

G protein-coupled receptors (GPCRs) are the largest family of signaling proteins expressed in every cell in the body and targeted by the majority of clinically used drugs [1]. GPCR signaling, including rhodopsin-drive phototransduction, is terminated by receptor phosphorylation followed by arrestin binding [2]. Genetic defects in receptor phosphorylation and excessive signaling by overactive GPCR mutants result in a wide variety of diseases, from retinal degeneration to cancer [3-6]. Here we tested whether arrestin1 mutants with enhanced ability to bind active unphosphorylated rhodopsin [7-10] can suppress uncontrolled signaling, bypassing receptor phosphorylation by rhodopsin kinase (RK) and replacing this two-step mechanism with a single step deactivation in rod photoreceptors. We show that in this precisely timed signaling system with single photon sensitivity [11], an “enhanced” arrestin1 mutant partially compensates for defects in rhodopsin phosphorylation, promoting photoreceptor survival, improving functional performance, and facilitating photoresponse recovery. These proof-of-principle experiments demonstrate the feasibility of functional compensation in vivo for the first time, which is a promising approach for correcting genetic defects associated with gain-of-function mutations. Successful modification of protein-protein interactions by appropriate mutations paves the way to targeted redesign of signaling pathways to achieve desired functional outcomes.
Results and Discussion
Enhanced arrestin1 mutant protects photoreceptors in the absence of rhodopsin phosphorylation
Wild type (WT) arrestins bind active unphosphorylated GPCRs with low affinity [2]. The activation of the “phosphate sensor” by mutagenesis significantly increases arrestin1 binding to unphosphorylated light-activated rhodopsin (Rh*) in vitro [7, 12]. To test whether “enhanced” mutants can compensate for the defects of rhodopsin phosphorylation in vivo, we generated transgenic mice expressing mouse arrestin1 (Arr1) with triple alanine substitution in the C-tail (3A) that demonstrates high binding to Rh* (Figure S1). Transgenic lines with high (3A-240) and moderate (3A-50) expression, corresponding to 240% and 50% of endogenous Arr1 in WT mice, respectively (Table S1), were bred onto Arr1 knockout (3A-50arr1-/- and 3A-240arr1-/-) and RK and Arr1 double knockout (3A-240arr1-/-rk-/- and 3A-50arr1-/-rk-/-) backgrounds.
Photoreceptors in RK-/- and Arr1-/- mice reared in cyclic light are disorganized and eventually degenerate [13, 14]. Dark rearing preserves normal retinal morphology, suggesting that excessive rhodopsin signaling in these animals damages photoreceptors. To test whether the mutant arrestin prevents light damage, the mice for morphological and electrophysiological studies were raised in cyclic light. We measured two morphological parameters: the length of the rod outer segments (OS), as a measure of overall photoreceptor health, and the thickness of the outer nuclear layer (ONL), which reflects the number of surviving rods (Figures 1, S2).
Figure 1. Phosphorylation-independent arrestin protects rod outer segments in the absence of rhodopsin phosphorylation.
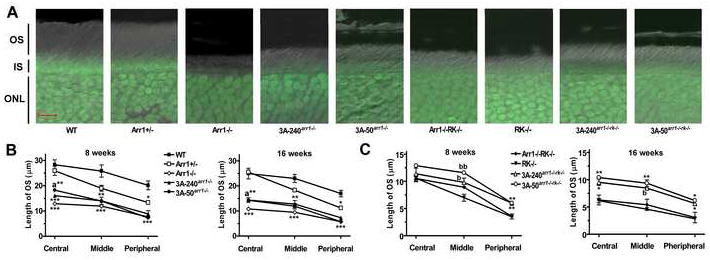
A. Combined DIC and green fluorescent Nissl images of the retina sections of 8 weeks old mice of indicated genotypes, enlarged to show OS more clearly. The positions of outer segments (OS), inner segments (IS), and outer nuclear layer (ONL) are shown on the left. B, C. The length of the OS measured in the Central, Middle, and Peripheral retina were the average of inferior and superior hemispheres. Means +/- SE from three animals per genotype are shown. The length of OS was compared separately for each age and retinal subdivision by one-way ANOVA with Genotype as main factor, followed by Scheffe’s post hoc test.
(B) * - p<0.05; ** - p<0.01; *** - p<0.001 as compared to WT; a – p<0.05 as compared to Arr1-/-. At 8 weeks in the central retina, OS in both transgenic strains are significantly shorter than in WT mice (p=0.0035 and p=0.0008, respectively), but in 3A-50 arr1-/- mice, OS are significantly longer than in Arr1-/- mice (p=0.037). In the middle retina, symbol ** applies to both transgenic lines, and in the peripheral retina symbol *** - to both transgenic and Arr1-/- lines. At 16 weeks in the central retina, OS in both transgenic strains are significantly shorter than in WT (p=0.0014 and 0.0015) but longer than in Arr1-/- mice (p=0.0213 and 0.0174, respectively for 3A-50 arr1-/- and 3A-240 arr1-/- lines). In the middle retina, symbol *** applies to both transgenic lines, and in the peripheral retina (***) - to both transgenic and Arr1-/- lines.
(C) * - p<0.05; ** - p<0.01; *** - p<0.001 as compared to both Arr1+/+RK-/- and Arr1-/-RK-/-. b – p<0.05; bb – p<0.01 as compared to Arr1+/+RK-/-. There were no significant differences in the length of OS between RK-/- and Arr1-/-RK-/- mice in any retinal subdivision at either age. High and moderate transgene expression similarly increased the length of the OS, as evidenced by the absence of significant differences between 3A-50 arr1-/-rk-/- and 3A-240 arr1-/-rk-/- lines across retinal subdivisions at both ages.
Healthy OS in WT mice maintain organized structure with lengths of ~20 μm in peripheral to 28 μm in the central retina (Figure 1A). The OS length progressively decreases in mice lacking arrestin, RK, or both (Figure 1, Table 1). The expression of mutant arrestin on the Arr1-/- background significantly increased the length of OS, although not to WT level (Figure 1B). The OS of RK-/- and Arr1-/-RK-/- were similar, indicating that WT arrestin has no beneficial effect in the absence of RK (Figure 1C). In contrast, 3A-240arr1-/-rk-/- and 3A-50arr1-/-rk-/- mice expressing mutant arrestin had significantly longer OS than RK-/- or Arr1-/-RK-/- animals (Figure 1C). The morphological protection afforded by the mutant was more evident at sixteen than at eight weeks, indicating that 3A arrestin slowed down progressive loss of OS in RK-deficient mice.
Table 1.
Morphological and functional characteristic of mice expressing WT arrestin1 and enhanced mutant in the presence and absence of rhodopsin kinase.
| Mouse line | Retinal morphology | ERG parameters | |||
|---|---|---|---|---|---|
| Length of the OS | Thickness of the ONL | Scotopic bmax (μV) | Scotopic b-wave I1/2 (logcd*s/m2) | Time of half recovery (thalf) (ms) | |
| WT | +++ | +++ | 370 ± 38 | -2.810 ± 0.044 | 378.8 ± 29.8 |
| Arr1+/- | ++a | +++ | 407 ± 73 | -3.223 ± 0.078 h | 423.9 ± 16.1 |
| Arr1-/- | - aa | + d | ND | ND | ND |
| 3A-240arr1-/- | + aa, bb | -- dd, f | 384 ± 82 | -2.522 ± 0.217 | 333.7 ± 13.9 |
| 3A-50arr1-/- | + aa, b | + dd, e | 518 ± 15 | -2.656 ± 0.093 | 372.2 ± 23.3 |
| RK-/- | - aa | -- dd | 105 ± 18 | -2.378 ± 0.200 | 18670.2 ± 3238.9 |
| Arr1-/-RK-/- | - aa | - dd, gg | ND | ND | ND |
| 3A-240arr1-/-rk-/- | + aa, c | + dd, g | 110 ± 21 | -2.478 ± 0.026 | 10200.4 ± 845.7 j |
| 3A-50arr1-/-rk-/- | + aa, c | + dd, g | 102 ± 3 | -3.015 ± 0.161i | 6571.6 ± 730.7 jj |
Morphological data were statistically analyzed by repeated measure ANOVA with Retinal Subdivision as a repeated measure and Genotype and Age as between group factors separately for Arr1-/- and RK-/- backgrounds. Since each morphological parameter is described by six measurements, here we qualitatively characterize them by symbols, as follows: +++, very healthy; ++, healthy; +, relatively healthy; -, unhealthy; --, very unhealthy. Statistical significance was obtained by Scheffe’s post hoc comparison of means for Genotype across ages and retinal subdivisions.
OS shorter than WT:
p<0.01;
p<0.0001;
OS longer than Arr1-/-:
p<0.05;
p<0.01;
OS longer than RK-/-:
p<0.001.
There were no significant differences in the effects of the mutant on OS at the two expression levels on Arr1-/- (p=0.87) or RK-/- (p=0.38) background.
ONL thinner than WT:
p<0.01;
p<0.0001;
ONL thicker than Arr1-/-:
p<0.05;
ONL thinner that Arr1-/-:
p<0.05;
ONL thicker than RK-/-:
p<0.01;
p<0.0001.
Light sensitivity (flash intensity necessary to elicit half-maximum rod-driven b-wave, I1/2) was analyzed by ANOVA with Genotype as main factor (F(6,13)-4.547; p=0.0106), followed by Scheffe post-hoc test with correction for multiple comparisons:
light sensitivity higher (I1/2 lower) than 3A-240arr1-/- (p=0.0287);
light sensitivity higher than RK-/- (p=0.0294).
Time of half-recovery of a-wave amplitude (thalf) shorter than in RK-/-:
p=0.0424;
p<0.0063.
Photoreceptor degeneration in Arr1-/- animals is reflected in progressive thinning of ONL with age (Table 1, Figure S2A). The effect of the mutant on the Arr1-/- background depended on its expression level. Moderate expression (3A-50arr1-/-) prevented photoreceptor loss, whereas high expression (3A-240arr1-/-) yielded significantly thinner ONL than other genotypes expressing RK (Figure S2A) (see supplemental discussion). All RK-deficient groups had significantly thinner ONL than WT animals (p<0.0001). Enhanced arrestin improved photoreceptor survival, as compared to RK-/- mice, in 3A-240arr1-/-rk-/- and 3A-50arr1-/-rk-/- lines across ages and retinal subdivisions (Table 1), indicating that in the absence of rhodopsin phosphorylation enhanced arrestin protects photoreceptor better than WT Arr1 (Figure S2B).
Thus, in animals with normal rhodopsin phosphorylation WT Arr1 affords healthier retinal morphology than the mutant. In contrast, in RK-deficient mice WT Arr1 does not protect photoreceptors, whereas enhanced mutant significantly improves their health and survival.
Functional rescue of RK-deficient photoreceptors by enhanced arrestin1 mutant
To compare the ability of WT arrestin and 3A mutant to inactivate rhodopsin we used suction electrodes [11] to record light-induced changes in membrane current in individual intact rods. Flash responses of RK-/- or Arr1-/- rods are abnormally prolonged [13-15]. Responses to flashes of varying strengths, as well as the amplitude of the single photon response, light sensitivity, and time constant of recovery of 3A-50arr1-/-, 3A-240arr1-/-, and WT rods were very similar (Figures 2A, S3A, Table S2).
Figure 2. Enhanced arrestin mutant is fully functional and improves photoreceptor performance in the absence of rhodopsin phosphorylation.
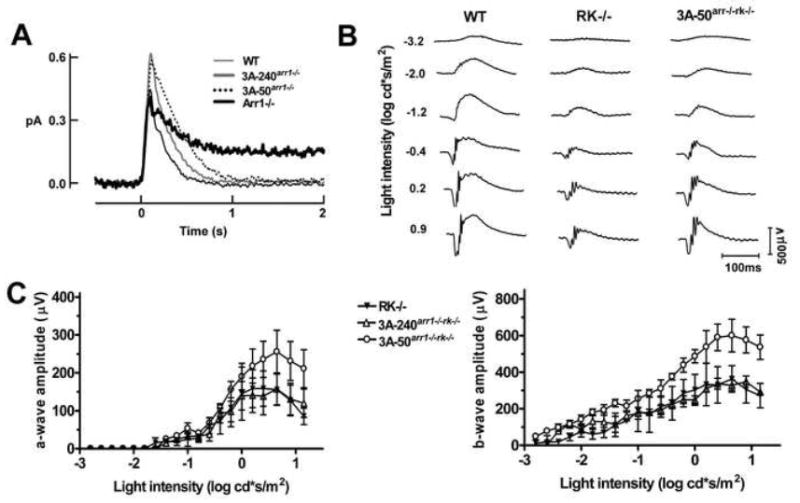
A. Single photon responses of individual rods from WT (n=13), 3A-240arr1-/- (n=19), and 3A-50arr1-/- (n=9) rods. The Arr1-/- trace shown for comparison (n=9) is from [33]. The parameters of these responses are reported in Table S2. B. Representative ERG traces of WT, RK-/-, and 3A-50arr1-/-rk-/- mice. C. Comparison of mouse lines expressing enhanced arrestin on Arr1-/-RK-/- background. Means ± SD for three animals per genotype are shown. ERG responses of 3A-50arr1-/-rk-/- mice at a given flash strength were significantly larger than those of RK-/- mice (p=0.0017), although they did not reach WT level (p=0.0002).
Moderately bright flash responses of 3A-240arr1-/-rk-/- rods were indistinguishable from those of RK-/- rods, but remained significantly slower than WT responses (Figure S3B). Dim flashes produced long-lasting step-like responses of variable durations in RK-deficient rods, reflecting the stochastic decay of individual Rh* molecules [13, 16-18]. The distributions of response durations fit single (τ=3.2 s for RK-/-) or double (τ1 = 4.1 s, τ2 = 24.3 s for RK-/-Arr1-/-) exponential functions, the time constants of which reflect the average lifetime of Rh* molecules [13, 16]. In RK-deficient rods, expression of the arrestin mutant did not alter the appearance of these step-like responses (Figure S4A). However, the distribution of response durations in 3A-240arr1-/-rk-/- rods lacked the second, slow time constant observed in Arr1-/-RK-/- rods (Figure S4B), indicating that the mutant could quench the longer-lived fraction of Rh*. The time constant of the 3A-240arr1-/-rk-/- distribution (τ=3.5 s) was similar to that of the RK-/- photoreceptors expressing WT arrestin1, and average single photon responses were indistinguishable (Figure S4C). Thus, in ex vivo single cell paradigm WT and mutant arrestin1 appear to inactivate Rh* similarly. Therefore, to elucidate functional differences underlying the stronger protective effect of the mutant in RK-deficient rods (Figures 1, S2), we used electroretinography (ERG). ERG is a non-invasive technique that allows quantitative characterization of the function of the whole retina in live animals. The early negative a-wave and subsequent positive b-wave (Figure 2B) reflect light-induced suppression of the circulating current in photoreceptors and the response of downstream cells to photoreceptor activation, respectively [19-21].
We compared mice with normal rhodopsin phosphorylation expressing WT and mutant arrestin1 by recording responses to a series of flashes and plotting the a- and b-wave amplitudes as a function of flash intensity (Figure S5). Compared to WT, light responses of the Arr1-/- mice are much smaller and detectable only at higher light levels, reflecting the contribution from cone photoreceptors [22]. 3A-50arr1-/- mice showed WT-like responses. The high expression line (3A-240arr1-/-) had lower a- and b-wave amplitudes, consistent with the decreased number of photoreceptors (Figure S2). However, maximum rod-driven b-wave (bmax) and light sensitivity (flash intensity producing half-maximum rod b-wave, I1/2) in both lines were similar and did not differ from WT (Table 1). The rate of photoreceptor recovery in vivo is determined in the double-flash paradigm [23], where the first flash desensitizes photoreceptors, and the amplitude of the response to the second flash is plotted as a function of time interval between flashes. It is characterized by halftime of recovery (thalf) [19]. The recovery kinetics of both 3A-50arr1-/- and 3A-240arr1-/- mice were indistinguishable from that of WT and Arr1+/- mice (Figure 3A,C), further confirming that the mutant successfully takes over the functions of missing WT Arr1.
Figure 3. Enhanced arrestin1 facilitates photoresponse recovery in rhodopsin kinase-deficient rods.
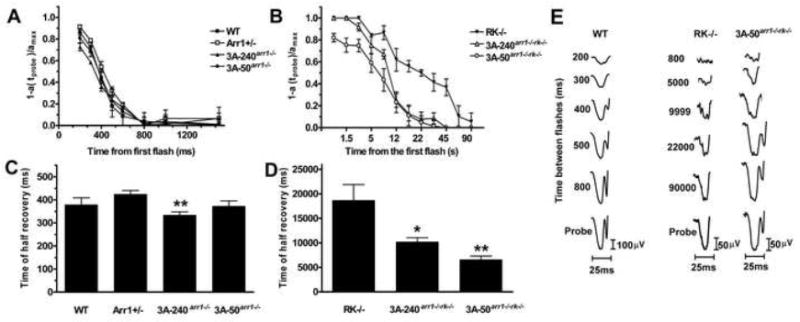
The intensities of the first (desensitizing) and second (probe) flash were 0.4 and 0.65 log cd*s/m2, respectively. The normalized amplitude of the probe flash a-wave was plotted as a function of time elapsed after the first flash (A, B) The interval between two flashes was varied from 200 to 1,500 ms for mice expressing RK (A) and from 800 to 90,000 ms for lines on RK-/- background (B). To calculate the time of half recovery, recovery kinetics were fitted by polynomial nonlinear regression, with R2>0.95, as described in methods. Means +/- SD are shown (n=4 for each genotype). The data were analyzed by one-way ANOVA with Genotype as a main factor (C, D), which was highly significant (F(6,21)=29.938; p<0.0001). C. The rates of recovery of 3A-240arr1-/- and 3A-50arr1-/- were not different from WT (p=0.9796 and p=0.9962, respectively). D. Both transgenic lines on Arr1-/-RK-/- background recovered significantly faster than RK-/-: 3A-240arr1-/-rk-/- (p=0.0424); 3A-50 arr1-/-rk-/- (p=0.0063).
To directly compare the ability of WT and mutant arrestin to turn off unphosphorylated rhodopsin, we performed similar experiments with RK-/- mice and lines expressing enhanced mutant on Arr1-/-RK-/- background. 3A-50arr1-/-rk-/- mice had significantly greater b-wave amplitude than RK-/- (p=0.0148), whereas 3A-240arr1-/-rk-/- mice were similar to RK-/- in this respect (Figure 2C). The light sensitivity of 3A-50arr1-/-rk-/- mice was comparable to that of WT animals and higher than RK-/- (p=0.0294). Most importantly, in RK-deficient animals the enhanced mutant ensured significantly faster recovery from bright flashes than WT arrestin1 (Figure 3B,D,E). The halftime of recovery was ~19s in RK-/- animals, but only ~10s in 3A-240arr1-/-rk-/- and ~6s in 3A-50arr1-/-rk-/- mice (Table 1). Thus, the enhanced mutant has a superior ability to deactivate unphosphorylated rhodopsin than WT arrestin, yielding faster photoresponse recovery in phosphorylation-deficient photoreceptors in vivo.
Biological implications of functional compensation in vivo
Both rhodopsin phosphorylation and Arr1 binding are necessary for the timely shutoff of the photoresponse [13, 14, 16, 24]. Due to rapid dissociation of the complex, the affinity of WT Arr1 for Rh* is low [25], but it can be significantly increased by “activating” mutations [7, 12, 25]. WT arrestin1 does not protect RK-deficient photoreceptors, whereas enhanced mutant improves their morphology and survival. Using ERG we found that enhanced mutant afforded significant functional rescue: rods in 3A-50arr1-/-rk-/- mice show more robust responses, normal light sensitivity, and recover three times faster than RK-/- animals (Table 1). These differences were not observed in single cell recordings assaying the activity of individual Rh* molecules, suggesting that the protective effect of the mutant Arr1 either is revealed only at higher flash intensities used in ERG, or that it requires the native photoreceptor environment. Our data indicate that in vivo the enhanced mutant outperforms WT arrestin1 in quenching unphosphorylated rhodopsin. Clear correlation between the functional rescue and morphological protection afforded by the mutant (Table 1) supports the idea that excessive rhodopsin signaling in the absence of phosphorylation drives photoreceptor demise in RK-/- mice [13].
Based on our understanding of the “inner workings” of the arrestin molecule we designed an enhanced mutant that compensates for the defects in receptor phosphorylation. However, engineered single step receptor inactivation yielded slower recovery than the normal two-step mechanism. The most likely reason for the partial rescue is that the complex of the mutant with Rh* is not as stable as the Arr1-P-Rh* complex. To construct an enhanced mutant that retains high-affinity for P-Rh*, we relieved a conformational constraint (Figure S1). The extensive receptor-binding surface of Arr1 [26, 27] carries eight positively charged residues [10, 12, 28, 29] that bind three or more phosphates on P-Rh* [16, 30]. Unphosphorylated rhodopsin cannot neutralize these charges, which may destabilize the arrestin-Rh* complex. The replacement of positive charges with neutral hydrophilic residues may improve the mutant’s performance. This approach must be tested experimentally, although it has an obvious downside: an “improved” mutant could become selective for the Rh*, losing affinity for P-Rh* due to loss of the phosphate-binding residues.
Defects in rhodopsin shutoff underlie several visual disorders. Patients lacking RK or arrestin suffer from Oguchi disease, a form of stationary night blindness [31, 32]. In these loss-of-function cases, replacement of the missing protein is the most straightforward strategy for the gene therapy. Unfortunately, when gain-of-function mutations underlie the pathology (e.g., rhodopsin lacking phosphorylation sites in retinitis pigmentosa [3-5]), WT proteins cannot solve the problem. Theoretically, mutant mRNA can be eliminated by an appropriately designed ribozyme. However, this approach is hardly practical: it would require unrealistic 100% efficiency of “bad” mRNA elimination (even low expression of phosphorylation-deficient rhodopsin is harmful [24]) without affecting the “good” one, which may differ by as little as one base. The introduction of an enhanced arrestin to quench the signaling of the overactive receptor is a logical alternative. Our data demonstrate that this “compensational” approach yields partial rescue in the most efficient GPCR-driven signaling system, rod phototransduction. Its high amplification yields unparalleled sensitivity, which makes photoreceptors particularly vulnerable: rods are the only cell type where the defects in GPCR shutoff result in cell death. Increased survival and improved functional performance of RK-deficient photoreceptors expressing enhanced arrestin1 are two sides of the same coin, independently demonstrating the compensational potential of the mutant. Partial rescue in the exceptionally demanding visual system suggests that similar approach would rein in excessive signaling by non-visual GPCRs in other cell types more efficiently.
Our results show that redesigning GPCR inactivation machinery in living animals is feasible. This approach paves the way to the development of compensational gene therapy for congenital disorders associated with gain-of-function mutations.
Acknowledgments
We are grateful to Drs. Arkady Lyubarsky and Edward N. Pugh, Jr. for expert advice on mouse ERG, Drs. C.-K. Chen, L. A. Donoso, R. S. Molday, and C.M. Craft for rhodopsin kinase knockout mice, monoclonal anti-arrestin and anti-rhodopsin antibodies, and mouse rod arrestin cDNA, respectively, and to Frances Y. Cheng and Tatiana Vishnivetskaya for technical assistance. Supported by NIH grants EY011500 (VVG), EY014047 (MEB), NS045117 (EVG), EY012155 (JC), Vanderbilt University Discovery Grant 1040659012 (VVG), Research to Prevent Blindness (MEB), and P30 core grant in vision research EY008126 (to Vanderbilt University).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Methods are described in the Supplement
References
- 1.Rompler H, Staubert C, Thor D, Schulz A, Hofreiter M, Schoneberg T. G protein-coupled time travel: evolutionary aspects of GPCR research. Mol Interv. 2007;7:17–25. doi: 10.1124/mi.7.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Gurevich VV, Gurevich EV. The molecular acrobatics of arrestin activation. TIPS. 2004;25:59–112. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 3.Apfelstedt-Sylla E, Kunisch M, Horn M, Ruther K, Gerding H, Gal A, Zrenner E. Ocular findings in a family with autosomal dominant retinitis pigmentosa and a frameshift mutation altering the carboxyl terminal sequence of rhodopsin. Br J Ophthalmol. 1993;77:495–501. doi: 10.1136/bjo.77.8.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim RY, al-Maghtheh M, Fitzke FW, Arden GB, Jay M, Bhattacharya SS, Bird AC. Dominant retinitis pigmentosa associated with two rhodopsin gene mutations. Leu-40-Arg and an insertion disrupting the 5’-splice junction of exon 5. Arch Ophthalmol. 1993;111:1518–1524. doi: 10.1001/archopht.1993.01090110084030. [DOI] [PubMed] [Google Scholar]
- 5.Restagno G, Maghtheh M, Bhattacharya S, Ferrone M, Garnerone S, Samuelly R, Carbonara A. A large deletion at the 3’ end of the rhodopsin gene in an Italian family with a diffuse form of autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1993;2:207–208. doi: 10.1093/hmg/2.2.207. [DOI] [PubMed] [Google Scholar]
- 6.Daaka Y. G proteins in cancer: the prostate cancer paradigm. Sci STKE. 2004;2004:re2. doi: 10.1126/stke.2162004re2. [DOI] [PubMed] [Google Scholar]
- 7.Vishnivetskiy SA, Paz CL, Schubert C, Hirsch JA, Sigler PB, Gurevich VV. How does arrestin respond to the phosphorylated state of rhodopsin? J Biol Chem. 1999;274:11451–11454. doi: 10.1074/jbc.274.17.11451. [DOI] [PubMed] [Google Scholar]
- 8.Kovoor A, Celver J, Abdryashitov RI, Chavkin C, Gurevich VV. Targeted construction of phosphorylation-independent b-arrestin mutants with constitutive activity in cells. J Biol Chem. 1999;274:6831–6834. doi: 10.1074/jbc.274.11.6831. [DOI] [PubMed] [Google Scholar]
- 9.Celver J, Vishnivetskiy SA, Chavkin C, Gurevich VV. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J Biol Chem. 2002;277:9043–9048. doi: 10.1074/jbc.M107400200. [DOI] [PubMed] [Google Scholar]
- 10.Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, Kono M, Navarro J, Gurevich VV. Crystal Structure of Cone Arrestin at 2.3Å: Evolution of Receptor Specificity. J Mol Biol. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 11.Baylor DA, Lamb TD, Yau KW. Responses of retinal rods to single photons. J Physiol. 1979;288:613–634. [PMC free article] [PubMed] [Google Scholar]
- 12.Vishnivetskiy SA, Schubert C, Climaco GC, Gurevich YV, Velez M-G, Gurevich VV. An additional phosphate-binding element in arrestin molecule: implications for the mechanism of arrestin activation. J Biol Chem. 2000;275:41049–41057. doi: 10.1074/jbc.M007159200. [DOI] [PubMed] [Google Scholar]
- 13.Chen CK, Burns ME, Spencer M, Niemi GA, Chen J, Hurley JB, Baylor DA, Simon MI. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Nat Acad Sci USA. 1999;96:3718–3722. doi: 10.1073/pnas.96.7.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu J, Dodd RL, Makino CL, Simon MI, Baylor DA, Chen J. Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature. 1997;389:505–509. doi: 10.1038/39068. [DOI] [PubMed] [Google Scholar]
- 15.Burns ME, Arshavsky VY. Beyond counting photons: trials and trends in vertebrate visual transduction. Neuron. 2005;48:387–401. doi: 10.1016/j.neuron.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 16.Mendez A, Burns ME, Roca A, Lem J, Wu LW, Simon MI, Baylor DA, Chen J. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron. 2000;28:153–164. doi: 10.1016/s0896-6273(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 17.Doan T, Mendez A, Detwiler PB, Chen J, Rieke F. Multiple phosphorylation sites confer reproducibility of the rod’s single-photon responses. Science. 2006;313:530–533. doi: 10.1126/science.1126612. [DOI] [PubMed] [Google Scholar]
- 18.Burns ME, Mendez A, Chen CK, Almuete A, Quillinan N, Simon MI, Baylor DA, Chen J. Deactivation of phosphorylated and nonphosphorylated rhodopsin by arrestin splice variants. J Neurosci. 2006;26:1036–1044. doi: 10.1523/JNEUROSCI.3301-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyubarsky AL, Pugh EN., Jr Recovery phase of the murine rod photoresponse reconstructed from electroretinographic recordings. J Neurosci. 1996;16:563–571. doi: 10.1523/JNEUROSCI.16-02-00563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robson JG, Frishman LJ. Response linearity and kinetics of the cat retina: the bipolar cell component of the dark-adapted electroretinogram. Vis Neurosci. 1995;12:837–850. doi: 10.1017/s0952523800009408. [DOI] [PubMed] [Google Scholar]
- 21.Robson JG, Frishman LJ. Dissecting the dark-adapted electroretinogram. Doc Ophthalmol. 1999;95:187–215. doi: 10.1023/a:1001891904176. [DOI] [PubMed] [Google Scholar]
- 22.Pugh EN, Jr, Falsini B, Lyubarsky AL. The origin of the major rod- and cone-driven components of the rodent electroretinogram, and the effect of age and light-rearing history on the magnitudes of these components. In: Williams TP, Thistle AB, editors. Photostasis and Related Phenomena. New York: Plenum Press; 1998. pp. 93–128. [Google Scholar]
- 23.Pepperberg DR, Birch DG, Hood DC. Photoresponses of human rods in vivo derived from paired-flash electroretinograms. Vis Neurosci. 1997;14:73–82. doi: 10.1017/s0952523800008774. [DOI] [PubMed] [Google Scholar]
- 24.Chen J, Makino CL, Peachey NS, Baylor DA, Simon MI. Mechanisms of rhodopsin inactivation in vivo as revealed by a COOH-terminal truncation mutant. Science. 1995;267:374–377. doi: 10.1126/science.7824934. [DOI] [PubMed] [Google Scholar]
- 25.Gurevich VV, Benovic JL. Visual arrestin interaction with rhodopsin: Sequential multisite binding ensures strict selectivity towards light-activated phosphorylated rhodopsin. J Biol Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- 26.Hanson SM, Francis DJ, Vishnivetskiy SA, Kolobova EA, Hubbell WL, Klug CS, Gurevich VV. Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc Natl Acad Sci U S A. 2006;103:4900–4905. doi: 10.1073/pnas.0600733103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vishnivetskiy SA, Hosey MM, Benovic JL, Gurevich VV. Mapping the arrestin-receptor interface: structural elements responsible for receptor specificity of arrestin proteins. J Biol Chem. 2004;279:1262–1268. doi: 10.1074/jbc.M308834200. [DOI] [PubMed] [Google Scholar]
- 28.Gurevich VV, Benovic JL. Visual arrestin binding to rhodopsin: diverse functional roles of positively charged residues within the phosphorylation-recignition region of arrestin. J Biol Chem. 1995;270:6010–6016. doi: 10.1074/jbc.270.11.6010. [DOI] [PubMed] [Google Scholar]
- 29.Hanson SM, Gurevich VV. The differential engagement of arrestin surface charges by the various functional forms of the receptor. J Biol Chem. 2006;281:3458–3462. doi: 10.1074/jbc.M512148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vishnivetskiy SA, Raman D, Wei J, Kennedy MJ, Hurley JB, Gurevich VV. Regulation of arrestin binding by rhodopsin phosphorylation level. J Biol Chem. 2007;282:32075–32083. doi: 10.1074/jbc.M706057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuchs S, Nakazawa M, Maw M, Tamai M, Oguchi Y, Gal A. A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat Genet. 1995;10:360–362. doi: 10.1038/ng0795-360. [DOI] [PubMed] [Google Scholar]
- 32.Yamamoto S, Sippel KC, Berson EL, Dryja TP. Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet. 1997;15:175–178. doi: 10.1038/ng0297-175. [DOI] [PubMed] [Google Scholar]
- 33.Chan S, Rubin WW, Mendez A, Liu X, Song X, Hanson SM, Craft CM, Gurevich VV, Burns ME, Chen J. Functional comparisons of visual arrestins in rod photoreceptors of transgenic mice. Invest Ophthalmol Vis Sci. 2007;48:1968–1075. doi: 10.1167/iovs.06-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]