

Abstract
It remains unclear whether γδ T cell receptors (TCRs) detect antigens in a manner similar to antibodies or αβ TCRs. Here we show that reactivity between G8 and KN6 γδ TCRs and the MHC class Ib molecule T22 can be transplanted, with retention of wild-type ligand affinity, after en bloc grafting of G8 and KN6 CDR3δ loops in place onto the CDR3α loop of an αβ TCR. We also find that a shared sequence motif within CDR3δ loops of all T22-reactive γδ TCRs binds T22 in energetically distinct fashions, and that T10d, which binds G8 with weak affinity, is converted into a high-affinity ligand by a single point mutation. These results demonstrate an unprecedented autonomy of a single CDR3 loop in antigen recognition.
Introduction
γδ T cells express a somatically rearranged T cell receptor (TCR) through which they recognize antigen. γδ TCRs are composed of a γ and δ chain, with a structure generally similar to that of the αβ TCR, or the Fab fragment of an antibody1, 2. Recognition of ligand results in a variety of effector outcomes from γδ T cells, some of which are similar to those seen in αβ T cells3–6, such as cytokine secretion and cytotoxic activity7–9, and others that are more unique to γδ T cells, such as wound-repair10, antigen presenting activity through MHC class II expression11 and immunosuppresion12. γδ T cells have been implicated as effectors in a variety of infectious and autoimmune diseases13 and some have the ability to spontaneously lyse tumor cells7, 14–18.
The molecular basis of γδ TCR recognition of ligand is currently not well understood19, 20. Is it more similar to that of an αβ TCR, whereby the germline-encoded variable regions interact with MHC through a co-evolved ‘bias’, and the CDR3 regions recombine uniquely to ‘readout’ different antigenic peptides?21, 22 Or is γδ TCR ligand recognition more similar to that seen in antibody-antigen interactions, where there is presumably no preferred antigen ‘bias’, and a combination of somatic hypermutation and CDR3 recombination enables reactivity with a universe of antigens?23 In both cases, interactions are usually mediated by simultaneous contact of multiple CDR loops of the antigen receptor with the antigen, with variable contributions of CDR involvement between different complexes. Also, in both cases the recombined CDR3-H and CDR3β loops exert a general dominance during ligand recognition by antibodies and αβ TCRs, respectively, as measured by the extent of contact.
There has been much effort spent trying to determine the ligands for γδ TCRs, yet only a few have been well defined. These include MHC Ib molecules (CD1c4, 24, T10 and T2225, 26, MICA27, H2 I-Ek 25), non-immune system-related proteins (F1 subunit of the ATP synthase28), and microbial proteins, such as the herpes simplex virus-1 (HSV-1) glycoprotein I (gI)29. The structural diversity of these ligands suggests that the antigen recognition process of γδ T cells is unlike that of conventional αβ T cells. Indeed, the genetics of γδ TCR rearrangement suggests that diversity of the CDR1 and CDR2 loops, encoded within the 6–10 variable segments of the δ chain and 8–12 variable segments of the γ chain is not extensive. Instead, sequence diversity is concentrated in the rearranged portion of the receptor, specifically the CDR3γ and CDR3δ loops. The ability to use multiple diversity (D) segments in the rearrangement of the δ chain30 endows this loop with the greatest amino acid diversity potential of all the rearranged receptors.
The molecular details of how this unique feature of γδ TCRs is used in antigen recognition remains unclear except for interactions between T10 and T22 and γδ TCRs. Approximately 0.1–1.0% of murine γδ T cells recognize the MHC class Ib molecules T10 and T2231. Of this population, G832 and KN633, 34 are the two best characterized γδ TCRs specific for T10 and T22. The three dimensional structure of G8 in complex with T22 1 provided structural insight into how γδ T cells “see” antigen, and revealed a recognition strategy substantially different from that seen in αβ TCR recognition of peptide-MHC (pMHC) complexes, or antibody recognition of antigen. G8 binds T22 almost exclusively through an extended CDR3δ loop, with minor contacts from the remaining CDR loops; the result is a tilted G8-T22 docking orientation. Other T22-binding γδ TCRs, despite using different Vδ and Vγ domains, are related to G8 through a shared six amino acid (W…EGYEL) motif in their CDR3δ loops35. TCRs using the same Vγ and Vδ but with variation in the number and composition of amino acids between the W and the EGYEL were shown, by tetramer decay, to bind T22 with different affinities35. In the case of G8, this CDR3δ motif forms extensive interactions with T22 in the complex interface, and thus may be the primary T22 recognition determinant. However, variation in length and amino acid composition of the intervening residues between the ‘W’ and ‘EGYEL’ motifs raise the question of how this variability, generated by the rearrangement process, is accommodated during recognition. The structures of an unliganded human Vδ3 domain36, and a human γδ TCR2 also revealed protruding CDR3 loops, but the structural ramifications for antigen recognition are not known.
In addition to the variation in the CDR3δ loop of these T10- and T22-reactive γδ TCRs, previous work has shown that allelic forms of T10 and T22 are recognized differently. T10 from H-2d mice weakly stimulates the G8 hybridoma, approximately ~5–10 fold less than T10 and T22 from H-2b-expressing mice. This demonstrates that amino acid variation within these loci can modulate γδ T cell reactivity. The parameters governing TCR binding to T10d that result in this weak stimulation of G8, and the identity of position(s) of variation between the H-2b and H-2d versions of T10 and T22 that are critical for stimulation are not known.
Here we undertook a structure-function study to better understand the molecular determinants of γδ TCR recognition of T10 and T22 molecules. We explored the question of whether the CDR3δ loop is the primary recognition determinant in T22 binding, and how variations in the W…EGYEL motifs across receptors can modulate binding to T22. We also defined the critical variable residues in T10 and T22 responsible for differential binding by and stimulation of T22-specific γδ TCRs. The conclusions of these studies point to a surprising structural autonomy of the TCR CDR3δ loop in ligand recognition, and a single amino acid substitution in T10 and T22 that is responsible for ligand specificity.
Results
G8 CDR3δ: minimal T22 recognition determinant
Previous results1, 35 revealed that γδ T cells that recognize T22 contain a variety of Vδ and Vγ domains in their rearranged γδ TCRs, indicating the presence of a variety of sequences in their CDR1 and CDR2 loops. However, the feature shared amongst these TCRs is a motif (W…SEGEL) assembled by exclusive use of the Dδ2 segment (EGYE) with partial contributions of the Vδ segment (W), the Dδ1 segment (W) and/or P nucleotides (L)35. This observation suggested that the primary recognition determinant for T22 recognition was localized to the CDR3δ loop. Structural elucidation of the complex of the G8 γδ TCR with T22 revealed that this CDR3δ loop comprised the majority of the recognition interface (67% of the total buried surface area) between G8 and T22 (Fig. 1). The sequence motif in this specific CDR3δ (‥WHISEGYEL‥) formed the principal contacts with the floor of the interhelical groove in T22 (Fig. 1). We also observed a substantial ‘wobble’ in the binding orientation of G8 when docked with T22. That is, superposition of the T22 molecules in the two complexes in the crystallographic asymmetric unit revealed a resultant difference of approximately 6Å between the two TCRs around a structurally invariant CDR3δ pivot point that formed the T22 contact. This suggested that the CDR3δ loop anchored the complex, allowing the ‘body’ of the TCR to adopt a range of different positions when bound to T22. That the CDR3δ appears to be the “glue” holding the complex together led us to hypothesize that the CDR3δ loop alone could endow binding of T22 when “grafted” onto an alternative structural scaffold.
Figure 1.

Three dimensional structure of the G8 γδ TCR in complex with the MHC class Ib molecule T22. The G8 δ-chain is shown in blue, and the G8 γ-chain in green; T22 is shown in yellow and the G8 CDR3δ loop in red. Inset, right, shows magnified view of the G8 CDR3δ loop (red) in contact with T22 (yellow). The residues of the W…GYEL motif found in all T22-binding γδ TCRs are labeled.
To test this hypothesis, we grafted the CDR3δ loops of two canonical T22-specific TCRs, G8 and KN6, onto a “naïve” TCR background, that of the 172.10 αβ TCR that binds the class II MHC I-Au bound to the peptide Ac1-1137, but not to T22. The availability of the coordinates for both G8 and 172.10 allowed us to design the grafting boundary based on superposition (<2Å R.M.S.D.) of the two structures (Fig. 2 and Supplementary Fig. 1, online). We used the structures to visually guide grafting of the CDR3δ loop in place of the 172.10 CDR3α loop, such that the graft junctions would be sterochemically reasonable, and that it would be presented in a sterically unobstructed manner in the context of the αβ TCR framework and binding site (Supplementary Fig. 1, online). In fact, the framework residues at the CDR3 graft junctions (f and g strands of the TCRs) superimposed closely with respect to register of the polypeptide chain and distance between the residues at the bases of the respective loops (Supplementary Fig. 1, online). The CDR3α of 172.10 is a short, tight turn, and we replaced the Asn95-Ser96-Gly97 at the apex of the loop with the respective CDR3δ sequences (Fig. 2 and Supplementary Fig. 1, online). The CDR3δ loops of G8 and KN6 represent two of the more divergent motifs within the population of TCRs that recognize T22 (Fig. 2). Both encode a W…SEGYEL, however the spacing between the W and EGYEL differs between them; G8 has a three amino acid residue spacer whereas KN6 has none.
Figure 2.

Transfer of CDR3δ onto CDR3α. a. Top, ribbon representation of the full-length G8 γδ TCR1 (grey, with the CDR3δ loop shown in sticks and colored red), the 172 αβ TCR scFc (blue), and the fusion in which the CDR3δ loop was grafted onto the CDR3α loop. Bottom, sequences of CDR3δ loops grafted from the G8 and KN6 γδ TCRs (red) onto the CDR3α (replaced residues are in blue).
Wild-type soluble full length γδ TCR heterodimers expressed in insect cells were used to measure affinities between wild-type TCRs and ligands (Fig. 3). To control for artifacts that might be introduced by differences in constructs and expression systems, we expressed the G8 and KN6 CDR3δ-172.10 fusions both in E.coli as single-chain Fv constructs (the form of 172.10 that was crystallized), and in insect cells as full-length, leucine-zippered ectodomain heterodimers. T22 was engineered to contain a C terminal site-specific biotinylation sequence, allowing for orientation-specific immobilization on streptavidin (SA) Biacore chips. Approximately 250RU of biotinylated T22 was immobilized on the chip surface. The wild-type and fusion TCRs were flowed as analyte at concentrations ranging from 31.25nM to 5uM depending on the analyte. These curves were fit with a 1:1 Langmuir binding isotherm and the affinities (KD) of wild-type G8 and KN6 were determined to be ~100nM and 15uM respectively, indicating that wild-type G8 and KN6, despite sharing a common, but non-identical CDR3δ motif, bind T22 with substantially different affinities and kinetics (Fig. 3). Each respective fusion protein specifically bound to T22 (although wild-type 172.10 did not bind) with slightly higher affinities than their wild-type counterparts (~50nM and ~4uM for the G8-172.10 and KN6-172.10 fusions, respectively) (Fig. 3). These results demonstrated that the CDR3δ loop is the primary recognition determinant of G8 and KN6 TCRs, and thus that this loop will likely play a dominant role in T22 recognition by other γδ TCRs bearing this motif. That the affinities of the fusion proteins are higher than those of wild-type TCRs suggest that the CDR1 and/or CDR2 loops, or the scaffold, of the wild-type γδ TCRs may impose a barrier to T22 binding and that the fusion proteins essentially unmask the ‘naked’ affinity of each of these CDR3δ sequences in an uninhibited context. In this fashion, the surrounding structural context of CDR3δ, can sensitively exert a modulatory role on the affinity of each γδ TCR for T22 by presenting more or less of an inhibitory role.
Figure 3.

CDR3δ grafted into αβ TCR retains wild-type affinity for T10 and T22 ligands. SPR measurements of binding between T22 and wild-type or fusion G8 and KN6 γδ TCRs. Columns (G8, left; KN6, right) show sensograms corresponding to the wild-type full-length γδ TCR (top), fusions of the CDR3δ loop with the single chain 172 αβ TCR (172sv) expressed in E. Coli or insect cells (middle), and the wild-type full-length 172 αβ TCR expressed in insect cells (bottom).
Energetically distinct modes of CDR3δ binding
The 100-fold affinity difference between the G8 and KN6 CDR3δ loops binding to T22 (as shown in both wild-type and fusion TCR measurements), and the different sequence contexts of the shared W‥EGYEL motif raises the question of whether these two divergent loops bind to T22 in similar or distinct fashions. To address this question, we performed alanine-scanning mutagenesis of the CDR3δ loops of these two TCRs to determine the energetically critical residues involved in T22 binding. SPR measurements were performed similarly to those of the fusion experiments, with T22 immobilized on the chip surface and eachmutant flowed as analyte. For the G8 CDR3δ loop, the residues that had the greatest energetic change (colored red) when mutated to alanine corresponded well with the residues that contacted the T22 surface, as seen in the three dimensional structure of G8 in complex with T22 (Fig. 4a,b). In particular, Trp93, Tyr99 and Leu101 most profoundly influenced free energy (mutation of Trp93 and Leu101 reduced binding to undetectable levels), whereas the remaining residues of the loop exerted either moderate (e.g. Ile95 which also contacts T22 and Gly98 and Gly102) or little to negative impact (His94, Ser96, Glu97, Glu100, and Thr103).
Figure 4.

Heterogeneous energetic landscapes of G8 and KN6 CDR3δ interactions with T10 and T22. a) Alanine scanning mutagenesis of the G8 (top) and KN6 (bottom) CDR3δ loops. +, mutations that resulted in immeasurable binding. Bars are colored spectrally red through blue based on the affect of the mutation on binding, with red indicating a large ΔΔG penalty on binding versus blue for little or no ΔΔG penalty. b) The interface between the G8 γδ TCR and T221 showing the residues of the CDR3δ loop color-coded according to their energetic contribution to binding as in a.
Surpringly, alanine scanning of the KN6 CDR3δ loop revealed substantial differences in energetically important residues. Whereas Trp93 was also critical for KN6 binding to T22, other residues energetically critical for KN6 CDR3δ loop binding to T22 appeared to be carrying out different energetic roles in G8 (Fig. 4a). Glu97 was required for KN6 binding, whereas it had essentially no effect on G8 binding to T22. Conversely, Tyr99 was critical for G8 binding but the KN6 Tyr residue had only a moderate affect on KN6 association with T22. These findings provide the first evidence that the γδ TCRs that bind to T22, whereas recognizing this ligand almost exclusively via a shared, simple motif (W‥EGYEL), can establish an interface with the ligand in substantially different ways. Perhaps this long, solvent extended motif has evolved to accommodate a variety of loop lengths, and therefore registers, in T22 recognition through CDR3δ structural plasticity.
Allele-specific γδ TCR binding
To understand the effect of allelic variation in T22 and T10 on γδ TCR binding and γδ T cell stimulation, we measured the affinity of G8 binding to T22 and T10b, which strongly stimulate G8 γδ TCR+ cells, and T10d, which weakly stimulates G8 γδ TCR+ cells (Fig. 5a). To do this, we expressed the G8 γδ TCR and each ligand in insect cells using a baculovirus expression system. The TCR was engineered with a specific biotinylation sequence at the C terminus that enabled orientation-specific immobilization on a streptavidin chip. Purified T22, T10b and T10d were flowed as analyte at concentrations varying from 31.25nM to 250nM, and the Kon and Koff were measured from these sensograms (Fig. 5b). Curves were fit with a 1:1 Langmuir binding and affinities (KD) were calculated. G8 bound to its strongly stimulatory ligands T22 and T10b with affinities of approximately 70 and 80nM respectively, similar to what was calculated in previous studies31 that used bacterially expressed and refolded T22. Surprisingly, G8 also bound to T10d, a significantly weaker (5–10 fold less potent) agonist than T22 even when presented in high concentrations. The affinity of G8 for T10d was approximately 10-fold weaker than that of G8 for T22; although G8 bound T22 and T10d with similar association rates, G8 bound T10d with a 10-fold faster dissociation rate (Fig. 5b).
Figure 5.

The G8 γδ TCR binds to strong and weak stimulatory ligands with different affinity. SPR measurements of T22, T10b and T10d binding to the G8 γδ TCR. KD and association and dissociation rates are listed in Table X.
To determine the amino acid positions responsible for the strong versus weak stimulatory activity of the distinct G8 ligands, we generated a series of gain of function and loss of function mutants of T22, T10b and T10d. Only those amino acids differing between the strong (T22 and T10b) and weak (T10d) ligands were examined (Fig. 6). We generated point mutations of such residues located in the α1 and α2 domains, as, according to the complex structure and functional data determined by others1, 38, these residues were most likely to affect recognition by the G8 TCR. For simplicity, both relevant amino acids in the α3 domain were mutated in a single molecule. Mutant ligands were expressed and purified from insect cell supernatants and used for SPR (as described above), and stimulation assays with the G8 γδ T cell hybridoma.
Figure 6.

Mutations responsible for differential G8 γδ TCR binding and stimulation by T22 and T10b versus T10d. SPR measurements of G8 γδ TCR binding to T22 mutants (left sensograms) and T10d mutants (right sensograms). T22 and T10d wild-type residues are indicated in blue and orange, respectively. The position of each point mutation and the corresponding calculated affinity (KD) are indicated in the upper right region of each sensogram. Structures in left column depict position of each point mutation in the structure of T2256.
Four of the five T22 and T10d point mutants replicated wild-type molecules with regard to affinity and stimulatory capacity (Fig. 6, 7a). However, molecules containing a point mutation at position 124 (a histidine in place of a proline in T22, and a proline in place of a histidine in T10d) reversed the affinity and stimulatory capacity of T22 and T10d (Fig. 6, 7a). This mutation is located at the edge of the binding interface in the complex structure of G8 and T221, and forms a main-chain hydrogen bond with the side chain of Trp93 of CDR3δ, as well as van der Waals contacts with the main chain at the apex of CDR1δ, in a ‘knob-in-holes’ type of interaction. Introduction of a histidine in this position would clearly alter this contact between T22 (and presumably T10b) and the TCR, as the main chain would be distorted after removing a proline, and a histidine side chain is likely too bulky to be accommodated within this tightly packed interface (Fig. 7b). As indicated in the comparison between the kinetics of binding between T22, T10b and T10d in Fig. 5, the difference in binding between the G8 TCR and wild-type and P124H mutant T22 is due almost exclusively to an increased off-rate approaching that of T10d (Fig. 6).
Figure 7.

A single amino acid substitution converts T10d into a strong G8 γδ TCR agonist. a) Graphs depict stimulation of the G8 hybridoma, as measured by activity of a NFAT-dependent alkaline phosphatase reporter, by the indicated wild-type or mutant versions of T22 (left; ◆=R35L, □=D58G, ▲=M89V, ■=P124H, 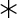 =α3, ●=T22 and
=α3, ●=T22 and 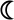 =negative control, m157) and T10d (right; ◆=L35R, □=G58D, ▲=V89M, ■=H124P,
=negative control, m157) and T10d (right; ◆=L35R, □=G58D, ▲=V89M, ■=H124P, 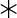 =α3, ●=T10d and
=α3, ●=T10d and 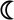 =negative control, m157). Graphs depicting P124H and H124P mutants are drawn in orange and blue, respectively. All stimulation assays were performed in triplicate. b) Location of position 124 in the interface between the G8 γδ TCR and T22. The proline is colored in orange, and is located on the edge of the platform domain of T22, in direct contact with the G8 γδ TCR. c) Melting temperatures (Tm) of wild-type and mutant T22 and T10d. At least two independent measurements were made for each sample; error bars represent the standard error of these measurements.
=negative control, m157). Graphs depicting P124H and H124P mutants are drawn in orange and blue, respectively. All stimulation assays were performed in triplicate. b) Location of position 124 in the interface between the G8 γδ TCR and T22. The proline is colored in orange, and is located on the edge of the platform domain of T22, in direct contact with the G8 γδ TCR. c) Melting temperatures (Tm) of wild-type and mutant T22 and T10d. At least two independent measurements were made for each sample; error bars represent the standard error of these measurements.
To test whether these mutations affect the stability of the T22 and T10d structures, we performed circular dichroism (CD) studies of each of the mutants to determine their stability at 37°C, the physiological temperature at which the stimulation studies are performed. Measurements were performed after one hour incubation at 37°C and an equilibration step for 20 minutes, which we have determined to be critical for a stable measurement of secondary structure parameters. The melting temperature values for the mutants, although differing slightly between the T22 and T10d alleles, did not correlate with the observed differences in stimulatory capacity (Fig. 7c and Supplementary Fig. 2, online). Most notable are the position 124 point mutants, which despite their markedly distinct stimulatory capacity, maintained closely the Tm of their wild-type counterparts. These experiments confirmed that the difference in stimulatory capacity between the mutants is due to the affect of the single point mutation, and not to disruption of the secondary structure or stability of the structural backbone. Combined, these data reveal that a single amino acid polymorphism at position 124 modulates the recognition of T22 and T10 molecules by the G8 γδ TCR.
Discussion
The molecular mechanisms by which γδ TCR engage ligand(s) still remain largely unknown for most γδ TCR-ligand systems. The three dimensional structure of the G8 γδ TCR in complex with the MHC class Ib molecule, T221, revealed that G8, instead of using an assemblage of multiple CDR loops to engage ligand as usually seen between αβ TCR and pMHC22, or antibody and antigen39, bound T22 almost exclusively through its CDR3δ loop with only minor contacts from other CDR loops. Whereas certain TCR-pMHC40, 41 and antibody-antigen42, 43 complexes show a preponderence of interaction via CDR3 regions, the extent of CDR3δ involvement in the G8-T22 complex, and the focus of this interaction on a seven amino acid motif (W…EGYEL) within CDR3δ, is highly unusual. In fact, the most similar structural analog derives from that of the single-chain shark antibodies (IgNAR), which have been shown to bind lysozyme through a long CDR3 that penetrates a canyon on the antigen surface44.
Here we showed through grafting experiments that the primary recognition determinant of the G8 and KN6 γδ TCRs for T22 is their CDR3δ loops. The ability to transfer reactivity through a single CDR loop has not been reported previously for TCRs or antibodies, and suggests that this recognition process has ‘innate-like’ features, as the majority of these residues are encoded in germline fragments rather than through the diversifying P and N nucleotide variation that encodes many of the CDR3 contact residues in αβ TCR-pMHC and antibody-antigen complexes. Importantly, however, the affinity of this germline segment for T22 is modulated by the P and N nucleotide variation bracketing the motif. The G8 γδ TCR, which was cloned from an alloreactive T cell line derived from a BALB/c (H-2d) nu/nu mouse stimulated with irradiated B10.BR (H-2k, T22 positive) splenocytes45, binds T22 with 100 fold greater affinity than the KN6 γδ TCR which was of syngenic origin. That we have been able to sample this range of affinities of γδ TCRs for T22 is likely due to their allogeneic versus syngeneic origins, as other syngeneic γδ TCRs specific for T22 also bind with affinities lower than that of G835. Surprisingly, our alanine-scanning mutagenesis of the CDR3δ loops from G8 and KN6, which differ in the length and “spacer” amino acids between the W and EGYEL portions of the motif, suggest that recognition of T22 has an additional layer of complexity. Our results reveal that the residues comprising the shared sequence motif contribute differently to T22 interaction in G8 and KN6, strongly implying that different registers of these amino acids are used for T22 recognition. It is difficult to understand why, or how, a shared motif would be used in different energetic and structural contexts by different TCRs recognizing an identical ligand. However, it is clear that in both cases, the TCR reactivity is endowed by the CDR3δ alone, so both loops share the same level of structural autonomy. The distinct T22-binding energetics of the W‥EGYEL motif in different γδ TCRs could endow these receptors with an additional level of flexibility in ligand recognition that would serve to enhance the repertoire of ligands recognized.
More broadly, we do not know of another example from a protein-protein interaction system, in the immune system or otherwise, where transfer of a single contiguous polypeptide loop sequence from one protein to a naïve protein transfers wild-type affinity to the hybrid protein. This suggests a evolutionary focusing of the binding energetics to a single short polypeptide stretch. A comparable example is that of in vitro evolution of antibodies and/or TCR using phage or yeast display methods. Examples exist where randomization and selection of single CDR loops results in substantially higher affinity of the antibody46 or TCR47 for its cognate ligand. However, even in those cases, the other CDR loops contributed to and were necessary for ligand recognition. For αβ TCRs, CDR grafting has so far not succeeded in reconstituting reactivity to a naïve TCR, even when multiple CDRs are replaced in parallel48. In cases of antibodies, where multiple CDR loops are transferred from mouse to human frameworks for antibody ‘humanization’, a substantial amount of protein engineering of the junctional and framework residues are usually required to reconstitute the affinity of the mouse monoclonal antibody in the humanized antibody49.
What might be the functional implications of such an autonomous and focused binding determinant? These observations might imply that, in this case, a germline-encoded element has a co-evolved bias for its ligand, in a similar fashion as seen for the αβ TCR V segment bias for MHC50. Thus, as there are only a fixed number of germline-encoded γδ TCR elements, the repertoire of γδ TCR ligands might be quite limited. The ligand specificities of the other germline elements remain unknown. However, we must also introduce the caveat that the G8-T22 system could be unusual, even for γδ TCRs, and that other γδ TCRs may more closely resemble the more canonical antigen receptor interactions where specificity is achieved through CDR3 recombination and a contribution of multiple CDR loops.
The MHC class Ib molecules T10 and T22 are polymorphic in their genomic and allelic forms, suggesting that there is selection to alter their molecular makeup. This is not unusual in ligands of the immune response, as these molecules are often targets of pathogen downregulation or modulation during immunoevasion52 and therefore are rapidly evolving. The polymorphisms outside of those required for the function of the protein might be due to a molecular “arms race” with the pathogen, affecting how microbial proteins interact with these ligands. To compensate, host receptors that recognize these ligands are either polymorphic (e.g. killer immunoglobulin like receptors and Ly49 receptors), or have an intrinsic diversifying process (e.g. somatically rearranged TCRs and BCRs).
Our results identify a single polymorphism (P124H) that modulates the kinetics and stimulatory capacity of the T10 and T22 molecules with the G8 γδ TCR, and show that other polymorphisms in these ligands do not play a major role in G8 engagement, nor do they affect ligand stability as measured by CD at physiological temperatures. The proline to histidine change at position 124, located at the edge of T22’s docking site, would introduce a steric clash to G8 binding as is evident from the three dimensional structure of G8 in complex with T221. Only weak van der Waals contacts are made with proline 124 by the CDR1 and HV4 loops and, signaling that this contact is likely nonspecific, there are evident shifts of these contacts between the two complexes in the asymmetric unit. This observation, along with the fact that T22 reactive γδ TCRs use a variety of Vδ (and in some cases Vα) domains in their rearrangement, strongly suggest that substantial binding specificity is not contributed by CDR1 or HV4. Instead, as suggested by our CDR3δ fusion experiments, the CDR1 and HV4 loops may be exerting an inhibitory affect on G8 and KN6 binding as transfer of the CDR3δ loops from these TCRs into a naïve background resulted in higher affinity of binding. Thus, the variation present in the T10 and T22 ligands, possibly a consequence of pathogen-driven selection, appears to be accommodated by the structural and sequence adaptability intrinsic to their rearranged receptors. We have shown one example of a ligand polymorphism that has a profound affect on TCR engagement and stimulation; specific details of ligand discrimination await study of TCRs specific for other allelic forms of these ligands.
The adaptability of receptors to rapidly evolving or novel ligands is the hallmark of the unique process of somatic V-D-J rearrangement. The intrinsic variation of these receptors and a carefully tuned selection process allows αβ TCRs to discriminate between allelic forms of MHC molecules and the variable peptides they bind and present, and antibodies to distinguish between novel protein and non-protein ligands such as those introduced in a bacterial or viral infections. We show here that γδ TCR recognition of T22 and T10 can be modulated by allelic variation, that the CDR3δ loop is an autonomous determinant of ligand recognition, and that the shared amino acid motif within this loop can have its ligand affinity “tuned” by different surrounding CDR3δ sequence contexts.
Methods
Protein expression and purification
T10b, T10d and T22 were expressed and purified as previously described1. In brief, the soluble ectodomains of these proteins were engineered for secretion into the supernatant of Hi5 insect cells via the baculovirus expression system. Hexa-histidine tags were engineered at the C terminus for Nickel agarose purification. Supernatants of infected cells were collected, concentrated and buffer exchanged with HBS (10mM Hepes, pH 7.2, 150mM NaCl, 0.02% Azide) and Nickel Agarose (Ni-NTA, Qiagen) was added at ~1/100-1/50 dilution, depending on the expression of the protein. Imidazole (pH 8) was added to ~20mM to reduce nonspecific protein binding to the Ni-NTA. Protein was eluted off the Nickel agarose resin by the addition of 200mM Imidazole in HBS. Protein was further purified by gel filtration over a Superdex S200 size exclusion column (GE Healthcare). The G8 and KN6 full length TCRs were expressed as previously described for the G8 TCR. In brief, these receptors were expressed using the baculovirus expression system (see above). The receptors were fused to acid and basic leucine zippers containing hexa-histidine tags at their C termini to aid in proper heterodimerization and disulphide pairing of the γ and δ chains. 3C Rhinovirus protease sites engineered between the TCR and zippers allowed for the easy removal of the zippers and His tags after Nickel agarose purification (as described above for T10 and T22). Protease digested TCRs were further purified by gel filtration over a Superdex S200 size exclusion column (GE Healthcare). TCRs were assayed for proper disulphide linked dimerization by non-reducing and reducing SDS page which revealed 100% efficiency of dimerization in the purified protein. The G8 and KN6 fusion proteins were engineered by superposition of the G81 and 17237 protein structures using the program Pymol (Delano Scientific) to best determine the point at which fusion of the CDR3δ loop with the CDR3α loop of 172 should begin and end. The α-chain was chosen as the fusion partner due to its close homology with the δ-chain (e.g. some Vα domains are used in lieu of Vδ in γδ TCR rearrangement; G8, for example, uses Vα11.3). To maintain the register of amino acids, the CDR3α loop (amino acids NSG) were swapped with the CDR3δ loops, starting with the W and ending with the T (Fig. 2). Fusions were made with overlapping PCR primers encoding the respective motifs of the G8 and KN6 loops. Sequences of all clones were verified. Expression constructs were made for insect expression (full-length, including V and C domains of both chains, producing disulphide linked, heterodimeric ectodomains) and E. coli (single chain, containing V domains only, linked with a 15 amino acid glycine/serine linker37). Full-length fusion TCRs were expressed using the baculovirus expression system as described above for the G8 and KN6 TCRs, and single-chain fusion TCRs were expressed via periplasmic secretion53. All proteins were purified via Nickel agarose chromatography and gel filtration over a Superdex S200 column (GE Healthcare). Alanine mutants were generated by overlapping PCR using the G8 and KN6 svTCR fusion proteins as templates. The sequence of each mutant was verified and mutants were expressed as described above for the fusion proteins via periplasmic secretion in E. coli. All purification proceeded as described above. All biotinylated constructs were engineered with a site-specific biotinylation sequence (GLNDIFEAQKIEWHE; Avidity) at the C terminus to allow for orientation-specific immobilization on a Strepavidin Biacore chip (see Surface Plasmon Resonance Methods section for details). A 3C Rhinovirus protease site and hexa-histidine tag were engineered after the biotinylation site for purification using Nickel agarose (Ni-NTA, Qiagen) chromatography. After Ni-NTA purification, the hexa-histidine tag was removed by 3C-Rhinovirus protease digestion overnight at 4°C. Biotinylation of these constructs was performed using the BirA Biotin Protein Ligase (Avidity) according to manufacturer’s protocols. Biotinylated products were purified over a Superdex S200 column (GE Healthcare) in HBS.
Circular dichroism Analysis
Circular dichroism (CD) data were collected on an Aviv 202-01 spectrometer equipped with a thermoelectric unit using a 1 mm path length cell. Protein samples were 15 µM in buffer containing 20 mM sodium phosphate pH 7.1, and 25 mM NaCl. Protein concentrations were determined by UV spectrophotometry. Thermal melts were monitored at 220 nm. Data were collected every 2°C with an equilibration time of 20 min and an averaging time of 20 s. The equilibration time for the thermal denaturations was chosen based on a kinetic scan. In the kinetic scan, thermal unfolding was initiated by temperature increase to 50°C (approximate midpoint of the thermal denaturation) and the circular dichroism signal was continuously monitored for 1 h. The protein appeared to reach equilibrium in approximately 15 min. Tm was determined by fitting the melting curves to a two-state model as described previously54.
Surface plasmon resonance
All surface plasmon resonance (SPR) measurements were performed at 25°C on a Biacore 3000 using SA-Chips (Biacore) for immobilizing ligand. HBS-P buffer (10mM Hepes, pH 7.4, 150mM NaCl and 0.005% surfactant P20, Biacore) was used for all measurements. In each experiment, approximately 200–400 RU of biotinylated ligand was immobilized to the chip, then blocked with free biotin (100nM) for 1 min. Concentrations of analyte varied from 10uM to 0.0625uM depending on the experiment. Sensograms were fit locally with the Biacore 3000 analysis software (BIAevaluation Version 3.1) with 1:1 Langumuir binding. Kon and Koff were determined from these concentration dependent fits and the dissociation constant (KD) calculated. ΔΔG values were calculated using the Gibbs free energy equation incorporating differences between mutant and wild-type (ΔΔG=RTln(KDmut/KDwt) where R is the gas constant and T is the temperature in Kelvin. KDmut =dissociation constant of the mutant; KDWT =dissociation contants of the wild-type.
Cell stimulation
Cell stimulation assays were performed using the G8 hybridoma transfected with an alkaline phosphatase reporter under the NFAT reporter as described previously55. Cells were maintained in RPMI 1640 supplemented with 10% FCS, L-glutamine, and the antibiotics penicillin and streptomycin (RPMI complete). 100µl of insect-expressed wild-type and mutant T22 and T10d diluted in PBS, pH 7.2 were plated at concentrations varying from 100µg/ml to o.1µg/ml in a flat bottom, polystyrene tissue culture plate and incubated overnight at 4°C. The following day, the plate was washed with PBS and 100µl of the G8 hybridoma in RPMI 1640 supplemented with 10% FCS, at a concentration of 1×106 cells/ml were added to each well and incubated at 37°C for 24 h in a 5% CO2 incubator. Endogenous phosphatase activity was inactivated by incubating the plate at 65°C for 1 h. 100µl of 4-methylumbelliferyl palmitate (4MUP) (Sigma) was added and incubated overnight at 37°C. Fluorescence was measured (excitation wavelength: 360nm, emisson wavelenth: 440nm). All measurements were performed in triplicate.
Supplementary Material
Acknowledgements
This work was supported by NIH RO1- AI65504 (K.C.G.), Howard Hughes Medical Institute (K.C.G.) and NIH RO1-AI073922 (E.J.A.).
References
- 1.Adams EJ, Chien YH, Garcia KC. Structure of a gammadelta T cell receptor in complex with the nonclassical MHC T22. Science. 2005;308:227–231. doi: 10.1126/science.1106885. [DOI] [PubMed] [Google Scholar]
- 2.Allison TJ, Winter CC, Fournie JJ, Bonneville M, Garboczi DN. Structure of a human gammadelta T-cell antigen receptor. Nature. 2001;411:820–824. doi: 10.1038/35081115. [DOI] [PubMed] [Google Scholar]
- 3.Battistini L, et al. Phenotypic and cytokine analysis of human peripheral blood gamma delta T cells expressing NK cell receptors. J Immunol. 1997;159:3723–3730. [PubMed] [Google Scholar]
- 4.Spada FM, et al. Self-recognition of CD1 by gamma/delta T cells: implications for innate immunity. J Exp Med. 2000;191:937–948. doi: 10.1084/jem.191.6.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrick DA, et al. Differential production of interferon-gamma and interleukin-4 in response to Th1- and Th2-stimulating pathogens by gamma delta T cells in vivo. Nature. 1995;373:255–257. doi: 10.1038/373255a0. [DOI] [PubMed] [Google Scholar]
- 6.Wen L, et al. Primary gamma delta cell clones can be defined phenotypically and functionally as Th1/Th2 cells and illustrate the association of CD4 with Th2 differentiation. J Immunol. 1998;160:1965–1974. [PubMed] [Google Scholar]
- 7.Brenner MB, et al. Identification of a putative second T-cell receptor. Nature. 1986;322:145–149. [PubMed] [Google Scholar]
- 8.Fisch P, et al. Gamma/delta T cell clones and natural killer cell clones mediate distinct patterns of non-major histocompatibility complex-restricted cytolysis. J Exp Med. 1990;171:1567–1579. doi: 10.1084/jem.171.5.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wright A, et al. Cytotoxic T lymphocytes specific for self tumor immunoglobulin express T cell receptor delta chain. J Exp Med. 1989;169:1557–1564. doi: 10.1084/jem.169.5.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jameson J, et al. A role for skin gammadelta T cells in wound repair. Science. 2002;296:747–749. doi: 10.1126/science.1069639. [DOI] [PubMed] [Google Scholar]
- 11.Brandes M, Willimann K, Moser B. Professional antigen-presentation function by human gammadelta T Cells. Science. 2005;309:264–268. doi: 10.1126/science.1110267. [DOI] [PubMed] [Google Scholar]
- 12.Kapp JA, Kapp LM, McKenna KC. Gammadelta T cells play an essential role in several forms of tolerance. Immunol Res. 2004;29:93–102. doi: 10.1385/IR:29:1-3:093. [DOI] [PubMed] [Google Scholar]
- 13.De Libero G. Tissue distribution, antigen specificity and effector functions of gamma delta T cells in human diseases. Springer Semin Immunopathol. 2000;22:219–238. doi: 10.1007/s002810000043. [DOI] [PubMed] [Google Scholar]
- 14.Bensussan A, Lagabrielle JF, Degos L. TCR gamma delta bearing lymphocyte clones with lymphokine-activated killer activity against autologous leukemic cells. Blood. 1989;73:2077–2080. [PubMed] [Google Scholar]
- 15.Di Fabrizio L, Kimura Y, Ware R, Rogozinski L, Chess L. Specific triggering of gamma, delta T cells by K562 activates the gamma, delta T cell receptor and may regulate natural killer-like function. J Immunol. 1991;146:2495–2503. [PubMed] [Google Scholar]
- 16.Maeurer MJ, et al. Human intestinal Vdelta1+ lymphocytes recognize tumor cells of epithelial origin. J Exp Med. 1996;183:1681–1696. doi: 10.1084/jem.183.4.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sturm E, et al. Human V gamma 9-V delta 2 T cell receptor-gamma delta lymphocytes show specificity to Daudi Burkitt's lymphoma cells. J Immunol. 1990;145:3202–3208. [PubMed] [Google Scholar]
- 18.Zocchi MR, Ferrarini M, Rugarli C. Selective lysis of the autologous tumor by delta TCS1+ gamma/delta+ tumor-infiltrating lymphocytes from human lung carcinomas. Eur J Immunol. 1990;20:2685–2689. doi: 10.1002/eji.1830201224. [DOI] [PubMed] [Google Scholar]
- 19.Allison TJ, Garboczi DN. Structure of gammadelta T cell receptors and their recognition of non-peptide antigens. Mol Immunol. 2002;38:1051–1061. doi: 10.1016/s0161-5890(02)00034-2. [DOI] [PubMed] [Google Scholar]
- 20.Chien YH, Jores R, Crowley MP. Recognition by gamma/delta T cells. Annu Rev Immunol. 1996;14:511–532. doi: 10.1146/annurev.immunol.14.1.511. [DOI] [PubMed] [Google Scholar]
- 21.Garcia KC, Adams EJ. How the T cell receptor sees antigen--a structural view. Cell. 2005;122:333–336. doi: 10.1016/j.cell.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 22.Rudolph MG, Wilson IA. The specificity of TCR/pMHC interaction. Curr Opin Immunol. 2002;14:52–65. doi: 10.1016/s0952-7915(01)00298-9. [DOI] [PubMed] [Google Scholar]
- 23.Davies DR, Cohen GH. Interactions of protein antigens with antibodies. Proc. Natl. Acad. Sci. U.S.A. 1996;93:7–12. doi: 10.1073/pnas.93.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porcelli S, et al. Recognition of cluster of differentiation 1 antigens by human CD4-CD8-cytolytic T lymphocytes. Nature. 1989;341:447–450. doi: 10.1038/341447a0. [DOI] [PubMed] [Google Scholar]
- 25.Schild H, et al. The nature of major histocompatibility complex recognition by gamma delta T cells. Cell. 1994;76:29–37. doi: 10.1016/0092-8674(94)90170-8. [DOI] [PubMed] [Google Scholar]
- 26.Weintraub BC, Jackson MR, Hedrick SM. Gamma delta T cells can recognize nonclassical MHC in the absence of conventional antigenic peptides. J Immunol. 1994;153:3051–3058. [PubMed] [Google Scholar]
- 27.Groh V, Steinle A, Bauer S, Spies T. Recognition of stress-induced MHC molecules by intestinal epithelial gammadelta T cells. Science. 1998;279:1737–1740. doi: 10.1126/science.279.5357.1737. [DOI] [PubMed] [Google Scholar]
- 28.Scotet E, et al. Tumor recognition following Vgamma9Vdelta2 T cell receptor interactions with a surface F1-ATPase-related structure and apolipoprotein A-I. Immunity. 2005;22:71–80. doi: 10.1016/j.immuni.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 29.Johnson RM, et al. A murine CD4-, CD8- T cell receptor-gamma delta T lymphocyte clone specific for herpes simplex virus glycoprotein I. J Immunol. 1992;148:983–988. [PubMed] [Google Scholar]
- 30.Elliott JF, Rock EP, Patten PA, Davis MM, Chien YH. The adult T-cell receptor delta-chain is diverse and distinct from that of fetal thymocytes. Nature. 1988;331:627–631. doi: 10.1038/331627a0. [DOI] [PubMed] [Google Scholar]
- 31.Crowley MP, et al. A population of murine gammadelta T cells that recognize an inducible MHC class Ib molecule. Science. 2000;287:314–316. doi: 10.1126/science.287.5451.314. [DOI] [PubMed] [Google Scholar]
- 32.Bluestone JA, Cron RQ, Cotterman M, Houlden BA, Matis LA. Structure and specificity of T cell receptor gamma/delta on major histocompatibility complex antigen-specific CD3+, CD4-, CD8- T lymphocytes. J Exp Med. 1988;168:1899–1916. doi: 10.1084/jem.168.5.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonneville M, et al. Recognition of a self major histocompatibility complex TL region product by gamma delta T-cell receptors. Proc Natl Acad Sci U S A. 1989;86:5928–5932. doi: 10.1073/pnas.86.15.5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ito K, et al. Recognition of the product of a novel MHC TL region gene (27b) by a mouse gamma delta T cell receptor. Cell. 1990;62:549–561. doi: 10.1016/0092-8674(90)90019-b. [DOI] [PubMed] [Google Scholar]
- 35.Shin S, et al. Antigen recognition determinants of gammadelta T cell receptors. Science. 2005;308:252–255. doi: 10.1126/science.1106480. [DOI] [PubMed] [Google Scholar]
- 36.Li H, et al. Structure of the Vdelta domain of a human gammadelta T-cell antigen receptor. Nature. 1998;391:502–506. doi: 10.1038/35172. [DOI] [PubMed] [Google Scholar]
- 37.Maynard J, et al. Structure of an autoimmune T cell receptor complexed with class II peptide-MHC: insights into MHC bias and antigen specificity. Immunity. 2005;22:81–92. doi: 10.1016/j.immuni.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 38.Moriwaki S, Korn BS, Ichikawa Y, van Kaer L, Tonegawa S. Amino acid substitutions in the floor of the putative antigen-binding site of H-2T22 affect recognition by a gamma delta T-cell receptor. Proc Natl Acad Sci U S A. 1993;90:11396–11400. doi: 10.1073/pnas.90.23.11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Padlan E. Anatomy of the antibody molecule. Molec.Immunol. 1994;31:169–217. doi: 10.1016/0161-5890(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 40.Garboczi DN, et al. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature. 1996;384:134–141. [PubMed] [Google Scholar]
- 41.Reiser JB, et al. A T cell receptor CDR3beta loop undergoes conformational changes of unprecedented magnitude upon binding to a peptide/MHC class I complex. Immunity. 2002;16:345–354. doi: 10.1016/s1074-7613(02)00288-1. [DOI] [PubMed] [Google Scholar]
- 42.Huang CC, et al. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science. 2007;317:1930–1934. doi: 10.1126/science.1145373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kodandapani R, Veerapandian B, Kunicki TJ, Ely KR. Crystal structure of the OPG2 Fab. An antireceptor antibody that mimics an RGD cell adhesion site. J Biol Chem. 1995;270:2268–2273. doi: 10.1074/jbc.270.5.2268. [DOI] [PubMed] [Google Scholar]
- 44.Stanfield RL, Dooley H, Flajnik MF, Wilson IA. Crystal structure of a shark single-domain antibody V region in complex with lysozyme. Science. 2004;305:1770–1773. doi: 10.1126/science.1101148. [DOI] [PubMed] [Google Scholar]
- 45.Matis LA, Cron R, Bluestone JA. Major histocompatibility complex-linked specificity of gamma delta receptor-bearing T lymphocytes. Nature. 1987;330:262–264. doi: 10.1038/330262a0. [DOI] [PubMed] [Google Scholar]
- 46.Lee CV, et al. High-affinity human antibodies from phage-displayed synthetic Fab libraries with a single framework scaffold. J Mol Biol. 2004;340:1073–1093. doi: 10.1016/j.jmb.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 47.Holler PD, et al. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc Natl Acad Sci U S A. 2000;97:5387–5392. doi: 10.1073/pnas.080078297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patten PA, et al. Transfer of putative complementarity-determining region loops of T cell receptor V domains confers toxin reactivity but not peptide/MHC specificity. J Immunol. 1993;150:2281–2294. [PubMed] [Google Scholar]
- 49.Presta LG, et al. Humanization of an antibody directed against IgE. J Immunol. 1993;151:2623–2632. [PubMed] [Google Scholar]
- 50.Feng D, Bond CJ, Ely LK, Maynard J, Garcia KC. Structural evidence for a germline-encoded T cell receptor-major histocompatibility complex interaction 'codon'. Nature immunology. 2007;8:975–983. doi: 10.1038/ni1502. [DOI] [PubMed] [Google Scholar]
- 51.Huseby ES, et al. How the T cell repertoire becomes peptide and MHC specific. Cell. 2005;122:247–260. doi: 10.1016/j.cell.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 52.Hewitt EW. The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology. 2003;110:163–169. doi: 10.1046/j.1365-2567.2003.01738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maynard J, et al. High-level bacterial secretion of single-chain alphabeta T-cell receptors. J Immunol Methods. 2005;306:51–67. doi: 10.1016/j.jim.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 54.Minor DL, Jr, Kim PS. Measurement of the beta-sheet-forming propensities of amino acids. Nature. 1994;367:660–663. doi: 10.1038/367660a0. [DOI] [PubMed] [Google Scholar]
- 55.Crowley MP, Reich Z, Mavaddat N, Altman JD, Chien Y. The recognition of the nonclassical major histocompatibility complex (MHC) class I molecule, T10, by the gammadelta T cell, G8. J Exp Med. 1997;185:1223–1230. doi: 10.1084/jem.185.7.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wingren C, Crowley MP, Degano M, Chien Y, Wilson IA. Crystal structure of a gammadelta T cell receptor ligand T22: a truncated MHC-like fold. Science. 2000;287:310–314. doi: 10.1126/science.287.5451.310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.