anti-mouse ultrasmall gold as the secondary antibody. Images were obtained under epipolarized light. The same sections were also stained with hematoxylin and eosin for visualization of lesion morphology under the light microscope.
Results
Macrophages From Transgenic Mice Secrete huLPL
We measured LPL protein, mRNA, and enzymatic activity in peritoneal macrophages from each of 4 transgenic mouse lines. Representative Western blot results for these 4 mouse lines are compiled in Figure 1A. For each huLPL+ animal, we also ran samples from an huLPL− littermate (sex matched) as a control. huLPL protein was abundant in the culture media of mouse thioglycollate-stimulated peritoneal macrophages from each of lines 3717/4, 4676/3, 4697/3, and 4699/1. The identity of huLPL was verified by running human milk LPL in an adjacent lane. Our results show that for each mouse line, the littermate with the huLPL− genotype failed to show a band for huLPL protein. moLPL was not detected because IgG-5D2 is specific for huLPL. The heavier band seen at the top edge of the panel in Figure 1A is the IgY heavy chain.
Figure 1.

huLPL protein and mRNA in macrophages from transgenic mice. A, LPL protein was immunoprecipitated from the culture media of thioglycollate-stimulated macrophages of huLPL+ and huLPL− transgenic mice. huLPL was identified with the specific antibody IgG 5D2. B, huLPL and moLPL mRNA in thioglycollate-stimulated macrophages isolated from huLPL+ and huLPL− littermates from each of 4 transgenic lines. C, Comparison of LPL mRNA in macrophages harvested before (resident) and 4 days after intraperitoneal injection of thioglycollate broth in huLPL+ and huLPL− littermates.
The Table shows the lipase activity in culture media of resident macrophages isolated from peritoneal cavities of these mice. Each pair consists of pooled cells obtained from 3 to 7 huLPL+ or huLPL− littermates. Results are averages of 3 separate cell cultures for each group. The lipase activity represents the total of endogenous moLPL and transgenic huLPL, both of which would be secreted into the media by these cells. Thus, we expected to see lipase activity in mice with either the huLPL+ or huLPL− genotypes, with increased activity in huLPL+ animals. As shown in the Table, the activity ranged from 3 to 7.5 mmol/L free fatty acid (FFA) per minute per milligram of cell protein for huLPL− mice and from 7 to 24 mmol/L FFA · min−1 · mg−1 cell protein for huLPL+ mice. In all cases, macrophages from huLPL+ mice exhibited 1.3- to 3.4-fold higher lipase activity than cells from their huLPL− littermates.
Lipase Activity in Culture Media of Peritoneal Macrophages
| Mouse Line | Genotype | Lipase Activity, mmol/L FFA · min−1 · mg−1 (n±3) | P |
|---|---|---|---|
| 3717/4 | huLPL− | 5.43±0.36 | <0.03 |
| huLPL+ | 7.12±0.78 | ||
| 4676/3 | huLPL− | 3.12±0.56 | <0.0001 |
| huLPL+ | 9.33±0.45 | ||
| 4697/3 | huLPL− | 7.49±0.08 | <0.0001 |
| huLPL+ | 14.86±0.86 | ||
| 4699/1 | huLPL− | 7.19±0.87 | <0.002 |
| huLPL+ | 24.34±4 |
FFA indicates free fatty acid.
Resident mouse macrophages were isolated from each mouse line. Cells from 3 to 7 mice with either huLPL+ or huLPL− genotype were pooled and cultured as in Figure 1. Lipase activity in culture media was determined as in Methods. Results are corrected for cell number by use of total cellular protein.
Expression of huLPL RNA by Transgenic Mice
We next examined the relative expression of huLPL and moLPL mRNA in all 4 lines by Northern blot analysis. Macrophages for this experiment were elicited by inoculation with thioglycollate. moLPL mRNA was abundant in all of the samples. huLPL mRNA was detected in the huLPL+ mice from all 4 lines, but none was seen in huLPL− littermates (Figure 1B). The level of expression of huLPL mRNA was different in the 4 lines, with the greatest levels seen in line 4676/3. In thioglycollate-stimulated macrophages from the other 3 lines, the level of huLPL mRNA was less than that of moLPL mRNA.
We hypothesized that the low ratio of huLPL to moLPL mRNA might result from induction of endogenous moLPL transcription by thioglycollate-mediated immunological activation of macrophages. We thus examined the pattern of huLPL and moLPL expression in resident macrophages. Representative results are shown in Figure 1C. Each lane represents total RNA isolated from 3 confluent 60-mm culture wells. The amount of RNA loaded per lane within each mouse line was comparable, as determined by the amount of 18S RNA (data not shown). Clearly, in resident macrophages, the level of huLPL mRNA was significantly greater than moLPL mRNA in all mouse lines. It was also evident that thioglycollate stimulation led to an induction of moLPL mRNA by severalfold.
Expression of huLPL Is Macrophage Specific
Next, we investigated whether huLPL expression driven by the SRA promoter was indeed specific to macrophages by examining the expression of huLPL in various mouse tissues. The results for all 4 of the mouse lines were similar. Figure 2 shows representative Northern blots for tissues from lines 4676/3 and 4699/1. The presence of comparable levels of total RNA in all of the tissue homogenates was verified by probing for 18S RNA (data not shown). We found significant amounts of moLPL mRNA in heart and adipose tissues, with spleen and muscle also presenting detectable levels. On the other hand, huLPL was not detected in any of these tissues (Figure 2). We were able to detect huLPL mRNA in macrophages isolated from spleen (data not shown). These experiments demonstrate that the expression of the huLPL transgene is specific for macrophages, and it is not transcribed in other cell types.
Figure 2.

huLPL mRNA transcription is macrophage specific. Macrophages (Mφ) were harvested 4 days after thioglycollate injection in huLPL+ transgenic mice. Animals were allowed to recover for 1 week and were then killed. Various tissues were removed and instantly frozen in liquid nitrogen. Total RNA was isolated from macrophages after 24 hours in culture or from frozen tissues and subjected to Northern blot analysis probing for LPL mRNA. Representative results are shown for mouse lines 4676/3 and 4699/1.
Effect of huLPL Transgene on Plasma Lipids and Aortic Lesions
We investigated the effect of macrophage-specific expression of huLPL on the phenotype. Initially, we studied transgenic mice with the C57BL/6 background. There was no effect of huLPL expression on plasma total cholesterol or triglyceride levels of C57BL/6 mice regardless of diet or sex (male mice on atherosclerotic diet: cholesterol (mmol/L), huLPL+ 3.73 ± 0.21, huLPL− 4.30±0.28, P = 0.13; triglyceride (mmol/L), huLPL+ 0.14±0.03, huLPL− 0.22±0.04, P = 0.17). In eighth-generation transgenic mice, the aortic sinus region and the proximal thoracic aorta were examined for the presence of atherosclerotic lesions by histochemistry with the Verhoeff–van Gieson staining technique. We found that even after 32 weeks on an atherosclerotic diet, the mice did not develop significant lesions. A few small lesions were seen, particularly at branch points, but they were present in the same frequency and intensity in both huLPL− and huLPL+ transgenic mice.
Next, we repeatedly crossed huLPL + mice with apoE KO mice to obtain expression of the transgene in the apoE KO background. The level of huLPL mRNA, protein, and activity in apoE KO transgenic mice was similar to that obtained in the C57BL/6 background. ApoE KO mice with huLPL+ or huLPL− genotype were maintained on normal chow or a Western diet for up to 17 weeks. Figure 3 shows plasma lipid levels after 11 to 13 weeks on a Western diet. The mice were separated into groups based on sex and diet. In each of the 4 groups, we found that there was no difference in either plasma total cholesterol or triglyceride between huLPL− and huLPL+ mice. As reported earlier for apoE KO mice, plasma cholesterol levels increased and triglyceride levels decreased after fat feeding.
Figure 3.
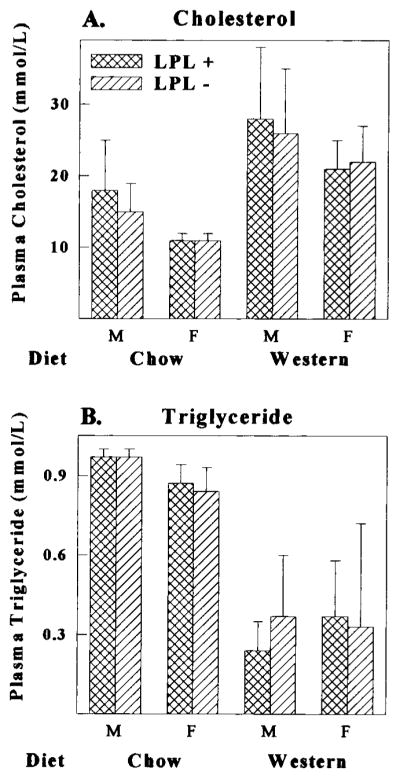
Plasma cholesterol and triglyceride levels in huLPL+ and huLPL− transgenic mice. Transgenic mice in apoE KO background were fed either a chow or Western diet for 11 to 13 weeks. Mice were divided into 4 groups based on diet and sex (M indicates males; F, females). EDTA plasma was collected after a 4-hour fast and assayed for total cholesterol (A) and triglyceride (B) levels as described in Methods. Results are averages of 8 to 12 animals in each group.
We also studied aortic lesions in huLPL+/apoE KO and huLPL−/apoE KO mice. Figure 4A shows typical photomicrographs of cross sections of the aorta through the aortic sinus region. The Verhoeff stain makes the elastin bands dark and clearly visible. Lesions were seen on the luminal side of the elastin, with obvious foam cell morphology in early lesions and cholesterol crystals in more advanced plaques. We used the University of Iowa Imaging Facility and their V-Trace software to trace the lesion and luminal regions and calculate the areas. Initially, we measured lesions in the aortic sinus region after mice had been fed a Western diet for 8 weeks. The results are shown in Figure 4B as a percentage of the luminal area. The extent of occlusion in the aortic sinus region of huLPL +/apoE KO mice was increased 51% (P<0.002) compared with huLPL−/apoE KO mice in males but not in females. Consistent with previous reports, female mice showed greater lesion area than males even in the absence of the transgene.
Figure 4.

Atherosclerotic lesions in huLPL+ and huLPL− mice. huLPL+/apoE KO and huLPL−/apoE KO mice were maintained on a Western atherogenic diet for 8, 13, or 17 weeks. A, Representative photomicrographs from aortic sinus region of huLPL+/apoE KO and huLPL−/apoE KO male mice they had been fed a Western diet for 13 weeks. Lesion and luminal areas were traced and the areas measured with imaging software. B, Lesion areas for males and females after being fed a Western diet for 8 weeks are represented as percentage of total luminal area of that particular section. Each point represents a unique section from the aortic sinus region of a different animal. C, Lesion area was measured in serial sections starting from the aortic sinus region and proceeding away from the heart. Results for male huLPL+/apoE KO (closed circles) or huLPL−/apoE KO (open squares) mice after 3 weeks on a Western diet are shown.
Lesion size is also dependent on the position in the aorta being largest at the aortic sinus and gradually shrinking toward the thoracic region. Thus, we stained serial sections starting at the sinus and proceeding away from the heart. For these studies, we killed mice after they had been fed a Western diet for 3, 8, or 13 weeks or a regular chow diet for 15 weeks. In all groups, male huLPL+/apoE KO mice showed significantly larger lesions than male huLPL−/apoE KO mice. Figure 4C shows the data for male mice after they had been fed a Western diet for 3 weeks. The difference in lesion size between the huLPL+ and huLPL− mice was greatest (50%) at the sinus, and the plots converged as the distance from the sinus increased. The data were subjected to a statistical significance test with a repeated-measures analysis. All analyses were performed with the software SAS/STAT, procedure MIXED (SAS version 8, SAS Institute Inc). Using this method, we tested for an overall difference in aortic lesion area between huLPL+ and huLPL− mice rather than differences at each specific distance from the sinus. The test for type main effect showed a difference in mean aortic lesion area (P= 0.093), with huLPL+ mice having an average mean aortic lesion area 4.4 ± 1.5 times that of the huLPL− mice. There was no significant difference in lesion size between female huLPL+ and huLPL− mice on a Western diet.
huLPL Protein Is Present in Aortic Lesions
We investigated whether huLPL protein was present in lesions of huLPL+ mice by immunohistochemistry, as described in Methods. Representative results are shown in Figure 5. huLPL was detected in lesions of huLPL+/apoE KO mice but not of huLPL−/apoE KO mice. The same sections were also stained with hematoxylin and eosin to visualize vessel morphology. huLPL was present in lesion areas closest to the lumen, which displayed a foam cell–like morphology.
Figure 5.

huLPL protein is present in aortic lesions of transgenic mice. Male apoE KO mice with transgenic huLPL+ (A and C) or huLPL− (B and D) genotype were maintained on a Western diet for 17 weeks. Aortic sections were processed for immunohisto-chemistry with an huLPL-specific monoclonal IgG 5D2 as the primary antibody. The secondary antibody used was goat anti-mouse ultrasmall gold. Images were obtained under epipolarized light (C and D). Arrows indicate staining for LPL. The same sections were also stained with hematoxylin and eosin for visualization of lesion morphology under the light microscope (A and B).
Discussion
Abnormal LDL and HDL cholesterol levels account for only 50% to 60% of clinically significant atherosclerotic events. Additional risk factors may involve cellular mechanisms that are localized to the vessel wall. Although the protective role of macrophage apoE has been well documented, the role of macrophage LPL is not clear. It has been demonstrated that macrophages are the major cells responsible for LPL synthesis in atheromas. 4,5 LPL may accelerate atherosclerosis by promoting receptor-mediated cholesterol accumulation.15,16 LPL-mediated lipolysis of large chylomicrons to progressively smaller particles may facilitate their penetration of the endothelial barrier.17 Lipolysis leads to the release of free fatty acids, which have been shown to increase endothelial permeability and transendothelial movement of lipoproteins in vitro.18 LPL may also decrease lipoprotein efflux from arteries by increasing lipoprotein retention in the subendothelial matrix.19 Clee et al10 demonstrated greater dyslipidemia but smaller lesions in LPL+/− mice than in LPL+/+ mice. On the basis of this evidence, we hypothesized that macrophage LPL might be atherogenic.
To directly test our hypothesis, we developed transgenic mice with macrophage-specific expression of huLPL. The presence of huLPL DNA and mRNA was confirmed by PCR amplification of genomic DNA and reverse transcriptase–PCR of total RNA (data not shown). In resident macrophages from all 4 transgenic mouse lines, the level of huLPL mRNA expression was significantly higher than that of moLPL. However, when inflammatory macrophages were isolated after stimulation by thioglycollate injection, the expression of moLPL mRNA was induced severalfold, resulting in higher levels than that of the transgene. This was in agreement with earlier studies by Goldberg and Khoo,20 who reported a 10-fold increase in LPL secretion when macrophages were elicited by thioglycollate injection. However, inflammatory macrophages did not induce expression of huLPL mRNA, which supports the suggestion that the SR promoter is not affected by thioglycollate.21 The physiological significance of this observation is not clear. It is well known that inflammation plays a role in atherogenesis.22 However, previous studies indicate that inflammatory cytokines such as tumor necrosis factor-α and interferon-γ downregulate LPL synthesis and secretion in in vitro assays with cultured macrophages. 23,24 Clearly, the mechanisms of thioglycollate-mediated inflammation are distinct from those triggered by inflammatory cytokines.
The macrophage specificity of the transgene is similar to that in previous studies by Horvai et al,25 who used the same promoter to express human growth hormone. They obtained a high level of transgene expression in macrophages without expression in other tissues. In the present studies, huLPL mRNA was not detectable in total RNA isolated from whole tissues including thymus, heart, lung, liver, spleen, muscle, and adipose. The liver, heart, spleen, lung, and thymus are expected to contain macrophages along with other cell types; however, the relative abundance of macrophages may be low enough that they do not contribute detectable levels of huLPL mRNA. When macrophages were isolated from the spleen, we were able to demonstrate the expression of huLPL mRNA. As expected, moLPL mRNA was detected in heart, muscle, and adipose tissues.
We were unable to detect huLPL in postheparinized plasma of transgenic mice. This was consistent with the fact that the transgene is specific to macrophages and is not expressed by monocytes. Also, the level of expression in macrophages is not high enough for the secreted protein to enter the circulation. As a result, plasma total cholesterol and triglyceride levels were similar between huLPL+ and huLPL− animals matched for sex and diet. Thus, the difference in atherogenicity between huLPL+ and huLPL− mice is independent of plasma lipid profiles and completely dependent on events at the macrophage cellular level. The demonstration of the presence of huLPL protein in the lesions is consistent with a localized effect of macrophage LPL on the vascular wall.
In C57BL/6 mice, we found only small atherosclerotic lesions in the thoracic aorta of both huLPL + and huLPL− male mice even after they had been fed an atherosclerotic diet for 32 weeks, whereas apoE KO mice displayed 30% to 40% occlusion of the aortic sinus region within 3 weeks of being fed a Western diet. The levels of huLPL mRNA expression, protein mass, and enzymatic activity were similar between C57BL/6 and apoE KO mice. Also, as described in Methods, the atherosclerotic diet fed to C57BL/6 has a total fat and cholesterol content higher than that in the Western diet fed to apoE knockout mice; thus, it is unlikely that the absence of lesions in C57BL/6 mice was due to the different diet. The plasma cholesterol and triglyceride levels of apoE KO mice on the Western diet were 3- to 6-fold greater than the levels in C57BL/6 mice on the atherosclerotic diet. We believe that this resistance to hyperlipidemia and atherosclerosis in C57BL/6 mice is because of the presence of apoE, which mediates clearance of lipoprotein particles from the plasma and promotes reverse cholesterol transport from vascular wall macrophages to the liver. It is also possible that during generation of the transgenic lines, the C57BL/6 genetic background became mixed with an atherosclerosis-resistant strain. It is well known that different mouse strains display differing susceptibilities to atherosclerosis. As is common for transgenesis, we generated our transgenic mice by microinjecting embryos from B6SJL parents. The B6SJL are a hybrid mice obtained by crossing C57BL/6 mice to SJL/J mice (Jackson Labs). The SJL/J strain is resistant to atherosclerosis. Even though we have studied sixth-to eighth-generation transgenic mice, it is possible that the genetic material from the SJL/J strain is not sufficiently diluted and contributes to atherosclerosis resistance in the C57BL/6 background. However, when apoE was absent, as in the apoE KO background, the extent of lesions dramatically increased, which confirms the protective role of macrophage apoE against atherosclerosis.
Our results indicate a proatherogenic role for macrophage LPL in apoE-deficient male mice. The presence of huLPL transgene increased atherosclerosis at all time points after a high-fat diet. There was some inherent variability in the size and weight of individual mice within each diet- and sex-matched group. Thus, in Figure 4B, luminal area was used as an internal control to correct for differences in animal size. When data were presented as a percentage of total luminal area, male huLPL + mice fed a Western diet for 8 weeks showed a significant 51% increase in lesion size compared with huLPL− mice. We also measured the absolute lesion area in progressive serial sections starting from the aortic sinus region in male mice after 3 weeks on a Western diet. When represented as absolute lesion size, there were larger error bars for each position on the aorta. We attribute this to differences between individual animals. It is also likely that the increased variability seen in Figure 4C is because the data represent early lesions seen after 3 weeks on the Western diet. At this point, the animals were young and growing and the lesions were still rapidly developing, and thus they displayed a wide range of lesion areas. However, in spite of the variability between animals, huLPL+/apoE KO mice consistently displayed larger lesions than huLPL−/apoE KO mice at all positions on the aorta. Our data were examined by a biostatistician using a repeated-measures analysis. The mean aortic lesion area for huLPL+ male mice was found to be 4.4±1.5 times the mean lesion area for huLPL− mice, with a probability value of 0.093. Normally a probability value >0.05 is not considered significant; however, in this case, because we were calculating the combined probability of all points on the plots to align such that the huLPL+ plot was higher than the huLPL− plot, a probability of 0.093 is still considered significant. Taken together, the data presented in Figures 4B and 4C demonstrate a proatherogenic role for macrophage LPL in male apoE KO mice.
There was no significant difference in lesion size between huLPL+ and huLPL− female mice. The reason for this is not clear. However, similar to several other reports in C57BL/6 mice,26–28 female mice had consistently larger lesions than males. This has been attributed to lower plasma HDL levels in female mice than in males.26 In the present study, female mice had 50% to 70% larger lesions than males, whereas in males, the presence of huLPL transgene increased lesion size by 50%. It is likely that LPL-independent proatherogenic factors in females render the contribution of macrophage LPL insignificant. There is precedence for sex-specific resistance of females to small alterations in LPL mass and activity. Male LPL+/− mice show larger lesions than male LPL +/+ mice, whereas there is no difference in lesion size between LPL+/+ and LPL+/− female mice.28 In humans, certain polymorphisms of the LPL gene alter plasma HDL cholesterol and triglyceride levels in men but not in women. The S447X mutation in the LPL gene protects men from cardiovascular disease due to an increase in plasma HDL cholesterol and a decrease in triglyceride levels, whereas female carriers of this mutation are unaffected.29 Similarly, the D9N polymorphism decreases plasma HDL cholesterol levels in male but not in female carriers.30
In a recent study, Babaev et al9 took the reverse approach and generated mice with macrophage-specific KO of LPL by fetal liver cell transplantation. They reported that the absence of macrophage LPL leads to a 55% reduction in aortic lesion area compared with wild-type controls. This result corroborates a proatherogenic role for macrophage LPL. Another macrophage enzyme, hormone sensitive lipase (HSL), is a cholesterol ester hydrolase. In a recent report, Escary et al31 demonstrated that macrophage-specific overexpression of HSL enhances atherosclerosis in C57BL/6 mice on an atherosclerotic diet. The mechanisms by which HSL overexpression enhances atherosclerosis are not clear, but certain downstream effects of lipolysis may be similar to those of LPL. Additional studies will help to clearly delineate macrophage LPL-mediated in vivo cellular mechanisms that may influence atherosclerosis.
Acknowledgments
This work was supported by grant HL49264 from the National Institutes of Health and a Grant-in-Aid award from the American Heart Association. Transgenic mice were generated at the University of Iowa Transgenic Animal Facility, supported in part by the College of Medicine and the Diabetes and Endocrine Research Center. We thank Norma Sinclair, Lucy Robbins, Patricia Lovell, and Kelly Andringa for production and genotyping of transgenic mice. We thank Kathy Walters and the University of Iowa Central Microscopy Research Facility for help with microscopy techniques and Steven Beck at the University of Iowa Image Analysis Facility for help with the V-Trace software.
References
- 1.Camps L, Reina M, Llobera M, Vilaro S, Olivecrona T. Lipoprotein lipase: cellular origin and functional distribution. Am J Physiol. 1990;258:C673–C681. doi: 10.1152/ajpcell.1990.258.4.C673. [DOI] [PubMed] [Google Scholar]
- 2.Chappell DA, Medh JD. Receptor-mediated mechanisms of lipoprotein remnant catabolism. Prog Lipid Res. 1998;37:393–422. doi: 10.1016/s0163-7827(98)00017-4. [DOI] [PubMed] [Google Scholar]
- 3.Santamarina-Fojo S, Dugi KA. Structure, function and role of lipoprotein lipase in lipoprotein metabolism. Curr Opin Lipidol. 1994;5:117–125. doi: 10.1097/00041433-199404000-00008. [DOI] [PubMed] [Google Scholar]
- 4.O’Brien KD, Gordon D, Deeb S, Ferguson M, Chait A. Lipoprotein lipase is synthesized by macrophage-derived foam cells in human coronary atherosclerotic plaques. J Clin Invest. 1992;89:1544–1550. doi: 10.1172/JCI115747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yla-Herttuala S, Lipton BA, Rosenfeld ME, Goldberg IJ, Steinberg D, Witztum JL. Macrophages and smooth muscle cells express lipoprotein lipase in human and rabbit atherosclerotic lesions. Proc Natl Acad Sci U S A. 1991;88:10143–10147. doi: 10.1073/pnas.88.22.10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corey JE, Zilversmit DB. Effect of cholesterol feeding on arterial lipolytic activity in the rabbit. Atherosclerosis. 1977;27:201–212. doi: 10.1016/0021-9150(77)90057-0. [DOI] [PubMed] [Google Scholar]
- 7.Paigen B, Morrow A, Brandon C, Mitchell D, Holmes P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis. 1985;57:65–73. doi: 10.1016/0021-9150(85)90138-8. [DOI] [PubMed] [Google Scholar]
- 8.Renier G, Skamene E, DeSanctis JB, Radzioch D. High macrophage lipoprotein lipase expression and secretion are associated in inbred murine strains with susceptibility to atherosclerosis. Arterioscler Thromb. 1993;13:190–196. doi: 10.1161/01.atv.13.2.190. [DOI] [PubMed] [Google Scholar]
- 9.Babaev VR, Fazio S, Gleaves LA, Carter KJ, Semenkovich CF, Linton MF. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in vivo. J Clin Invest. 1999;103:1697–1705. doi: 10.1172/JCI6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clee SM, Bissada N, Miao L, Marais AD, Henderson HE, Steures P, McManus J, McManus B, LeBoeuf RC, Kastelein JJP, Hayden MR. Plasma and vessel wall lipoprotein lipase have different roles in atherosclerosis. J Lipid Res. 2000;41:521–531. [PubMed] [Google Scholar]
- 11.Kirchgessner TG, Svenson KL, Lusis AJ, Schotz MC. The sequence of cDNA encoding lipoprotein lipase: a member of a lipase gene family. J Biol Chem. 1987;262:8463–8466. [PubMed] [Google Scholar]
- 12.Wion KL, Kirchgessner TG, Lusis AJ, Schotz MC, Lawn RM. Human lipoprotein lipase complementary DNA sequence. Science. 1987;235:1638–1641. doi: 10.1126/science.3823907. [DOI] [PubMed] [Google Scholar]
- 13.Deeb SS, Peng RL. Structure of the human lipoprotein lipase gene [published erratum appears in Biochemistry 1989;28:6786] Biochemistry. 1989;28:4131–4135. doi: 10.1021/bi00436a001. [DOI] [PubMed] [Google Scholar]
- 14.Iverius PH, Ostlund-Lindqvist AM. Preparation, characterization, and measurement of lipoprotein lipase. Methods Enzymol. 1986;129:691–704. doi: 10.1016/0076-6879(86)29099-0. [DOI] [PubMed] [Google Scholar]
- 15.Edwards IJ, Goldberg IJ, Parks JS, Xu H, Wagner WD. Lipoprotein lipase enhances the interaction of low density lipoproteins with artery-derived extracellular matrix proteoglycans. J Lipid Res. 1993;34:1155–1163. [PubMed] [Google Scholar]
- 16.Saxena U, Goldberg IJ. Endothelial cells and atherosclerosis: lipoprotein metabolism, matrix interactions, and monocyte recruitment. Curr Opin Lipidol. 1994;5:316–322. doi: 10.1097/00041433-199410000-00002. [DOI] [PubMed] [Google Scholar]
- 17.Stender S, Zilversmit DB. Transfer of plasma lipoprotein components and of plasma proteins into aorta of cholesterol-fed rabbits: molecular size as a determinant of plasma lipoprotein influx. Arteriosclerosis. 1981;1:38–49. doi: 10.1161/01.atv.1.1.38. [DOI] [PubMed] [Google Scholar]
- 18.Hennig B, Shasby DM, Spector AA. Exposure to fatty acid increases human low density lipoprotein transfer across cultured endothelial monolayers. Circ Res. 1985;57:776–780. doi: 10.1161/01.res.57.5.776. [DOI] [PubMed] [Google Scholar]
- 19.Saxena U, Klein MG, Vanni TM, Goldberg IJ. Lipoprotein lipase increases low density lipoprotein retention by subendothelial cell matrix. J Clin Invest. 1992;89:373–380. doi: 10.1172/JCI115595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldberg DI, Khoo JC. Regulation of lipoprotein lipase secretion by mouse peritoneal macrophages. Biochem Biophys Res Commun. 1987;142:1–6. doi: 10.1016/0006-291x(87)90443-8. [DOI] [PubMed] [Google Scholar]
- 21.Lau W, Devery JM, Geczy CL. A chemotactic S100 peptide enhances scavenger receptor and Mac-1 expression and cholesteryl ester accumulation in murine peritoneal macrophages in vivo. J Clin Invest. 1995;95:1957–1965. doi: 10.1172/JCI117879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hajjar DP, Nicholson AC. Atherosclerosis. Am Sci. 1995;83:460–467. [Google Scholar]
- 23.Friedman G, Chajek-Shaul T, Gallily R, Stein O, Shiloni E, Etienne J, Stein Y. Modulation of lipoprotein lipase activity in mouse peritoneal macrophages by recombinant human tumor necrosis factor. Biochim Biophys Acta. 1988;963:201–207. doi: 10.1016/0005-2760(88)90281-0. [DOI] [PubMed] [Google Scholar]
- 24.Querfeld U, Ong JM, Prehn J, Carty J, Saffari B, Jordan SC, Kern PA. Effects of cytokines on the production of lipoprotein lipase in cultured human macrophages. J Lipid Res. 1990;31:1379–1386. [PubMed] [Google Scholar]
- 25.Horvai A, Palinski W, Wu H, Moulton KS, Kalla K, Glass CK. Scavenger receptor A gene regulatory elements target gene expression to macrophages and to foam cells of atherosclerotic lesions. Proc Natl Acad Sci U S A. 1995;92:5391–5395. doi: 10.1073/pnas.92.12.5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paigen B, Holmes PA, Mitchell D, Albee D. Comparison of atherosclerotic lesions and HDL-lipid levels in male, female, and testosterone-treated female mice from strains C57BL/6, BALB/c and C3H. Atherosclerosis. 1987;64:215–221. doi: 10.1016/0021-9150(87)90249-8. [DOI] [PubMed] [Google Scholar]
- 27.Purcell-Huynh DA, Farese RVJ, Johnson DF, Flynn LM, Pierotti V, Newland DL, Linton MF, Sanan DA, Young SG. Transgenic mice expressing high levels of human apolipoprotein B develop severe atherosclerotic lesions in response to a high-fat diet. J Clin Invest. 1995;95:2246–2257. doi: 10.1172/JCI117915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Semenkovich CF, Coleman T, Daugherty A. Effects of heterozygous lipoprotein lipase deficiency on diet-induced atherosclerosis in mice. J Lipid Res. 1998;39:1141–1151. [PubMed] [Google Scholar]
- 29.Gagne SE, Larson MG, Pimstone SN, Schaefer EJ, Kastelein JJP, Wilson PWF, Ordovas JM, Hayden MR. A common truncation variant of lipoprotein lipase (ser447X) confers protection against coronary heart disease: the Framingham offspring study. Clin Genet. 1999;55:450–454. doi: 10.1034/j.1399-0004.1999.550609.x. [DOI] [PubMed] [Google Scholar]
- 30.Kastelein JJP, Ordovas JM, Wittekoek ME, Pimstone SN, Wilson PWF, Gagne SE, Larson MG, Schaefer EJ, Boer JMA, Gerdes C, Hayden MR. Two common mutations (D9N, N291S) in lipoprotein lipase: a cumulative analysis of their influence on plasma lipids and lipoproteins in men and women. Clin Genet. 1999;56:297–305. doi: 10.1034/j.1399-0004.1999.560407.x. [DOI] [PubMed] [Google Scholar]
- 31.Escary JL, Choy HA, Reue K, Wang XP, Castellani LW, Glass CK, Lusis AJ, Schotz MC. Paradoxical effect on atherosclerosis of hormone-sensitive lipase overexpression in macrophages. J Lipid Res. 1999;40:397–404. [PubMed] [Google Scholar]